 Dyllon G. Randall
Dyllon G. Randall Antoine Brison2,3
Antoine Brison2,3 Kai M. Udert
Kai M. Udert- 1Department of Civil Engineering, University of Cape Town, Cape Town, South Africa
- 2Eawag, Swiss Federal Institute of Aquatic Science and Technology, Dübendorf, Switzerland
- 3ETH Zürich, Institute of Environmental Engineering, Zürich, Switzerland
Human urine is rich in valuable nitrogen which can easily be lost due to biological urea hydrolysis and subsequent ammonia volatilization. While this enzymatic reaction can be prevented by alkalizing the urine, recent studies suggest that chemical urea hydrolysis can result in substantial nitrogen losses when drying alkalinized urine at high temperatures. Furthermore, it was previously suggested that CO2 dissolution from the air used to evaporate water from alkalinized urine could result in a pH decrease to values which allows for biological urea hydrolysis and subsequent ammonia losses. This study aimed to determine the kinetics of chemical urea hydrolysis in alkalinized human urine and confirm the effect of CO2 dissolution with controlled laboratory experiments. We measured the change in urea concentration at different temperatures and pH values for real human urine and determined the corresponding rate constants for chemical urea hydrolysis. We showed that the rate constant increases as a function of temperature and that pH has a negligible effect on the rate of chemical urea hydrolysis in the high pH range of alkalized urine (>11). The rate constants for chemical urea hydrolysis in a saturated calcium hydroxide solution were found to be 0.00147 d−1, 0.00595 d−1, 0.0204 d−1 and 0.0848 d−1 for temperatures of 25°C, 40°C, 55°C and 70°C, respectively. The effect of CO2 dissolution on urea hydrolysis was determined by aerating human urine alkalinized with calcium hydroxide (Ca(OH)2). In order to represent biological urea hydrolysis, urease was added to the solution. The computer simulations of the experimental results showed that CO2 dissolution and the subsequent dissociation of carbonic acid to carbonate ions, bicarbonate ions and protons is the main cause of the pH decrease, but CaCO3 precipitation, and NH3 volatilization foster the pH decrease. However, biological urea hydrolysis prevents the pH from decreasing below 9. Residual undissolved Ca(OH)2 was shown to substantially delay the pH decrease. Overall, this work provides valuable insights into the mechanisms of urea hydrolysis in alkalinized urine during dehydration, which can be used to design more efficient decentralized sanitation systems and minimize nitrogen losses.
1 Introduction
The diversion and subsequent separate treatment of human excreta offers many benefits in that valuable nutrients can be recovered (Randall and Naidoo, 2018) while limiting the load on existing wastewater treatment facilities (Larsen and Gujer 1996). For example, (Wilsenach and van Loosdrecht, 2003) showed that biological nutrient removal processes would benefit from the implementation of urine separation and treatment while (Ishii and Boyer, 2015) showed source-separation of urine, coupled with the subsequent recovery of struvite, offers the smallest environmental burden when compared to a centralized wastewater treatment facility with no nutrient recovery. In addition, on-site sanitation is an alternative to centralized wastewater treatment, where a sewer network cannot be established or is very costly. However, the provision of on-site sanitation systems call for more efficient treatment processes because of the different properties of urine and feces (Udert et al., 2015).
Fresh urine is rich in nutrients, containing over 50% of the phosphorus and over 75% of the nitrogen found in domestic wastewater (Larsen and Gujer, 1996). Approximately 90% of the nitrogen in urine is present as urea, which is readily converted into ammonia and bicarbonate ions in the presence of urease (Udert et al., 2003). The total nitrogen found in urine is typically 9.2 ± 1.8 g L−1 (Udert et al., 2006). Other major nitrogenous compounds found in urine include, creatinine, hippuric acid and uric acid (Maggi and Daly, 2013). These nitrogenous compounds are also eventually converted to ammonia (Maggi and Daly, 2013) through a range of complex decomposition reactions that are difficult to determine. Considering creatine and creatinine as one example, researchers have shown that it is challenging to determine the equilibrium between these compounds because of the occurrence of side reactions (Edgar and Shiver, 1925). These side reactions would be more pronounced in complex solutions such as urine. However, the researchers did manage to show that creatinine is at least partially converted into creatine in alkaline conditions. Some researchers have also shown that bacteria discovered in human urine are capable of degrading creatine to creatinine (Wyss and Kaddurah-Daouk, 2000), while other researchers reported that allantoin was produced during the decomposition of uric acid in alkaline solutions (Brooks and Lant, 1976). Ellerbe et al. (1988) reported that there is some evidence that urea is the product from uric acid decomposition. These reactions ultimately change the pH of the solution where the pH of the urine is increased to values around 9 after urea hydrolysis (Udert et al., 2003). This results in free ammonia losses by volatilization, which can be substantial. For example, Siegrist et al. (2013) reported ammonia losses of up to 47% in a urine separating toilet system with rising pipes open to the atmosphere.
The hydrolysis of the major nitrogenous compound, urea, can be prevented by inhibiting urease through acidification (Hellström et al., 1999) and various compounds (Svane et al., 2020). However, this approach has limitations because of safety concerns with handling of an acid and also because exact dosing equipment is required (Randall et al., 2016). Other methods include adding inhibitors such as the natural compounds allium (typically found in garlic, onions and leeks) to inhibit urease activity (Olech et al., 2014).
We have previously shown that increasing the pH of urine by calcium hydroxide addition is a cheap, simple and reliable method for preventing biological urea hydrolysis (Randall et al., 2016), while Vasiljev et al. (2022) have shown that magnesium hydroxide or magnesium oxide could also be used to increase the pH of fresh urine. The method works by pre-dosing (10 g L−1) a urine collection container, such as a urinal (Flanagan and Randall, 2018), with calcium hydroxide which dissolves with each new urine addition, regardless of the composition of the urine. This passive dosing system creates a saturated solution and keeps the pH of the urine high. We have termed this process “urine stabilization” which simply refers to the inhibition of urease activity in urine and the prevention, or at least limitation, of nitrogen loss. The stabilized urine solution can then be dehydrated on-site (Riechmann et al., 2021). Besides stabilizing urine, calcium hydroxide dosage can also be used for almost complete phosphorus recovery from fresh urine by producing calcium phosphate (Flanagan and Randall, 2018). This calcium phosphate derived from urine has also been shown to be an effective fertilizer (Meyer et al., 2018). We also proved with our experiments that urea hydrolysis was not significant for a period of 27 days after calcium hydroxide addition (Randall et al., 2016), however nitrogen losses occurred during dehydration (Riechmann et al., 2021). This was due to CO2 dissolution from the air used during the dehydration process (Simha et al., 2018; Riechmann et al., 2021). In addition, higher operating temperatures could also increase nitrogen losses.
With our previous work, we showed that biological urea hydrolysis can be inhibited effectively at pH values around 12.5 and at room temperature. However, literature has shown that for synthetic urea solutions, if temperatures are substantially high and pH values are above 13, chemical urea hydrolysis could be substantial (Warner, 1942). While previous researchers have focused on chemical urea hydrolysis in different urea solutions for a pH range of 0–14 (Warner, 1942), temperature range of 5–50°C (Chin and Kroontje, 1963) and a pharmaceutical solution (Panyachariwat and Steckel, 2014), and many researchers have also looked at biological urea hydrolysis in urine (Udert et al., 2003; Kabdasli et al., 2006; Randall et al., 2016), none have determined the rate constants for chemical urea hydrolysis in human urine. In addition, in most studies with urea solutions, the investigated temperatures and pH values were not within the range we are interested in for urine stabilization processes (Table 1).

TABLE 1. pH and temperature conditions for various urea systems.
Interactions with the atmosphere can also have a strong influence on the composition of urine. In work on dehydration of alkalized urine, Riechmann et al. (2021) and Simha et al. (2018) have suggested that for urine stabilized at high pH values, CO2 exchange with air can be critical. Strong absorption of CO2 from the atmosphere results in the conversion of Ca(OH)2 to CaCO3, a process which we refer to as carbonation (López-Periago et al., 2013). Carbon dioxide absorption in stabilized urine is problematic because CO2 dissolution and CaCO3 formation will result in a pH decrease (Morel and Hering, 1993).
As discussed, both high temperatures and lower pH values, due to CO2 adsorption, can lead to urea hydrolysis, but in two different ways: high temperatures, i.e. above 40°C, foster chemical urea hydrolysis, but low pH values, i.e. below 11, can allow for biological urea hydrolysis.
In this study, we looked at both influences, temperature and CO2 adsorption independently, because both can result in urea hydrolysis, but in different ways.
The first part of the study focused on temperature-dependent chemical urea hydrolysis. We determined the reaction rate constants of stabilized urine at 25°C, 40°C, 55°C and 70°C and at three different pH solutions (13, 12.5 and saturation with Ca(OH)2). The high temperatures were chosen to produce kinetic constants for interpolation. At temperatures below 25°C, the kinetics of urea hydrolysis were too low to determine accurate kinetic constants in the time that was available for this study. We used the determined reaction rate constants for pH values at saturation with Ca(OH)2 to model two urine stabilization scenarios in order to investigate if the process is feasible in terms of urea losses while still retaining excess Ca(OH)2 in the system at all times, but with no CO2 exchange and dehydration.
In the second part of study, we operated aerated batch reactors with urine, partially spiked with urease, to assess the effect of CO2 and NH3 gas exchange on biological urea degradation. A computer model was used to identify the main physical, chemical and biological processes, which leads to biological urea hydrolysis.
2 Materials and methods
2.1 Fresh urine solution
The urine used for the experiments was collected anonymously from both men and women working at Eawag in Dübendorf, Switzerland. The fresh urine was used within 3 days of collection. The fresh urine was stored overnight in a refrigerator kept at 4°C during the collection period.
2.2 Stabilization of fresh urine
A dosage of 10 g Ca(OH)2 (calcium hydroxide, Merck) per litre of fresh urine was used to ensure the solution was saturated with respect to Ca(OH)2 (Randall et al., 2016). The solution was then continuously stirred with a magnetic stirrer for 3 hours to ensure complete dissolution as per standard recommendations for dissolution processes (Myerson, 2002). After 3 hours, the solution was filtered in stages using a 1 L vacuum filtration setup with filter paper of pore size 180 µm (5892, Schleicher and Schuell) to remove excess Ca(OH)2 and any solids that had precipitated from the urine. The filtrate was collected and stored at room temperature for each of the subsequent gas exchange experiments. The average compositions of the fresh urine samples used for the carbonation and reaction rate experiments are shown in Table 2.
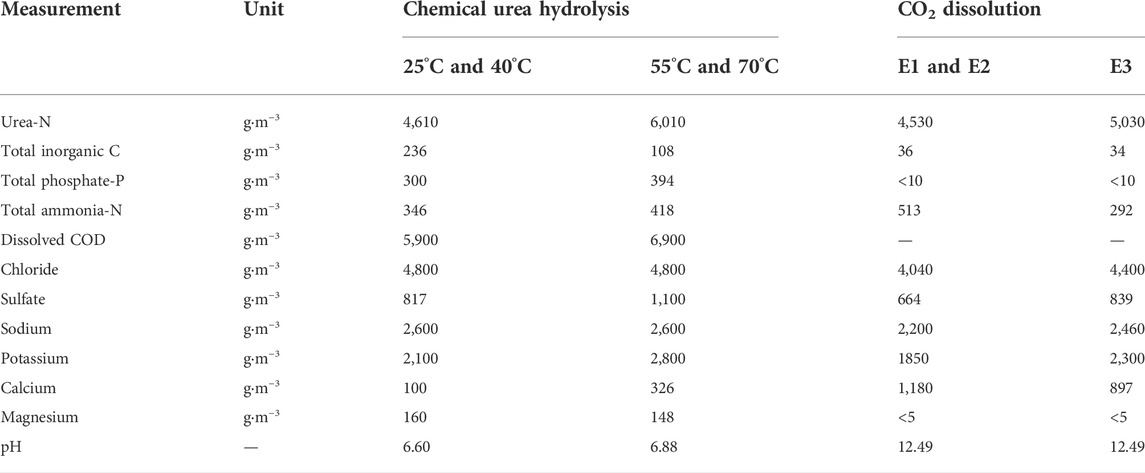
TABLE 2. Compositions of fresh urine used for chemical urea hydrolysis experiments and of stabilized urine used for the CO2 experiments (E1, E2 and E3). Experiment E1 was stabilized but had no excess Ca(OH)2 while experiment E2 was stabilized and had excess Ca(OH)2. Experiment E3 had no excess Ca(OH)2.
2.3 Chemical urea hydrolysis
Approximately 150 g of stabilized urine were distributed to three 50 ml Schott bottles in equal quantities. Three bottles for each experimental condition were positioned in a jacketed glass crystallizer (Schmizo AG) maintained at the respective temperatures using a circulation thermostat (F32, JULABO GmbH). Water was added directly to the crystallizer and gently mixed using a magnetic stirrer set at 300 rpm to help with the heat distribution to the three Schott bottles. The experiments were performed in triplicate and the mean concentrations were used for determination of the rate constants for the urea degradation. Samples were taken after 8 h, 1 day, 3 days, 9 days, 15 days and 21 days and the concentration of total ammonia was measured using cuvette tests (LCK304, Hach-Lange). Experiments were conducted at three different pH values (13, 12.5 and the pH at saturation with Ca(OH)2) and temperatures of 25°C, 40°C, 55°C and 70°C. We chose these pH values because at lower pH values urea hydrolysis is negligible (Warner, 1942) and we were specifically focused on pH values relevant to a solution saturated with calcium hydroxide. The rate constants were assumed to be first order with respect to urea. This assumption is consistent with the findings of (Chin and Kroontje, 1963).
2.4 CO2 dissolution: Effect of residual Ca(OH)2 on pH decrease
In order to investigate how the availability of residual Ca(OH)2 affects the pH decrease due to CO2 dissolution, two experiments (E1 and E2) were conducted simultaneously. The composition of the urine used for the experiments is given in Table 2. In experiment E1, the undissolved Ca(OH)2 was removed using the same filtration setup as described in Section 2.2 while for experiment E2, the excess Ca(OH)2 remained in the solution. In experiment E3, the excess Ca(OH)2 was also removed.
The two solutions (E1 and E2) with 1 L each were added to two identical jacketed glass reactors (DN 100/DW 100, Schmizo, Oftringen, Switzerland). The solutions of the reactors were mixed using a magnetic stirrer while air was bubbled through each of the solutions using a rubber diffuser connected to an rotameter which enabled the air flow rate to be controlled to approximately 0.1 m3 h−1 (range ± 20%) at ambient conditions. A circulation thermostat (F32, Julabo, Seelbach, Germany) was connected to the reactors and used to maintain the temperatures in the reactors at 25°C. A pH-temperature probe (CPS11D - 7AA21, Endress and Hauser, Reinach, Switzerland) was fixed in each reactor and connected to a multi-parameter transmitter (Liquiline CM444, Endress and Hauser, Reinach, Switzerland) for process monitoring and control. The transmitter itself was connected to a memograph (RSG40, Endress and Hauser, Reinach, Switzerland), which logged the continuous pH measurements during the experiments in 5 min intervals.
2.5 CO2 dissolution: Effect on biological urea hydrolysis
In order to model the effect of CO2 dissolution on biological urea hydrolysis, a separate experiment (E3) was conducted using the same procedure as mentioned in Section 2.3. The key difference was that 70 µL Jack bean urease (116,493 Urease, Merck, Darmstadt, Germany) was added to the urine solution after 8.5 h when the pH had dropped to a value of 9.7.
A precipitation model, developed and described in detail by (Udert et al., 2003), was used as basis for simulating the effect of CO2 dissolution on biological urea hydroylsis. The model allows for calculating a thermodynamic equilibrium state while considering solubility equilibria, acid-base and complex formation reactions. Chemical urea hydrolysis was not considered in the model because the model was run at a temperature of 25°C, at which chemical urea hydrolysis is very slow when compared to biological urea hydrolysis. The following four dynamic processes were adapted and adjusted to simulate the carbonation experiments: CO2 dissolution, calcite precipitation and urea hydrolysis. The complete gas exchange equation (Benjamin and Lawler, 2013) for a batch reactor was implemented as follows in order to model the CO2 dissolution:
Where:V: liquid volume in the reactor [L].
In this study, the simplification was made that the same Henry constant applies to water and urine. HCO2 was set to 1.2 molg/molaq at 25°C (Stumm and Morgan, 1996).
Since NH3 is a well-soluble gas, the following simplified equation (Benjamin and Lawler, 2013) was used to model the NH3 stripping rate:
Where:
Also, for NH3, it was assumed that the same Henry constant applies to water and urine. HNH3 was set to 0.00071 molg/molaq at 25°C (Stumm and Morgan, 1996).
The rate of calcite precipitation represents the carbonation process and was modelled according to kinetic expressions describing surface-controlled crystal growth (Udert et al., 2003):
Where:XCaCO3: concentration of calcite [mol L−1].kcalcite: rate constant for calcite equilibrium [mol L−1 d−1].{Ca2+}: activity of calcium ions [-]{CO32-}: activity of carbonate ions [-]KSp,calcite: solubility product of calcite [-]p: exponent depending on the crystal growth mechanism.
The activities were determined according to the Davies approach as described in Udert et al. (2003). It should be noted that ion activities are by definition unitless (Stumm and Morgan, 1996). The rate constant kcalcite was fitted and a value of 10–8.48 was chosen for KSp,calcite for 25°C (Stumm and Morgan, 1996). The simulations revealed that p = 1 was an optimal value to describe CaCO3 precipitation in our experiments.
Finally, the pH-dependent rate of biological urea hydrolysis induced by Jack bean urease was implemented using the following equation (Fidaleo and Lavecchia, 2003):
Where:
The model was fitted using data from experiment E3. The measured initial concentrations of the ions, the dissolved TIC, the ammonium and the urea along with the experimental air flow served as an input for the model. The parameters were then fitted as follows:
1)
2) µmax was fitted on the experimentally observed urea concentration profiles with a minimum value of 0.23 mol NH3·Lreactor−1·d−1 according to manufacturer’s information (see below).
3) kcalcite was fitted on the experimentally determined dissolved calcium concentration.
4) Finally, step 1 was repeated.
In experiment E3, 70 μL of urease (Merck, No 11643) was dosed, which had a minimum activity of 1,000 mmol NH3,released·mL−1·min−1. at 25°C and pH 8. The reactor volume was 1 L and the temperature 25°C. Using the kinetic expression of Equation 4, a minimum μmax of 0.23 mol NH3·Lreactor·d−1 was calculated.
2.6 Determination of gas exchange parameters
The dissolution experiments served to determine the overall mass transfer rate constant (KLa) of O2 in water and stabilized urine. The latter was used to estimate the KLa of CO2 in both media. Since the KLa of CO2 in urine was a parameter that was fitted during the calibration of the precipitation model, the experimentally determined KLa was needed to have a starting value and to know which order of magnitude to expect.
The experimental setup was identical to the one of the carbonation experiments, except that O2 probes (WTW Oxi 340i) were installed in the reactors. One reactor contained stabilized urine without excess Ca(OH)2, while the other one contained nanopure water. At first, both reactors were bubbled with nitrogen gas in order to strip the oxygen down to a concentration of 2 mg L−1 or less. Subsequently, both reactors were aerated with ambient air at a flow rate of 100 L h−1 for 4 minutes and the oxygen concentration profiles were recorded at a 5 s resolution. The saturation concentrations in both media were experimentally determined by averaging the measured values in the last minute of the aeration, when the oxygen concentration was constant. The simplified gas exchange equation for a batch experiment (derived from (Benjamin and Lawler, 2013)) was used to determine the KLa of O2 in nanopure water and urine:
Where:KLaO2: mass transfer coefficient for oxygen [d−1].
For the determination of the
The oxygen transfer difference between urine and water was calculated as:
It was assumed that the CO2 transfer difference between urine and water is governed by the same α -value. The following relationship allowed determining the KLa of CO2 in water:
Where:Dw: diffusion coefficient in water of the corresponding gas [m2 s−1].
The relationship is based on the assumption that the relationship of KLa and DW is best described by the penetration theory (Benjamin and Lawler, 2013).
Literature values for the diffusion coefficients as well as the formula were adapted from (Lide, 2009). The
2.7 Simulation of urea loss in CSTR and batch mode
Matlab (version 7.10.0) was used to simulate the urea concentration profiles for two different scenarios: one with 10 L day−1 influent urine in a continuous flow stirred tank reactor (CSTR) and one with no urine inflow or outflow (batch).
2.8 Analytical methods
The following dissolved compounds were determined with ion chromatography (930 Compact IC Flex for cations and 881 Compact IC Pro for anions, Metrohm): Ca2+, Na+, Mg2+, K+, Cl−, SO42- and PO43-. NH4+-N concentrations were measured with cuvette tests (Hach-Lange). The dissolved total inorganic carbon (TIC) was measured applying the 680°C combustion catalytic oxidation method (TOC-L analyzer, SHIMADZU). In order to measure urea, 0.5 mL of the filtered sample were diluted 1:20 in nanopure water. The diluted sample was then acidified with 1 mol L−1 HCl to ensure a pH between 7 and 8. After measuring the NH4+-N concentration, 40 μL of Jack bean urease was added to the solution. The sample was left for 1 hour in order to ensure all urea would be hydrolyzed, where after the NH4+-N concentration was measured again. Subtracting the initial NH4+-N concentration of the sample allowed determining the urea concentration. The standard deviation for all wet chemistry was typically below 5%.
3 Results and discussion
3.1 Fast chemical urea hydrolysis in urine at high temperatures
3.1.1 Temperature dependency of chemical urea hydrolysis at saturation with Ca(OH)2
The rate constants for chemical urea hydrolysis in urine determined in this study show that temperature is the primary factor influencing the rate (Figure 1). At pH 13, the rate constants for the saturated urine-Ca(OH)2 solutions (see Table 3 for pH values at each temperature) are similar to the rate constants at a pH of 12.5, while the rate constants at a pH of 13 are only slightly higher. This is expected based on the pH dependency described by (Warner, 1942) at values greater than 13.
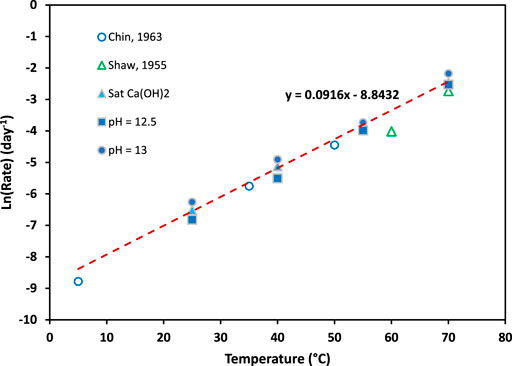
FIGURE 1. Reaction rate constants for urea in stabilized urine (compositions are given in Table 2) at different temperatures and pH values of 13, 12.5 and saturated with Ca(OH)2. The values and conditions are given in Table 3.
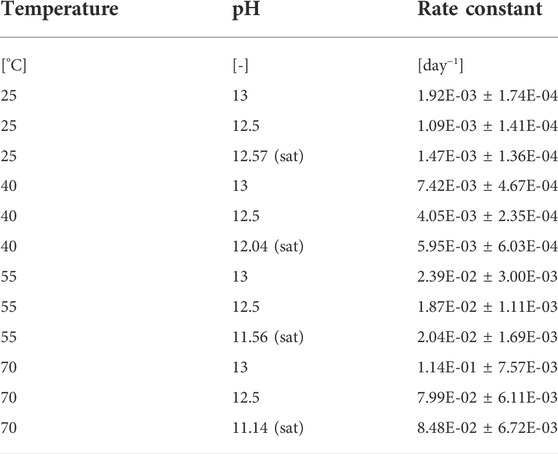
TABLE 3. Reaction rate constants at different concentration factors, temperatures and pH values for stabilized urine.
Chemical urea hydrolysis in real human urine increases as the temperature of the solution increases and is similar to synthetic urea hydrolysis as reported in literature (Warner, 1942; Shaw and Bordeaux, 1955; Chin and Kroontje, 1963; Panyachariwat and Steckel, 2014). Therefore, the rate constants are expected to be similar regardless of urine composition or types of urea solutions. The rate constants were found to be 0.00109 days−1, 0.00147 days−1 and 0.00192 days−1 at 25°C for pH values of 12.5, 12.57 (saturated with calcium hydroxide) and 13.0 respectively and significantly higher at 70°C.
The activation energy for urine saturated with Ca(OH)2 was calculated to be 74.9 ± 6.9 kJ mol−1, which compares well with the value determined by (Chin and Kroontje, 1963) for a synthetic urea solution (72 kJ mol−1). The activation energies for the higher pH values of 13 and 12.5 were similar: 76 ± 4.7 kJ mol−1 and 81 ± 3.9 kJ mol−1 respectively. The standard deviation of the measured error for each reaction rate constant was less than 25% with the lowest error being 6.5% and the majority of errors being less than 10% (see Table 3). The error was assumed to be proportional to the measured values.
3.1.2 Operating temperature is more critical than pH
In 2016 we reported that a precautionary lower temperature limit of 14°C should be utilized in the urine stabilization process with calcium hydroxide because of concerns that the higher pH values at saturation would increase the rate of chemical urea hydrolysis (Randall et al., 2016). The more detailed kinetics from this study show that a higher pH of 13 does result in slightly faster reaction kinetics but it is negligible in comparison to the effect temperature has on the kinetics of the process. The urine stabilization process can therefore run effectively at temperatures lower than 14°C as this would slow down the kinetics of the process. For example, operating the process at 14°C results in 71% slower kinetics and 90% at 5°C when compared to the reaction rate constants at 25°C. However, lower operating temperatures would reduce the rate of water evaporation during urine dehydration processes. The results also show that operating a system at higher temperatures, to increase the rate of water evaporation, will lead to higher rates of chemical urea hydrolysis and these aspects need to be carefully considered when designing urine dehydration systems.
3.1.3 Urea losses are higher in batch mode than in CSTR mode
The reaction rate constants for chemical urea hydrolysis determined in this study were used to model urea losses as a function of operating times for two scenarios (A and B; see Section 2.7). Scenario A had a constant urine inflow of 10 L day−1 (CSTR mode, Figure 2A), while scenario B had no urine inflow (Figure 2B) and so was essentially stored urine. We see that as the operating temperature increases, the loss of urea increases significantly for both scenarios, but that the urea loss is less for scenario A. In 30 days, 64% of the urea is lost at a temperature of 70°C while 25% is lost at 55°C, 8.4% at 40°C and 2.2% at 25°C for scenario A. In contrast, 92% of the urea is lost at 70°C, while 46% is lost at 55°C, 16% at 40°C and 4.3% at 25°C for scenario B. The loss of urea for scenario A is lower than for scenario B because the contents of the urine tank are constantly resupplied with urea from the influent. Importantly, these simulations did not include water evaporation during dehydration, but the rate constants still provide useful design parameters, especially for dehydration reactors where urine is collected and concentrated over extended periods of time.
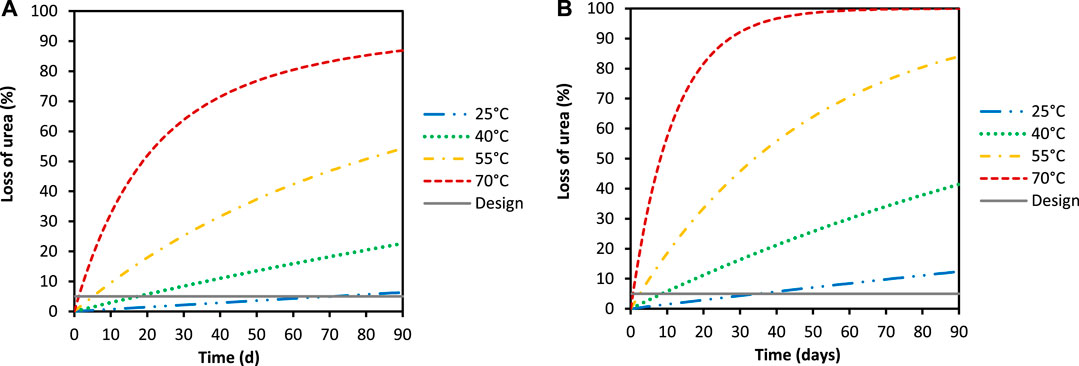
FIGURE 2. Loss of urea for a saturated Ca(OH)2-urine solution at different operating temperatures and two hypothetical scenarios: scenario (A) has constant 10 L day−1 inflow to the urine stabilization tank (CSTR mode) while scenario (B) has no urine inflow (batch mode). There was no CO2 exchange in the model and no urine dehydration. The solid grey design line depicts a 5% loss that is an arbitrary percentage urea loss, while the blue lines are for urea losses at 25°C, green is for 40°C, orange is for 55°C and red is for 70°C.
If the design specification for acceptable urea loss is set to an arbitrary 5%, it would mean the urine stabilization process could be effectively operated under 40°C for a period of approximately 17 days. At 25°C, the urea losses can be considered negligible, with a 5% loss only being reached after 70 days for scenario A. Even for scenario B, the time required to reach a 5% urea loss at 25°C is low (34 days). Scenario B can also essentially be used to determine the urea loss during the storage of stabilized urine. For example, if we wanted to store the stabilized urine before transporting it to a decentralized resource recovery plant, the results from this scenario would need to be used.
3.2 Carbon dioxide dissolution fosters biological urea hydrolysis
3.2.1 Residual Ca(OH)2 delays, but does not prevent a pH decrease
Residual Ca(OH)2 strongly delays the pH decrease. Two experiments were run to test the importance of residual Ca(OH)2 (Figure 3): one experiment looked at the dissolution of CO2 in stabilized urine with no excess solid Ca(OH)2 (E1) while the second experiment focused on the dissolution of CO2 in stabilized urine with excess solid Ca(OH)2 (E2). The rate of dissolution was increased by bubbling air through both solutions. The experimental results show that there is a slow but immediate pH decrease in the reactor without Ca(OH)2 excess during the first 4 h of aeration as shown in Figure 3. Within the next 4 h, the pH decreases and reaches a constant value of 8.2. In the reactor with excess Ca(OH)2, the pH stays constant for approximately 11 h and the start of the rapid pH decrease is delayed by 12 h. The presence of excess Ca(OH)2 therefore buffers the pH and keeps it at values greater than 12.
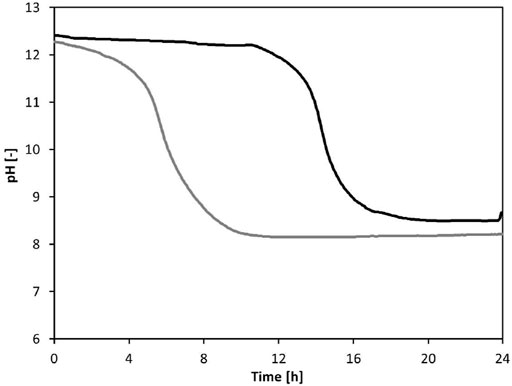
FIGURE 3. pH profiles for both reactors where E1 ( ) represents the experiment without excess calcium hydroxide while E2 (
) represents the experiment without excess calcium hydroxide while E2 ( ) represents the experiment with excess calcium hydroxide.
) represents the experiment with excess calcium hydroxide.
3.3 The pH behavior can be explained with four processes
Four chemical and physical processes influence the pH in fresh urine stabilized with Ca(OH)2, which ultimately influence the solution pH: the dissolution and dissociation of CO2 (Equation 9), the precipitation of CaCO3 (Equation 10), ammonia volatilization (Equation 11) and urea hydrolysis (Equation 12):
The involvement of all four processes can be derived from the measurements during experiment E3, when CO2 was bubbled into fresh urine, which was previously stabilized with Ca(OH)2, but from which excess Ca(OH)2 was removed (Figure 4). The selected compounds are meant to be indicators for the processes of interest. Total inorganic carbon is influenced by three processes: CO2 dissolution (Equation 9), CaCO3 precipitation (Equation 10) and urea hydrolysis (Equation 12). Since ammonia is liberated during urea hydrolysis, the NH4+ concentration curve was included on the graph. Finally, the Ca2+ concentration behavior is directly linked to the carbonation process since it gives an indication of CaCO3 precipitation.
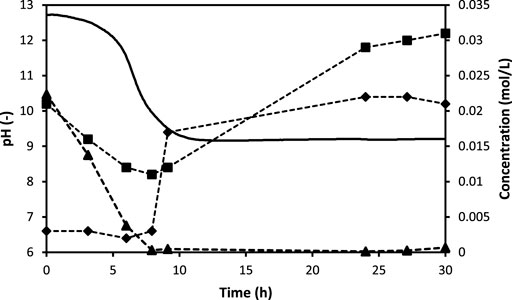
FIGURE 4. Experimentally measured Ca2+ (▲), TIC (♦), NH4+-N (■) concentrations and pH (–) during experiment E3.
The concentration of Ca2+ ions was below the detection limit of 0.0003 mol L−1 within the first 8 hours of the experiments. For the other two parameters, TIC and NH4+, the 8-h mark seems to be a turning point in their behaviour as well. Starting at 0.021 mol N L−1, the ammonium concentration exhibits a decrease throughout the first 8 hours before rising as the pH stays constant to a value of 0.031 mol L−1 after 30 h.
The TIC is constantly low during the first 8 hours (about 0.003 mol C L−1) and then rises rapidly to 0.017 mol C L−1. Starting at 12.5, and after a fluctuating decrease throughout the first 9 hours, the pH stabilizes at a constant value of approximately 9.1.
Figure 4 shows the observed pH during experiment E3 compared to the simulated pH with the precipitation model (Figure 5). For the simulation, apart from all relevant chemical equilibria, only the processes of carbon dioxide dissolution, urea hydrolysis, calcite precipitation (carbonation) and ammonia volatilization were considered.
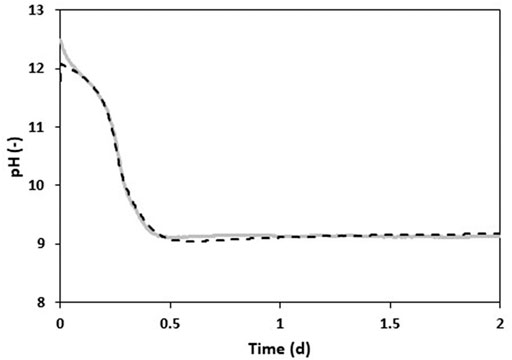
FIGURE 5. Simulated pH (╍) for experiment E3. All parameters were fitted in respect to the experimentally measured pH (–).
The simulated pH was obtained with the following set of fitted parameters:
KLa CO2 3,890 d−1 (starting value 3,300 d−1, calculated).
Qair 2900 L d−1 (starting value 2400 L d−1, measured).
initial TIC 0.0144 mol L−1 (starting value 0.003 mol L−1, measured).
initial OH− concentration 0.0165 mol L−1 (starting value 0.031 mol L−1, according to measured pH value).
µmax 0.353 mol NH3·Lreactor·d−1 (starting value 0.23 mol NH3·Lreactor·d−1, calculated based on manufacturer’s information).
kcalcite 48 mol L−1 d−1 (no measurement and estimation available).
The model allows for the following explanation: The production of protons by CO2 dissolution decreases the solution pH unless OH− ions are present to neutralize them. As soon as the OH− ions are depleted the concentration of H+ will dramatically increase, which explains the abrupt observed pH decrease seen in Figure 3. In the case where the stabilized urine contains excess Ca(OH)2, the latter dissolves in order to balance out the depletion of OH− ions, which explains why the pH remained constant over an extended period of 11 h (Figure 3). Excess Ca(OH)2 therefore increases the pH buffer capacity of the solution. Although CO32- ions are the main carbonate compound produced by CO2 dissolution (Equation 9) at pH values greater than 10.3 (Stumm and Morgan, 1996), the dissolved TIC remains constant during the first 8 h (see Figure 4). An explanation for this is provided by the carbonation process where CO32- ions are consumed at the same rate as they are produced by CO2 dissolution. The strong depletion of Ca2+ ions during the first 8 h of experiment E3 underlines the involvement of the carbonation process. The decrease of the NH4+ ion concentration during that period is likely due to ammonia stripping and flattens out as the pH decreases and the pKa of NH3 is reached. Since no CaCO3 precipitation occurs after hour 8 for experiment E3, the CO32- ions produced by CO2 dissolution are no longer consumed and this explains the subsequent increase of dissolved TIC. Additionally, as biological urea hydrolysis starts after 8.5 h, HCO3− ions are formed (Equation 12) and reinforces the dissolved TIC increase. At the same time, the ammonia liberated during the urea hydrolysis results in the subsequent increase of NH4+ ions. The simulations do not consider chemical urea hydrolysis because the experiments were operated at room temperature and chemical urea hydrolysis was considered negligible.
In addition, another process can decrease the pH buffer capacity of excess Ca(OH)2: the establishment of an impermeable layer of CaCO3 on the surface of the solid Ca(OH)2, which would prevent Ca(OH)2 dissolution (Van Balen, 2005). The work by (Ruiz-Agudo et al., 2013) showed that the carbonation of Ca(OH)2 is in fact governed by a complex interface-coupled dissolution-precipitation process in an aqueous solution. More Ca(OH)2 will therefore be coated with an impermeable layer of CaCO3 when the concentration of CO32- ions in solution increases, thus depleting the use of excess Ca(OH)2 for further neutralization and stabilization. However, it is difficult to establish a relationship between the amount of excess Ca(OH)2 and the additional pH buffer capacity, but this should be investigated further.
In order to further investigate the effect of the four processes dissolution and dissociation of CO2, the precipitation of CaCO3, ammonia volatilization and urea hydrolysis, four different process scenarios were simulated. Figure 6A shows a scenario where only CO2 dissolution is switched on. The simulated pH that can be observed in Figure 6B was obtained by turning off urea hydrolysis. Figure 6C was generated by switching off CaCO3 precipitation and Figure 6D was obtained by turning off ammonia volatilization in the simulation.
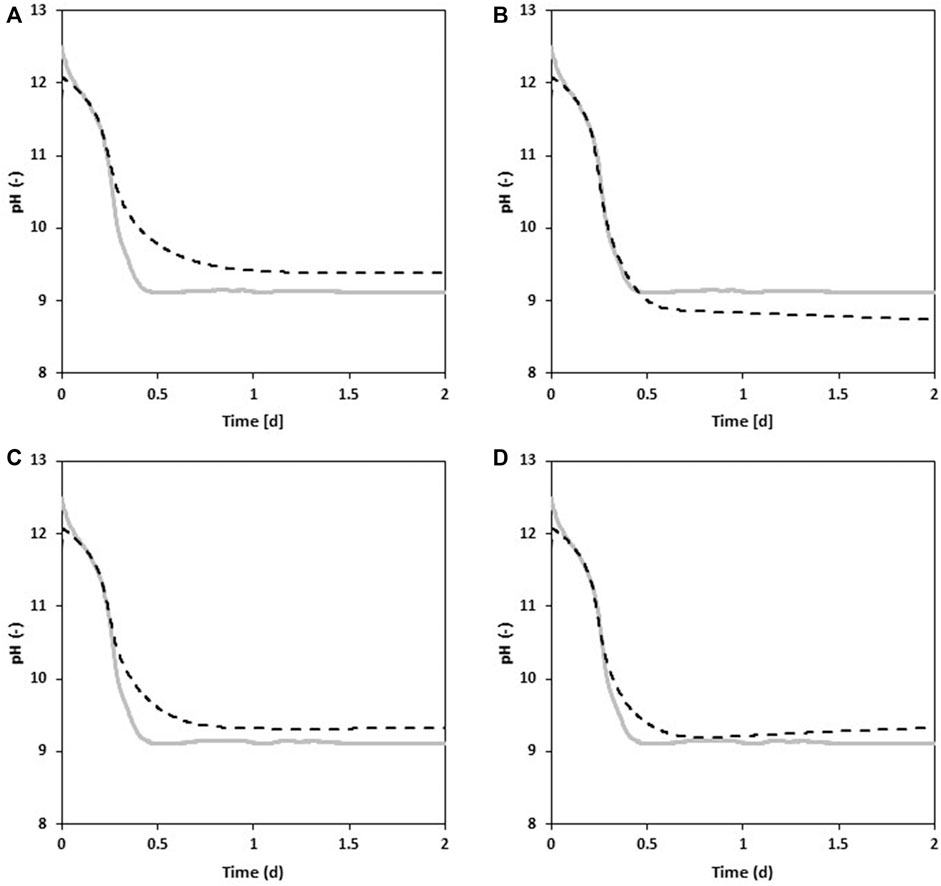
FIGURE 6. Simulated and experimental results where only CO2 dissolution is turned on in the model (A), urea hydrolysis is turned off (B) CaCO3 precipitation is turned off (C) and ammonia volatilization is turned off (D).
Figure 6A shows that CO2 dissolution is the key process responsible for the pH decrease of stabilized urine during aeration. This is consistent with findings by (Courtney et al., 2021), who measured and modelled CO2 dissolution in Ca(OH)2 stabilized urine, but for the purpose of removing excess calcium ions prior to reverse osmosis treatment. The pH decreases from 12.2 to approximately 9.4 within 1 day after which it remains constant. The fact that the pH stagnates, means that the saturation pH of CO2 predicted by the model is at 9.4 because no more CO32- ions are supplied to the system. If only CO2 dissolution is considered, the pH decrease is flattened by the end of the simulation compared to the observed one.
When urea hydrolysis is switched off (Figure 6B), but the other three processes are switched on, the simulated pH constantly decreases after dropping below the observed pH during the initial decrease. The ammonia volatilization and the CaCO3 precipitation allows for a more pronounced initial pH decrease. In addition, after the Ca2+ ions are all depleted, the slow continuous decrease after the initial drop is only due to ammonia volatilization. Since the ammonia concentration is low to begin with, and the stripping rate is directly proportional to it, it is safe to assume that the ammonia stripping process causes a slight pH decrease over the entire period of the simulation, while CaCO3 precipitation is responsible for the steep initial decrease.
Switching off CaCO3 precipitation (Figure 6C) and considering CO2 dissolution, ammonia stripping and urea hydrolysis, leads to a similar behaviour as scenario A where only CO2 dissolution is turned on. The key difference is that the pH stabilizes at 9.3 instead of 9.4. The ammonia stripping which causes a constant pH decrease is missing because it is buffered out by the urea hydrolysis process. This is expected since ammonia volatilization depends mainly on the NH3 concentration and the only source of ammonia into the system is from urea hydrolysis. Thus, at pH values close to the pKa of NH3, the ammonia stripping rate will be re-adjusted to the rate of urea hydrolysis.
Finally, when ammonia volatilization is switched off (Figure 6D), the pH constantly increases after a pronounced initial decrease. Figure 6D provides evidence that urea hydrolysis leads to a pH increase if ammonia stripping is not occurring. Also, the initial pH decrease is stronger compared to the scenario where CaCO3 precipitation is turned off (Figure 6C). This reinforces the fact that CaCO3 precipitation is responsible for the initial pH decrease as a result of CO2 dissolution. As CaCO3 precipitation depletes the carbonate ions produced by the CO2 dissolution, the equilibrium is shifted to the right-hand side of Equation 9, so that more CO2 can dissolve which ultimately leads to a stronger pH decrease.
3.4 Preventing nitrogen losses during volume reduction
The treatment of source-separated urine should ideally encompass on-site treatment technologies (Larsen et al., 2021) that also reduce the volume of urine. This will increase the storage capacity of any product that forms while also reducing the transport costs of these products. Perhaps the simplest method of volume reduction involves evaporating the water from urine (Simha et al., 2020a; Riechmann et al., 2021), although reverse osmosis (Courtney and Randall, 2022) and forward osmosis (Patel et al., 2020) could also be used. We have shown that the rates of chemical urea hydrolysis are slow at temperatures less than 40°C, which means the stabilized urine could be evaporated at ambient conditions to reduce the volume, but the dehydration rate would be slow. When operating dehydration systems at higher temperatures, the evaporation rates need to be very fast to prevent substantial chemical urea hydrolysis (Simha et al., 2020b).
However, considering biological urea hydrolysis, the solution should always have excess calcium hydroxide to maintain a high operating pH. This is because the dissolution of CO2 can also take place during dehydration by convection which results in a pH decrease and therefore urea loss by biological urea hydrolysis (Riechmann et al., 2021). The rate of carbonation of the excess calcium hydroxide should be slower than the rate of evaporation since this would also negatively affect the pH of the solution. The CO2 present in the air could be removed prior to the dehydration process by means of a CO2 scrubber in order to reduce any carbonation issues. For example, (Alabadi et al., 2015) showed that activated carbon prepared from biomass was very effective at removing CO2, having an adsorption capacity of 7.49 mmol g−1 at 0°C. The concentration of stabilized urine by evaporation would ultimately result in a solid fertilizer product rich in urea.
4 Conclusion
• Chemical urea hydrolysis is a strong function of temperature, with the reaction rate constants of urea in human urine increasing significantly as the temperature increases. The activation energy of chemical urea hydrolysis was found to be 75 ± 6.9 kJ mol−1 for a solution saturated with Ca(OH)2.
• Chemical urea hydrolysis does increase at higher pH values, but it is not as significant as the effect of temperature. A pH dependency does not have to be considered in the pH range of interest for urine stabilization with Ca(OH)2, that is up to a pH value of 13.
• Carbon dioxide dissolution from ambient air is the major reason for a pH decrease in the calcium hydroxide urine stabilization process.
• Besides CO2 dissolution, other processes such as urea hydrolysis, calcite precipitation and ammonia volatilization also affect the pH during CO2 exchange with the atmosphere, but to a lesser extent.
• The presence of excess solid Ca(OH)2 can buffer any pH decrease due to CO2 dissolution and thus keep the urine stabilized, especially during volume reduction by dehydration with ambient air.
Data availability statement
The datasets presented in this study will be made available by the corresponding author upon request.
Author contributions
DR and KU conceptualized the study and designed the experiments. DR performed the chemical urea hydrolysis experiments and AB performed the carbon dioxide dissolution experiments. AB and KU modelled the experiments. KU supervised the study, provided resources and acquired funding. DR wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This work was carried out in the context of the Blue Diversion AUTARKY-Project, funded by the Bill and Melinda Gates Foundation (OPP1111293).
Acknowledgments
The input from Karin Rottermann, Claudia Bänninger and Brian Sinnet from Eawag for the chemical analysis is also gratefully acknowledged. Last but not least, thanks goes to the numerous urine donors from the Process Engineering and Urban Water Management departments at Eawag.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alabadi, A., Razzaque, S., Yang, Y. W., Chen, S., and Tan, B. (2015). Highly porous activated carbon materials from carbonized biomass with high CO2 capturing capacity. Chem. Eng. J. 281, 606–612. doi:10.1016/j.cej.2015.06.032
Benjamin, M. M., and Lawler, D. F. (2013). Water quality engineering: Physical/chemical treatment processes. New Jersey: John Wiley & Sons.
Brooks, B. A., and Lant, A. F. (1976). Chemical instability of [2-14C] uric acid in alkaline solution: The effect on observed kinetics of urate transport in human erythrocytes. Anal. Biochem. 75 (2), 640–645. doi:10.1016/0003-2697(76)90120-2
Chin, W., and Kroontje, W. (1963). Urea hydrolysis and subsequent loss of ammonia. Soil Sci. Soc. Am. J. 27 (3), 316–318. doi:10.2136/sssaj1963.03615995002700030030x
Courtney, C., Brison, A., and Randall, D. G. (2021). Calcium removal from stabilized human urine by air and CO2 bubbling. Water Res. 202, 117467. doi:10.1016/j.watres.2021.117467
Courtney, C., and Randall, D. G. (2022). Concentrating stabilized urine with reverse osmosis: How does stabilization method and pre-treatment affect nutrient recovery, flux, and scaling? Water Res. 209, 117970. doi:10.1016/j.watres.2021.117970
Edgar, G., and Shiver, H. E. (1925). The equilibrium between creatine and creatinine, in aqueous solution. The effect of hydrogen ion. J. Am. Chem. Soc. 47, 1179–1188. doi:10.1021/ja01681a040
Ellerbe, P., Cohen, A., Welch, M. J., and White, E. (1988). The stability of uric-acid in ammonium hydroxide. Clin. Chem. 34 (11), 2280–2282. doi:10.1093/clinchem/34.11.2280
Fidaleo, M., and Lavecchia, R. (2003). Kinetic study of enzymatic urea hydrolysis in the pH range 4-9. Chem. Biochem. Eng. Q. 17 (4), 311–318.
Flanagan, C. P., and Randall, D. G. (2018). Development of a novel nutrient recovery urinal for on-site fertilizer production. J. Environ. Chem. Eng. 6 (5), 6344–6350. doi:10.1016/j.jece.2018.09.060
Hellström, D., Johannson, E., and Grennberg, K. (1999). Storage of human urine: Acidification as a method to inhibit decomposition of urea. Ecol. Eng. 12 (3-4), 253–269. doi:10.1016/s0925-8574(98)00074-3
Ishii, S. K., and Boyer, T. H. (2015). Life cycle comparison of centralized wastewater treatment and urine source separation with struvite precipitation: Focus on urine nutrient management. Water Res. 79, 88–103. doi:10.1016/j.watres.2015.04.010
Kabdasli, I., Tunay, O., Islek, C., Erdinc, E., Huskalar, S., and Tatli, M. B. (2006). Nitrogen recovery by urea hydrolysis and struvite precipitation from anthropogenic urine. Water Sci. Technol. 53 (12), 305–312. doi:10.2166/wst.2006.433
Larsen, T. A., and Gujer, W. (1996). Separate management of anthropogenic nutrient solutions (human urine). Water Sci. Technol. 34 (3-4), 87–94. doi:10.2166/wst.1996.0420
Larsen, T. A., Riechmann, M. E., and Udert, K. M. (2021). State of the art of urine treatment technologies: A critical review. Water Res. X 13, 100114. doi:10.1016/j.wroa.2021.100114
López-Periago, A. M., Fraile, J., López-Aranguren, P., Vega, L. F., and Domingo, C. (2013). CO2 capture efficiency and carbonation/calcination kinetics of micro and nanosized particles of supercritically precipitated calcium carbonate. Chem. Eng. J. 226, 357–366. doi:10.1016/j.cej.2013.04.071
Maggi, F., and Daly, E. (2013). Decomposition pathways and rates of human urine in Soils. J. Agric. Food Chem. 61 (26), 6175–6186. doi:10.1021/jf401212n
Meyer, G., Frossard, E., Mäder, P., Nanzer, S., Randall, D. G., Udert, K. M., et al. (2018). Water soluble phosphate fertilizers for crops grown in calcareous soils – An outdated paradigm for recycled phosphorus fertilizers? Plant Soil 424 (1), 367–388. doi:10.1007/s11104-017-3545-x
Morel, F. o., and Hering, J. G. (1993). Principles and applications of aquatic chemistry. New York: Wiley.
Olech, Z., Zaborska, W., and Kot, M. (2014). Jack bean urease inhibition by crude juices of Allium and Brassica plants. Determination of thiosulfinates. Food Chem. 145, 154–160. doi:10.1016/j.foodchem.2013.08.044
Panyachariwat, N., and Steckel, H. (2014). Stability of urea in solution and pharmaceutical preparations. J. Cosmet. Sci. 65 (3), 187–195.
Patel, A., Mungray, A. A., and Mungray, A. K. (2020). Technologies for the recovery of nutrients, water and energy from human urine: A review. Chemosphere 259, 127372. doi:10.1016/j.chemosphere.2020.127372
Randall, D. G., Krahenbuhl, M., Kopping, I., Larsen, T. A., and Udert, K. M. (2016). A novel approach for stabilizing fresh urine by calcium hydroxide addition. Water Res. 95, 361–369. doi:10.1016/j.watres.2016.03.007
Randall, D. G., and Naidoo, V. (2018). Urine: The liquid gold of wastewater. J. Environ. Chem. Eng. 6 (2), 2627–2635. doi:10.1016/j.jece.2018.04.012
Riechmann, M. E., Ndwandwe, B., Greenwood, E. E., Reynaert, E., Morgenroth, E., and Udert, K. M. (2021). On-site urine treatment combining Ca(OH)2 dissolution and dehydration with ambient air. Water Res. X 13, 100124. doi:10.1016/j.wroa.2021.100124
Ruiz-Agudo, E., Kudlacz, K., Putnis, C. V., Putnis, A., and Rodriguez-Navarro, C. (2013). Dissolution and carbonation of portlandite [Ca(OH)2] single crystals. Environ. Sci. Technol. 47 (19), 11342–11349. doi:10.1021/es402061c
Sahu, J. N., Mahalik, K., Patwardhan, A. V., and Meikap, B. C. (2008). Equilibrium and kinetic studies on the hydrolysis of urea for ammonia generation in a semibatch reactor. Ind. Eng. Chem. Res. 47 (14), 4689–4696. doi:10.1021/ie800481z
Shaw, W. H. R., and Bordeaux, J. J. (1955). The decomposition of urea in aqueous media. J. Am. Chem. Soc. 77 (18), 4729–4733. doi:10.1021/ja01623a011
Siegrist, H., Laureni, M., Udert, K. M., Larsen, T., Udert, K., and Lienert, J. (2013). Transfer into the gas phase: Ammonia stripping.” in Source Separation and Decentralization for Wastewater Management. London: IWA Publishing, 337
Simha, P., Friedrich, C., Randall, D. G., and Vinnerås, B. (2020a). Alkaline dehydration of human urine collected in source-separated sanitation systems using magnesium oxide. Front. Environ. Sci. 8, 286. doi:10.3389/fenvs.2020.619901
Simha, P., Lalander, C., Nordin, A., and Vinnerås, B. (2020b). Alkaline dehydration of source-separated fresh human urine: Preliminary insights into using different dehydration temperature and media. Sci. Total Environ. 733, 139313. doi:10.1016/j.scitotenv.2020.139313
Simha, P., Senecal, J., Nordin, A., Lalander, C., and Vinnerås, B. (2018). Alkaline dehydration of anion–exchanged human urine: Volume reduction, nutrient recovery and process optimisation. Water Res. 142, 325–336. doi:10.1016/j.watres.2018.06.001
Stumm, W., and Morgan, J. J. (1996). Aquatic chemistry chemical equilibria and rates in natural waters. New York: Wiley.
Svane, S., Sigurdarson, J. J., Finkenwirth, F., Eitinger, T., and Karring, H. (2020). Inhibition of urease activity by different compounds provides insight into the modulation and association of bacterial nickel import and ureolysis. Sci. Rep. 10 (1), 8503. doi:10.1038/s41598-020-65107-9
Udert, K. M., Buckley, C. A., Wachter, M., McArdell, C. S., Kohn, T., Strande, L., et al. (2015). Technologies for the treatment of source-separated urine in the eThekwini Municipality. Water sa. 41 (2), 212–221. doi:10.4314/wsa.v41i2.06
Udert, K. M., Larsen, T. A., Biebow, M., and Gujer, W. (2003). Urea hydrolysis and precipitation dynamics in a urine-collecting system. Water Res. 37 (11), 2571–2582. doi:10.1016/s0043-1354(03)00065-4
Udert, K. M., Larsen, T. A., and Gujer, W. (2006). Fate of major compounds in source-separated urine. Water Sci. Technol. 54 (11-12), 413–420. doi:10.2166/wst.2006.921
Van Balen, K. (2005). Carbonation reaction of lime, kinetics at ambient temperature. Cem. Concr. Res. 35 (4), 647–657. doi:10.1016/j.cemconres.2004.06.020
Vasiljev, A., Simha, P., Demisse, N., Karlsson, C., Randall, D. G., and Vinnerås, B. (2022). Drying fresh human urine in magnesium-doped alkaline substrates: Capture of free ammonia, inhibition of enzymatic urea hydrolysis & minimisation of chemical urea hydrolysis. Chem. Eng. J. 428, 131026. doi:10.1016/j.cej.2021.131026
Warner, R. C. (1942). The kinetics of the hydrolysis of urea and of arginine. J. Biol. Chem. 142 (2), 705–723. doi:10.1016/s0021-9258(18)45072-7
Wilsenach, J., and van Loosdrecht, M. (2003). Impact of separate urine collection on wastewater treatment systems. Water Sci. Technol. 48 (1), 103–110. doi:10.2166/wst.2003.0027
Keywords: calcium phosphate, fertilizer, sanitation, source separation, urea hydrolysis
Citation: Randall DG, Brison A and Udert KM (2022) High temperatures and CO2 dissolution can cause nitrogen losses from urine stabilized with base. Front. Environ. Sci. 10:998743. doi: 10.3389/fenvs.2022.998743
Received: 20 July 2022; Accepted: 06 October 2022;
Published: 20 October 2022.
Edited by:
Zifu Li, University of Science and Technology Beijing, ChinaReviewed by:
Yaohuan Gao, Dalhousie University, CanadaArvind Kumar Mungray, Sardar Vallabhbhai National Institute of Technology, India
Copyright © 2022 Randall, Brison and Udert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dyllon G. Randall, RHlsbG9uLnJhbmRhbGxAdWN0LmFjLnph