
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci. , 28 August 2023
Sec. Interdisciplinary Climate Studies
Volume 11 - 2023 | https://doi.org/10.3389/fenvs.2023.1204690
This article is part of the Research Topic Innovations in Climate Resilience View all 6 articles
 Bruce Elmegreen1*
Bruce Elmegreen1* Hendrik F. Hamann1
Hendrik F. Hamann1 Benjamin Wunsch1Theodore Van Kessel1
Benjamin Wunsch1Theodore Van Kessel1 Binquan Luan1Tonia Elengikal1Mathias Steiner2Rodrigo Neumann Barros Ferreira2Ricardo Luis Ohta2Felipe Lopes Oliveira2James L. McDonagh3†Breanndan O’Conchuir3Stamatia Zavitsanou3Alexander Harrison3Flaviu Cipcigan3Geeth de Mel3Young-Hye La4
Binquan Luan1Tonia Elengikal1Mathias Steiner2Rodrigo Neumann Barros Ferreira2Ricardo Luis Ohta2Felipe Lopes Oliveira2James L. McDonagh3†Breanndan O’Conchuir3Stamatia Zavitsanou3Alexander Harrison3Flaviu Cipcigan3Geeth de Mel3Young-Hye La4 Vidushi Sharma4
Vidushi Sharma4 Dmitry Yu Zubarev4
Dmitry Yu Zubarev4There is a growing urgency to discover better materials that capture CO2 from air and improve battery performance. An important step is to search large databases of materials properties to find examples that resemble known carbon capture agents or electrolytes and then test them for effectiveness. This paper describes novel computational tools for accelerated discovery of solvents, nano-porous materials, and electrolytes. These tools have produced interesting results so far, such as the identification of a relatively isolated location in amine configuration space for the solvents with known carbon capture use, and the demonstration of an end-to-end simulation and process model for carbon capture in MOFs.
An essential part of climate change mitigation is CO2 capture from ambient air (Lackner et al., 1999; Keith et al., 2006; Budinis et al., 2020; Shi et al., 2020). Capture from power plants is important too (Adams, 2010; Songolzadeh et al., 2014; Bhattacharyya and Miller, 2017; Jiang et al., 2019; Chao et al., 2020) if the global conversion from fossil fuels to renewable energy is slow (Tong et al., 2019). CO2 capture involves separation from other gases, which takes energy for the reduction of entropy (Lackner, 2013; Castel and Favre, 2018) regardless of the mechanism, and it involves material that attracts CO2 more the other gases, or selectively filters it out, and additional energy in the final stages of compression and long-term storage, which is presumably underground (Rubin et al., 2015; Bui et al., 2018; Daniel et al., 2022).
The cost of CO2 separation is a key reason there are so few capture sites today. According to Steyn et al. (2022), there are only 30 operational sites worldwide, 11 under construction and 153 planned as of 2022, compared to ∼2,700 coal and ∼4,000 natural gas power plants in the world, as listed in the World Resources Institute Global Power Plant Database1. There is a far smaller number of sites experimenting with direct air capture, e.g., by Climeworks2 and Carbon Engineering3. Cost reduction from better CO2 capture materials is essential to global scale-out, and this means better materials with higher CO2/N2 selectivities and higher volumetric rates for capture and release.
Electrifying transportation systems to reduce our dependence on fossil fuels is another critical priority to address climate change. More powerful and affordable batteries are key to accelerate the adoption of zero-emission electric vehicles. Current battery technologies need to seek ecofriendly and cost-effective materials that demonstrate high energy density, power density, and longer cycle life, which calls for targeted material discovery and innovation.
Electrolytes are one of the most primary aspects of batteries as they have a strong influence on safety and performance, including rate kinetics and cycle life. Most battery failures are due to electrolyte decomposition or unstable interfaces between the electrolyte and the electrodes. Electrode dissolution and the formation of dendrites on lithium metal electrodes are problems for future battery technologies that could be addressed with adequate selection of electrolytes. As battery technologies are undergoing continuous innovation, the primary challenges are associated with the discovery and selection of electrolytes with overall high-performance, long-term stability, low-cost, and safety.
The goal of IBM’s MDLab is to accelerate discovery of better materials for targeted applications. MDLab contains chemical, topological, and structural informatics and simulation tools that allow rapid screening of solvents, electrolytes, and nano-porous solids using large databases. This review summarizes MDLab. More detailed publications are in McDonagh et al. (2022, 2023), Neumann et al. (2022), and Sharma et al. (2023).
The most mature technology for CO2 capture is absorption by amines, such as monoethanolamine (MEA) in water (Rochelle, 2009; Luis, 2016; Bernhardsen and Knuutila, 2017; Wu et al., 2020). Proprietary solvents4, water-lean solvents (Jiang et al., 2023), and ionic liquids (Wang et al., 2011; Ramdin et al., 2012; Mota-Lima et al., 2021) are also in use. Chemical Informatics techniques have been applied to search for better solvents before (Li et al., 2014; Papadopoulos et al., 2016; Yang et al., 2017; Puxty and Maeder, 2020; Orlov et al., 2022).
For our analysis, MDLab begins with an identification of the most important and relevant structural characteristics of the known solvents and uses these to identify other molecules that may show promise for carbon capture. We identified 167 unique amines with CO2 capture properties in the literature (McDonagh et al., 2022) and extracted string representations of these molecules from PubChem (Kim et al., 2021) and ChemSpider (Pence and Williams, 2010). From this set of molecules, we carried out a chemical space analysis (McDonagh et al., 2022). This analysis applied tools from chemical informatics, such as sub-structure searching with RDKit (Rational Discovery Kit, Landrum, 2014) and topological data analysis (Wasserman, 2018; Zhou et al., 2021), to identify similarities and differences in the molecular graphs of carbon capture amines compared to a set of 20,938 commercially available amines from the ZINC database (Irwin et al., 2012). From this analysis we identified 72 structural features, which we codified as a binary chemical “CCS” fingerprint related to the capability of a molecule to act as a carbon capture solvent. The code to generate these fingerprints can be accessed at github5. Datasets references in this paper are listed in Table 1.
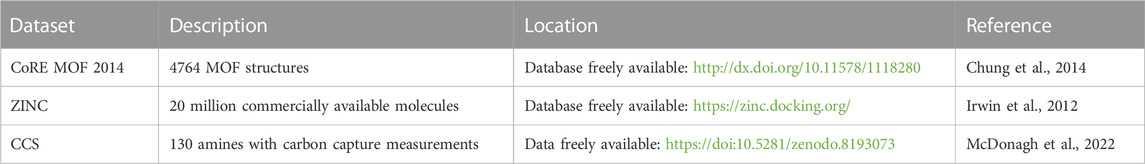
TABLE 1. Datasets for solvents and nanoporous solids used for this study.
Noting that information on carbon capture amines was sparse in the literature, we generated a new dataset from a single experimental source. We applied computational methods to select molecules that make up this data set and predict molecular performances for carbon capture. The data set is 130 molecules in total. We have provided these data to the community with the molecules represented by IUPAC (International Union of Pure and Applied Chemistry) name, InChI (International Chemical Identifier), InChIKey (a 27 character digital representation of the InChI) and SMILES (Simplified Molecular-Input Line-Entry System), making the set amenable to chemical informatics modelling. This dataset can be found at Zenodo6.
We further utilized our CCS fingerprint as a representation for Machine Learning (ML) models. To do this, we separated our data into binary classes and trained models to predict whether the molecules are promising or not. The concept here is to provide a high throughput virtual screen that enables prioritization of molecules for experimental tests. We trialed a range of ML models that are computationally inexpensive to train and are in widely available Python packages such as scikit-learn and xgboost. Promising performance was found with the Extra Trees Classifier (Geurts et al., 2006), the Ada Boost Classifier (Freund and Schapire, 1997; Hastie et al., 2009), Logistic regression (Yu et al., 2011; Defazio et al., 2014; Schmidt et al., 2017) and the Gaussian Process Classifier (Williams and Rasmussen, 2006). We judged the ability of these classifiers using a range of metrics including overall accuracy and the accuracy of classification of the positive and negative classes separately. Our initial work compares the performance of the CCS fingerprint to other commonly applied fingerprints such as MACCS keys (Molecular ACCess System; Durant et al., 2002) and 2D chemical descriptors from the Mordred engine (Moriwaki et al., 2018). Our most recent work outlines a combined computational and experimental discovery cycle that identifies several new candidate molecules (McDonagh et al., 2023; private comm). The development of these methods is shown in McDonagh et al. (2022).
Nano-porous solids are another class of materials that capture CO2, mostly by physisorption (Oschatz and Antonietti, 2018; Khandaker et al., 2020) but also by chemisorption if the surfaces are coated with reactants like amines (Emerson et al., 2018). Nano-porous solids include zeolites, metal-organic frameworks (MOF), covalent-organic frameworks (COF), zeolitic imidazolate frameworks (ZIF), and porous polymer networks (PPN), among other things. There are many varieties of these materials, both hypothetical and real, amounting to 105–106 of them in databases (Moghadam et al., 2017; Boyd et al., 2019) that allow machine learning tools for screening (Wilmer et al., 2012a). The MDLab results reported here use three materials from the database CoRE MOF 2014 (Computation-Ready, Experimental Metal–Organic Frameworks; Chung et al., 2014), and place these data, the screening workflows, and a Jupyter Lab interface in containers on a cloud-based computer cluster (Neumann et al., 2022) to facilitate user interaction and workload management. A more extensive study than what is reported here is in Oliveira et al. (2023).
The screening workflow starts by querying the database for a material or class of materials to get the corresponding Crystallographic Information File (CIF) files. These files are input to applications like Zeo++ (Willems et al., 2012) and RASPA (Dubbeldam et al., 2016), which return geometric and molecular adsorption figures-of-merit. From the material structure, several geometric properties are calculated, such as accessible and non-accessible surface area, volume, and volume fraction, along with density, and the diameters of the largest free sphere, the largest included sphere, and the largest included sphere along a free path. The code used to generate the geometric figures-of-merit can be accessed at GitHub7. Figure 1 illustrates these diameters.
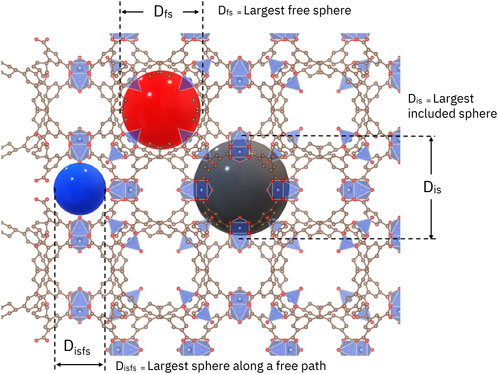
FIGURE 1. Geometric figures-of-merit overlaid on a sample crystal structure (from Neumann et al., 2022).
The CIF file for the unit cell is also used to calculate partial atomic charges in the method of EQeq (Wilmer et al., 2012b), which is saved as a new CIF file for input to RASPA to calculate an energy grid for CO2, N2 and H2O molecules (Luan et al., 2022). With this energy grid, several Grand Canonical Monte Carlo (GCMC) simulations are run for 10,000 cycles, one for each temperature and pressure under consideration. The result, added to the database, is molecular uptake capacity as a function of pressure at each temperature, i.e., the standard isotherm for adsorption. The code used to generate the adsorption figures-of-merit can be accessed at GitHub8. A schematic of the GCMC method is shown in Figure 2.
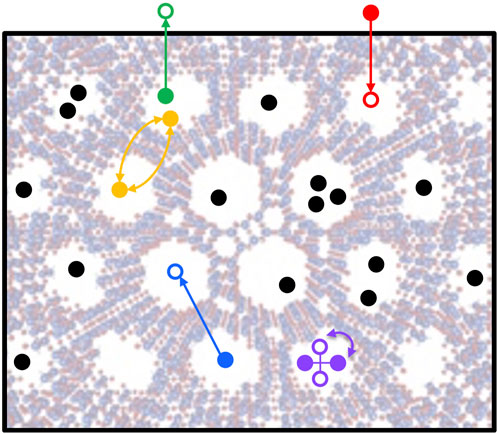
FIGURE 2. Grand Canonical Monte Carlo method showing molecule insertion (red), deletion (green), translation (blue), rotation (purple) and swap (yellow) moves, where the acceptance of these moves depends on the system energy variation (from Neumann et al., 2022).
Adsorption properties were determined from a time-dependent, one-dimensional dynamic model (see Eq. 1) in which multi-component gas enters one end of an adsorption bed and exits the other end. The bed model was run with solid adsorption capacity (qi) and gas concentration (cg,i) for component i as a function of time (t) and space (x,z) using the molecular-level adsorption isotherms as input to model the uptake as a function of temperature. The axial dispersion coefficient is represented by Dax, the gas interstitial velocity by ug, the density of the solid by ρs and the bed void fraction by ε. The model assumed local equilibrium between gas and solid phases from the GCMC results, and at the end of the absorption bed recorded the CO2 purity and recovery fraction, the specific energy consumption in units of MJ per kg CO2, and the volumetric productivity, in units of kg CO2 per day per Liter of sorbent. The result was a rank ordering of nano-porous solids for CO2 separation. Water was assumed to be an equal contaminant for all the sorbents and to not affect the rank order. Thus, water was not included in the input stream, which was 4.1% CO2 and the rest N2, but is assumed to be adsorbed with the same affinity as CO2. Although this simplification neglects the relative water affinity between materials, it does capture the competitive nature of CO2 and H2O adsorption, which affects the final CO2 capacity metrics. See Neumann et al. (2022) for details.
Liquid electrolytes in modern energy storage devices typically contain one or more organic solvents, one or more inorganic salts, and may also include supplementary additives (Jie et al., 2020). Discovery of electrolyte constituents for new battery technologies using advanced computational techniques has been a key research topic. Accelerated electrolyte discovery workflow by JCESR (Joint Center for Energy Storage Research; Qu et al., 2015; Cheng et al., 2015) directs the selection of electrolyte constituents by a high-throughput screening process that focusses on a set of essential domain properties such as stability in an electrochemical window, solubility of salts, and ionic conductivities (Cheng et al., 2015). However, the subsequent development of optimum electrolyte compositions usually requires extensive experimental follow-up because the problem has a large chemical space (Benayad et al., 2022). Our electrolyte discovery framework extends beyond the computational funneling of electrolyte molecules and incorporates advanced deep learning models to optimize the composition of new electrolytes for improved battery performance. Our simulation-AI experiment synergistic framework for battery electrolyte discovery emphasizes the three aspects shown in Figure 3: (a) identification of component molecules, (b) optimization of electrolyte formulations, and (c) in silico characterization of the solid electrolyte interface.
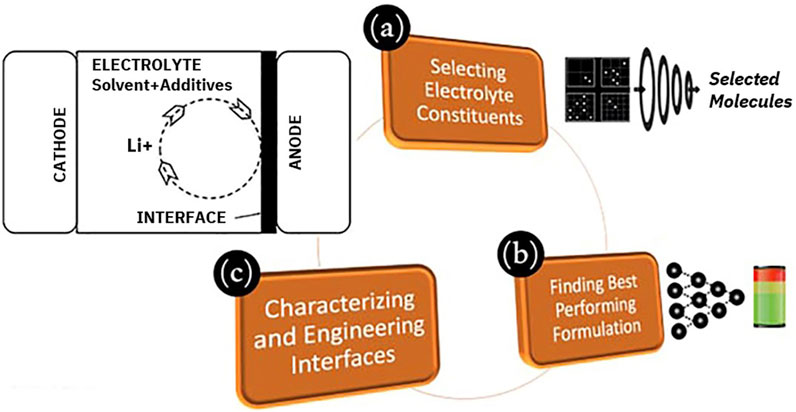
FIGURE 3. IBM’s accelerated electrolyte discovery workflow emphasizing constituent-to-performance and end-to-end electrolyte discovery using a stack of computational tools.
The electrolyte discovery toolkit stack consists of AI and simulation modules that facilitate constituent-to-performance and end-to-end application of the battery electrolyte discovery framework. IBM’s AI enriched simulation workflow (Vassiliadis et al., 2022), which enables high throughput computation of molecular properties, allows screening of electrolyte constituents with desirable properties for targeted battery chemistries. In subsequent discovery steps, we utilize a formulation-based deep learning prediction model to shortlist electrolyte compositions made of candidates that predictively demonstrate target performance (Sharma et al., 2023). These models are infused with domain physical-chemical knowledge and are trained on a small set of data (representing electrolyte formulations and their battery performances – about 100 data points). Here we demonstrate the use-case of the AI models for discovering new electrolyte solvent candidates for the oxygen-assisted lithium Iodine (OALI) battery we have recently developed (Giammona et al., 2023).
Using the 72 component CCS fingerprint, we compared the structures of the 167 amines which had been reported in the literature with carbon capture performance information, to the structures of 20,938 amines in a sub-set of the ZINC database. The fractional occurrences of each of the 72 sub-structure graphs in the two datasets are compared in Figure 4. We see from Figure 4 that, despite the differences in size of the data sets, on normalized axes there are some chemical functionalities that stand out as over or under-represented in the carbon capture molecules (shown in blue) and the background commercially available amines (shown in red). For example, we see carbonyl and halocarbons are common among the background group but very uncommon in the carbon capture group. This figure gives us an indication of the organic molecule functionalities that have been associated with carbon capture solvents. It suggests the functionalities common in the background group and uncommon in the carbon capture group are either untested or unreported for carbon capture or may lead to poor carbon capture performance in these solvent molecules. In either case this helps us determine the chemical space for innovation in these solvents, either to target the underexplored functionalities or exploit the known groups which have led to promising carbon capture performance already.
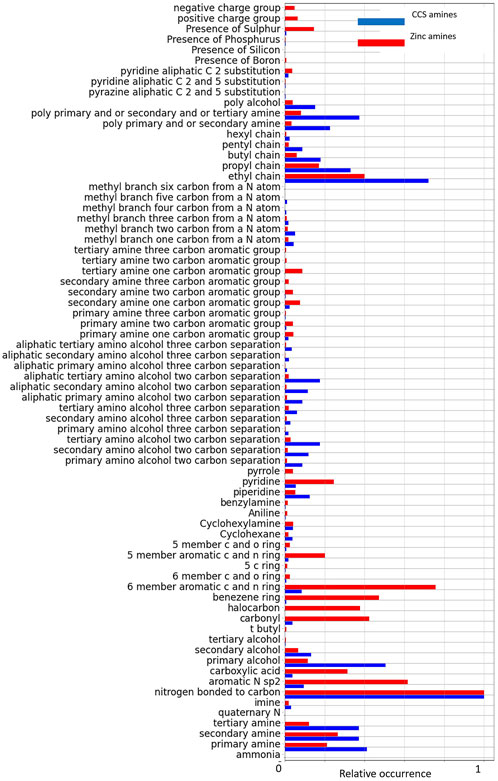
FIGURE 4. Fingerprint comparisons over two amine data sets consisting of 20,938 commercially available amines and a combination of 167 with carbon capture properties in the literature and 130 that have been tested in our own lab (from McDonagh et al., 2022).
Figure 5 shows the chemical space using Tree Map (TMAP). TMAP is a computationally efficient method to analyze and visualize chemical spaces of large datasets. Here we have represented the molecules using MHFP6 (MinHash fingerprint, up to six bonds) fingerprints. TMAP uses these fingerprints to index the molecules and generate an undirected c-approximate k-nearest neighbor graph. A Minimum Spanning Tree (MST) is constructed upon this graph. A projection of this MST is plotted in the figure. The molecules come from a sub-set of the ZINC database, plus the 167 amines we identified as having been tested for carbon capture in the literature, and the 130-molecule dataset generated in McDonagh et al. (2022).
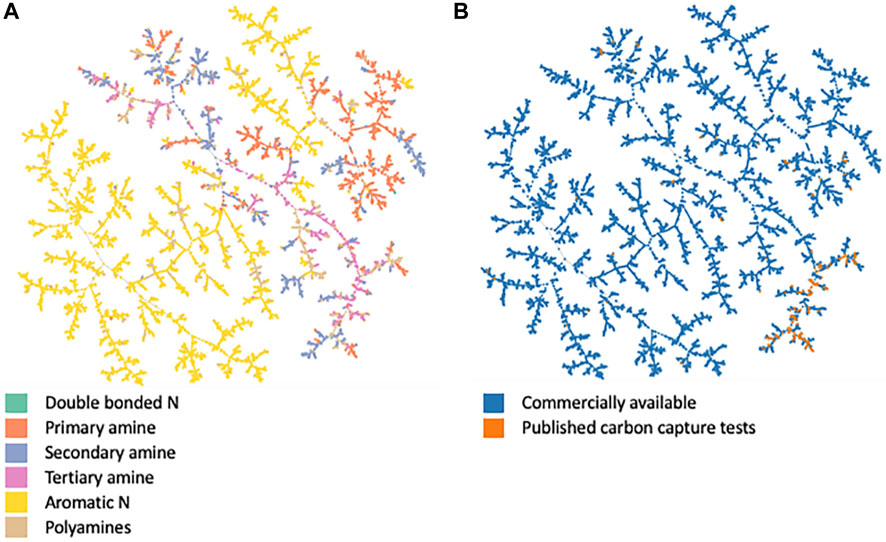
FIGURE 5. TMAP representation of chemical space. (A) is colored by the type of nitrogen environment present in the molecule. (B) is colored by the dataset, i.e., whether the molecule is part of the commercially available sub-set of amines from the ZINC database or from the set of molecules with published carbon capture metrics.
Figure 5 suggests that only a sub-space of the amine chemical space has published carbon capture metrics despite the wider chemical space being commercially available. Figure 5B highlights that the tested molecules broadly exist in one branch of the chemical space depicted here. That branch is dominated by molecules with more than one nitrogen atom, such as primary, secondary, tertiary, or poly-amines. However, there are many other branches dominated by these functionalities, as shown in Figure 5A, but molecules which make up these other branches have not been reported in terms of their carbon capture capabilities to our knowledge. This hints at a potential for innovation in the wider amine chemical space. Alternatively, some essential features of the known capture amines may not be captured by the MHFP6 fingerprints, as would be the case for any dimensionality reduction. Work in McDonagh et al. (2022), shows an additional detailed analysis of these chemical spaces.
We also looked for correlations between molecular graphs and CO2 absorption capacity. 98 molecules with known absorption capacity were divided into those with high (Class 1) and low (Class 0) capacities. This ranking took into account that primary and secondary amines have a theoretical capacity of one-half CO2 per amine group, while tertiary amines have a theoretical capacity of one CO2 per amine group. Because the sample had an imbalance of high and low-capacity molecules (27 vs. 71, resp.), additional sampling points were created for the high-absorption class using the Synthetic Minority Over-Sampling Technique (Smote, Chawla et al., 2002). In this method, the neighborhood of each high-capacity molecule was searched for its five nearest other high-capacity molecules, and one of these other molecules was selected randomly. A synthetic molecule to supplement the minority group was then inserted into the sample, having properties represented by a point on the inter-connecting line between the two molecules in the feature graph. After this additional sampling, the study contained the original 98 molecules and an additional 44 synthetic molecules with theoretically high capacities, making the high and low-capacity samples equal in size.
Machine Learning models were constructed based on carbon capture capacity and molecular features from the Mordred descriptor engine, MACCS fingerprints, and CCS fingerprints. The ML models generated are based on Ada Boost, logistic regression, and Gaussian Process classification methods. All descriptors made relatively good classifications for all models, with >70% accuracy. The use of the CCS fingerprint extended this accuracy to >80% for all models. This result suggests that common, shallow-learning algorithms can predict the classes of both high and low absorption capacities reasonably accurately. The best predictor used the CCS fingerprint and the logistic regression classifier, which predicted the high absorber class with 70% accuracy and the low absorber class with 89% accuracy.
We evaluated the feature importance of the logistic regression model built on the CCS fingerprint features. Figure 6 provides a plot of the feature importance based on the values of the logistic regression coefficients. We have highlighted the five features with the largest coefficients as the most important. It is important not to over-interpret these values. The model is purely statistical and there is no underlying theory to justify the reason for these features’ importance. However, we can see logic in the majority of the most important features. Three of the features reference aspects of the nitrogen atoms’ environments. Another references the presence of alcohol functionalities. The presence of the butyl chain is difficult to justify chemically but may suggest some size criterion. In McDonagh et al. (2022) we provide feature importance analysis for all feature sets using the logistic regression models. We have additionally explored SHAP analyses (SHapley Additive exPlanations; Lundberg and Lee, 2017) for these classifiers. We provide this analysis in the Supporting Material of McDonagh et al. (2022). The SHAP analysis is a finer grained method to evaluate the impact of different features on the ML models.
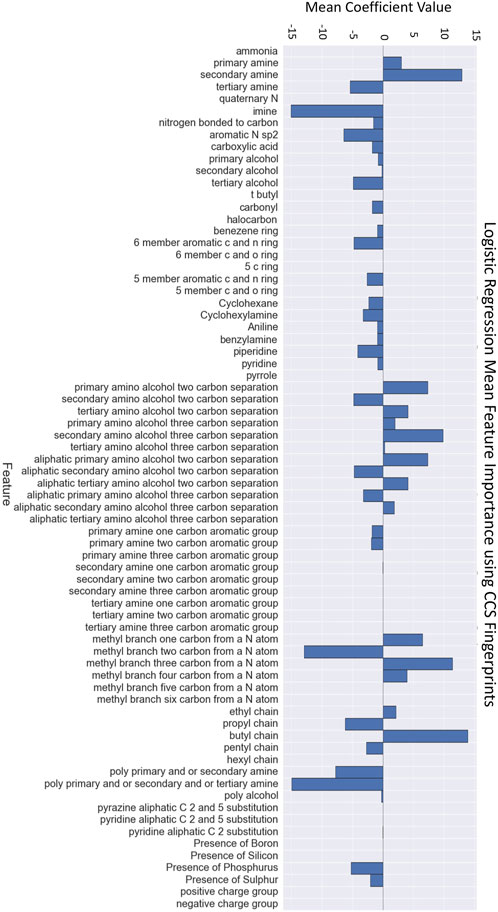
FIGURE 6. Feature importance values for each element of the CCS fingerprint from logistic regression classifier (from McDonagh et al., 2022).
These studies from MDLab suggest that liquid amine solvents which show a high capacity for carbon capture are somewhat isolated in chemical space, but they do have commercially available neighbors in this space that may be candidates for new CO2 capture materials or additives to blends. The molecular component analyses combined with chemical space also highlights certain chemical groups that are relatively less common in carbon capture materials, although they could still be found important in some untested molecules.
Grand Canonical Monte Carlo simulations were used to calculate isotherms for adsorption of CO2, N2, and H2O in three nano-porous materials, HKUST-1, Mg-MOF74, and ZIF-8.
The isotherms were calculated for single component adsorptions and fit to multi-component Langmuir models, as in Eqs 2, 3. Here, ΔHi,k and ΔSi,k are enthalpy and entropy of adsorption for species i on site k, qsat,k is the saturation capacity of site k, independent of species, pi is the partial pressure of species i, T is the temperature and R is the gas constant. The fits were used to determine the enthalpy of adsorption, which was then used in the dynamic adsorption model to determine optimum process cycle times and the corresponding purity and recovery fractions of CO2 at the output, along with the energy requirements and volumetric rates.
The results for Mg-MOF-74 are shown in Figure 7. The molecular structure is on the left, the GCMC isotherms with multi-component Langmuir fits are in the middle (the fit is better for CO2 and N2 than for H2O, not shown). On the right is the calculated purity versus recovery fraction for CO2.
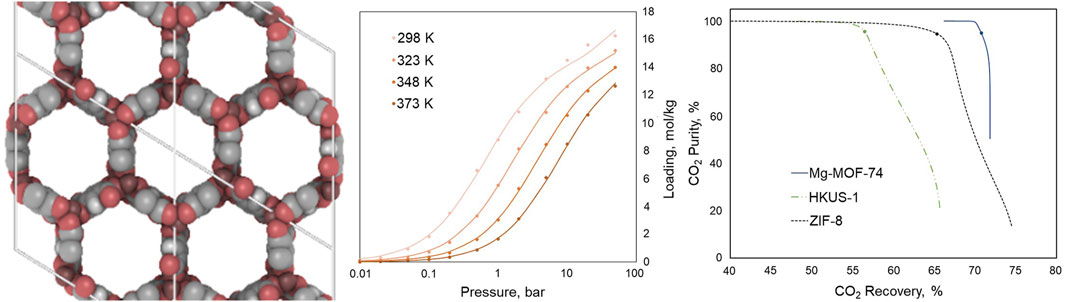
FIGURE 7. Sample results for Mg-MOF-74, with the molecular structure on the left, the CO2 isotherms from multi-component Langmuir fits to the GCGC calculations in the middle (points are the GCMC results) and the Pareto-optimal purity versus recovery fraction of CO2 from a one-dimensional process model on the right, compared to other adsorbers (from Neumann et al., 2022).
The process model shows residual H2O at the end of the desorption cycle for Mg-MOF-74 because of the high affinity for both CO2 and H2O. Nevertheless, the CO2 adsorbed and desorbed for Mg-MOF-74 is much higher than for the others, amounting to about 1.19 kg CO2 per day per liter of sorbent at 95% purity for Mg-MOF-74 compared to 0.41 and 0.21 kg CO2 per day per liter for HKUST-1 and ZIF-8, respectively. The specific energy is also much less for Mg-MOF-74: 11.7 MJ/kg compared to 79.1 MJ/kg and 196.4 MJ/kg for HKUST-1 and ZIF-8, again at 95% purity. Figure 7 also shows higher recovery fractions for Mg-MOF-74. These trends are in agreement with literature expectations for dry CO2/N2 separation (Liu et al., 2012).
Next-generation batteries based on lithium (Li) metal anodes typically use ether-based electrolytes due to their outstanding compatibility with Li metal. Our OALI battery uses an electrolyte based on 1,3-dioxalane (DOL) and 1,2-dimethoxyethane (DME) mixed solvent system that contains two or more inorganic lithium salts such as lithium bis (trifluoromethyl) sulfonylimide (LiTFSI) and lithium nitrate (LiNO3) (Giammona et al., 2023). While the OALI battery utilizing this DOL/DME-based electrolyte has shown excellent rate capability (<50 mA/cm2) and areal capacity (>12 mAh/cm2), there is room for further enhancing cyclic stability, especially at higher active cathode loading (>60 w/w%). To achieve this improvement, a superior electrolyte design is a must.
For discovering a high-performance electrolyte, we begin by screening solvent candidates based on key properties such as low molecular weight (molecules containing <20 non-H atoms), commercial availability, toxicity, thermal properties, salt solubilities and electrochemical stability toward Li metal. Low molecular weight solvents would form a favorable solvation structure with Li ions, facilitating efficient ionic transport. Special emphasis is given to hazard labels and thermal properties of solvents to avoid any “regrettable” substitution in terms of sustainability. For thermal properties such as boiling point (b.p.), flash point (f.p.), and melting point (m.p.), a three tier criteria is defined as described in Figure 8A to make an informed decision for solvent selection.
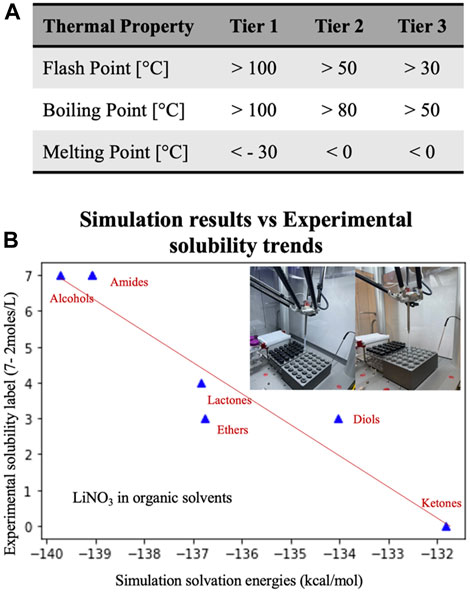
FIGURE 8. (A) Thermal properties extracted from PUBCHEM database for solvents as per three Tiers based on boiling point, flash point and melting point. (B) Solubility trends from solvation simulations for LiNO3 salt validated using high throughput solubility assessment tool experimentally (shown in figure inset).
United States Environmental Protection Agency (EPA) has established a Program for Assisting the Replacement of Industrial Solvent (PARIS-III) tool and a database which contains approximately 4,000 commercially available solvents that have low environmental impact (Harten et al., 2014). PARIS-III database solvents were extracted, followed by screening using data mining and simulation toolkits. It is important to highlight here that criteria priority in the down selecting process can impact the resulting outcomes. Here, we demonstrate the advantage of using simulation and AI synergistic workflows to discover new mixed materials such as electrolytes for batteries by following two screening Routes (Figure 9): Route-1 demonstrates application of simulation toolkits for quick screening of molecular candidates with target properties; and Route-2 shows that machine learning models trained with battery electrolyte datasets can better assist in selecting molecules for formulation by considering complex interactions between all its constituent components rather than their individual behavior.
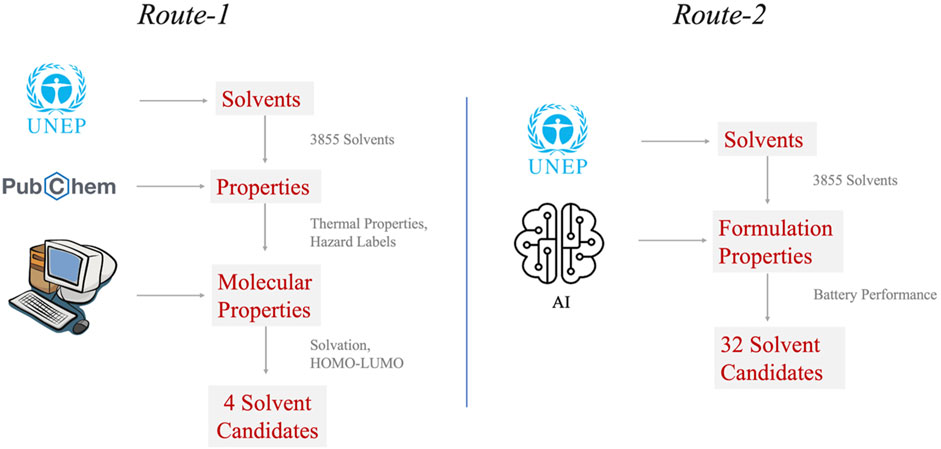
FIGURE 9. Virtual screening routes for discovery of new solvent systems for next-generation battery. Route-1 demonstrates application of simulation toolkits for quick screening of molecular candidates with target properties; and Route-2 shows that machine learning models trained with battery electrolyte datasets can better assist in selecting molecules for formulation by considering complex interactions between all its constituent components rather than their individual behavior.
In Route-1 screening, we first screened 2,871 solvents based on their molecular weight (<20 non-H atoms) from the PARIS-III dataset. Extracting thermal properties like boiling point (b.p.), flash point (f.p.), and melting point (m.p.) from PUBCHEM using API, we further down selected solvents to 107 count in Tier-3 criteria (b.p. > 50°C; f.p. > 30°C; m.p. < 0°C). Next, we deployed virtual experiments based on Density Functional Theory (DFT) to calculate energy levels (HOMO-LUMO levels) of shortlisted solvents and their solvation free energy for primary salts in electrolyte (LiNO3, LiI and LiBOB) using GAMESS software with functional B3LYP and basis set 6–311 G (d,p). For electrochemical stability toward Li metal, solvent LUMO level should be higher than Li/Li + potential. LUMO of DOL and DME are only slightly above Li/Li + potential and are therefore used as comparative standard for current simulation dataset. Solubility trends from the solvation simulations were validated experimentally using a high throughput solubility assessment setup as shown in Figure 8B. Passing through these screening steps, the potential electrolyte solvent candidates were further narrowed down to 4 solvents: tetraglyme, diglyme, 1,4-dioxane and tetrahydrofuran. LiNO3 solubility was the major limiting factor in the down selection process. For experimental validation, all 4 solvents were tested as solvents and co-solvents for OALI electrolytes in coin cells. Among them, tetraglyme (TG) mixed with DOL demonstrated the best cycling stability at 60 wt% active cathode loading, however, the specific capacity decreased by 10% in comparison with the previously reported value (Giammona et al., 2023).
We next developed a deep learning model to map molecular structures, compositions of constituent molecules, to battery performance such as specific capacity and Coulombic efficiency (Sharma et al., 2023). As mentioned in the experimental section, the model was trained with the dataset of OALI battery’s electrolyte formulations and their corresponding battery performance, which was generated in our lab. The trained model was used to screen solvent candidates in Route-2 based on the specific capacity prediction of electrolyte formulations. As a result, 32 solvents emerged as potential high performing electrolyte solvents from PARIS-III dataset. The solvents were now further shortlisted based on electrochemical stability over Li metal and thermal properties. Upon further testing in the lab, the results showed that adding 3-Methoxybutyraldehyde dimethyl acetal (MBDA) as a co-solvent to the DOL:TG system enhances the specific capacity to the highest value (ca. 148 mAh/g) that was observed by DOE:DME system, while demonstrating even longer cycling stability (>250 cycles). Meanwhile, this new electrolyte system (DOL:TG:MBDA) exhibits better safely quotient than DOL:DME system since both TG and MBDA have higher flash points than DME (f.p. = 136°C, 46°C and 21°C, respectively).
MDLab is a toolkit for screening and simulating liquid solvents, solid adsorbents, and electrolytes. This paper summarizes some the methods and preliminary results from our initial use of these tools.
The chemical space of carbon capture amines was compared with that of commercially available amines using several molecular descriptors or fingerprints. The capture amines were found to inhabit the periphery of the chemical space with few neighbors, although there are some neighbors that are close enough to warrant lab analyses. Conversely, there are chemical functional groups in amines that are relatively less common in CO2 absorbers, suggesting possible inhibition or incomplete testing in the larger sample.
MDLab also contains an end-to-end computational screening framework for solid sorbents that capture CO2, as tested on three well-known porous materials, Mg-MOF-74, HKUST-1, and ZIF-8. Monte Carlo Molecular Dynamics simulations were used to determine single-component isotherms, which were then combined into multi-component Langmuir models. These models were then used in a one-dimensional process simulator that calculates the adsorption of CO2 and N2 as functions of time and position. The results allowed a ranking of the material in terms of CO2 purity, recovery fraction, and average adsorption rate per liter of sorbent under optimal conditions. Mg-MOF-74 was found to be better than the other two in these important figures of merit, in agreement with expectations from the literature.
Screening and simulation workflows were used to downselect new solvents to achieve long cycling stability in next-generation OALI battery with high active cathode loading. This was followed by AI-assisted screening of formulation constituents using formulation models pre-trained with OALI battery data. The resulting new electrolyte solvent system (DOL:TG:MBDA) demonstrated longer cycling stability (>250 cycles) while maintaining high capacity (150 mAh/g) for high active cathode material loading. Additionally, new solvents have higher flash points and therefore have better safely and sustainability quotient than conventional electrolytes system. These results are excellent examples of how simulation and AI synergistic workflows can be used to discover new materials for complex systems like batteries. While simulation toolkits allow quick screening of molecular candidates with target properties, machine learning models can better assist in selecting molecules for mixed materials like formulation. With the right approach, formulation models trained with electrolyte data consider complex interactions between all its constituent components rather than their individual behaviors.
We encourage the carbon capture community to make more laboratory data available in machine readable form, even the null results. Combining computational and experimental work with machine learning tools can lead to significant insight. The computation can screen and rank materials for further lab studies, and the lab results can fine-tune the screening for better predictions in the next step. This is a proven model for “Accelerated Discovery” that requires as a first step the availability of data and a suite of material descriptors that can be combined with machine learning tools.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/IBM/Carbon-capture-fingerprint-generation https://github.com/st4sd/nanopore-geometry-experiment https://github.com/st4sd/nanopore-adsorption-experiment.
BE wrote the manuscript helped conceptualize the projects. HH was project manager and research lead for all projects. MS was project manager, conceptualizer, and research contributor for adsorbents. FC, JM were project leads, conceptualizers, and research contributors for solvents. RN was project lead, conceptualizer, and research contributor for adsorbents. BL, TE, RO, FO, and BC conceptualized and contributed to the adsorbent research. Y-HL and VS. conceptualized and contributed to the electrolyte and storage research. BW and TV generated experimental solvent data for machine learning. SZ trained machine learning models to predict solvent capacities. AH was software engineer helping with compute infrastructure. GM and DZ contributed to the text data analysis. All authors contributed to the article and approved the submitted version.
The authors are grateful to Ashish B. Mhadeshwar, Jayashree Kalyanaraman, Anantha Sundaram, Joseph M. Falkowski, Jonathan R. Szlachta, and Yogesh V. Joshi for contributions to this project. Some of the contents of this paper are in the on-line preprints by McDonagh et al. (2022) and Sharma et al. (2023). We are grateful to two referees for suggestions that improved this manuscript.
Author JM, was employed by the Ladder Therapeutics Doing Business as Serna Bio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1https://datasets.wri.org/dataset/globalpowerplantdatabase (accessed Feb. 5, 2023).
2https://climeworks.com/roadmap/orca
3https://carbonengineering.com/
4https://en.wikipedia.org/wiki/Rectisol, https://en.wikipedia.org/wiki/Selexol, https://chempedia.info/info/fluor_solvent_process/
5https://github.com/IBM/Carbon-capture-fingerprint-generation
6https://zenodo.org/record/7828286
7https://github.com/st4sd/nanopore-geometry-experiment
8https://github.com/st4sd/nanopore-adsorption-experiment
Adams, D. (2010). Flue gas treatment for CO2 capture. London, UK: IEA Clean Coal Centre. https://usea.org/sites/default/files/062010_Flue%20gas%20treatment%20for%20co2%20capture_ccc169.pdf.
Benayad, A., Diddens, D., Heuer, A., Krishnamoorthy, A. N., Maiti, M., Cras, F. L., et al. (2022). High-throughput experimentation and computational freeway lanes for accelerated battery electrolyte and interface development research. Adv. Energy Mater. 12, 2102678. doi:10.1002/aenm.202102678
Bernhardsen, I. M., and Knuutila, H. K. (2017). A review of potential amine solvents for CO2 absorption process: absorption capacity, cyclic capacity and pKa. Int. J. Greenh. Gas Control 61, 27–48. doi:10.1016/j.ijggc.2017.03.021
Bhattacharyya, D., and Miller, D. C. (2017). Post-combustion CO2 capture technologies— a review of processes for solvent-based and sorbent-based CO2 capture. Curr. Opin. Chem. Eng. 17, 78–92. doi:10.1016/j.coche.2017.06.005
Boyd, P. G., Chidambaram, A., García-Díez, E., Ireland, C. P., Daff, T. D., Bounds, R., et al. (2019). Data-driven design of metal-organic frameworks for wet flue gas CO2 capture. Nature 576, 253–256. doi:10.1038/s41586-019-1798-7
Budinis, S. (2020). IEA 2020, direct air capture. Paris, France: IEA. https://www.iea.org/reports/direct-air-capture.
Bui, M., Adjiman, C. S., Bardow, A., Anthony, E. J., Boston, A., Brown, S., et al. (2018). Carbon capture and storage (CCS): the way forward. Energy Environ. Sci. 11, 1062–1176. doi:10.1039/c7ee02342a
Castel, C., and Favre, E. (2018). Membrane separations and energy efficiency. J. Membr. Sci. 548, 345–357. doi:10.1016/j.memsci.2017.11.035
Chao, C., Deng, Y., Dewil, R., Baeyens, J., and Fan, X. (2021). Post-combustion carbon capture. Renew. Sustain. Energy Rev. 138, 110490. doi:10.1016/j.rser.2020.110490
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). Smote: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357. doi:10.1613/jair.953
Cheng, L., Assary, R. S., Qu, X., Jain, A., Ong, S. P., Rajput, N. N., et al. (2015). Accelerating electrolyte discovery for energy storage with high-throughput screening. J. Phys. Chem. Lett. 6, 283–291. doi:10.1021/jz502319n
Chung, Y. G., Camp, J., Haranczyk, M., Sikora, B. J., Bury, W., Krungleviciute, V., et al. (2014). Computation-ready, experimental metal–organic frameworks: A tool to enable high-throughput screening of nanoporous crystals. Chem. 26, 6185–6192. doi:10.1021/cm502594j
Daniel, T., Masini, A., Milne, C., Nourshagh, N., Iranpour, C., and Xuan, J. (2022). Techno-economic analysis of direct air carbon capture with CO2 utilization. Carbon Capture Sci. Technol. 2, 100025. doi:10.1016/j.ccst.2021.100025
Defazio, A., Bach, F., and Lacoste-Julien, S. (2014). Saga: a fast incremental gradient method with support for non-strongly convex composite objectives. https://arxiv.org/abs/1407.0202.
Dubbeldam, D., Calero, S., Ellis, D. E., and Snurr, R. Q. (2016). Raspa: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 42, 81–101. doi:10.1080/08927022.2015.1010082
Durant, J. L., Leland, B. A., Henry, D. R., and Nourse, J. G. (2002). Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280. doi:10.1021/ci010132r
Emerson, A. J., Chahine, A., Batten, S. R., and Turner, D. R. (2018). Synthetic approaches for the incorporation of free amine functionalities in porous coordination polymers for enhanced CO2 sorption. Coord. Chem. Rev. 365, 1–22. doi:10.1016/j.ccr.2018.02.012
Freund, Y., and Schapire, R. E. (1997). A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 55, 119–139. doi:10.1006/jcss.1997.1504
Geurts, P., Ernst, D., and Wehenkel, L. (2006). Extremely randomized trees. Mach. Learn. 63, 3–42. doi:10.1007/s10994-006-6226-1
Giammona, M. J., Kim, J., Kim, Y., Medina, P., Nguyen, K., Bui, H., et al. (2023). Oxygen assisted lithium-iodine batteries: Towards practical iodine cathodes and viable lithium metal protection strategies. Adv. Mater. Interfaces 10, 2300058. doi:10.1002/admi.202300058
Harten, P. F. (2014). Program for assisting the replacement of industrial solvents (PARIS III). 18th Annual Green Chemistry & Engineering Conference, North Bethesda, MD, USA.
Hastie, T., Rosset, S., Zhu, J., and Zou, H. (2009). Multi-class AdaBoost. Statistics Its Interface 2, 349–360. doi:10.4310/SII.2009.v2.n3.a8
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S., and Coleman, R. G. (2012). Zinc: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768. doi:10.1021/ci3001277
Jiang, L., Gonzalez-Diaz, A., Ling-Chin, J., Roskilly, A. P., and Smallbone, A. J. (2019). Post-combustion CO2 capture from a natural gas combined cycle power plant using activated carbon adsorption. Appl. Energy 245, 1–15. doi:10.1016/j.apenergy.2019.04.006
Jiang, Y., Mathias, P. M., Zheng, R. F., Freeman, C. J., Barpaga, D., Malhotra, D., et al. (2023). Energy-effective and low-cost carbon capture from point-sources enabled by water-lean solvents. J. Clean. Prod. 388, 135696. doi:10.1016/j.jclepro.2022.135696
Jie, Y., Ren, X., Cao, R., Cai, W., and Jiao, S. (2020). Advanced liquid electrolytes for rechargeable Li metal batteries. Adv. Funct. Mater. 30, 1910777. doi:10.1002/adfm.201910777
Keith, D. W., Ha-Duong, M., and Stolaroff, J. K. (2006). Climate strategy with CO2 capture from the air. Clim. Change 74, 17–45. doi:10.1007/s10584-005-9026-x
Khandaker, T., Hossain, M. S., Dhar, P. K., Rahman, M. S., Hossain, M. A., and Ahmed, M. B. (2020). Efficacies of carbon-based adsorbents for carbon dioxide capture. Processes 8, 654. doi:10.3390/pr8060654
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2021). Pubchem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395. doi:10.1093/nar/gkaa971
Lackner, K. (2013). The thermodynamics of direct air capture of carbon dioxide. Energy 50, 38–46. doi:10.1016/j.energy.2012.09.012
Lackner, K., Ziock, H.-J., and Grimes, P. (1999). Carbon Dioxide extraction from air: Is it an option? in 24th Annual Technical Conference on Coal Utilization & Fuel Systems. Clearwater, Florida, Los Alamos National Laboratory, https://digital.library.unt.edu/ark:/67531/metadc715467/m2/1/high_res_d/770509.pdf.
Landrum, G. (2014). RDKit: Open-source cheminformatics release 2014.03.1. https://zenodo.org/record/10398.
Li, H. C., Chai, J.-D., and Tsai, M.-K. (2014). Assessment of dispersion-improved exchange-correlation functionals for the simulation of CO2 binding by alcoholamines. Int. J. Quantum Chem. 114, 805–812. doi:10.1002/qua.24670
Liu, J., Thallapally, P. K., McGrail, B. P., Brown, D. R., and Liu, J. (2012). Progress in adsorption-based CO2 capture by metal-organic frameworks. Chem. Soc. Rev. 41, 2308–2322. doi:10.1039/C1CS15221A
Luan, B., Neumann Barros Ferreira, R., Giro, R., Elengikal, T., Oliveira, A., Liu, S., et al. (2022). The role of partial charges in data-driven analytics of gas adsorption simulations. APS March Meeting Minneapolis, Minnesota https://meetings.aps.org/Meeting/MAR22/Session/K01.5.
Luis, P. (2016). Use of monoethanolamine (MEA) for CO2 capture in a global scenario: Consequences and alternatives. Desalination 380, 93–99. doi:10.1016/j.desal.2015.08.004
Lundberg, S. M., and Lee, S.-I. (2017). A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 30, 4765–4774. doi:10.5555/3295222.3295230
McDonagh, J. L., Zavitsanou, S., Harrison, A., Zubarev, D. Y., Wunsch, B. H., van Kessel, T., et al. (2022). Chemical space analysis and property prediction for carbon capture amine molecules. https://chemrxiv.org/engage/chemrxiv/article-details/62e110cbadb01e653cae19f4.
Moghadam, P. Z., Li, A., Wiggin, S. B., Tao, A., Maloney, A. G., Wood, P. A., et al. (2017). Development of a cambridge structural database subset: A collection of metal-organic frameworks for past, present, and future. Chem. Mater. 29, 2618–2625. doi:10.1021/acs.chemmater.7b00441
Moriwaki, H., Tian, Y.-S., Kawashita, N., and Takagi, T. (2018). Mordred: A molecular descriptor calculator. J. Cheminformatics 10, 4. doi:10.1186/s13321-018-0258-y
Mota-Lima, A., Alcantara, M. L., Pérez-Sanz, F. J., Bazito, R. C., Vidinha, P., Alves, R. M. B., et al. (2021). Review—high-pressure carbon dioxide separation using ionic liquids: A CO2-electrocatalysis perspective. J. Electrochem. Soc. 168, 86502. doi:10.1149/1945-7111/ac085d
Neumann Barros Ferreira, R., O Conchuir, B., Elengika, T., Luan, B., Ohta, R. L., Oliveira, F. L., et al. (2022). Cloud-based, high-throughput, end-to-end computational screening of solid sorbent materials for carbon capture. 16th International Conference on Greenhouse Gas Control Technologie. GHGT 2022, Lyon, France, https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4275135.
Oliveira, F. L., Cleeton, C., Neumann Barros Ferreira, R., Luan, B., Farmahini, A. H., Sarkisov, L., and Steiner, M. (2023). Crafted: An exploratory database of simulated adsorption isotherms of metal-organic frameworks. Sci. Data 10, 230. doi:10.1038/s41597-023-02116-zOrlov
Orlov, A. A., Valtz, A., Coquelet, C., Rozanska, X., Wimmer, E., Marcou, G., et al. (2022). Computational screening methodology identifies effective solvents for CO2 capture. Commun. Chem. 5, 1–7. doi:10.1038/s42004-022-00654-y
Oschatz, M., and Antonietti, M. (2018). A search for selectivity to enable CO2 capture with porous adsorbents. Energy Environ. Sci. 11, 57–70. doi:10.1039/C7EE02110K
Papadopoulos, A. I., Badr, S., Chremos, A., Forte, E., Zarogiannis, T., Seferlis, P., et al. (2016). Computer-aided molecular design and selection of CO2 capture solvents based on thermodynamics, reactivity and sustainability. Mol. Syst. Des. Eng. 1, 313–334. doi:10.1039/C6ME00049E
Pence, H. E., and Williams, A. (2010). Chemspider: An online chemical information resource. J. Chem. Educ. 87, 1123–1124. doi:10.1021/ED100697W
Puxty, G., and Maeder, M. (2020). Using chemometrics tools to gain detailed molecular information on chemical processes. J. Chemom. 34, 3207. doi:10.1002/cem.3207
Qu, X., Jain, A., Rajput, N., Cheng, L., Zhang, Y., Ong, S. P., et al. (2015). The electrolyte genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 103, 56–67. doi:10.1016/j.commatsci.2015.02.050
Ramdin, M., de Loos, T. W., and Vlugt, T. J. H. (2012). State-of-the-Art of CO2 capture with ionic liquids. Ind. Eng. Chem. Res. 51, 8149–8177. doi:10.1021/ie3003705
Rochelle, G. T. (2009). Amine scrubbing for CO2 capture. Science 325, 1652–1654. doi:10.1126/science.1176731
Rubin, E. S., Davison, J. E., and Herzog, H. J. (2015). The cost of CO2 capture and storage. Int. J. Greenh. Gas Control 40, 378–400. doi:10.1016/j.ijggc.2015.05.018
Schmidt, M., Le Roux, N., and Bach, F. (2017). Minimizing finite sums with the stochastic average gradient. Math. Program. 162, 83–112. doi:10.1007/s10107-016-1030-6
Sharma, V., Giammona, M., Zubarev, D., Tek, A., Nugyuen, K., Sundberg, L., et al. (2023). Formulation graphs for mapping structure-composition of battery electrolytes to device performance. https://arxiv.org/abs/2307.03811.
Shi, X., Xiao, H., Azarabadi, H., Song, J., Wu, X., Chen, X., et al. (2020). Sorbents for the direct capture of CO2 from ambient air. Angew. Chem. 59, 6984–7006. doi:10.1002/anie.201906756
Songolzadeh, M., Soleimani, M., Ravanchi, M. T., and Songolzadeh, R. (2014). Carbon dioxide separation from flue gases: a technological review emphasizing reduction in greenhouse gas emissions. Sci. World J. 2014, 828131. doi:10.1155/2014/828131
Steyn, M., Oglesby, J., Turan, G., Zapantis, A., Gebremedhin, R., Amer, N. A., et al. (2022). The global status of CCS: 2022. Global CCS Institute. Melbourne, Australia, https://status22.globalccsinstitute.com/wp-content/uploads/2022/12/Global-Status-of-CCS-2022_Download_1222.pdf.
Tong, D., Zhang, Q., Zheng, Y., Caldeira, K., Shearer, C., Hong, C., et al. (2019). Committed emissions from existing energy infrastructure jeopardize 1.5°C climate target. Nature 572, 373–377. doi:10.1038/s41586-019-1364-3
Vassiliadis, V., Johnston, M. A., and McDonagh, J. L. (2022). Fast, transparent, and high-fidelity memoization cache-keys for computational workflows. 2022 IEEE International Conference on Services Computing (SCC), Barcelona, Spain, pp. 174–184. doi:10.1109/SCC55611.2022.00036
Wang, C., Luo, X., Luo, H., Jiang, D-e., Li, H., and Dai, S. (2011). Tuning the basicity of ionic liquids for equimolar CO2 capture. Angew. Chem. 123, 5020–5024. doi:10.1002/ange.201008151
Wasserman, L. (2018). Topological data analysis. Annu. Rev. Statistics Its Appl. 5, 501–532. doi:10.1146/annurev-statistics-031017-100045
Willems, T. F., Rycroft, C. H., Kazi, M., Meza, J. C., and Haranczyk, M. (2012). Algorithms and tools for high throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 149, 134–141. doi:10.1016/j.micromeso.2011.08.020
Williams, C. K., and Rasmussen, C. E. (2006). Gaussian processes for machine learning, 2. Cambridge, MA, USA: MIT press. doi:10.7551/mitpress/3206.001.0001
Wilmer, C. E., Kim, K. C., and Snurr, R. Q. (2012b). An extended charge equilibration method. J. Phys. Chem. Lett. 3, 2506–2511. doi:10.1021/jz3008485
Wilmer, C. E., Leaf, M., Lee, C. Y., Farha, O. K., Hauser, B. G., Hupp, J. T., et al. (2012a). Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 4, 83–89. doi:10.1038/nchem.1192
Wu, X., Wang, M., Liao, P., Shen, J., and Li, Y. (2020). Solvent-based post-combustion CO2 capture for power plants: A critical review and perspective on dynamic modelling, system identification, process control and flexible operation. Appl. Energy 257, 113941. doi:10.1016/j.apenergy.2019.113941
Yang, X., Rees, R. J., Conway, W., Puxty, G., Yang, Q., and Winkler, D. A. (2017). Computational modeling and simulation of CO2 capture by aqueous amines. Chem. Rev. 117, 9524–9593. doi:10.1021/acs.chemrev.6b00662
Yu, H.-F., Huang, F.-L., and Lin, C.-J. (2011). Dual coordinate descent methods for logistic regression and maximum entropy models. Mach. Learn. 85, 41–75. doi:10.1007/s10994-010-5221-8
Keywords: carbon capture, chemical informatics, amines, computational chemistry, nanoporous materials, electrolytes
Citation: Elmegreen B, Hamann HF, Wunsch B, Van Kessel T, Luan B, Elengikal T, Steiner M, Neumann Barros Ferreira R, Ohta RL, Oliveira FL, McDonagh JL, O’Conchuir B, Zavitsanou S, Harrison A, Cipcigan F, de Mel G, La Y-H, Sharma V and Zubarev DY (2023) MDLab: AI frameworks for carbon capture and battery materials. Front. Environ. Sci. 11:1204690. doi: 10.3389/fenvs.2023.1204690
Received: 19 April 2023; Accepted: 15 August 2023;
Published: 28 August 2023.
Edited by:
Jill A. Engel-Cox, National Renewable Energy Laboratory (DOE), United StatesReviewed by:
Jan Steckel, National Energy Technology Laboratory (DOE), United StatesCopyright © 2023 Elmegreen, Hamann, Wunsch, Van Kessel, Luan, Elengikal, Steiner, Neumann Barros Ferreira, Ohta, Oliveira, McDonagh, O’Conchuir, Zavitsanou, Harrison, Cipcigan, de Mel, La, Sharma and Zubarev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce Elmegreen, YmdlQHVzLmlibS5jb20=
†Present address: James L. McDonagh, Ladder Therapeutics Doing Business as Serna Bio, Stevenage, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.