
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci., 30 March 2023
Sec. Biogeochemical Dynamics
Volume 11 - 2023 | https://doi.org/10.3389/fenvs.2023.1110169
This article is part of the Research TopicWomen in Biogeochemical Dynamics Research: 2022View all 10 articles
 Maria Vila-Costa1*
Maria Vila-Costa1* Daniel Lundin2
Daniel Lundin2 Maria-Carmen Fernández-Pinos1†
Maria-Carmen Fernández-Pinos1† Jon Iriarte1
Jon Iriarte1 Xavier Irigoien3,4
Xavier Irigoien3,4 Benjamin Piña1
Benjamin Piña1 Jordi Dachs1
Jordi Dachs1Background and chronic pollution by organic pollutants (OPs) is a widespread threat in the oceans with still uncharacterized effects on marine ecosystems and the modulation of major biogeochemical cycles. The ecological impact and toxicity of this anthropogenic dissolved organic carbon (ADOC) is not related to the presence of a single compound but to the co-occurrence of a myriad of synthetic chemicals with largely unknown effects on heterotrophic microbial communities. We have analyzed the metabolic capacity of metagenome-assembled genomes (MAGs) of natural oceanic communities from the north Pacific (Costa Rica dome) and Atlantic oceans challenged with environmentally relevant levels of ADOC. In the Atlantic, ADOC-exposed MAGs responded transcriptionally more strongly compared to controls than in the Pacific, possibly mirroring the higher relevance of ADOC compounds as carbon source in oligotrophic environments. The largest proportions of transcripts originated from MAGs belonging in the families Rhodobacteraceae and Flavobacteriaceae, known to play a role on consumption of several OPs. In the Atlantic, archaeal Poseidoniales showed the highest transcription levels after 2 h of ADOC exposure, although no increase of relative abundances in the DNA pool was recorded after 24 h, whereas Methylophaga showed the opposite pattern. Both taxa are suggested to be actively involved in the consumption of biogenic alkanes produced by cyanobacteria. We observed similar gene expression profiles of alkane degradation and methylotrophy signature genes. These findings, plus the chemical degradation of alkanes measured in the experiments, provides experimental evidence of the consumption of anthropogenic hydrocarbons and synthetic chemicals at the low concentrations found in the ocean, and modulation of microbiomes by ADOC.
Anthropogenic dissolved organic carbon (ADOC) is among the most under-studied anthropogenic perturbation in the environment under the current scenario of global change (Vila-Costa et al., 2020), specifically in terms of its influence on the major biogeochemical cycles, and thus the functioning of the Earth system. ADOC is composed by the mixture of the thousands of synthetic organic compounds used by humans, as well as hydrocarbons from incomplete combustion of fossil fuels, released to the environment. This pool of organic carbon includes flame retardants, pesticides and herbicides, plasticizers and other leachates from plastics, hydrocarbons, surface active substances, among other chemicals used in a myriad of industrial, agricultural and domestic applications. Currently, there are 300,000 synthetic chemicals in use (Wang et al., 2020) with an unquantified release to the marine environment. Even though the absolute concentrations of ADOC may account for a small percentage of the overall dissolved organic carbon (DOC), this pool is important due to their persistence, which allow them to distribute in the global oceans, and eventually inducing an effect on cells due to their accumulation in lipids at concentrations many orders of magnitude above the seawater levels (Lohmann et al., 2007; Vila-Costa et al., 2020). Nevertheless, some components of ADOC, such as hydrocarbons or organophosphate esters are degraded in the environment, and thus used as a source of C and P (González-Gaya et al., 2019; Vila-Costa et al., 2019). The subtle influence of ADOC modulating the major biogeochemical cycles remains uncharacterized, even though it is presumably increasing in pace with social and economic development, as more and more synthetic chemicals are used by the contemporary society.
Organic pollutants contributing to ADOC are ubiquitous in the ocean, including the remote regions of the Pacific, Indian, Southern and Atlantic oceans (Galbán-malagón, 2012; González-Gaya et al., 2014; González-Gaya et al., 2016; Casal et al., 2019; Muir and Miaz 2021). Many of the organic pollutants reaching the ocean are semivolatile, such as polycyclic aromatic hydrocarbons (PAHs), alkanes, organophosphate esters, organochlorine compounds, etc. These chemicals reach the ocean through atmospheric transport and subsequent deposition. However, some chemicals such as perfluoroalkyl substances are prone to oceanic transport due to their low volatility and high solubility in water (Muir and Miaz 2021). The transport mechanism, as well the biogeochemistry of the pollutant once in the water column, determines their spatial and vertical distribution. The oceanic sinks of ADOC are accounted mainly by the biological pump and degradation (Galbán-malagón, 2012; González-Gaya et al., 2019). In coastal waters, strong gradients and high concentrations of pollutants can be found locally. On the contrary, low variability of pollutant levels is found in the large open oceanic regions as atmospheric transport can re-distribute pollutants over large regions and to the low lability of these chemicals. Indeed, highly labile pollutants do not reach open sea waters as they are degraded during their transport from land. Nevertheless, there are some global trends, such as generally lower concentrations in the open Pacific than the Atlantic, or higher concentrations in the northern hemisphere than the southern hemisphere (González-Gaya et al., 2016; Muir and Miaz 2021; Xie et al., 2022). Importantly, some alkanes with lower carbon number can have biogenic sources, especially from cyanobacteria (Dachs et al., 1998; Love et al., 2021). A cryptic but ubiquitous and fast consumption cycle for these biogenic alkanes has been suggested (Love et al., 2021). Whether or not the same genetic machinery is used for the degradation of both biogenic and anthropogenic alkanes, remains unknown.
Even though there are several previous studies assessing the influence of given individual or families of organic pollutants on phytoplankton and bacteria (Echeveste et al., 2016; Cerro-Galvez et al., 2019; Martinez-Varela et al., 2021), very few of these works have addressed the influence of the overall ADOC on microbiomes. For phytoplankton, it has been shown that ADOC concentrations slightly above the background oceanic levels induce a decrease of the abundance of Prochlorococcus and Synechococcus, their viability, as well as that the expression of genes related to carbon fixation, such as Rubisco (Echeveste et al., 2016; Fernández Pinos et al., 2017). In these works, in order to simulate the influence of ADOC, microbial communities were exposed to extracts of seawater, following the standard procedures usually used for the analysis of a wide range of organic pollutants, which concentrates the hydrophobic and non-ionic fraction of DOC. Similar effects of mixtures of organic pollutants on cyanobacteria have been described for leachates from microplastics (Tetu et al., 2020). The influence of ADOC on heterotrophic prokaryotes (Bacteria and Archaea) have received much less attention. Recent works suggested a range of subtle effects of low concentrations of ADOC on Arctic, Antarctic and Mediterranean Bacteria (Cerro-Gálvez et al., 2019, 2020). In these works, metagenomic and metatranscriptomic approaches allowed to identify cellular to small variations in exposure ADOC concentrations. High-throughput sequencing is a useful tool to access the genomic repository of the uncultured Bacteria and Archaea that account for most species in the oceans, and how it respond to environmental stressors. The current exponential number of works reconstructing high-quality metagenome-assembled genomes (MAGs) from oceanic microbiomes is expanding the knowledge on microbial metabolisms, enzymatic capabilities and habitat distributions (e.g., Tully et al., 2018), including MAGs retrieved from sites influenced by oil spilled accidents (Karthikeyan et al., 2021). However, no attempts have been made to reconstruct MAGs in manipulated experiments to analyze responses to specific conditions, for instance, to exposure of background concentrations of pollutants.
The objectives of this work were 1) to explore the subtle effects of small variations in concentrations of ADOC (less than a factor of 2) by exposing microbial communities to extracts of hydrophobic compounds in seawater 2) prove the utility of assessing metagenome assembled genomes (MAGs) resolved from exposure experiments as indicators of subtle effects of ADOC on microbiomes, 3) to provide evidences on the interactions between ADOC, trophic status and microbiomes for some sectors of the North Pacific and Atlantic oceans.
The experiments were done during the Malaspina circumnavigation cruise in 2011. Four experiments were performed, two in the North Pacific, on the Costa Rica dome off-shore central America. The other two experiments were performed in the North Atlantic (Figure 1, Supplementary Table S1). Experiments consisted in spiking 1 L of seawater from the deep chlorophyll maximum (DCM) with a mixture of hydrophobic organic pollutants obtained by concentrating the hydrophobic DOC on a XAD-2 column. Control experiments using the same seawater, but with no addition of organic pollutants, were carried out in parallel to the treatments. Details of the preparation of these complex mixtures are given elsewhere (Fernández-Pinos et al., 2017). This mixture contained 50.9 ng/L of PAHs, 137 pg/L of polychlorinated biphenyls (PCBs), 8 pg/L of hexachlorobenzene, and 3.2 pg/L of hexachlorocyclohexanes. In addition to these chemicals, the mixture also contained other pollutants such as n-alkanes and chlorinated-alkanes, that were at tens of ng/L, and polybrominated diphenyl ethers (PBDEs), polychlorinated dibenzo-p-dioxins and dibenzofurans, dechlorane plus, chlorinated naphthalene at concentrations between 1 and 50 pg/L, as well the unresolved complex mixture of aliphatic and aromatic hydrocarbons at concentrations of few ug/L These chemical mixtures also contain unknown POPs which have not been described yet in the literature due to lack of appropriate analytical methods (Muir and Howard, 2006), and which can contribute significantly to the overall mixture toxicity as found in other studies (Tang et al., 2013). PAHs and n-alkanes were analyzed in the treatments and controls for the initial and final time (24 h, Figure 1, Supplementary Figure S1). The addition of this mixture of pollutants to seawater, resulted in a final concentration less than 2-fold increase with respect to the in-situ concentrations (comparison of concentration in treatments versus controls). During the course of experiments, there was no apparent degradation of PAHs, but a reduction of n-alkane concentrations to half of the initial values (Figure 1, Supplementary Figure S1 and details in Supplementary Table S3 in Fernández-Pinos et al., 2017). The incubations were performed in baked 1-L glass bottles. The pollutant mixtures were added to the treatment bottles and the solvent (acetone) to the control bottles 1 h before adding the seawater to allow for evaporation and avoid potential toxic effects of the solvents used to prepare the POPs mixtures. After addition of seawater from the DCM, the bottles were manually stirred and immediately placed in an incubator located on the deck of the vessel that maintained the surface temperature (Supplementary Table S1). No further stirring was performed. The light radiation at the sampled depth was simulated using a net covering the bottles. All experiments started between 10 and 12:30 h local time and we used one pair of treatment and control for each time point. After 30 min, 2 h and 24 h, we collected samples for DNA and mRNA.
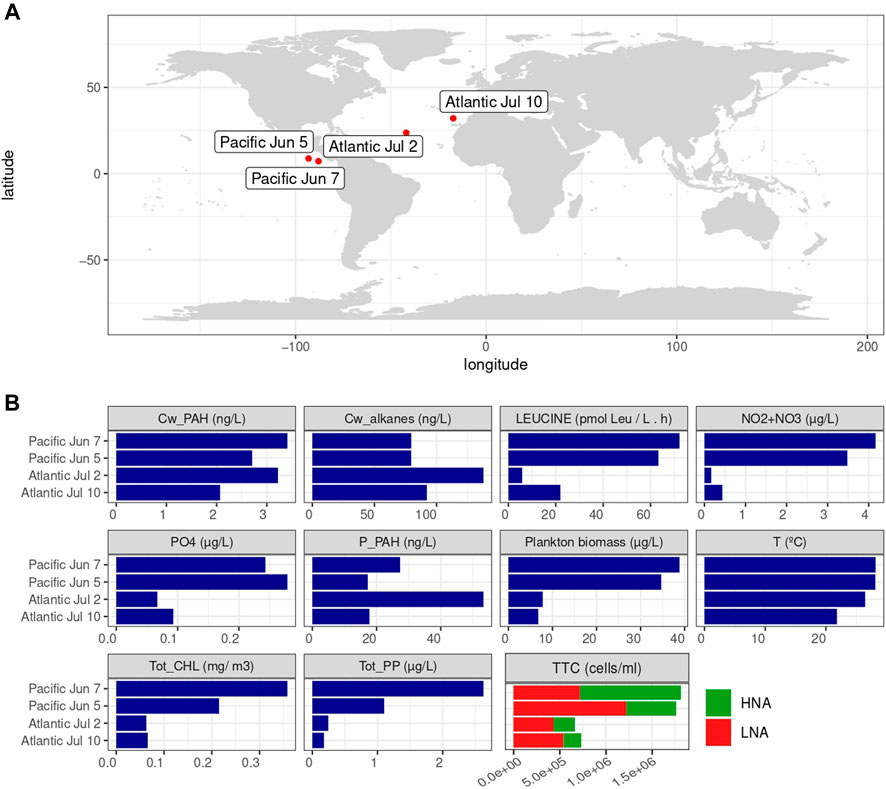
FIGURE 1. Water sampling stations and in situ measurements. (A) Stations where samples were taken for the experiments. (B) Measurements of nutrients, chlorophyll, cell abundances and pollutants. “Cw_PAH”, “p_PAH”: PAH concentrations in the dissolved phase (ng/L) and plankton (ng/g) respectively, “Cw_alkanes”: alkane concentrations in the dissolved phase (ng/L), “LEUCINE”: leucine incorporation rates (pmol Leu/L h), “NO2+NO3″ and “PO4”: concentrations of NO2− + NO3− and PO4 respectively (µg/L), “Plankton biomass” (µg/L), “Tot_CHL”: total chlorophyll A (mg/m3) and “Tot_PP”: total primary production (mg C/m3 h), “TCC”: total cell counts, that include “HNA” and “LNA”: high and low nucleic acid content bacteria (cells/ml).
After 0.5, 2 h and 24 h incubations, seawater from experimental bottles was pre-filtered through a 3 µm pore-size 47 mm diameter polytetrafluoroethylene filter and bacterial cells were collected onto a 0.2 µm pore-size 47 mm polytetrafluoroethylene filter under low vacuum pressure. The duration of the filtration step was no longer than 15 min to minimize RNA degradation. Each filter was cut in two-halves, one was placed in 1 ml RNAlater (Sigma-Aldrich, Saint Louis, MO) and the other one into 1 ml lysis buffer (50 mM Tris HCl, 40 mM EDTA, 0.75 M Sucrose) and stored at -80°C to preserve RNA and DNA respectively. DNA extraction was performed following the protocol described in Cerro-Gálvez et al. (2019). mRNA was extracted and amplified as described in Poretsky et al. (2010) with the modification of the use of mirVana isolation kit (Ambion) to extract the total RNA. Resulting DNA and amplified RNA were sequenced at the National Center for Genomic Analysis (CNAG, Barcelona, Spain) using Illumina high output mode HS200 2 × 100 bp v4.
DNA and cDNA sequences were quality trimmed and internal standard sequences removed using the ERNE mapping program (v. 2.1.1, Del Fabbro et al., 2013) using default parameters (--fast mode, --min-size 25, --min-mean-phred-quality 20, --errors-rate 15, --indels-max). For cDNA sequences, ERNE was run twice, to also remove stable RNA sequences using an in-house database. After quality trimming and removal of sequences, an average of 4.00 e + 07 reads remained per sample (Supplementary Table S2).
Genomes were constructed from DNA reads to create metagenome assembled genomes (MAGs) using the MetaWrap tool (v. 1.0, Uritskiy et al., 2018)), with MEGAHIT (v. 1.1.2, default settings, i.e. 21, 29, 39, 59, 79, 99, 119 and 141 kmer sizes, Li et al., 2015) for assembly and Metabat2 (v. 2.12.1, Kang et al., 2019) as the binning tool. MAGs were quality checked with CheckM (v. 1.0.12, Parks et al., 2015) (Supplementary Table S3), taxonomy determined with GTDB-Tk (v. 1.5.1, Chaumeil et al., 2020) using release r06-rs202 of the GTDB database (Parks et al., 2018) and functionally annotated with Prokka (v. 1.12, Seemann 2014), all using default settings. MAG abundances in the samples were calculated by mapping DNA reads back to the MAGs using Bowtie 2 (v. 2.3.4.1, Langmead and Salzberg 2012) and Samtools to create idxstats files (v. 1.7, Li et al., 2009). Bowtie 2 was run with default settings, i.e., in paired end “end-to-end” mode to map the full length of reads to contigs. Genes were quantified using the Bowtie 2 mapping results and feature Count from the Subread package (v. 2.0.1, Liao et al., 2014) with gff files from the Prokka annotation. An average of 52% of reads mapped to contigs, and 22% of reads mapped to MAGs. Transcripts per million (tpm) was used as the unit of abundance of genes and MAGs. This was calculated by taking the ratio of counts per gene/genome to the length of the gene/genome and dividing that by the sum of the ratio over the sample. Finally, the number was multiplied by a million. The proportion of reads mapping to MAGs made up on average 42% of reads mapping to contigs (SupplementaryTable S1). Selected specific signature genes coding for enzymes involved in the interaction between ADOC and microorganisms were searched for against genes in the MAGs using HMMER (v. 3.1. b. 2, Eddy, 2011) with a set of inhouse and Pfam (Finn et al., 2016; Table 1) profiles. In-house alkB profile can be found in Supplemental Material. R (v. 4.1.2, R Core Team. 2021), RStudio (v. 2022.7.2.576, RStudio Team 2022) and the Tidyverse (v. 1.3.0, Wickham et al., 2019) were used to create all figures, except for the Anvi’o (v. 7.1, Eren et al., 2021) figures.
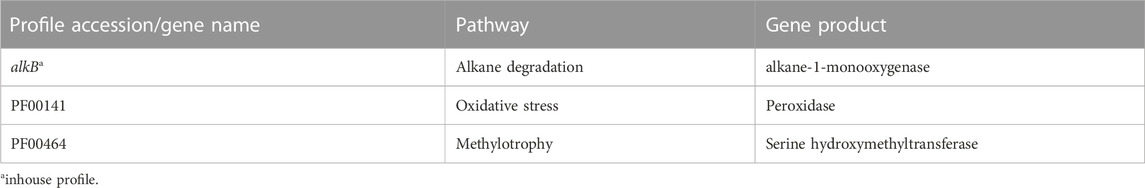
TABLE 1. Profiles used to identify signature genes. Gen alkB (alkane-1-monooxygenase) was selected as surrogates of genes associated with the degradation of mid-chain alkanes. File of alkB profile can found in Supplemental Material.
To analyze the effects of treatments (controls vs. ADOC addition), experiments and ocean basin on both community structure (DNA relative abundances) and community transcriptional activities (RNA relative abundances), a Permanova using a Bray–Curtis distance matrix (Anderson, 2001) was performed. Variance in community structure and transcription described by Bray–Curtis dissimilarities was visualized using principal component analysis in R.
Several families of organic compounds were analyzed in seawater and plankton at the sites and in the regions, where experiments were carried out, as reported previously (González-Gaya et al., 2014; Morales et al., 2015; Casal et al., 2017; González-Gaya et al., 2019), which include PAHs, PCBs, dioxins, and perfluoroalkyl substances. Here we use 64 PAHs (González-Gaya et al., 2019) and alkanes ranging from 21 to 35 carbon chains (previously unpublished, see Materials and Methods) as surrogates or chemical indicators of hydrophobic ADOC to characterize the level of pollution at each sampling site. Concentrations of PAH and alkanes in the dissolved and plankton phase (Figure 1, Supplementary Figure S1) were similar at the four experiment sites. Even though the North Pacific Ocean is generally less polluted than the North Atlantic for a wide range of pollutants (González-Gaya et al., 2014, 2019; Muir and Miaz, 2021; Xie et al., 2022), the similarity in concentrations is consistent with the fact that experiments in the Pacific Ocean were performed on the Coastal Rica Dome, which is influenced by a regional upwelling, while the experiments in the Atlantic were performed in oligotrophic regions remote from primary sources. Concentrations of pollutants in surface seawater in the Costa Rica dome are comparable to those reported for the North Atlantic, probably due to the influence of proximate continental sources and the regional upwelling re-mobilizing previously deposited chemicals in deep waters (Casal et al., 2017; González-Gaya et al., 2019). The more eutrophic conditions of this upwelling region were mirrored by the higher nutrient concentrations at Pacific sites compared to North Atlantic sites (Figure 1). In consequence, this location harbors dynamic biological communities. Consistently, total Chl a concentration, primary production, bacterial production and concentration of heterotrophic cells were significantly higher in the Pacific than in the Atlantic sites. The two experiments in the Pacific ocean were carried out under identical surface seawater temperatures (28°C) which were above those in the subtropical Atlantic Ocean experiments (26°C and 22°C respectively).
From the DNA samples, we constructed 976 metagenome assembled genomes (MAGs) representing both Archaea and Bacteria (Supplementary Table S2). The majority were of low quality, following Bowers et al. classification (Bowers et al., 2017), and nearly half of them lacked sufficient markers for taxonomy classification. MAGs were quite fragmented (Supplementary Table S3) with a median number of contigs of 159, 331 and 355 for high, medium and low quality respectively (excluding MAGs without taxonomy). The corresponding median N50 values were 27,831, 8,161 and 9,678. Nevertheless, we found 24 archaeal and 208 bacterial MAGs of medium or high quality, i.e. with an estimated degree of contamination less than 10%. In addition, we used 25 archaeal and 209 bacterial low-quality MAGs that were less than 10% contaminated and were possible to assign taxonomically. While incomplete and not suitable for analysis of their full genetic potential, these low-quality MAGs could still provide taxonomic information about key functional responses in our experiments.
We recovered several species known to be important in marine surface waters, such as the unicellular Cyanobacteria Synechococcus spp. and Prochlorococcus spp, as well as the alphaproteobacterial Pelagibacteraceae spp. (“SAR11”; all unfortunately of low quality) and Rhodobacteraceae spp, the gammaproteobacterial SAR86 order, Alteromonadaceae spp. and Pseudomonadales spp, as well as several Flavobacteriales (Sunagawa et al., 2015). The recently named Poseidonales order of Archaea, formerly known as “Marine Group II Archaea” (Rinke et al., 2019), was also well represented by MAGs.
In terms of relative abundances of MAGs, both from DNA and RNA samples, communities were dominated by a few phyla: the archaeal Thermoplasmatota, and the bacterial Bacteriodota, Cyanobacteria, Patescibacteria, Proteobacteria and the automatically named phylum T1SED10-198M, recently named as Candidatus Gulusiota (Pallen et al., 2022) (Figure 2 and Supplementary Table S4). A few MAGs from Thermoplasmatota, particularly from the order Poseidoniales, dominated gene expression in the two experiments performed in the Atlantic, but not for the Pacific sites. The proportion of reads mapping to contigs from taxonomy-assigned MAGs, was considerably smaller in the RNA than in the DNA samples, suggesting that our MAGs were representative of the active part of the community.
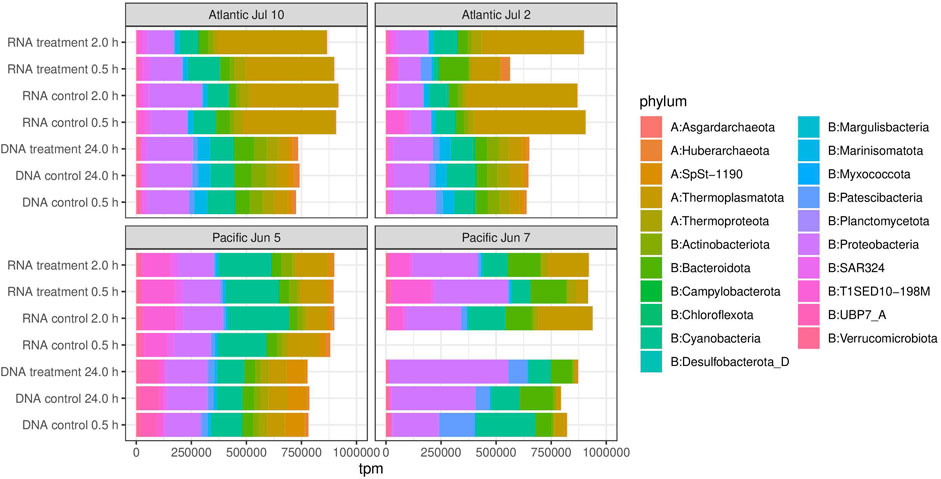
FIGURE 2. Overall taxonomic composition of microbial communities. Transcripts per million (tpm) were used as the unit of abundance. See materials and methods for details on tpm calculation.
To analyze populations that were transcriptionally responding to exposure to ADOC, we identified 331 MAGs that contributed at least eight-fold more, or less, to the total RNA in the treatment than in the control sample for at least one experiment and sampling time (Figure 3; Table 2, Supplementary Table S4). In general, we detected a higher number of responding MAGs in the Atlantic than in the Pacific, although the number varied considerably between experiments and time points, from 0 to 5 Pacific June 5 experiments, to 107 after half an hour at the Atlantic July 2 experiment and around 80 after 2 h of exposure in both of the Atlantic experiments. The balance between increasing and decreasing MAGs also varied considerably between experiments and time points. In the Atlantic experiments, most of the transcriptionally responding MAGs belonged to Proteobacteria, Thermoplasmatota, Actinobacteriota and Bacteroidota (Supplementary Table S5). In the Pacific experiments, transcriptionally responding MAGs were from the Bacteroidota and the Proteobacteria. The higher number of transcriptionally responding MAGs in the Atlantic than in the Pacific experiments can be related to the more oligotrophic status of Atlantic waters, where ADOC addition represented a more relevant pulse of carbon than in the Pacific. This is consistent with the higher sensitivity of oligotrophic systems to external anthropogenic inputs (Shai et al., 2021). Our results indicate that this is also true at ADOC concentrations relevant for the open ocean, as previously reported for coastal Mediterranean waters (Cerro-Gálvez et al., 2021). The changes in community structure using DNA relative abundances of all MAGs assessed using a Permanova test significantly differed between experiments (p < 0.01) and oceans (p < 0.01). The same pattern was observed for transcriptional activities (Supplementary Table S6). The microbial responses in Atlantic experiments were more similar to each other than to either of the Pacific experiments consistent with a minor effect of temperature on the microbial responses to ADOC additions (Supplementary Figure S2).
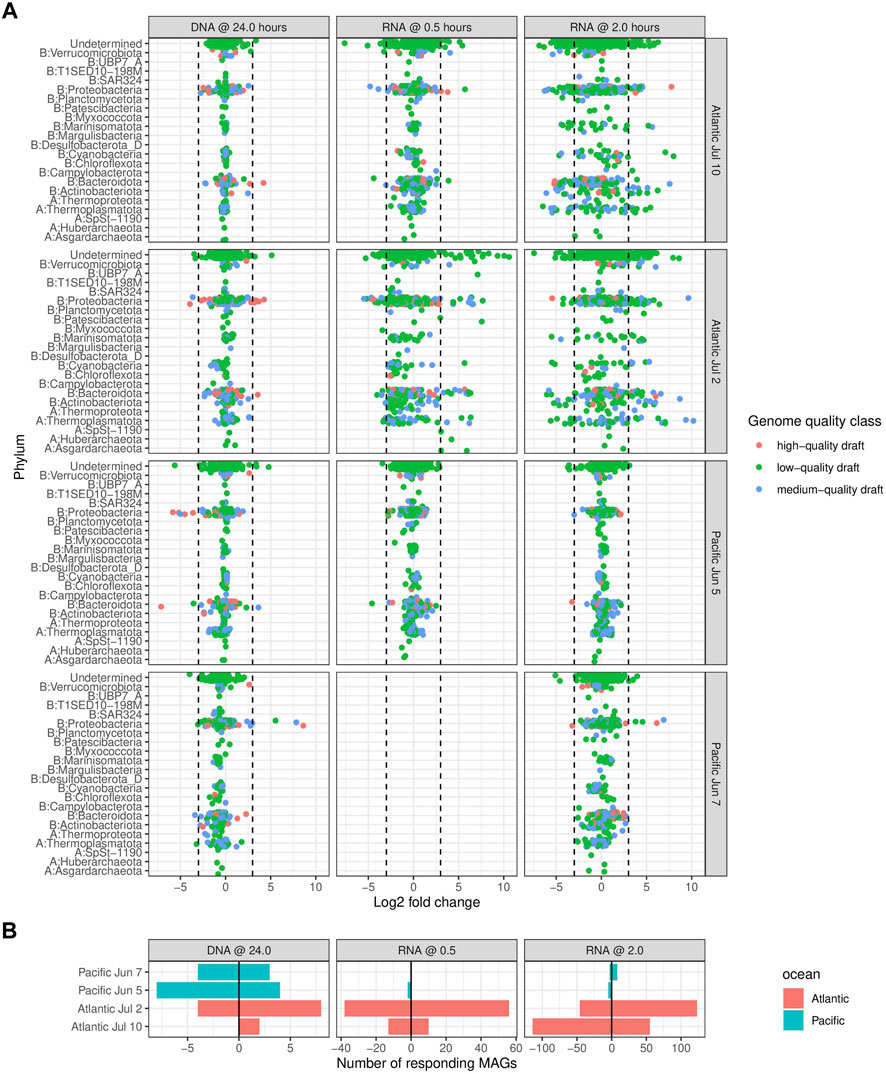
FIGURE 3. Log 2 fold changes of relative abundances of MAGs between treatments and controls in the DNA pool (indicating enrichment) and in the RNA pool (indicating transcriptional activity) in each experiment. To be included, a MAG needed to be present both in the treatment and control samples. Phyla are prepended with an (A, B) to denote Bacteria and Archaea respectively. Dashed lines at log 2 fold changes of -3 and 3, corresponding to eight-fold decrease and increase respectively, indicate limits for inclusion in further analyses below. MAGs appearing to the left of the leftmost or right of the rightmost line were considered as “responders” to treatment.
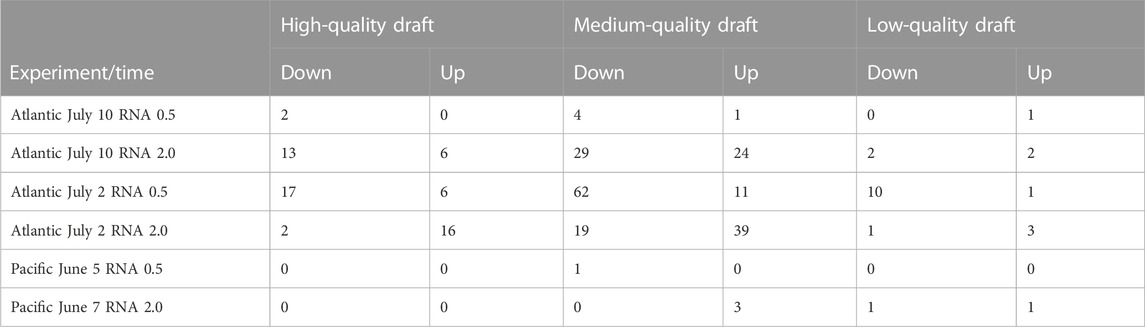
TABLE 2. Number of transcriptionally responding MAGs per experiment and time point. Transcriptionally responding was defined as MAGs being at least eight-fold more or less abundant in the treatment than in the control. Only low-quality draft genomes with at most 10% contamination and determined taxonomy included.
Among all the transcriptionally more active MAGs, up to 86 had not only a fold-change in gene expression higher than eight between treatments and controls, but also a quality classification of at least “medium-quality draft”. These included 77 Bacteria (9 Actinobacteriota, 18 Bacteroidota, four Synechococcus, 2 Marinisomatota, 22 Alphaproteobacteria, 17 Gammaproteobacteria and five Verrucomicrobiota) and nine Archaea (8 Thermoplasmatota and one Thermoproteota) (Supplementary Table S5). We labeled them as “responding MAGs” and selected them for deeper gene content and transcriptional analyses in ADOC exposed communities. In the Pacific experiments, we observed some high contributions of specific taxa to the DNA and RNA pool. These were, for instance, MAGs from Cyanobacteria and some from Flavobacteriales and Rhodobacterales. The Atlantic experiments had a more equal distribution of contributions from several bacterial groups. Three archaeal MAGs belonging to Thalassarchaeaceae within the Thermoplasmatota phylum contributed considerably to the DNA and RNA pools. (Figure 4).
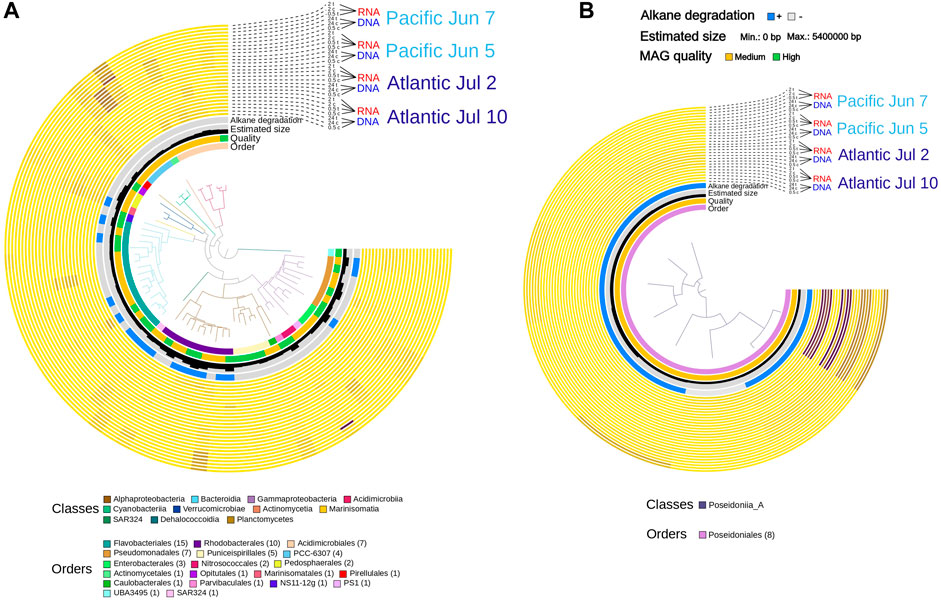
FIGURE 4. Phylogeny and abundance of strongly responding MAGs per sample together with genome statistics as well as presence and expression of signature genes for alkane oxidation. (A) bacteria, (B) archaea. Branches are colored according to taxonomic Classes. Abundances were normalized by calculating square root of tpm values. The color scale was set from 0 (min) to 290 (max).
Contrasting how individual MAGs responded to ADOC addition in terms of transcriptional activity and increase of contribution to the DNA pool (called enrichment hereafter) respectively, we detected different patterns: 1) increased transcription in treatments compared to controls during the first 2 h of exposure but not a concurrent growth after 24 h, 2) stable or increased transcription with growth after 24 h, 3) stable transcription and no enrichment, 4) decreases in community size, i.e., DNA contribution, after 24 h, and v) no clear responses neither in transcription nor enrichment. In the Atlantic Ocean, the highest number of MAGs fell into the “transcription increase, no enrichment” and “no clear response” categories, whereas in the Pacific most MAGs were in the “stable in both” category and, a lower number, in “decreases in community size”. The top responding MAGs within all categories belonged to the families Rhodobacteraceae and Flavobacteriaceae but with an ocean dependent pattern, as these contributed the most to the active transcription categories (with and without growth) in Atlantic experiments, whereas they were stable in the Pacific experiments (Figure 5, Supplementary Figure S7). These two families of bacteria are generalists, known for their copiotrophic lifestyle and opportunistic activation of transcription after inputs of organic matter, including ADOC compounds, and thus capable of rapid growth in carbon-rich environments (Yooseph et al., 2010; Cerro-Gálvez et al., 2021). The different responses in contributions to the DNA pool in each ocean might be related to the importance of ADOC respect to the total pool of DOC, that was probably less labile and more limited in the oligotrophic Atlantic than in the Costa Rica Dome in the Pacific.
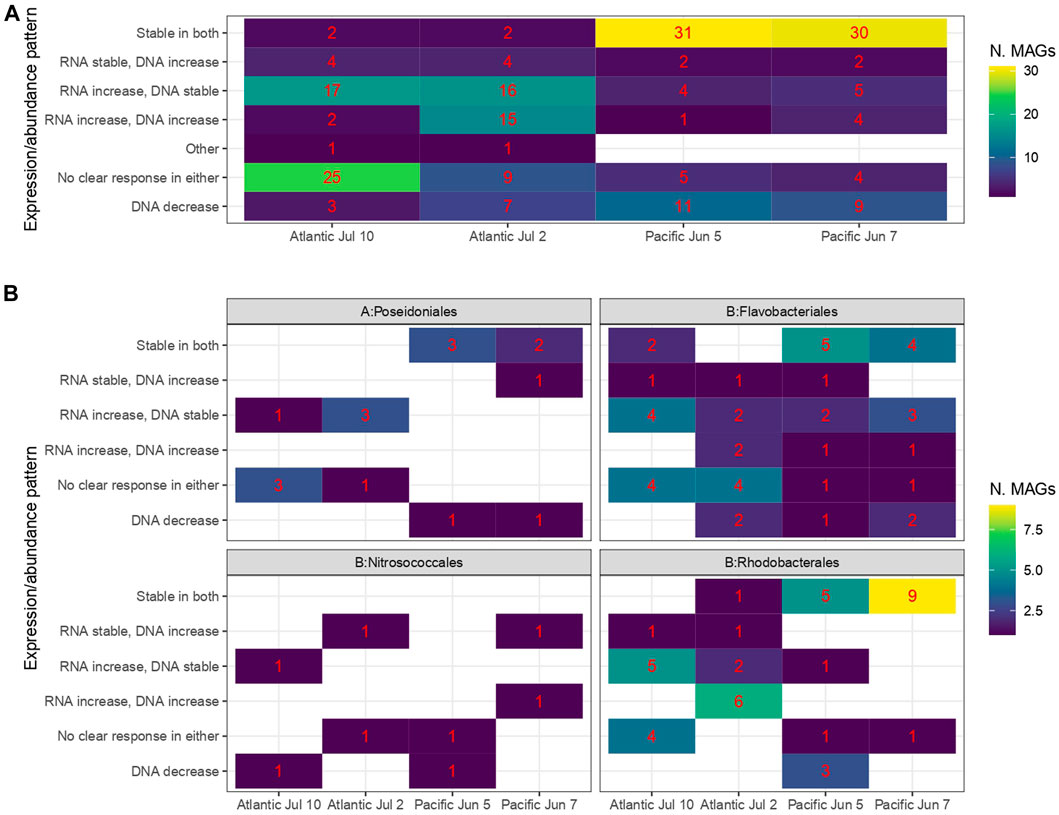
FIGURE 5. Overall patterns in RNA expression and DNA abundance in MAGs increasing at least eight-fold in at least one sample pair. The RNA 0.5, RNA 2.0 and DNA 24 h sample pairs were classified as increasing in the treatment if the treatment was at least two-fold higher than the control. Individual patterns were then classified in classes. (A) Overall, (B) Selected orders.
Several MAGs belonging to the archaeal Poseidoniales order were found to have increasing transcriptional activity after 2 h of exposure to ADOC, although they did not get significantly enriched. This is a pattern already observed in Alteromonadales in coastal Mediterranean waters after exposures to low levels of ADOC concentrations (Cerro-Gálvez et al., 2021). Poseidoniales is the most abundant archaeal group in surface seawaters, with different genera present in both oligotrophic and eutrophic waters (Rinke et al., 2019). We observed similar behavior of two families, Poseidoniaceae and Thalassarchaeaceae, with a higher activity in the Atlantic experiments than in the Pacific. Importantly, this group has been found to commonly harbor alkB-like monooxygenase genes in the North Atlantic, and a role in degradation of biogenic medium chain alkanes has been suggested (Love et al., 2021). The activation of gene expression but lack of significant enrichment, however, suggests that Poseidoniales are able to quickly respond to ADOC presence, but that this capacity, at least in oligotrophic waters, does not always result in population growth likely due to nutrient limitation. This is consistent with an oligotrophic lifestyle, among other things characterized by expression of high-affinity transporters effective at low substrate concentrations (Lauro et al., 2009).
Interestingly, the responding MAGs that showed the highest increase in relative contributions to the DNA pool after 24 h (independent of the transcription patterns) in Atlantic July 2 and Pacific June 7 experiments belonged to Methylophagaceae (order Nitrosococcales). The capacity of species in the Methylophagaceae to degrade aliphatic hydrocarbons has been shown under oil spill scenarios (Mishamandani et al., 2014; 2016; Gutierrez and Aitken 2014; Kleindienst et al., 2016), as well for consumption of biogenic alkanes, such as those from cyanobacteria (Love et al., 2021). Although Methylophagaceae in most marine environments belong in the “rare biosphere”, i.e., present, but at low abundances, this group was observed to bloom in polluted coastal Mediterranean waters after a pulse of ADOC at background concentrations (Cerro-Gálvez et al., 2021). A similar result was found in both the Pacific and the Atlantic experiments, providing field evidence on the high adaptation of Methylophagaceae to growth on ADOC chemicals at the low concentrations found in the global oceans.
On average, between 2 and 26 genes from each MAG from the four most characteristic orders of strongly responding MAGs—Poseidonales, Flavobacteriales, Nitrosococcales and Rhodobacterales—contributed at least two-fold more to the total RNA pool in treatments than in controls (Figure 6). Except for in Rhodobacterales, the number of responding genes varied considerably between experiments and for different time points. In order to assess the nature of the responses, these individual genes were classified into nine relevant classes—ATP synthase (energy conversion), DNA polymerase (replication), RNA polymerase (transcriptional activity), enzymes (overall metabolism; genes not included in more specific classes but having an EC number), stress, translation, transporters, hypothetical proteins and others. We observed a low contribution of transporters, quite a small but consistently present contribution of genes involved in transcription and translation and conspicuously low numbers of genes involved in replication (Figure 6). We found a consistently high number of hypothetical genes in the Poseidonales consistent with the relatively recent discovery of the group and lack of cultures for phenotypic characterization of the group (Rinke et al., 2022).
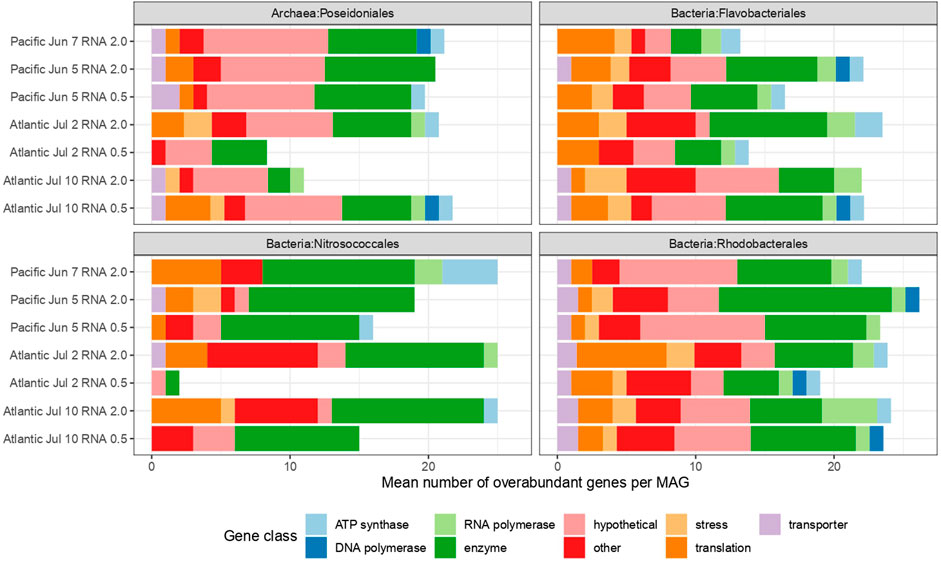
FIGURE 6. Mean number of overabundant genes in treatments compared with controls for selected orders. Genes reaching abundances in treatments at least two-fold higher than in controls. Genes were classified based on product names in the Prokka annotation.
To look for patterns of consistent gene expression between different populations of the same phylum, we searched for genes increasing at least two-fold in any of the experiments and counted the number of different MAGs that expressed genes with the same product name. Any genes occurring in the list at least twice were considered (Supplementary Table S8). Furthermore, we weighted the gene list by the product of each product’s highest relative abundance and its highest fold-change. At the top of this list, appearing in five different MAGs at relative abundances between 0.2 and 230, and log 2 fold-changes between 1.0 and 3.7, we found “Serine hydroxymethyltransferase”, an important enzyme in one-carbon metabolism or methylotrophy (Supplementary Table S8) (Rao et al., 2003). The species were representatives of the Alteromonadaceae, Rhodobacteraceae and SAR86 families. In the actinobacterial families MedAcidi-G1, TK06 we found “Putative formate dehydrogenase”, also involved in one-carbon metabolism (Ferry 1990), represented in two genomes. In alkane metabolism, “Alkane 1-monooxygenase” (3 species from Flavobacteriaceae, Schleiferiaceae and UBA10066 families of Bacteroidota), “Alkanesulfonate monooxygenase” (3 species from the actinobacterial families MedAcidi-G1, MedAcidi-G1 and TK06) and “Haloalkane dehalogenase” (3 species from the proteobacterial families Pseudohongiellaceae, Rhodobacteraceae and SAR86), were discovered. Other enzymes expressed in more than one species, and with a potential physiological role after addition of ADOC included the oxidative stress-related “Superoxide dismutase” (five species from Alteromonadaceae, Rhodobacteraceae and Methylophagaceae), “Catalase-peroxidase” (nine species from Flavobacteriaceae, Schleiferiaceae, Alteromonadaceae, Pseudohongiellaceae, Puniceispirillaceae and Rhodobacteraceae), “putative peroxiredoxin” (24 species from Actinobacteriota, Bacteroidota, Cyanobacteria and Proteobacteria) and, in electron transport, a number of cytochrome c components (45 species from the phyla Actinobacteriota, Bacteroidota, Marinisomatota and Proteobacteria). Lastly, different components of ribonucleotide reductase, the enzyme catalyzing the only known de novo pathway for production of deoxyribonucleotides, building blocks of DNA (Torrents et al., 2008), was found to be highly expressed by 17 MAGs from the archaeal Thermoplasmatota phylum and the bacterial Bacteroidota and Proteobacteria phyla.
Whereas generic functional annotation of genomes is helpful for the identification of patterns without strong presumptions about which pathways are potentially affected by ADOC addition, the fact that alkane families are important components of hydrophobic ADOC suggests a more targeted approach. We screened the MAGs for the potential degradation of alkanes with sequence searches using profiles for the gene coding for the enzyme that catalyzes the committed step in degradation “alkane-1-monooxygenase” (alkB). Since PAH concentrations did not change significantly during our experiments and because it has been shown difficult to exactly identify the key enzyme involved in PAH degradation (CITE)—Aromatic ring-hydroxylating dioxygenase—we did not analyze our MAGs for the presence or expression of this gene. The highest number of presence of alkane degrading genes in Bacteria were found in Alphaproteobacteria (encoding genes for both pathways, four only for alkanes) and Gammaproteobacteria (with both pathways, six only alkanes) (Figure 4). These results are consistent with the reported geographical ubiquity of n-alkane degradation genes in the open ocean (Love et al., 2021).
For the study of differentially expressed genes in key metabolisms, we included specific markers for oxidative stress (“peroxidase”) and methylotrophy (“Serine hydroxymethyltransferase”). The former because of the toxicity of these compounds on cells (Cerro-Gálvez et al., 2019), and the latter because methylotrophy has been implicated in ADOC degradation, probably alkyl PAHs and alkanes (Gutierrez and Aitken 2014; Cerro-Gálvez et al., 2019). We detected many responding signature genes for the four pathways with at least an eight-fold higher abundance in treatments than in controls, primarily from the experiments in the Atlantic and mostly in samples taken after 2 h incubation (Figure 7, Supplementary Table S9). Among MAGs that decreased or had a stable contribution to the RNA pool, we detected roughly equal numbers of signature genes going up as going down (Figure 7), whereas in MAGs that increased their contribution only increases, baring one exception, were observed (Figure 7; Table 3). In particular, several MAGs expressed genes for alkane degradation and methylotrophy more abundantly in treatments than in control, but only in the Atlantic experiments. This supports previous findings of the higher lability of alkanes within the ADOC pool, thanks to its simpler chemical structure, higher transcription of alkB genes in oil spills and widespread occurrence and divergence of alkB-harboring bacteria in marine microbiomes (Mason et al., 2012; Rivers et al., 2013; Karthikeyan et al., 2021; Love et al., 2021). The increasing alkane degradation signature genes in increasing MAGs were expressed by species from the archaeal Poseidoniales order, the actinobacterial Acidimicrobiales order, the Bacteroidia order Flavobacteriales and the proteobacterial Rhodobacteraceae, Saccharospirillaceae and Methylophagaceae families. Methylotrophy was expressed by the same groups plus the archaeal Nitrosopumilaceae family, the cyanobacterial Cyanobiaceae family, the Marinisomatota phylum and the gammaproteobacterial Puniceispirillales order (Figure 7 and Supplementary Table S9). The taxonomical connection between alkane oxidation on the one hand, and methylotrophy on the other hand, reflects the proposed role of methylotrophy in degradation of hydrocarbons in the ocean that has been analyzed under oil spill scenarios, but it is here evidenced at low oceanic concentrations. For instance, methylotrophs were enriched in the oil plume in late stages of the Deepwater Horizon oil spill (Gulf of Mexico) after the initial bloom of opportunistic hydrocarbonoclastic bacteria (Kessler. et al., 2011). Direct evidence of Methylophaga in consumption of hexadecane was obtained in experiments using stable isotope probing (Mishamandani et al., 2014). Further, Methylophaga have the capacity to remove methyl-groups from alkylated hydrocarbons, for example methyl phenanthrenes, that are generally abundant in the ADOC pool (Gutierrez and Aitken 2014; Cerro-Gálvez et al., 2019). Overall, our gene expression profiles support a fast consumption of alkanes, along with an activation of methylotrophy, at the low concentrations of ADOC ubiquitous in oceanic waters.
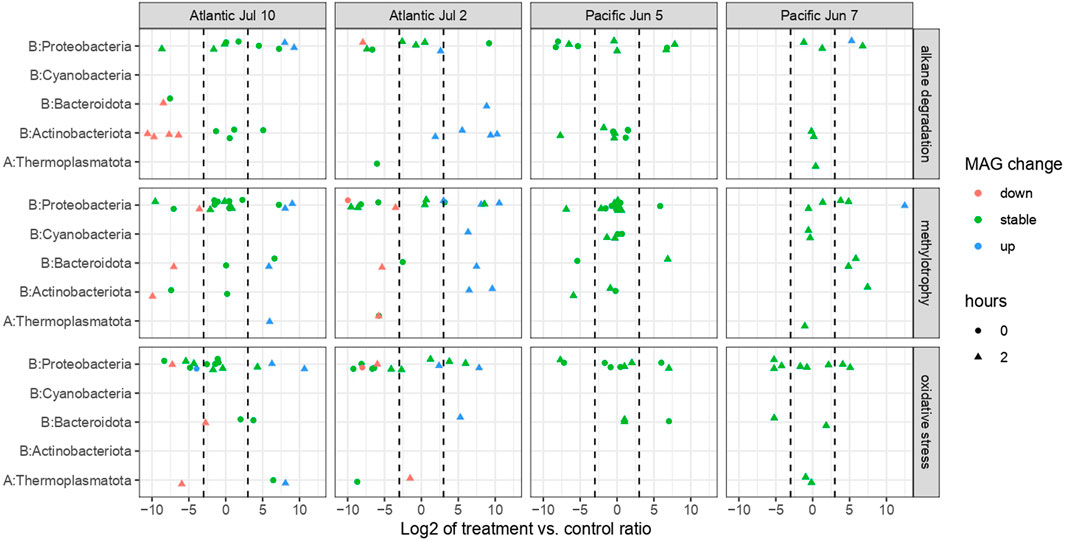
FIGURE 7. Expression of signature genes for alkane degradation, methylotrophy and oxidative stress in MAGs responding with growth or decrease in at least one experiment (panel (A)) and averaged changes in relative concentrations (treatment/controls) of PAHs and alkanes in the experiments after 0.5 and 24 h of incubation (panel (B)). On the top panel, the x-axis values show gene log 2 fold-changes, while the MAGs’s response is encoded with color. When calculating ratios, a zero in either the treatment or control was replaced with 0.001. Specific changes in relative concentration ratios of each chemical within PAH and alkanes families is described in Supplementary Figure S1.
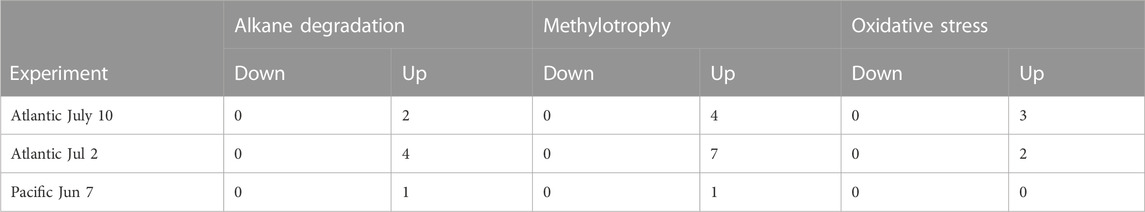
TABLE 3. Number of genes increasing or decreasing in responding MAGs more than eight-fold between treatment and control. Responding MAGs were defined as those being at least eight-fold more abundant in treatments than in controls. Similarly, genes were defined as up or down if they were eight-fold more or less respectively abundant in treatments than in controls.
The signature gene for oxidative stress was enriched in a subset of the taxa that also expressed signature genes for the other three processes: Poseidoniales, Acidimicrobiales, Flavobacteriales, Puniceispirillales, Rhodobacterales and Pseudomonadales. Similar correlations between metabolic responses at gene level to ADOC addition has been reported previously (Cerro-Gálvez et al., 2019, 2021). The higher metabolic rate that might be a consequence of oxidative metabolism of ADOC, either as a carbon source or as a defense mechanism against toxicity, can be expected to result in higher concentrations of reactive oxygen species, and hence trigger transcription of defense systems.
We performed different ADOC exposure experiments at background concentrations in the mesotrophic tropical Pacific (Costa Rica Dome) and the oligotrophic subtropical North Atlantic. During these experiments, there was degradation of n-alkanes with 21–35 carbons that are thought to be from fossil fuels (petroleum, coal), and thus have anthropogenic origin (Fernández-pinos et al., 2017). The most active MAGs responding to ADOC belonged to the families Rhodobacteraceae and Flavobacteriaceae. Recently, it has been suggested a fast cryptic cycle of C15 and C17 alkanes produced by cyanobacteria and consumed by some heterotrophic bacteria (including specialist hydrocarbonoclastic bacteria such as Alcanivorax and methylotrophs such as Methylophaga spp.) and archaeal Poseidoniales. Whether or not the recycling of anthropogenic and other biogenic n-alkanes is similar to that of the cryptic cycle remains unknown. In this study, besides high activities of MAGs from families Rhodobacteraceae and Flavobacteriaceae responding to ADOC after short-term exposures, we observed a high transcription activity of Poseidonales and growth of Methylophaga, and similar patterns of methylotrophy and alkane degradation gene expression. These observations, along with the efficient degradation of C21-C35 alkanes (Fernández-pinos et al., 2017) support the high lability of alkanes within the ADOC pool and the suggested role of several taxa consuming medium high chain alkanes. This can possibly be related to their capacity to outcompete other opportunistic bacteria at low alkane concentrations (and fail at high concentrations such as in initial stages of oil spill accidents), similar to the concentrations of alkanes produced by cyanobacteria. As the experiments were performed exposing the microbial communities to changes in ADOC concentrations by a factor of two, the results suggest that anthropogenic carbon introduction in the ocean could play a role modulating microbiomes, which will require extensive research efforts in the coming years.
All sequence data presented here can be downloaded from the European Nucleotide Archive (ENA) under the project accession PRJEB60184. Metagenome samples have accessions ERS14678684-ERS14678695 and metatranscriptome samples have ERS14678669-ERS14678683. Accessions for metagenome bins and MAGs (better quality bins) that were possible to annotate taxonomically and were less than 10% contaminated can be found in Supplementary Table S11.
MV-C, DL, and JD wrote sections of the manuscript. M-CF-P, JD, and BP did the conceptualization of the experiments. M-CF-P performed the experiments. M-CF-P and MV-C processed DNA and RNA samples. DL and MV-C did the computational and bioinformatic analyses. JI did anvio analyses. All the authors contributed to provide feedback to the manuscript and approved the submitted version.
This work was supported by the Spanish Ministry of Science and Innovation through projects MIQAS (PID2021-128084OB-I00) and PANTOC (PID2021-127769NB-I00). The research group of Global Change and Genomic Biogeochemistry receives support from the Catalan Government (2017SGR800). IDAEA-CSIC is a Centre of Excellence Severo Ochoa (Spanish Ministry of Science and Innovation, Project CEX2018-000794-S). DL was supported by the Faculty of Health and Life Sciences at Linnaeus University and the Swedish Research Council FORMAS Strong Research environment Eco Change.
We thank the staff of the Marine Technology Unit (UTM-CSIC) for their logistical support during the sampling Malaspina campaign.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2023.1110169/full#supplementary-material
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi:10.1038/nbt.3893
Casal, P., Casas, G., Vila-Costa, M., Cabrerizo, A., Pizarro, M., Jiménez, B., et al. (2019). Snow amplification of persistent organic pollutants at coastal Antarctica. Environ. Sci. Technol. 53, 8872–8882. doi:10.1021/acs.est.9b03006
Casal, P., Zhang, Y., Reardon, F., Martin, J. W., and Dachs, J. (2017). Accumulation of perfluoroalkylated substances in oceanic plankton. Environ. Sci. Technol. 51, 2766–2775. doi:10.1021/acs.est.6b05821
Cerro-Gálvez, E., Dachs, J., Lundin, D., Fernández-Pinos, M. C., Sebastián, M., and Vila-Costa, M. (2021). Responses of coastal marine microbiomes exposed to anthropogenic dissolved organic carbon. Environ. Sci. Technol. 55, 9609–9621. doi:10.1021/acs.est.0c07262
Cerro-Gálvez, E., Sala, M. M., Marrasé, C., Gasol, J. M., Dachs, J., and Vila-Costa, M. (2019). Modulation of microbial growth and enzymatic activities in the marine environment due to exposure to organic contaminants of emerging concern and hydrocarbons. Sci. Total Environ. 678, 486–498. doi:10.1016/j.scitotenv.2019.04.361
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2020). GTDB-tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi:10.1093/bioinformatics/btz848
Dachs, J., Bayona, J. M., Fowler, S. W., Miquel, J. C., and Albaigés, J. (1998). Evidence for cyanobacterial inputs and heterotrophic alteration of lipids in sinking particles in the Alboran Sea (SW Mediterranean). Mar. Chem. 60, 189–201. doi:10.1016/s0304-4203(97)00105-9
Del Fabbro, D., Giorgi, F. M., Scalabrin, C., Morgante, S., and Giorgi, M. M. (2013). An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One 8, 85024. doi:10.1371/journal.pone.0085024
Echeveste, P., Galbán-Malagón, C., Dachs, J., Berrojalbiz, N., and Agustí, S. (2016). Toxicity of natural mixtures of organic pollutants in temperate and polar marine phytoplankton. Sci. Total Environ. 571, 34–41. doi:10.1016/j.scitotenv.2016.07.111
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7 7, e1002195. doi:10.1371/journal.pcbi.1002195
Eren, A. M., Kiefl, E., Shaiber, A., Veseli, I., Miller, S. E., Schechter, M. S., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6. doi:10.1038/s41564-020-00834-3
Fernández-Pinos, M. C., Vila-Costa, M., Arrieta, J. M., Morales, L., González-Gaya, B., Piña, B., et al. (2017). Dysregulation of photosynthetic genes in oceanic Prochlorococcus populations exposed to organic pollutants. Sci. Rep. 7, 8029. doi:10.1038/s41598-017-08425-9
Ferry, J. G. (1990). Formate dehydrogenase. FEMS Microbiol. Rev. 87, 377–382. doi:10.1111/j.1574-6968.1990.tb04940.x
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi:10.1093/nar/gkv1344
Galbán-malagón, C., Berrojalbiz, N., Ojeda, M-J., and Dachs, J. (2012). The oceanic biological pump modulates the atmospheric transport of persistent organic pollutants to the Arctic. Nature Communications 3, doi:10.1038/ncomms1858
González-Gaya, B., Dachs, J., Roscales, J. L., Caballero, G., and Jiménez, B. (2014). Perfluoroalkylated substances in the global tropical and subtropical surface oceans. Environ. Sci. Technol. 48, 13076–13084. doi:10.1021/es503490z
González-Gaya, B., Fernández-Pinos, M. C., Morales, L., Méjanelle, L., Abad, E., Piña, B., et al. (2016). High atmosphere-ocean exchange of semivolatile aromatic hydrocarbons. Nat. Geosci. 9, 438–442. doi:10.1038/ngeo2714
González-Gaya, B., Martínez-Varela, A., Vila-Costa, M., Casal, P., Cerro-Gálvez, E., Berrojalbiz, N., et al. (2019). Biodegradation as an important sink of aromatic hydrocarbons in the oceans. Nat. Geosci. 12, 119–125. doi:10.1038/s41561-018-0285-3
Gutierrez, T., and Aitken, M. D. (2014). Role of methylotrophs in the degradation of hydrocarbons during the Deepwater Horizon oil spill. ISME J. 8, 2543–2545. doi:10.1038/ismej.2014.88
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. doi:10.7717/peerj.7359
Karthikeyan, S., Hatt, J. K., Kim, M., Spain, J. C., Huettel, M., Kostka, J. E., et al. (2021). A novel, divergent alkane monooxygenase (alkB) clade involved in crude oil biodegradation. Environ. Microbiol. Rep. 13, 830–840. doi:10.1111/1758-2229.13018
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923
Lauro, F. M., McDougald, D., Thomas, T., Williams, T. J., Egan, S., Rice, S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. U. S. A. 106, 15527–15533. doi:10.1073/pnas.0903507106
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinforma. Appl. NOTE 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Lohmann, R., Breivik, K., Dachs, J., and Muir, D. (2007). Global fate of POPs: Current and future research directions. Environ. Pollut. 150, 150–165. doi:10.1016/j.envpol.2007.06.051
Love, C. R., Arrington, E. C., Gosselin, K. M., Reddy, C. M., Van Mooy, B. A. S., Nelson, R. K., et al. (2021). Microbial production and consumption of hydrocarbons in the global ocean. Nat. Microbiol. 6, 489–498. doi:10.1038/s41564-020-00859-8
Martinez-Varela, A., Cerro-Gálvez, E., Auladell, A., Sharma, S., Moran, M. A., Kiene, R. P., et al. (2021). Bacterial responses to background organic pollutants in the northeast subarctic Pacific Ocean. Environ. Microbiol. 23, 4532–4546. doi:10.1111/1462-2920.15646
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S. G., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 6, 1715–1727. doi:10.1038/ismej.2012.59
Mishamandani, S., Gutierrez, T., and Aitken, M. D. (2014). DNA-based stable isotope probing coupled with cultivation methods implicates Methylophaga in hydrocarbon degradation. Front. Microbiol. 5, 76–79. doi:10.3389/fmicb.2014.00076
Mishamandani, S., Gutierrez, T., Berry, D., and Aitken, M. D. (2016). Response of the bacterial community associated with a cosmopolitan marine diatom to crude oil shows a preference for the biodegradation of aromatic hydrocarbons. Environ. Microbiol. 18, 1817–1833. doi:10.1111/1462-2920.12988
Morales, L., Dachs, J., Fernándezfernández-Pinos, M.-C., Berrojalbiz, N., Mompean, C., Belén Gonzaíez-Gaya, B., et al. (2015). Oceanic sink and biogeochemical controls on the accumulation of polychlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyls in plankton. Environ. Sci. Technol. 49, 13853–13861. doi:10.1021/acs.est.5b01360
Muir, D. C. G., and Howard, P. H. (2006). Are there other persistent organic pollutants? A challenge for environmental chemists. Environ. Sci. Technol. 40, 7157–7166. doi:10.1021/es061677a
Muir, D., and Miaz, L. T. (2021). Spatial and temporal trends of perfluoroalkyl substances in global ocean and coastal waters. Cite This Environ. Sci. Technol. 55, 9527–9537. doi:10.1021/acs.est.0c08035
Pallen, M. J., Rodriguez-R, L. M., and Alikhan, N.-F. (2022). Naming the unnamed: Over 65,000 Candidatus names for unnamed archaea and bacteria in the genome taxonomy database. Int. J. Syst. Evol. Microbiol. 72, 5482. doi:10.1099/ijsem.0.005482
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi:10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi:10.1101/gr.186072.114
Poretsky, R. S., Sun, S., Mou, X., and Moran, M. A. (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ. Microbiol. 12, 616–627. doi:10.1111/j.1462-2920.2009.02102.x
Rao, N. A., Ambili, M., Jala, V. R., Subramanya, H. S., and Savithri, H. S. (2003). Structure-function relationship in serine hydroxymethyltransferase. Biochim. Biophys. Acta - Proteins Proteomics 1647, 24–29. doi:10.1016/s1570-9639(03)00043-8
Rinke, C., Rubino, F., Messer, L. F., Youssef, N., Parks, D. H., Chuvochina, M., et al. (2019). A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J. 13, 663–675. doi:10.1038/s41396-018-0282-y
Rivers, A. R., Sharma, S., Tringe, S. G., Martin, J., Joye, S. B., and Moran, M. A. (2013). Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill. ISME J. 7, 2315–2329. doi:10.1038/ismej.2013.129
RStudio Team (2022). RStudio: Integrated development environment for R. RStudio, PBC, Boston, MA URL.
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi:10.1093/bioinformatics/btu153
Shai, Y., Rubin-Blum, M., Angel, D. L., Sisma-Ventura, G., Zurel, D., Astrahan, P., et al. (2021). Response of oligotrophic coastal microbial populations in the SE Mediterranean Sea to crude oil pollution; lessons from mesocosm studies. Estuar. Coast. Shelf Sci. 249, 107102. doi:10.1016/j.ecss.2020.107102
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Ocean plankton. Structure and function of the global ocean microbiome. Sci. (80-. ). 348, 1261359. doi:10.1126/science.1261359
Tang, J. Y. M., McCarty, S., Glenn, E., Neale, P. A., Warne, M. S. J., and Escher, B. I. (2013). Mixture effects of organic micropollutants present in water: Towards the development of effect-based water quality trigger values for baseline toxicity. Water Res. 47, 3300–3314. doi:10.1016/j.watres.2013.03.011
Tetu, S. G., Sarker, I., Schrameyer, V., Pickford, R., Elbourne, L. D. H., Moore, L. R., et al. (2020). Plastic leachates impair growth and oxygen production in Prochlorococcus, the ocean’s most abundant photosynthetic bacteria. Commun. Biol. 184. doi:10.1038/s42003-019-0410-x
Torrents, E., Sahlin, M., and Sjöberg, B. (2008). The ribonucleotide reductase family: Genetics and genomics. Undefined.
Uritskiy, G. V., Diruggiero, J., and Taylor, J. (2018). MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 158. doi:10.1186/s40168-018-0541-1
Vila-Costa, M., Cerro-Gálvez, E., Martínez-Varela, A., Casas, G., and Dachs, J. (2020). Anthropogenic dissolved organic carbon and marine microbiomes. ISME J. 14, 2646–2648. doi:10.1038/s41396-020-0712-5
Vila-Costa, M., Sebastián, M., Pizarro, M., Cerro-Gálvez, E., Lundin, D., Gasol, J. M., et al. (2019). Microbial consumption of organophosphate esters in seawater under phosphorus limited conditions. Sci. Rep. 9, 233–311. doi:10.1038/s41598-018-36635-2
Wang, Z., Walker, G. W., Muir, D. C. G., and Nagatani-Yoshida, K. (2020). Toward a global understanding of chemical pollution: A first comprehensive analysis of national and regional chemical inventories. Environ. Sci. Technol. 54, 2575–2584. doi:10.1021/acs.est.9b06379
Wickham, H., Averick, M., Bryan, J., Chang, W., McGowan, L., François, R., et al. (2019). Welcome to the Tidyverse. J. Open Source Softw. 4, 1686. doi:10.21105/joss.01686
Xie, Z., Wang, P., Wang, X., Castro-Jiménez, J., Kallenborn, R., Liao, C., et al. (2022). Organophosphate ester pollution in the oceans. Nat. Rev. Earth Environ. 3, 309–322. doi:10.1038/s43017-022-00277-w
Keywords: organic pollutants, surface seawater, anthropogenic organic matter, alkane biodegradation, polycyclic aromatic hydrocarbon degradation, Poseidonales, Methylophaga
Citation: Vila-Costa M, Lundin D, Fernández-Pinos M-C, Iriarte J, Irigoien X, Piña B and Dachs J (2023) Responses to organic pollutants in the tropical Pacific and subtropical Atlantic Oceans by pelagic marine bacteria. Front. Environ. Sci. 11:1110169. doi: 10.3389/fenvs.2023.1110169
Received: 28 November 2022; Accepted: 14 March 2023;
Published: 30 March 2023.
Edited by:
Claudia Cosio, Université de Reims Champagne-Ardenne, FranceReviewed by:
Eleanor Arrington, University of California, Santa Barbara, United StatesCopyright © 2023 Vila-Costa, Lundin, Fernández-Pinos, Iriarte, Irigoien, Piña and Dachs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Vila-Costa, bWFyaWEudmlsYUBpZGFlYS5jc2ljLmVz
†Present address: Maria-Carmen Fernández-Pinos, Ministry for the Ecological Transition and the Demographic challenge of Spain, Madrid, Spain
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.