 Shuzhen Zou
Shuzhen Zou Tan Lu1†
Tan Lu1† Dayong Li
Dayong Li- 1Key Laboratory of Southwest China Wildlife Resources Conservation of Ministry of Education, China West Normal University, Nanchong, China
- 2Key Laboratory of Conservation Biology of Rhinopithecus Roxellana at China West Normal University of Sichuan Province, China West Normal University, Nanchong, China
There are frequent exchanges of antibiotic-resistant bacteria and their antibiotic resistance genes (ARGs) between the external environment or livestock and wild animals. Grazing disturbance (GD) is a major factor that causes dramatic changes in China’s nature reserves. Studying the risk of ARGs in China’s nature reserves under GD has great significance for assessing the health of the habitats where wild animals live. In our study, the potential ecological risks of ARGs in soil microorganisms of wild animal habitats under GD were analyzed. Our results showed that the diversities of the ARGs in GD were higher than those in check control (CK) that not be disturbed by grazing, and 46 types of ARGs were only checked in GD. The ARGs were only carried by plasmids, and their relative abundances and the numbers of ARGs carried by plasmids were increased by GD, six types of transferred open reading frames (ORFs) carrying ARGs were detected only in GD. GD increased the relative abundances of drug-resistant pathogens, and the pathogen, Acinetobacter baumannii ACICU, that was only found in GD. Our results indicated that GD increased the risk of ARGs to the habitat of wild animals by reducing the total soil microbial species, increasing the numbers, species and mobility of ARGs in soil microorganisms and the species and abundance of drug-resistant pathogens in soil, as well as the ARG carrying capacity of microorganisms. Multidrug resistance genes (MRGs) posed the highest risk in the habitats of wild animals, and GD increased its risk because the largest proportion of ARGs carried by microorganisms were MRGs, which were also the most abundant ARGs carried by plasmids, and the highest proportion of ARGs carried by Proteobacteria (the dominant host bacterium of ARGs) were MRGs. Thus, GD may cause microorganisms in the habitats of wild animals become resistant to many antibiotics. MRGs in soil microorganisms could be used as an indicator for predicting the risk of GD in the habitats of wild animals.
1 Introduction
In China, a great deal of money and resources have been spent to build nature reserves containing wild animals, and they have achieved great success (Xu et al., 2022). However, most of the nature reserve areas are mountainous, their economic development level is limited, the cultural level of the indigenous residents is low, and animal husbandry is the economic source for the people to survive in these nature reserves (Jiang et al., 2018). Human disturbances are the main factors affecting the survival of wild animals in nature reserves because the fundamental contradiction between humans and wild animals in using nature reserves cannot be reconciled. Results showed that the number of livestock in 2015 increased by 43.37 percent compared to 2003 (Wang et al., 2019), and there were 46% and 56% of the roaming areas of horses and cattle are distributed in the habitat of wild animals (Lia et al., 2017). Among the various human disturbances, grazing disturbances (GD) have the highest occurrence rate in the habitats of wild animals in Southwest China (State Forestry Administration, 2021). Previous studies have shown that the feces produced by grazing animals could be used as an index to evaluate the nutrition of soil in the wild animal habitat because grazing could affect the characteristics of the physical, chemical and microbial communities in the soil through excrement discharged into the soil, which affects the microecological environment of the soil in nature reserves (Orellana et al., 2019). However, due to changes in the human living environment, the extensive use of antibiotics in animal husbandry has led to an increase in the abundances of antibiotic resistance genes (ARGs) in microorganisms in livestock manure and has polluted the surrounding environment (Jadeja & Worrich, 2022).
ARGs can be accumulated in the food chain and then affects the composition, structure and function of the intestinal microorganisms in the animals. Because ARGs can be carried by mobile genetic elements (MGEs) of microorganisms and will transfer along with the food chain by plant microbiome, the plant microbiome represents a major pathway by which animals are exposed to microbes and genes consumed with food, and affect the treatment of animal diseases (Cerqueira et al., 2020; Mei et al., 2021). To further demonstrate the transfer of ARGs in the food chain, the Folsomia candida and Hypoaspis aculeifer were used as a model to study the transfer of ARGs in food chain, which showed that ARGs were transferred from the microbial community of soil to Hypoaspis aculeifer. Eventually, the ARGs accumulate in the intestinal microorganisms of Hypoaspis aculeifer and their characteristics of intestinal microbes were changed (Zhu et al., 2019). Livestock, wild animals, humans, and their food have extensive and rapid exchanges of ARGs, and they are interconnected sources of ARGs (Brealey et al., 2021). The nature reserves of wild animals are special ecosystems that are relatively less disturbed by humans than human ecosystems, and the ARGs in the intestinal microorganisms of wild animals could be used as an ecological indicator to indicate the ARG pollution in the habitats of wild animals (Brealey et al., 2021). That is, when the habitats of wild animals are polluted by ARGs from grazing, the risk posed by ARGs to wild animals may increase. Our previous study also showed that the total abundance of ARGs in the intestinal microorganisms of wild animals in GD was significantly higher than that in the control check (CK), which was not disturbed by grazing (Xia et al., 2022). Therefore, it is necessary to detect the characteristics of ARGs in the habitats of wild animals under GD to evaluate the potential ecological risks of ARGs to wild animals. Previous results have already directly addressed the ARGs in intestinal microorganisms of wild animals to reflect the risk of contamination from antibiotics and their ARGs to wild animals under human interference (Huang et al., 2022). Unfortunately, there are few studies that use ARGs from soil microorganisms in wild animal habitats to assess the risks of ARGs to their survival under GD.
The accessibility, mobility, pathogenicity and clinical availability are the factors affecting ARGs’ ecological risk, which indicated that whether the ARGs can be accessed, the ARGs can be moved, or whether the host bacteria with ARGs is pathogenic bacteria and the danger of pathogenic bacteria must be considered (Zhang et al., 2021; Zhang et al., 2022). Thus, it is difficult to assess the risks of ARGs in the habitats of wild animals under GD. Large amounts of microbial information can be reflected by metagenomic analysis (Guo et al., 2017). Recently, researchers have used metagenome sequencing technology not only to obtain the species and abundances of ARGs and calculate the ability of ARGs to be transferred by MGEs but also to analyze pathogenic genes in soil microorganisms (Ju et al., 2019). If a VF and a ARG occurrence in one open reading frame (ORF) of the contigs, the host bacteria of the ORF is defined as potential drug-resistant pathogens, and it represent the highest risk of ARGs (Zhang et al., 2021). The species, abundances and the ability of ARGs together with pathogenic genes could be used to evaluate the ecological risk of ARGs (Liu et al., 2021). Thus, the ecological risks of ARGs could be comprehensively evaluated by metagenome sequencing technology. Additively, microorganisms are the hosts of ARGs, and it is necessary to consider the ability of microorganisms to transfer and store ARGs when their risks in ecosystems containing wild animals are evaluated under GD.
In conclusion, a hypothese be put forward that ARGs in soil microorganisms under GD is one of the potential factors that may threat the survival of wild animals. In this study, we investigated the ARGs of soil in GD and CK using 16S rRNA sequencing technology and metagenomic sequencing, with the aim of 1) analyze the characteristics and horizontal transfer potential of ARGs; 2) study the pathogenicity of their host bacteria in the soil microorganisms of the wild animal habitats under GD and CK; 3) research the ability of soil microorganisms to transfer and store ARGs. Our results could reflect the risk of ARGs in the soil microorganisms of the habitats under GD, which could provide a theoretical basis for supervising this risk in the habitats of wild animals and to formulate emergency measures for health monitoring of wild animals.
2 Materials and methods
2.1 Sample collection
Baihe National Nature Reserve is a habitat for endangered wild animals such as giant pandas (Ailuropoda melanoleuca) and snub-nosed golden monkeys (Rhinopithecus roxellana) in China, but it is not included in China’s Giant Panda National Park, so there are some problems such as lax management and control at present (Liu et al., 2021). The original residents of the nature reserve depended on animal husbandry for their livelihoods, and 19.69% of the total income of the original residents came from animal husbandry, but the feeding and management methods of animal husbandry are extensive (Yuan, 2018). In the nature reserve, the grazing traces even overlap with the home ranges of the Sichuan snub-nosed monkeys. Therefore, our study was conducted in the Baihe National Nature Reserve.
In this study, soil samples were taken from the habitats of Xiapingdi monkey groups with GD and Tongjiashan monkey groups without GD (CK), and the soil samples were selected according to Figure 1. The specific sampling method was as follows: First, survey lines were set up in the area under GD and CK, and the starting and end points of the sample lines referred to the results of Ran et al. (2003). After that, five large quadrats of 20 m × 20 m were set up in each sample line, and three soil samples in each large quadrat were collected randomly; then, the litter layer on the soil surface was removed. After that, a sieve with a pore size of 2 mm was used to remove roots and stones. Lastly, the treated soils were stored at −80°C for DNA extraction.
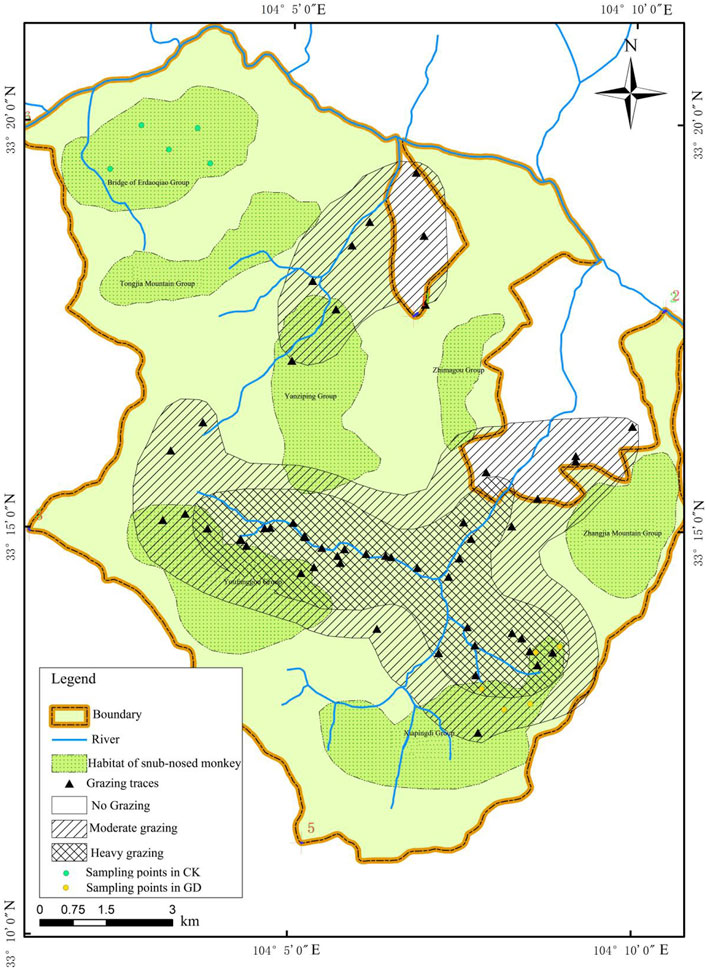
FIGURE 1. Baihe National Nature Reserve and sampling points.
2.2 DNA extraction and sequencing
Frozen soil (2 g) was used to extract DNA using the FastDNA® SPIN Kit for Soil (MP Biomedicals, United States) according to the manufacturer’s protocols. The DNA concentrations and purity were determined with a NanoDrop 2000 instrument (Thermo Fisher Scientific, Wilmington, DE, United States). The three DNA extracts of the three soils from each large quadrat were mixed for the following sequencing step. The V3-V4 hypervariable regions of 16S rRNA genes were amplified by PCR using the barcoded primer set 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACCAGGGTATCTAAT-3′). Purified amplicons of 16S rRNA genes were sequenced using the Illumina MiSeq platform (Majorbio Company in Shanghai) with the PE250 (paired-end sequencing 250 × 2) strategy. The data were deposited into the NCBI Sequence Read Archive (SRA) database under accession number PRJNA896787. To compare the sOTU abundance between different samples, the sequencing depth of each sample was normalized to 41723 reads. The taxonomy analyses of sOTUs were conducted using the feature-classifier plugin in QIIME2 with the Greengenes database (v. 13_8) at 97% shared similarity (Xia et al., 2022).
Shotgun metagenomic sequencing of DNA extracted from soil samples was performed simultaneously (Chen et al., 2021). In brief, approximately 1 μg of DNA was used to construct a library with a 300 bp insert size, followed by sequencing using the Zhu et al., 2021 platform (Majorbio Company in Shanghai) with the PE150 (paired end sequencing 150 × 2) strategy. The quality control method used on the raw data is described in detail in (Chen et al., 2021). There were 384443 reads of 10 soil samples. The read data were deposited into the NCBI Sequence Read Archive (SRA) database under accession number PRJNA906922. Following quality control, the clean reads were assembled using IDBAUD (Chen et al., 2021), and the assembled contigs longer than 500 bp were reserved. We obtained 7635863 contigs with an average length of 828.42 bp each from 10 metagenomic samples. The (ORFs) of the contigs were predicted using Prodigal v2.6.3 with a meta model. The coverage of each contig was calculated by mapping clean reads to the contigs using bbmap (https://sourceforge.net/projects/bbmap/) with the default parameters (Ju et al., 2019).
2.3 Identification of ARG-like ORFs and drug-resistant pathogens
The ORFs were identified against the CARD database (https://card.mcmaster.ca/) using DIAMOND with an E-value ≤10–10 (Buchfink et al., 2015), and the results that identified ≥80% were identified as ARG-like ORFs (Ma et al., 2015).
To compare coverage between different samples, the coverage of ARG-like ORFs was normalized using the data size of each sample (copies/Gb). The calculation formula was as follows:
where n is the total number of ARG-like ORFs in one sample, N is the number of clean reads mapped to ARG-like ORFs, 150 is the length of clean reads, L is the length of the target ARG-like ORFs, and G is the data size (Gb) of clean reads per sample (Ma, et al., 2015; Xiong, et al., 2018).
The ORFs were detected using VFanalyzer on VFDB in setB (http://www.mgc.ac.cn/VFs/) with an E-value ≤10–10 to identify the virulence factors (VFs), and the results that identified ≥80% were identified as ARG-like ORFs. If ARG-like ORFs also had VFs, ORFs were drug resistance genes that could reflect the risk of drug-resistant pathogens (Liu et al., 2018).
2.4 Risk of ARGs in transferring and spreading
2.4.1 Ability of MGEs to transfer ARGs
To estimate the potential horizontal gene transfer of ARGs in soil samples, the ORFs were searched against the MGE database (http://aclame.ulb.ac.be/) using BLASTP with an e-value ≤10–5, and the results with a shared identity ≥80% were identified as MGEs. The ORF annotation results were used to identify the MGEs on plasmids, chromosomes and viruses. To compare the MGE abundance among different samples, the coverage of the MGE-like ORFs was also normalized by the data size of each sample (copies/Gb) according to Eq. 1. If the ARG-like ORFs also contained MGEs, the ARGs could be moved by the MGEs.
2.4.2 Ability of microorganisms to transfer ARGs
Network analysis was used to explore the potential correlations between bacteria and ARGs (Chen et al., 2021). Based on the hypothesis that a significant correlation between sOTUs and ARG subtypes was observed, this observation indicated that the bacterial community structure determined the distribution of the resistome and that the horizontal gene transfer of ARGs was not sufficient to obscure their association with bacterial genomes. The abundance and species of the host bacteria carrying ARGs could also be used to indicate the ability of microorganisms to transfer the ARGs. The higher the abundance and species of host bacteria carrying ARGs was, the stronger the ability of microorganisms to transfer ARGs.
The ORF sequences that carried ARGs were searched against the NCBI RefSeq database using BLASTP with an e-value ≤10–5. The results were parsed using MEGAN (version 5) (Huson et al., 2016), and the ORFs were annotated as the taxon if more than 50% of the ORFs were annotated as the same taxon; the taxons are the host bacteria of ARGs (Ishii et al., 2013).
2.5 Statistical analysis
The diversities and relative abundances of the soil microorganisms and their ARGs as well as the abundances of MGEs were their mean values ± standard error in GD or CK. The differences in the diversities and relative abundances of the soil microorganisms and their ARGs as well as the abundances of MGEs between GD and CK were analyzed by a t-test with a significance level of p < 0.05. The average abundances of OTUs were higher than 0.1% in 10 soil samples, and the relative abundances of all types of ARGs were used to calculate the correlation coefficients (ρ) according to Spearman between soil microorganisms and ARGs using the vegan package in R4.2.1. If ρ ≥ 0.8 and p value ≤0.01, the relationship of OTUs and ARGs was used to perform a network analysis using the igraph package in R4.2.1 and then visualized in Gephi (version 0.9.5).
3 Results and discussion
3.1 Effects of GD on the characteristics of ARGs in soil microorganisms
Microbial diversity has a positive effect on the cycling of nutrients, such as the cycling of carbon and nitrogen (Zhang H et al., 2020). Simpson’s index gives more weight to the more abundant species in a sample. It takes into account the number of species present, as well as the abundance of each species, while the ACE diversities shows more species present in a sample (Schmidt et al., 2019). Supplementary Figure S1 shows that the Simpson’s diversities of soil microorganisms in GD were significantly higher than that of the CK (p = 0.032), while the ACE diversities in GD were significantly lower than that of the CK (p = 0.012), which indicated that GD reduced the total soil microbial species, but increased the dominance of dominant microbial species. GD reduced the total soil microbial species, which could be explained by the fact that GD reduces the diversity of plants and the content of humic substances in wildlife habitats (Zhang T et al., 2020), which results in the reduction of mineralization rates of C and N in soil and the decrease of available nutrients for microbial reproduction by GD (Wu et al., 2022). However, the relative abundances of dominant microorganisms were significantly increased by GD, they were Chloroflexi and Verrucomicrobia (p = 0.001 and p = 0.049), while GD had no significant effect on the abundances of other microorganisms (p > 0.05), thus the soil Simpson’s diversities were increased by GD, even resulted in a rise in the function of dominant microbial species in the soil of wild animal habitats. Chloroflexi and Verrucomicrobia are associated with the carbon and nitrogen contents of the soil. Chloroflexi is a photosynthetic bacterium that can synthesize carbon and nitrogen (Singleton et al., 2020), while the abundance of Verrucomicrobia is also significantly positively correlated with the total carbon and nitrogen contents in soils (Freitas et al., 2012). Thus, our results indicated that the cycling nutrients in the soils of the wildlife habitat were reduced by microorganisms under GD (Supplementary Figure S1C).
A total of 650 ORFs containing ARGs were detected in the soil microorganisms, and 291 types of ARGs were classified into 18 kinds of resistance classes in GD and CK (Figure 2A), which indicated that there was a potential risk from ARGs in the habitats of wild animals. Because ARGs in the surroundings of the habitat can affect the health of wild animal (Xu et al., 2020) and the accessibility of ARGs to animals is one of the factors that affect the risk from ARG (Zhang et al., 2022). Further analysis showed there were no significant difference in the total relative abundances of ARGs between the GD and CK (p > 0.05, Supplementary Figure S2A). However, the Simpson indexes of ARGs were increased significantly by GD (p < 0.05, Supplementary Figure S2B). Simpson’s diversity is a measure of diversity which takes into account both species and the relative abundance of the different species (Schmidt et al., 2019), thus combined with our analysis, we found two reasons for the increased the diversity of ARGs: First, there were 287 and 245 types of ARGs in GD and CK. Among them, 46 types of ARGs and triclosan and bicyclomycin were checked in GD but not in CK (Figures 2A, D), which showed that GD could increase types of ARGs. Second, the relative abundances of MLS and mupirocin in GD were higher than those in CK (p < 0.05, Figure 2B). The diversity of ARGs is higher, the risk from ARGs is greater (Yang et al., 2019). Thus, our result indicated that the risk of the ARGs was increased by increasing types and the abundances of ARGs under GD. ARGs in the surroundings of the habitat can affect the health of wild animal (Xu et al., 2020), as well as humans or other animals could promote the transfer of ARGs, and increases the relative abundance of ARGs (Li, et al., 2018; Lu, et al., 2022). Thus, we concluded that the wild animals may have a risk of ARGs in acessibility of ARGs in the habitat, especially, the ARGs of MLS, mupirocin, triclosan and bicyclomycin that only be checked in GD but not in CK. MLS, mupirocin, triclosan and bicyclomycin, can be used to treat infections caused by a variety of sensitive bacteria (Patteson, et al., 2017). We speculated that if the wild animals who lives in the habitats under GD are infected by pathogenic bacteria, the risk of treatment to wild animals will increase, for the pathogenic bacteria have increased their resistance to the four classes ARGs of MLS, mupirocin, triclosan and bicyclomycin under GD (Figure 2).
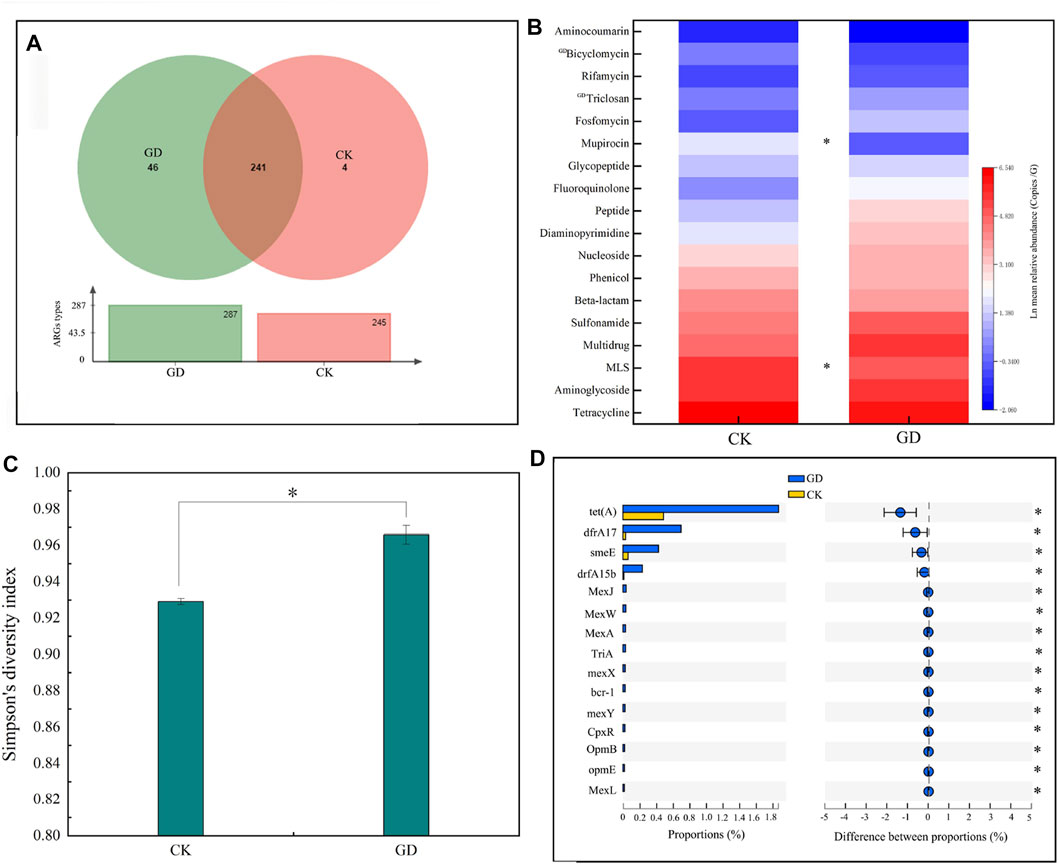
FIGURE 2. Effects of GD on characteristics of ARGs in soil microorganisms. Note: (A–C) represent the types of ARGs, the relative abundance of ARGs classes, and Simpson indexes of ARGs in GD and CK, respectively. (D) shows the relative abundances of ARGs with significant differences between GD and CK. * means that the Simpson indexes of ARGs or the relative abundance of the ARGs are significant difference between CK and GD (p < 0.05), “GD” means the ARGs only be checked in GD.
3.2 Effects of GD on the horizontal transfer potential of ARGs in soil microorganisms
MGEs play an important role in the horizontal transfer of ARGs, the higher the abundance of MGEs is in microorganisms, the stronger the ability of ARGs to be carried by MGEs, and the greater the risk of horizontal transfer of ARGs (Lu et al., 2022). Our results showed that there were 12160 MGE-like ORFs in both CK and GD, the relative abundances of MGEs in GD were higher than those in CK (Figure 3B; p = 0.000162), which indicated that GD could increase the horizontal transfer ability of MGEs to ARGs. Gut microbiome could acquire exogenous ARGs via horizontal gene transfer (Mei et al., 2021). Table 1 also shows that there are ARG-like ORFs carried by plasmids only be checked in GD, namely, tetM, MexD, ceoB, MuxC, sul1 and catII, which indicated that if the carried ARGs are transferred to gut microbiome of wild animals under GD, the wild animals they will develop resistance to some antibiotics.
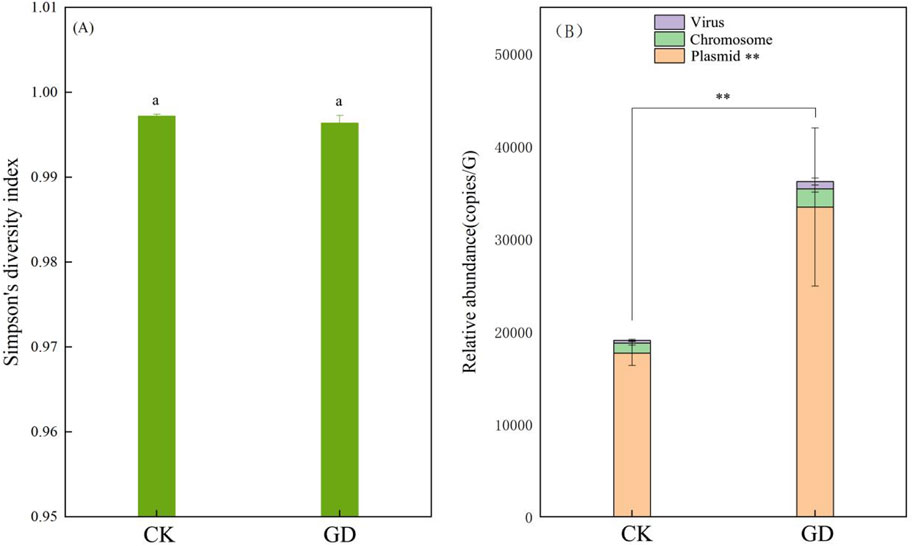
FIGURE 3. Effects of GD on the characteristics of MGEs in soil microorganisms. Note: (A, B) show the Simpson’s indexes and relative abundances of MGEs of soil microorganisms, respectively. The same lowercase indicates there are no significant difference between CK and GD (p ≥ 0.05).** means that the relative abundance of the MGEs are significant difference between CK and GD (p < 0.01).
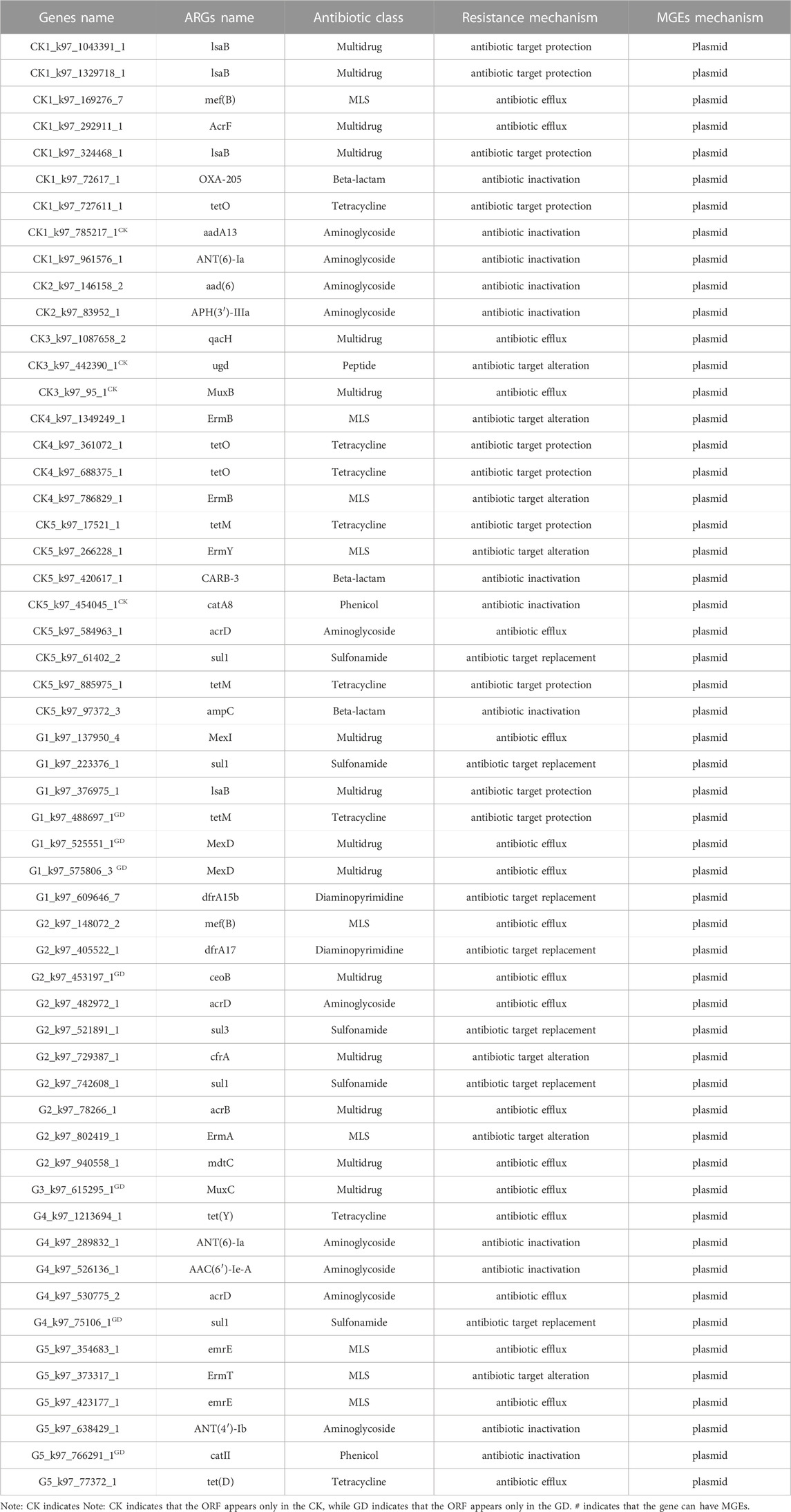
TABLE 1. The characteristics of ORFs that carrying ARGs and MGEs in CK and GD.
The relative abundances of MGEs carried by plasmid, prophage and virus were as follows: plasmid>prophage>virus, and the relative abundances of plasmids in the GD were significantly higher than those in the CK (Figure 3B; p = 0.000160), while there were no significant differences in the relative abundances of MGEs carried by prophages and virus between the CK and GD (Figure 3B; p > 0.05). If both ARGs and MGEs were present in one ORF, the ARGs could be moved by MGEs (Ju et al., 2019). Table 1 shows that 55 ARG-like ORFs could be carried by plasmids, and the total number of ORFs-like plasmids in GD was significantly higher than that in CK (Supplementary Figure S3; p = 0.034). Combined with Figure 3B; Table 1, we found that plasmids had the strongest ability to carry ARGs and GD increased their ability to carry ARGs in wildlife habitats. In nature, ARGs carried by plasmids are the main method by which ARGs are transferred horizontally, studies have shown that the ARGs in the microorganisms of water, activated sludge and soil are mainly carried by plasmids (Che et al., 2019; Fan et al., 2019; Li et al., 2020; Dai et., 2022), which supports our results.
3.3 Effects of GD on drug-resistant pathogens
If both an ARG and a virulence factor (VF) are present in one ORF, the bacteria in the ORF are called drug-resistant pathogens that reflected the highest level of risk of ARGs (Zhang et al., 2021). There were 26 ARG-like ORFs that were detected in three types of pathogenic bacteria: Klebsiella pneumoniae subsp. pneumoniae NTUH-K2044, Acinetobacter baumannii ACICU and Neisseria meningitidis MC58. The total number of the 26 types of ORFs in GD was significantly higher than that in CK (p = 0.032, Supplementary Figure S4). Among them, six types of ORFs were detected only in GD but not in CK (Supplementary Table S1), they were found to be two types of pathogenic bacteria: Klebsiella pneumoniae NTUH-K2044 and Acinetobacter baumannii ACICU. Thus, not only the total abundance but also the types of drug-resistant pathogens were increased by GD. Previous studies have shown that the wide application of animal manure could introduce a large number of ARGs and drug-resistant pathogen into the soil surroundings (Duan et al., 2017; Liu et al., 2017; Zhang Y et al., 2020), because antibiotic stress in animal manure under GD can not only increase the abundance and species of ARGs but also increase the abundance of VFs due to the adaptive evolution of pathogenic bacteria (Durrant et al., 2020). So, we speculate that once drug-resistant pathogens are transferred to wild animals, health of the wild animals are endangered. For the acquisition of antibiotic resistance in clinically relevant pathogens diminishes the effectiveness of antibiotics in treating bacterial infections (Zhu et al., 2017).
3.4 Effects of GD on the host bacteria of ARGs
Although these findings have demonstrated that the risk of ARGs was increased by GD in the habitats of wildlife, as the host, the microbial community determines the pattern and environmental behavior of ARGs, and the transfer of ARGs is also related to microorganisms (Bing et al., 2015). The co-occurrence network correlation between ARGs and bacterial taxa is regarded as an effective way to track potential ARG hosts in various environments (Hu et al., 2017). To investigate the association between ARGs and the bacterial community, a correlation analysis was performed between the relative abundance of ARGs and bacteria. There were 271 points and 520 edges in the CK, of which 56 edges were significantly negative and 462 edges were significantly positive (|r|>0.8, p < 0.01), while there were 305 points and 1,150 edges in the GD, of which 60 edges were significantly negative and 1,090 edges were significantly positive (|r|>0.8, p < 0.01; Figures 4A, B). If there is a significant correlation between the abundance of ARGs and the abundance of microorganisms, the microorganisms can be considered potential hosts of ARGs (Zhu et al., 2018), and the network relationship between ARGs and microorganisms can explain the transfer potential of microorganisms to ARGs in the environment to some extent (Lu et al., 2022). Thus, our results indicated that the bacterial community structure determined the distribution of the resistome and that the horizontal transfer of ARGs was not sufficient to obscure their association with bacterial genomes. The species and numbers of the host bacteria of ARGs were increased by GD, which indicated that the carrying capacity of ARGs in soil microorganisms to move was increased by GD.
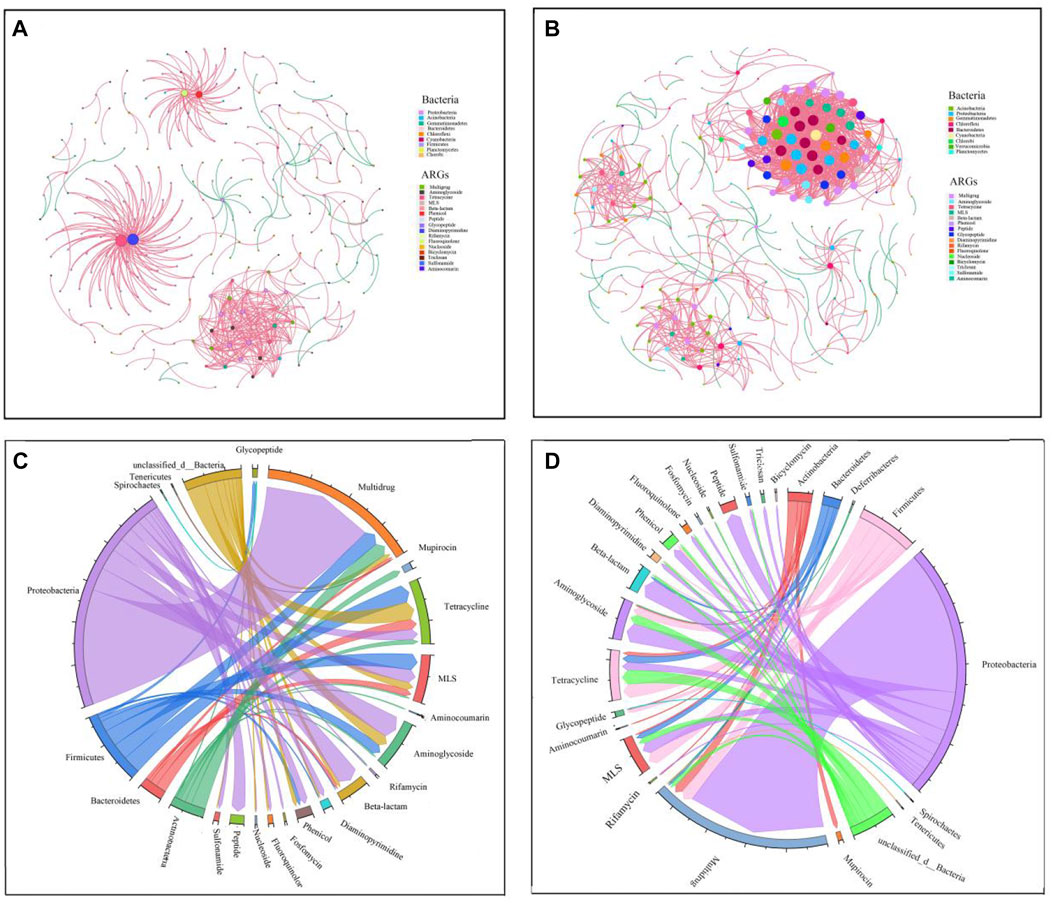
FIGURE 4. Host bacteria of ARGs and relationships between ARGs and soil microorganisms. Note: (A, B) were the network of spearman correlation relationships between ARGs calasses and OTUs in soil microorganisms in CK and GD, while (C, D) were the host bacteria of ARGs in CK and GD, respectively. The red lines indicated that there were significant positive correlations between the relative abundances of ARGs and the relative abundances of dominant OTUs (p < 0.01; r > 0.8). The green line indicated that the relative abundances of ARGs were significantly negatively correlated with the relative abundances of dominant OTUs (p < 0.01; r < −0.8).
Although the network relationship results indicated a significant correlation between ARGs and bacteria in the habitats of wild animals, they could not reflect the ability of microorganisms to carry ARGs. If a microorganism and ARG are on one same ORF, the microorganism can carry the ARGs to transfer (Zeng et al., 2019). Figure 4 shows that there were 232 types of ARG-like ORFs that were detected in seven types of bacteria, namely, Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, Spirochaetes, Tenericutes and unclassified bacteria, they carried 5.70%, 17.34%, 8.08%, 54.63%, 23.75%, 23.75% and 13.78% of the ARG-like ORFs in the CK, respectively. While there were 242 types of ARG-like ORFs that were detected in eight types of bacteria, namely, Actinobacteria, Bacteroidetes, Deferribacteres, Firmicutes, Proteobacteria, Spirochaetes, Tenericutes and unclassified bacteria, they carried 6.02%, 4.47%, 1.94%, 14.56%, 63.69%, 1.94%, 1.94% and 10.87% of the ARG-like ORFs in the GD, respectively. Among the ARG-like ORFs, 18 types and 16 antibiotic classes could be carried by bacteria in GD and CK, respectively, and triclosan and bicyclomycin (79 types of ARG-like ORFs were not detected in the CK) were not carried in CK. Results have showed that the bacterial hosts of ARGs were preferably distributed in Proteobacteria (Zeng et al., 2019), which was consistent with our research results that Proteobacteria was the main host of ARGs in the habitats of wild animals. Proteobacteria is one of the largest groups of bacteria in nature, and it includes many pathogenic bacteria (Rizzatti et al., 2017). In our results, the highest proportion (98.73%) of the host of the new ARGs in GD was Proteobacteria, and GD increased the ability of Proteobacteria to carry ARGs (Figures 4A, B). We concluded that the ecological risk of ARGs carried by Proteobacteria in the habitats of wild animals is the largest, which was increased by GD, as supported by the results of Supplementary Table S2 because Supplementary Table S2 shows that all three types of drug-resistant pathogens were Proteobacteria, including the two types of new drug-resistant pathogens in GD.
3.5 Effects of GD on the risk of multi-resistance genes (MRGs)
Figure 5 showed that the Simpson’s diversity of the MRGs was higher than that of the other ARG classes, and GD increased the Simpson’s diversity of the MRGs, as well as the MRGs accounted for the largest proportion in the ARG-like ORFs that were only checked in the GD (Figures 5A, B), which indicated that the risk of MRGs was the greatest, and GD increased this risk. MRGs was the most abundant and types and human disturbance increases the risk of MRGs in soil samples (Mei et al., 2021), which supported our result. Our result showed that the MRGs-like ORFs were the largest proportion that could be transferred by plasmids and they were increased by GD (Figure 5C; p = 0.007), the largest proportions of ARGs that the host bacteria carried were MRGs, GD increased this proportions (Figures 4C, D), as well as the MRGs had the maximum distribution degree in the network, GD increased their degree distribution, (Supplementary Table S2). Our results indicated that GD increased mobility of MRG and the storage capacity of MRGs in multidrug-resistant bacteria, which could be explained by this reason that multidrug-resistant bacteria are mainly generated by transfer of MRGs in MGEs and the accumulating MRGs in microorganisms (Kim et al., 2021). The risk of MRGs favors the development of multidrug-resistant bacteria through horizontal gene transfer (Kang et al., 2022), thus we concluded that GD increased the risk of MRGs by increasing the mobility of MRGs and the storage capacity of microorganisms to MRGs.

FIGURE 5. Effects of GD on the MRGs. Note: (A) is ORFs numbers of different calssses ARGs in GD, (B) are Simpson indexes of different calssses ARGs in CK and GD. (C) was MRG-like ORFs carried by plasmids, and (D) was MRG-like ORFs in pathogenic bacteria. The same lowercase indicates there are no significant difference between CK and GD (p ≥ 0.05), while the same lowercase indicates there are significant difference between CK and GD (p < 0.05). “**” and “**” means there are significant difference between CK group and GD on 0.01 level and 0.05 level.
MRGs present in microorganisms, which could reflect the risk level of ARGs in a region (Zeng et al., 2019). Today, infection caused by multidrug-resistant bacteria is a major threat to ecosystem health (Fiona and Brion, 2013). The bad thing is that, GD increased the abundance of multidrug-resistant pathogens (Figure 5D; p = 0.033), and the multidrug-resistant pathogens-like ORFs of Klebsiella pneumoniae NTUH-K2044 only check in GD. As early as 2008, result had shown that Acinetobacter baumannii ACICU could cause a variety of bacterial infectious diseases, and it had resistance to some (Iacono et al., 2008). The propensity of this bacterium to rapidly acquire antibiotic resistance could lead to the emergence and spread of Acinetobacter baumannii strains with MRGs, and could develop resistance to latest antibiotics (Law and Tan, 2022), which supported our findings that the ARGs in Acinetobacter baumannii ACICU are MRGs, it could develop resistance to a variety of antibiotics (Supplementary Table S1). More seriously, there was one MGE in the Acinetobacter baumannii ACICU under GD, it was pGMI1000MP, which indicated that the Acinetobacter baumannii ACICU could be moved by plasmid. Acinetobacter baumannii is a pathogen responsible for several serious infections, including pneu-monia, sepsis, and meningitis (Law and Tan, 2022). Thus, we concluded that GD increased the risk of MRGs, multidrug-resistant pathogens may appear in the habitats of wild animals after GD, there is also a risk of horizontal transfer of drug-resistant pathogens to wild animals, which has a profound impact on survival of wild animals.
4 Conclusion
The occurrence, characteristics and transfer risk of soil microorganism ARGs were studied under GD, which demonstrated that ARGs could be essential indicators for the risk of GD in the habitats of wild animals since GD had great influences on the characteristics, mobility, pathogenicity and microbial storage capacity of ARGs in the habitats of wild animals. GD increased the diversity of ARGs by increasing new types of ARGs, GD increased the carrying capacity of MGEs to ARGs by increasing the species and abundance of the MGEs, and the ARGs in the habitats of wild animals could only be transferred horizontally through plasmids. GD increased the risk of host bacteria pathogens by increasing the variety and abundance of drug-resistant pathogens, and Acinetobacter baumannii ACICU was the new drug-resistant pathogen after GD. GD increased the storage capacity of microorganisms, especially the dominant microorganism storage of ARGs, rather than increasing the microbial diversity in the soil. All three types of drug-resistant pathogens, Klebsiella pneumoniae subsp. pneumoniae NTUH-K2044, Acinetobacter baumannii ACICU and Neisseria meningitidis MC58, were in the Proteobacteria, and the ecological risk of ARGs carried by Proteobacteria in the habitats of wild animals was the largest, which was increased by GD. The risk of MRGs was the highest, and GD increased this risk. Moreover, multidrug-resistant pathogens may have a risk of transfer to wild animals. We worry that ARGs may spread widely in wildlife or be transferred to the gut microbiome of wildlife, which will use different resistance mechanisms to develop resistance to different antibiotics and increase the risk of disease treatment in wildlife under GD. We suggest that to protect the health of wild animals and their habitats, it is necessary to control the scale of grazing and monitor drug-resistant pathogens from GD.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceptualization, DL; Formal analysis, SZ; Funding acquisition, SZ; Investigation, TL and CH; Supervision, DL; Writing–original draft, SZ and TL; Writing–review and editing, DL. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (32200403 and 32270548), and National Natural Science Foundation of Sichuan Province (2023NSFS520).
Acknowledgments
We appreciate the workers for their assistance during sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2023.1109298/full#supplementary-material
Abbreviations
ARGs, Antibiotics resistance genes; GD, Grazing disturbance; CK, Check control; OFR, Open reading frame; MGEs, Mobile genetic elements; MRGs, Multidrug-resistant genes; VFs, Virulence factors.
References
Bing, C., Lin, F., and Zhang, T. (2015). Profile and fate of bacterial pathogens in sewage treatment plants revealed by high-throughput metagenomic approach. Environ. Sci. Technol. 49 (17), 10492–10502. doi:10.1021/acs.est.5b02345
Brealey, J. C., Leitao, H. G., Hofstede, T., Kalthoff, D. C., and Guschanski, K. (2021). The oral microbiota of wild bears in Sweden reflects the history of antibiotic use by humans. Curr. Biol. 31 (20), 4650–4658.e6. doi:10.1016/j.cub.2021.08.010
Buchfink, B., Chao, H., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. methods 12, 59–60. doi:10.1038/nmeth.3176
Cerqueira, F., Christou, A., Despo, F. K., Maria, V. C., Maria, B., Josep, M. B., et al. (2020). Effects of prescription antibiotics on soil- and root-associated microbiomes and resistomes in an agricultural context. J. Hazard. Mater. 400, 123208. doi:10.1016/j.jhazmat.2020.123208
Che, Y., Xia, Y., Liu, L., Li,Yang, A. D. Y., and Zhang, T. (2019). Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 7 (1), 44–13. doi:10.1186/s40168-019-0663-0
Chen, J., Yang, Y., Jiang, X., Ke, Y., He, T., and Xie, S. (2021). Metagenomic insights into the profile of antibiotic resistomes in sediments of aquaculture wastewater treatment system. Acta Sci. Circumstantiae 113 (3), 345–355. doi:10.1016/j.jes.2021.06.026
Dai, D. J., Brown, C., Burgmann, H., Larsson, D. G., Nambi, I., Zhang, T., et al. (2022). Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 10 (1), 20–16. doi:10.1186/s40168-021-01216-5
Duan, M., Li, H., Gu, J., Tuo, X., Sun, W., Qian, X., et al. (2017). Effects of biochar on reducing the abundance of oxytetracycline, antibiotic resistance genes, and human pathogenic bacteria in soil and lettuce. Environ. Pollut. 224 (MAY), 787–795. doi:10.1016/j.envpol.2017.01.021
Durrant, M. G., Li, M. M., Siranosian, B. A., Montgomery, S. B., and Bhatt, A. S. (2020). A bioinformatic analysis of integrative mobile genetic elements highlights their role in bacterial adaptation. Cell host microbe 27 (1), 140–153.e9. doi:10.1016/j.chom.2019.10.022
Fan, X. T., Li, H., Chen, Q. L., Zhang, Y. S., Ye, J., Zhu, Y. G., et al. (2019). Fate of antibiotic resistant Pseudomonas putida and broad host range plasmid in natural soil microcosms. Front. Microbiol. 10 (1), 194. doi:10.3389/fmicb.2019.00194
Fiona, W., and Brion, D. (2013). The culturable soil antibiotic resistome: A community of multi-drug resistant bacteria. PLoS ONE 8, e65567. doi:10.1371/journal.pone.00655678(6)
Freitas, S., Hatosy, S., Fuhrman, J. A., Huse, S. M., Welch, D., Sogin, M. L., et al. (2012). Global distribution and diversity of marine Verrucomicrobia. ISME J. 6 (8), 1499–1505. doi:10.1038/ismej.2012.3
Guo, J. H., Li, J., Chen, H., Bond, P. L., and Yuan, Z. G. (2017). Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 123, 468–478. doi:10.1016/j.watres.2017.07.002
Hu, H. W., Wang, J. T., Li, J., Shi, X. Z., Ma, Y. B., Chen, D., et al. (2017). Long-term nickel contamination increases the occurrence of antibiotic resistance genes in agricultural soils. Environ. Sci. Technol. 51 (2), 790–800. doi:10.1021/acs.est.6b03383
Huang, G., Qu, Q., Wang, M., Huang, M., Zhou, W., and Wei, F. (2022). Global landscape of gut microbiome diversity and antibiotic resistomes across vertebrates. Sci. Total Environ. 838, 156178. doi:10.1016/j.scitotenv.2022.156178
Huson, D. H., Beier, S., Flade, I., Gorska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12 (6), e1004957. doi:10.1371/journal.pcbi.1004957
Iacono, M., Villa, L., Fortini, D., Bordoni, R., Imperi, F., Bonnal, R. J. P., et al. (2008). Whole-genome pyrosequencing of an epidemic multidrug-resistant Acinetobacter baumannii strain belonging to the European clone II group. Antimicrob. Agents Chemother. 52 (7), 2616–2625. doi:10.1128/aac.01643-07
Ishii, S., Suzuki, S., Norden-Krichmar, T. M., Tenney, A., Chain, P. S., Scholz, M. B., et al. (2013). A novel metatranscriptomic approach to identify gene expression dynamics during extracellular electron transfer. Nat. Commun. 4 (3), 1601. doi:10.1038/ncomms2615
Jadeja, N. B., and Worrich, A. (2022). From gut to mud: Dissemination of antimicrobial resistance between animal and agricultural niches. Environ. Microbiol. 24 (8), 3290–3306. doi:10.1111/1462-2920.15927
Jiang, G. S., Wang, G. M., Marcel, H., Yu, Q., Jia, X. B., Guan, Y., et al. (2018). Land sharing and land sparing reveal social and ecological synergy in big cat conservation. Biol. Conserv. 211, 142–149. doi:10.1016/j.biocon.2017.05.018
Ju, F., Beck, K., Yin, X., Maccagnan, A., McArdell, C. S., Singer, H. P., et al. (2019). Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 13 (2), 346–360. doi:10.1038/s41396-018-0277-8
Kang, M., Yang, J., Kim, S., Park, J., Kim, M., and Park, W. (2022). Occurrence of antibiotic resistance genes and multidrug-resistant bacteria during wastewater treatment processes. Sci. Total Environ. 811 (10), 152331. doi:10.1016/j.scitotenv.2021.152331
Kim, M., Park, J., Kang, M., Yang, J., and Park, W. (2021). Gain and loss of antibiotic resistant genes in multidrug resistant bacteria: One health perspective. J. Microbiol. 59 (6), 535–545. doi:10.1007/s12275-021-1085-9
Law, S., and Tan, H. S. (2022). The role of quorum sensing, biofilm formation, and iron acquisition as key virulence mechanisms in acinetobacter baumannii and the corresponding anti-virulence strategies. Microbiol. Res. 260, 127032. doi:10.1016/j.micres.2022.127032
Li, B., Chen, Z., Zhang, F., Liu, Y., and Yan, T. (2020). Abundance, diversity and mobility potential of antibiotic resistance genes in pristine Tibetan plateau soil as revealed by soil metagenomics. FEMS Microbiol. Ecol. 96 (10), fiaa172–10. doi:10.1093/femsec/fiaa172
Li, L. G., Yin, X. L. Q., and Zhang, T. (2018). Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome 6 (1), 93–12. doi:10.1186/s40168-018-0480-x
Lia, B. V., Pimmb, S. L., Li, S., Zhao, L. J., and Luo, C. P. (2017). Free-ranging livestock threaten the long-term survival of giant pandas. Biol. Conserv. 216, 18–25. doi:10.1016/j.biocon.2017.09.019
Liu, P., Jia, S., He, X., Zhang, X., and Ye, L. (2017). Different impacts of manure and chemical fertilizers on bacterial community structure and antibiotic resistance genes in arable soils. Chemosphere 188, 455–464. doi:10.1016/j.chemosphere.2017.08.162
Liu, S., Wang, P.-F., Wang, C., Wang, X., and Chen, J. (2021). Anthropogenic disturbances on antibiotic resistome along the Yarlung Tsangpo River on the Tibetan Plateau: Ecological dissemination mechanisms of antibiotic resistance genes to bacterial pathogens. Water Res. 202, 117447. doi:10.1016/j.watres.2021.117447
Liu, T., Zhang, A. N., Wang, J. W., Liu, S. F., Jiang, X. T., Dang, C. Y., et al. (2018). Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 6 (1), 16–14. doi:10.1186/s40168-017-0388-x
Lu, L., He, Y., Peng, C., Wen, X., Ye, Y., Ren, D., et al. (2022). Dispersal of antibiotic resistance genes in an agricultural influenced multi-branch river network. Sci. Total Environ. 830, 154739. doi:10.1016/j.scitotenv.2022.154739
Ma, L. P., Xia, Y., Li, B., Yang, Y., Li, L. G., Tiedje, J. M., et al. (2015). Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ. Sci. Technol. 50, 420–427. doi:10.1021/acs.est.5b03522
Mei, Z., Xiang, L. L., Wang, F., Xu, M., Fu, Y. H., and Wang, Z. Q., (2021). Bioaccumulation of Manure-borne antibiotic resistance genes in carrot and its exposure assessment. Environ. Int. 157, 106830. doi:10.1016/j.envint.2021.106830
Orellana, C., Parraguez, V. H., Arana, W., Escanilla, J., Zavaleta, C., and Castellaro, G. (2019). Use of fecal indices as a non-invasive tool for nutritional evaluation in extensive-grazing sheep. Anim. (Basel) 10 (1), 46. doi:10.3390/ani10010046
Patteson, J., Cai, W., Johnson, R. A., Maria, K. S., and Li, B. (2017). Identification of the biosynthetic pathway for the antibiotic bicyclomycin. Biochemistry 57 (1), 61–65. doi:10.1021/acs.biochem.7b00943
Ran, J. H., Liu, S. Y., Wang, H. J., Sun, Z. Y., Zeng, Z. Y., and Liu, S. C. (2003). Effect of grazing on giant pandas'habitat in yele nature reserve. Acta Theriol. Sin. 23 (4), 289–294. doi:10.16829/j.slxb.2003.04.003
Rizzatti, G., Lopetuso, L. R., Gibiino, G., Binda, C., and Gasbarrini, A. (2017). Proteobacteria: A common factor in human diseases. Biomed Res. Int. 2017, 1–7. doi:10.1155/2017/9351507
Schmidt, E., Mykytczuk, N., and Schulte-Hostedde, A. I. (2019). Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 13 (5), 1293–1305. doi:10.1038/s41396-019-0345-8
Singleton, D. R., Furlong, M. A., Peacock, A. D., White, D. C., Coleman, D. C., and Whitman, W. B. (2020). Solirubrobacter pauli gen. nov., sp. nov., a mesophilic bacterium within the Rubrobacteridae related to common soil clones. Int. J. Syst. Evol. Microbiol. 53, 485–490. doi:10.1099/ijs.0.02438-0
State Forestry Administration, China (2021). Report of the fourth national survey of giant pandas. Sci. Press.
Wang, X., Huang, J. Y., Connor, T. A., Bai, W. K., Zhang, J. D., Wei, W., et al. (2019). Impact of livestock grazing on biodiversity and giant panda habitat. J. Wildl. Manag. 83 (7), 1592–1597. doi:10.1002/jwmg.21743
Wu, Y., Chen, D., Delgado-Baquerizo, M., Liu, S., Wang, B., Wu, J., et al. (2022). Long-term regional evidence of the effects of livestock grazing on soil microbial community structure and functions in surface and deep soil layers. Soil Biol. Biochem. 168, 108629. doi:10.1016/j.soilbio.2022.108629
Xia, W. C., Liu, G. Q., Wang, D. L., Chen, H., Zhu, L. F., and Li, D. Y. (2022). Functional convergence of Yunnan snub-nosed monkey and bamboo-eating panda gut microbiomes revealing the driving by dietary flexibility on mammal gut microbiome. Comput. Struct. Biotechnol. J. 20, 685–699. doi:10.1016/j.csbj.2022.01.011
Xiong, W. G., Wang, Y. L., Sun, Y. X., Ma, L. P., Zeng, Q. L., Jiang, X. T., et al. (2018). Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 6 (1), 34–11. doi:10.1186/s40168-018-0419-2
Xu, C. Y., Lv, Z. Q., Shen, Y. B., Liu, D. J., Fu, Y. L., Zhou, L., et al. (2020). Metagenomic insights into differences in environmental resistome profiles between integrated and monoculture aquaculture farms in China. Environ. Int. 144, 106005. doi:10.1016/j.envint.2020.106005
Xu, Y., Yang, B., Dai, Q., Pan, H., Zhong, X., Ran, J., et al. (2022). Landscape-scalegiant panda conservation based onmetapopulations within China’snational park system. Sci. Adv. 8, eabl8637. doi:10.1126/sciadv.abl8637
Yang, Y. Y., Liu, G. H., Ye, C., and Liu, W. Z. (2019). Bacterial community and climate change implication affected the diversity and abundance of antibiotic resistance genes in wetlands on the Qinghai-Tibetan Plateau. J. Hazard. Mater. 361, 283–293. doi:10.1016/j.jhazmat.2018.09.002
Yuan, S. (2018). Comprehensive scientific investigation report of sichuan Baihe nature reserve. Sci. Press.
Zeng, J., Pan, Y., Yang,Hou, J. M., Zeng, Z., and Xiong, W. (2019). Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ. Int. 126, 346–354. doi:10.1016/j.envint.2019.02.052
Zhang, A. N., Gaston, J. M., Dai, C. L., Zhao, S., Poyet, M., Groussin, M., et al. (2021). An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commucications 12, 4765. doi:10.1038/s41467-021-25096-3
Zhang, H., Zhang, Q., Song, J., Zhang, Z., Fang, H., Long, Z., et al. (2020). Tracking resistomes, virulence genes, and bacterial pathogens in long-term manure-amended greenhouse soils. J. Hazard. Mater. 396, 122618–122712. doi:10.1016/j.jhazmat.2020.122618
Zhang, T., Li, F. Y., Shi, C., Li, Y., and Baoyin, T. (2020). Enhancement of nutrient resorption efficiency increases plant production and helps maintain soil nutrients under summer grazing in a semi-arid steppe. Agric. Ecosyst. Environ. 292, 106840. doi:10.1016/j.agee.2020.106840
Zhang, Y., Wang, J., Lu, J., and Wu, J. (2020). Antibiotic resistance genes might serve as new indicators for wastewater contamination of coastal waters: Spatial distribution and source apportionment of antibiotic resistance genes in a coastal bay. Ecol. Indic. 114, 106299. doi:10.1016/j.ecolind.2020.106299
Zhang, Z. Y., Zhang, Q., Wang, T. Z., Xu, N. H., Lu, T., Hong, W. J., et al. (2022). Assessment of global health risk of antibiotic resistance genes. Nat. Commun. 13, 1553. doi:10.1038/s41467-022-29283-8
Zhu, B., Chen, Q. L., Chen, S. C., and Zhu, Y. G. (2017). Does organically produced lettuce harbor higher abundance of antibiotic resistance genes than conventionally produced? Environ. Int. 98, 152–159. doi:10.1016/j.envint.2016.11.001
Zhu, D., Chen, Q. L., Ding, J., Wang, Y. F., Cui, H. L., and Zhu, Y. G. (2019). Antibiotic resistance genes in the soil ecosystem and planetary health: Progress and prospect. Sci. Sin. 49 (12), 1652–1663. doi:10.1360/ssv-2019-0267
Zhu, D., Chen, Q. L., Li, H., Yang, X. R., Christie, P., Ke, X., et al. (2018). Land use influences antibiotic resistance in the microbiome of soil collembolans orchesellides sinensis. Environ. Sci. Technol. 52, 14088–14098. doi:10.1021/acs.est.8b05116
Keywords: grazing disturbance, nature reserves, wild animals, antibiotics resistance gene, pathogenic bacteria
Citation: Zou S, Lu T, Huang C, Wang J and Li D (2023) Grazing disturbance increased the mobility, pathogenicity and host microbial species of antibiotic resistance genes, and multidrug resistance genes posed the highest risk in the habitats of wild animals. Front. Environ. Sci. 11:1109298. doi: 10.3389/fenvs.2023.1109298
Received: 27 November 2022; Accepted: 20 February 2023;
Published: 06 March 2023.
Edited by:
Lei Deng, Northwest A&F University, ChinaReviewed by:
Liguan Li, The University of Hong Kong, Hong Kong SAR, ChinaWei Li, Northwest A&F University, China
Copyright © 2023 Zou, Lu, Huang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dayong Li, OTgwMTE5bHNjQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship