 Wenhuan Yang1
Wenhuan Yang1 Jie Ma
Jie Ma Weiying Feng
Weiying Feng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci. , 22 November 2022
Sec. Toxicology, Pollution and the Environment
Volume 10 - 2022 | https://doi.org/10.3389/fenvs.2022.994104
This article is part of the Research Topic Biodiversity Protection and Pollution Control on the Mongolian Plateau View all 7 articles
Microbial phosphorus metabolism in sediments and the biogeochemical cycling of phosphorus have been central issues in lake ecosystems, but the analysis of microbial community characteristics and influential factors of phosphorus metabolic processes in sediments from highland saline lakes have not been well documented. In this study, metagenome sequencing technology was used to analyze the diversity of community characteristics and the relationship between nitrogen and phosphorus functional genes of microorganisms involved in phosphorus metabolic processes in Daihai sediments under high saline conditions, as well as the response of microorganisms involved in phosphorus metabolic processes to changes in environmental factors, including salinity, pH, and the N/P ratio. The results showed that 1) salinity had positive correlations with Bacteroidetes involved in the major metabolic pathways (ko00030, ko00562, and ko00190) and positively correlated with the specific dominant bacteria involved in the minor pathways (ko00564 and ko00440); 2) norB and nirS denitrification genes were involved in a major component of phosphorus metabolism, and these functional genes of nitrogen metabolism had significant (p < 0.05) effects on phytate hydrolysis, phosphate hydrolysis, and polyphosphate synthesis and decomposition; and 3) environmental factors influenced the diversity of the bacterial community characteristics in the order of salinity > N/P ratio > pH. This study provides new insights into the analysis of the causes of eutrophication and the current state of imbalance in the hydroecological structure of saline lakes on plateaus, as well as an indication of the interactions between the global lake nitrogen and phosphorus cycles.
Sediments are an important component of lake ecosystems and an important constituent in controlling phosphorus cycling and nutrient bioavailability in lakes (Ding et al., 2018; Wang 2019). Phosphorus in lake sediments greatly promotes water eutrophication. In general, phosphorus generally accumulates in sediments in lake waters through processes that include precipitation and adsorption. When conditions are suitable, phosphorus is released into the overlying water body through dissolution and desorption into the sediment interstitial water, causing the phosphorus concentration in the water body to rise, resulting in secondary pollution (Huang et al., 2011; Feng et al., 2020). In recent years, although there have been many studies on phosphorus in sediments, these studies are concentrated on freshwater lakes such as Lake Dianchi, Chaohu Lake, and Taihu Lake, while research progress on plateau salinized lakes is inadequate (Xie 2012; Yang 2015; Wang 2019). Simultaneous occurrence of eutrophication and salinity increase in Lakes, causing changes in the physical and chemical structure of the water ecosystem, drastic changes in biological composition, and a decline or even disappearance of biological communities; this results in an imbalance of the water ecological structure, causing a sharp decline in the biomass of submerged plants and planktonic algae in lakes, the extinction of large fish, and the reduction of benthic organisms (Xu et al., 2020; Yang 2020; Pang et al., 2021; Zhang et al., 2021).
Saline lakes are extreme environments in terms of sustaining life, yet they are inhabited by rich, diverse, and functionally specific microbial communities (Wang 2019; Jiang et al., 2022; Yang 2015; Xie 2012). Studying the composition of microbial populations in natural environments can contribute to the understanding of the ecological functions of microbial communities. It is notable that extensive research has been devoted to the study of microbial community composition and its impact on biogeochemical cycle processes (Ren et al., 2017; Luo et al., 2020; Kinsman et al., 2017). In line with previous studies, these microorganisms perform degradation and biogeochemical cycling of nutrients such as carbon, nitrogen, phosphorus, and sulfur in water ecosystems through metabolic processes such as assimilation and dissimilation, and play a crucial role in sediment phosphorus cycling (Nelson et al., 2016; Kuypers et al., 2018; Li et al., 2022). In addition, Ren et al. (2017) have shown that changes in the soil microbial community composition were significantly correlated with the N/P ratio and microbial biomass, while the response of microbial activity to environmental changes was also associated with ecological resource imbalance. In the experiment of Huang et al. (2011), authors investigated the effect of microbial activity on sediment phosphorus through culture experiments and concluded that sediment microorganisms play an important role in lake phosphorus cycling and that ecological restoration can be conducted through microorganisms to subsequently restore lake water quality. Nevertheless, the microbial structure of sediments and their functions are influenced by environmental factors. Salinity is a key environmental factor affecting the microbial activity and community composition in natural environments, and with increasing salinity, sediment microbial activity decreases and community composition varies significantly (Lozupone and Knight. 2007). Yang et al. (2019) believed that salinity has a significant impact on microbial communities in different ecosystems, and the effect of salinity increases the connectivity and complexity of microbial networks, providing a valuable perspective on microbial population interactions (Tao et al., 2021). Recent experiments in this area suggested that environmental factors influence the physiological state of microorganisms at the level of species abundance, thus altering the structural composition of microbial communities (Ouyang et al., 2020; Zhang et al., 2022; Cao et al., 2016). However, the great diversity of extant microbial communities at the functional level makes the study of microbial ecological functions challenging (Jiang et al., 2010; Yang et al., 2020; Tao et al., 2021). At present, research on microbial communities and microbially driven biogeochemical cycles is still in its infancy (Kinsman et al., 2017; Banerjee et al., 2018; Zhu et al., 2021). Several studies involving soil, lake, and human symbiotic microbial communities have shown that although some specific metabolic functions are associated with environmental factors, there does not appear to be spatial and temporal specificity in the metabolic characteristics of most microorganisms (Campos et al., 2021; Hongxia et al., 2021; Zhang et al., 2021a).
Daihai Lake basin is an important barrier for ecological and environmental security in northern China and sensitive to environmental turbulence. It is a typical saline lake in plateau area, which is great of significance for the study of phosphorus metabolic processes and microbial community. Therefore, this study applied metagenome sequencing technology to sediment samples collected from five sample sites in the Daihai Lake basin. In this study, we hypothesized that salinity has a strong influence on the functional genes of nitrogen and phosphorus metabolism. And also different environmental factors (such as pH and the N/P ratio) are closely related to changes in the structure of the microbial communities involved in phosphorus metabolism. Therefore, we focused on 1) analyzing the changes in the microbial community characteristics of microorganisms involved in phosphorus metabolic processes in Daihai sediments under the influence of salinity, 2) comparing the variability of biomass at different sample sites under changes in different environmental factors, and 3) analyzing the response of environment factors to phosphorus metabolism microbial community structure and functional genes for nitrogen and phosphorus. These results will help to determine whether the changes in the community structure of microorganisms involved in the phosphorus metabolic process in the sediments of salinized lakes are closely related to changes in environmental factors and whether the response between salinity and other environmental factors is related to the current state of imbalance in the ecological water structure of Daihai Lake.
Daihai Lake (112°10′–112°59′E, 40°11′–40°55′N) is located in Liangcheng County, Ulanqab City, Inner Mongolia Autonomous Region, China. It is surrounded by mountains and is a typical closed inland saltwater lake. The annual rainfall in the basin is 350–450 mm. The main methods by which water is supplied to the lake are precipitation, surface runoff, and underground recharge, which provide insufficient water replenishment, resulting in increased salinity and continuous deterioration of water quality in Daihai Lake. The water surface area of the lake has shrunk sharply. The water surface area of Daihai Lake dropped from 115.94 km2 in 1989 to only 48.3 km2 in 2020 (Pang et al., 2021; Zhang et al., 2021). Daihai Lake is the third largest inland lake in Inner Mongolia. Together with Hulun Lake and Lake Ulansuhai, these water bodies are collectively referred to as “One Lake, Two Seas” and have important roles in climate regulation, ecological restoration, and water conservation, and also form an important part of China’s northern ecological security barrier (Yang 2020; Pang et al., 2021; Zhang et al., 2021).
In this study, sediments in Daihai Lake were collected in August 2020 according to the lake pollution characteristics to investigate the microbial population structure. We used a grab bucket to collect surface sediment samples. Furthermore, three parallel samples were taken from each point, mixed thoroughly, and stored in a sterilized polyethylene ziplock bag to ensure a sterile environment. Five sampling points (DH1, DH2, DH3, DH4, and DH5) were deployed from different locations of the lake (Figure 1). The collected samples were divided into two parts. One part was stored in dry ice and sent to Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) for metagenomics testing to analyze the microbial community characteristics and diversity. The other part of the sample was naturally air-dried, ground into a powder, and passed through a 100-mesh sieve for laboratory testing of various physicochemical indexes.

FIGURE 1. Sampling point locations in the Daihai watershed.
The physicochemical indexes of sediments included pH, electric conductivity (EC), salinity, total nitrogen (TN), total phosphorus (TP), ammonia nitrogen (NH4+-N), and nitrate nitrogen (NO3−-N). The specific detection methods and detection limits are shown in Table 1. All the experiments were performed in triplicate. Determination of pH: Water was used as extractant, the ratio of water to soil was 2.5:1, the temperature of the sample was controlled at (25 ± 1) °C, and the difference between the temperature of the sample and the temperature of the standard buffer solution should not exceed 2°C. The indicator electrode and the reference electrode (or pH composite electrode) were immersed in the soil suspension. The electrode probe was immersed in 1/3–2/3 of the vertical depth of the suspension under the liquid surface, and the sample was gently shaken. After the reading was stable, the pH value was recorded.

TABLE 1. Sediment physical and chemical indicator testing methods and detection limits.
Determination of EC: Natural air-dried soil samples were taken, water was added in a ratio of 1:5 (m/V), and shocked at 20 ± 1°C. The electrical conductivity of the extract was measured at 25 ± 1°C. When two electrodes are inserted into the extracting solution, the resistance between the two electrodes can be measured. When the temperature is constant, the resistance value R is inversely proportional to the conductivity K, namely R = Q/K. When the conductivity cell constant Q is known, the conductivity can be obtained by measuring the resistance of the extracting solution.
Salinity determination: The ratio of water to soil was 5:1. The sample with dry weight of 6 g was placed in a 50 ml centrifuge tube, and 30 ml of ultrapure water was added. After shaking for 3 min, the supernatant was taken after centrifugation, filtered with a filter membrane with a pore size of 0.45 μm, and the salinity of the soil was determined by a salinometer.
Determination of TN: The principle is to take the sample 0.2000g–1.0000 g (about 1 mg nitrogen) into the Kjeldahl nitrogen digestion bottle, under the action of sodium thiosulfate, concentrated sulfuric acid, perchloric acid and catalyst, all converted into ammonium nitrogen by redox reaction. The ammonia distilled from the alkaline solution after digestion was absorbed by boric acid, and titrated with standard hydrochloric acid solution (c = 0.01 mol/L). The total nitrogen content in soil was calculated according to the amount of standard hydrochloric acid solution.
Determination of TP: The principle is that the phosphorus-containing minerals and organic phosphorus compounds in the sample are all converted into soluble orthophosphate by sodium hydroxide melting, and react with molybdenum antimony anti-colorant to form phosphomolybdate blue under acidic conditions. Take 10.0 ml in a 50 ml stopper colorimetric tube, then add two to three drops of 2,4-dinitrophenol indicator, and then adjust the pH value to about 4.4 with 1 + 1 sulfuric acid solution and 2 mol/L sodium hydroxide solution to make the solution yellowish, and then add 1.00 ml ascorbic acid and mix well. After 30 s, 2.0 ml molybdate solution was added and placed at 20–30°C for 15 min. The absorbance was measured at a wavelength of 700 nm.
Determination of NH4+-N: 10.0 ml of the sample was taken and put into a 100 ml stopper colorimetric tube, 40 ml of sodium nitroprusside-phenol chromogenic agent was added to it, fully mixed, and stood for 15 min, then 1.00 ml of sodium dichloroisocyanurate chromogenic agent was added to it, fully mixed, and stood for at least 5 h at 15–35°C. Based on the principle that under alkaline conditions, the ammonia ion in the potassium chloride extract reacts with phenol in the presence of hypochlorite ion to produce blue indophenol dye, it has the maximum absorption at 630 nm wavelength.
Determination of NO3−-N: Take 1.00 ml of the sample to the reduction column, add 10 ml of ammonium chloride buffer solution, then open the piston, pass through the reduction column at a flow rate of 1 ml/min, and collect the eluent with a 50 ml stopper colorimetric tube. When the liquid level reaches the top of the cotton, 20 ml ammonium chloride buffer solution is added to collect all the effluent and remove the colorimetric tube. Finally, the reduction column was cleaned with 10 ml ammonium chloride buffer solution. Add 0.2 ml chromogenic agent to the above colorimetric tube, fully mixed, and stand at room temperature for 60 min to 90 min. The absorbance was measured at 543 nm wavelength with water as reference.
Sample DNA extraction was performed using the E. Z.N.A.® Soil DNA Kit (Omega Bio-tek, United States). After DNA extraction, DNA concentration and purity were determined, and DNA integrity was examined using 1% agarose gel electrophoresis. DNA was fragmented by using a Covaris M220 ultrasonicator (Gene Corporation, China). Fragments of about 400 bp were screened, and PE libraries were constructed using the NEXTFLEX Rapid DNA-Seq Library Prep kit (PerkinElmer, United States). Major Bio-Pharm Technology Co., Ltd. (Shanghai, China) was commissioned to perform sequencing, statistics, and data quality control on the samples using the Illumina sequencing platform. After quality control, the optimized sequences (clean data) were single-spliced and assembled using the software MEGAHIT (Li et al., 2015) (https://github.com/voutcn/megahit, version 1.1.2), and contigs ≥300 bp were selected as the final assembly results among the spliced results. The open reading frames of contigs in the splicing results were predicted using Prodigal (Hyatt et al., 2010) and MetaGene (Noguchi et al., 2006) (http://metagene.cb.k.u-tokyo.ac.jp/), and genes with nucleic acid lengths greater than or equal to 100 bp were selected and translated into amino acid sequences. The amino acid sequences of the non-redundant gene sets were compared with the NR database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/) using Diamond (Buchfink et al., 2015) (http://www.diamondsearch.org/index.php, version 0.8.35), and species annotations were obtained from the taxonomic information database corresponding to the NR database. The sum of gene abundances corresponding to the species was used to calculate the species abundance, which was used to obtain the microbial community composition in sediment samples and their diversity. Finally, the raw data sequences were deposited in the NCBI GenBank Sequence Read Archive (SRA) under accession numbers: PRJNA877385.
The raw data were preprocessed by Excel (2019) (Microsoft, United States) and drew with OriginPro 2021 (Origin Lab, United States) and ArcGIS Desktop 10.8 (Environment System Research Institute, United States). Data was checked for normality and skewed data was log transformed prior to analysis. Statistical comparisons of differences among different sampling sites in the three groups of repeated experiments were analyzed by One-way ANOVA (Analysis of variance) and post-hoc tests (Tukey’s HSD) using SPSS 26.0 software (IBM, United States). And SPSS 26.0 software (IBM, United States) was also used to perform Spearman correlation analysis to analyze the correlation between environmental factors and species as well as functional genes. The significance was set at p < 0.05. Redundancy analysis (RDA) was performed using Canoco 5.0 and Monte Carlo tests were used to analyze the significant differences between sediment parameters and microbial communities. Metabolic pathway map download from KEGG database.
There were differences in the sediment nutrient salt levels at different sampling points in the Daihai basin (Figure 2), which is consistent with the results of some studies that have shown that physicochemical factors such as pH, salinity, and N/P ratios in different environments can influence the distribution and composition of different microbial communities (Wu 2019; Peng et al., 2021; Zhang 2021). The pH of the entire lake ranged from 8.95 to 9.25 and was overall alkaline, with the highest alkalinity at point DH5 in the lake center. The salinity ranged from 14.11 to 20.32 g/kg, with DH4 having the highest salinity content and DH2 the lowest. The EC of the whole lake was in the range of 3490–6730 μs/cm, and the EC of DH3, DH4, and DH5 was significantly higher than that of DH1 and DH2. The TN content ranged from 1,632.18 to 2,975.64 mg/kg, with DH5 and DH2 being slightly higher than the other sampling sites, with significant differences between sites (p < 0.05). The TP levels ranged from 786.28 to 998.32 mg/kg, which were lower than the TN levels, and the DH5, DH2, and DH4 levels were significantly higher than the DH1 and DH3 levels (p < 0.01). The NH4+-N and NO3−-N levels were generally low in the sediment, both of which were highest in DH4 and lowest in DH3. The changing trend of the N/P ratio at each point was very obvious (p < 0.05). The ratio of DH4 was the lowest, and the ratio of DH5 was the highest. The N/P ratio of DH1, DH2, and DH3 was consistent. Overall, the present findings confirm there was some spatial variation in the physical and chemical indicators in the horizontal direction. The effect of pH on the sediments of the five sample sites in the Daihai basin was generally consistent, suggesting that the response to pH changes were available throughout the lake, and also reflected the important role of pH in the management of the water environment in the Daihai basin. The remaining physicochemical factors showed variability across geographic locations, and therefore the effect on microorganisms varied between sample sites in the Daihai sediments. A long-standing study on the diversity and structure of microbial communities in lakes in different regions found that environmental factors have an impact on microorganisms and lake ecology, with salinity being an important environmental factor in determining the distribution of lake microbial communities, with pH and the N/P ratio also playing an important role in influencing the characteristics of the lake microbial communities (Jackson and Vallaire 2009; Yang et al., 2020; Huang 2021).
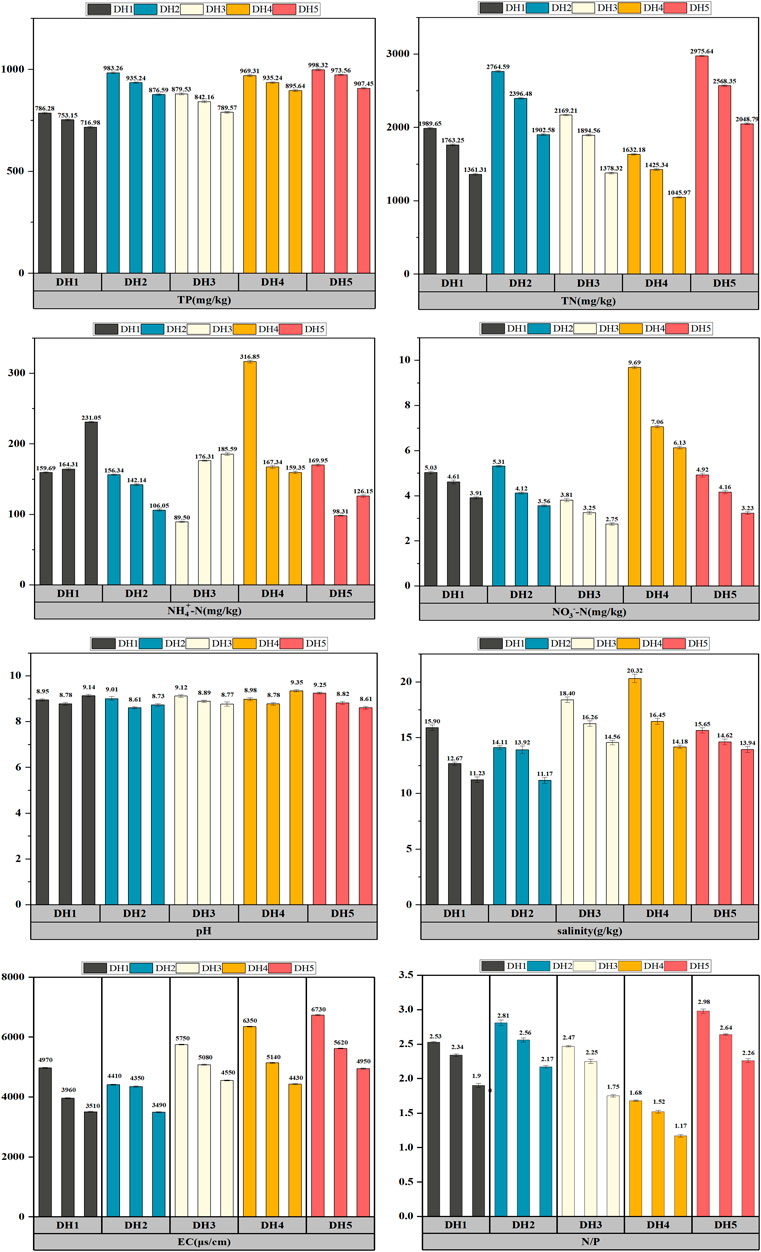
FIGURE 2. Characteristics of the variation of physicochemical factors in the sediments of Daihai Lake.
Species annotation statistics of metagenome sequences indicated that the sediment microbial communities in Daihai Lake had a rich species composition. In terms of microbial composition, all samples were dominated by Bacteria, with relative abundances ranging from 62.5% to 76.4%, followed by Archaea, with relative abundances ranging from 7.0% to 10.0%, significantly lower than those of Bacteria (Figure 3A). During the research process, the microbial composition at the phylum and genus levels was regular and representative across samples. Therefore, the top 10 microorganisms at the phylum level in terms of relative abundance were selected to characterize the community composition of microorganisms involved in the process of phosphorus metabolism (Figure 3B). As shown in Figure 4, the main metabolic pathways involved in phosphorus metabolism were identified as the pentose phosphate pathway (ko00030), inositol phosphate metabolism pathway (ko00562), glycerophospholipid metabolism pathway (ko00564), phosphonate and phosphinate metabolism pathway (ko00440), and oxidative phosphorylation pathway (ko00190). The community composition of microorganisms involved in phosphorus metabolism in all of the samples at the phylum level consisted mainly of Proteobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Firmicutes, and Euryarchaeota, with the remaining bacterial phyla classified as “Others.” Proteobacteria was the most dominant phylum of the microbial community. The ko00030 and ko00562 pathways, which are the basic metabolic pathways for phosphate, also included Cyanobacteria, Nitrospirae, and Planctomycetes as the most dominant phyla in these pathways. The remaining pathways for the synthesis of cellular substances also had their own unique composition of species that provided most of the activity in that pathway, also referred to as “key species” for a given metabolic pathway (Banerjee et al., 2018). For example, ko00564, which is involved in lipid metabolism, also included Cyanobacteria, Nitrospirae, and Verrucomicrobia; ko00190, which is involved in energy metabolism, also included Spirochaetes and Planctomycetes; and ko00440, which is involved in other amino acid metabolism, also included Nitrospirae, Planctomycetes, and Nitrospinae. In summary, Proteobacteria, Chloroflexi, Firmicutes, Actinobacteria, and Bacteroidetes were the most important phyla in phosphorus metabolism, which is consistent with the results of other domestic and international studies (Cao et al., 2016; Yang et al., 2020; Zhou et al., 2020; Zhang et al., 2022; Zhang et al., 2021b). In general, the relative abundances of the dominant phyla in the phosphorus metabolic process were relatively uniform among the different sampling points, and only the ko00440 pathway showed a large spatial variation.
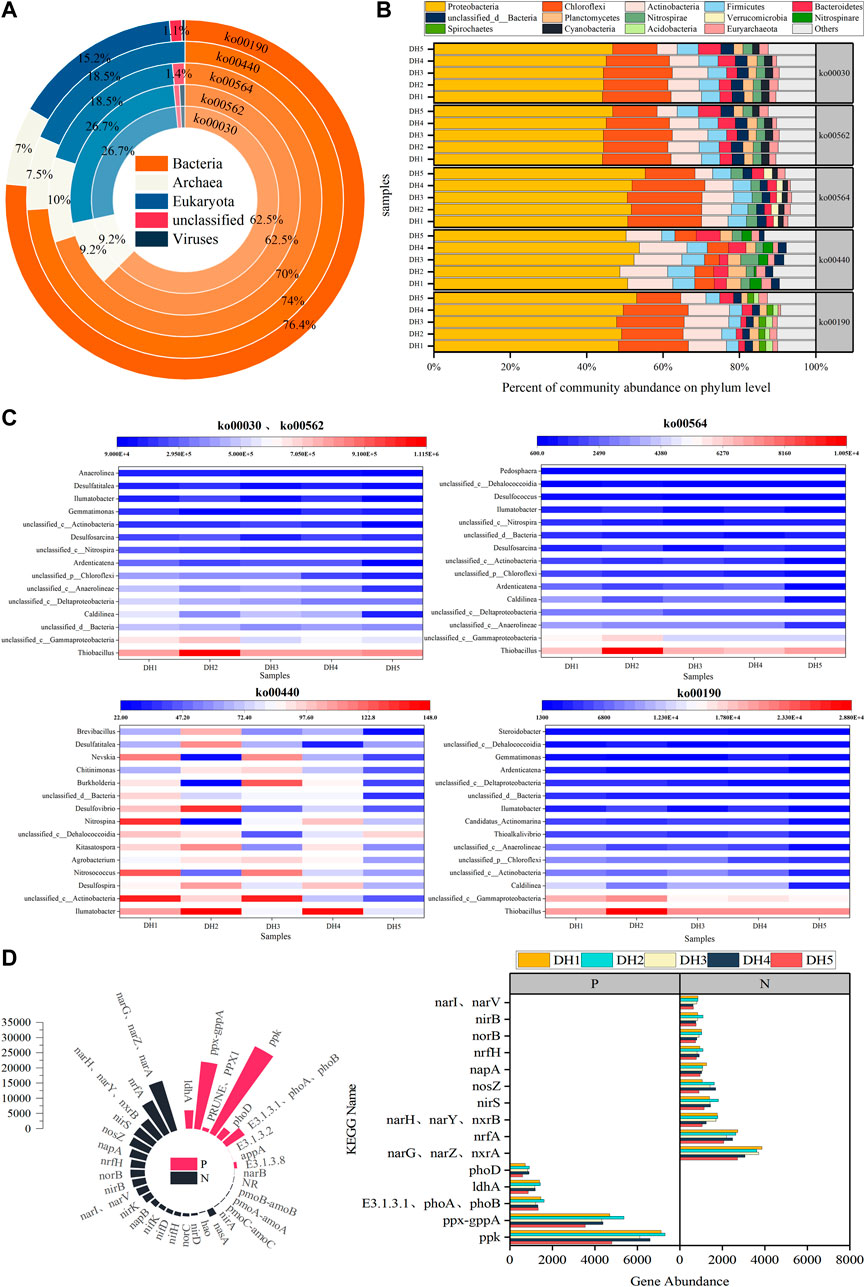
FIGURE 3. Distribution of the microbial community composition during phosphorus metabolism in Daihai sediments at the (A) domain level, (B) phylum level, and (C) genus level. (D) Distribution of nitrogen and phosphorus functional genes during phosphorus metabolism.
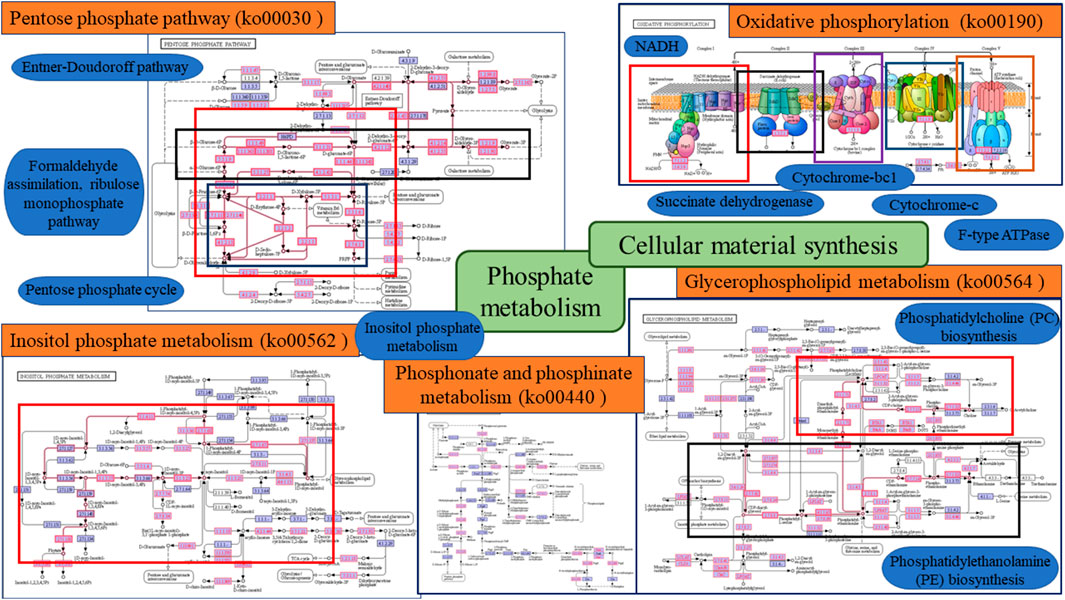
FIGURE 4. Diagram of phosphorus metabolic pathway in Daihai sediments.
Based on the results of metagenome species annotation, the bacterial community composition at the genus level was mapped for the top 15 genera belonging to Proteobacteria, Chloroflexi, Actinobacteria, Nitrospirae, Gemmatimonadetes, Verrucomicrobia, and Firmicutes in the five phosphorus metabolism-related pathways (Figure 3C). The results showed that the ko00030 and ko00562 pathways contributed the most to phosphorus metabolism and also had a consistent species composition, with unclassified Gammaproteobacteria, unclassified Deltaproteobacteria, Thiobacillus, and unclassified Bacteria as the dominant genera at each point in the pathway, while unclassified Anaerolineae and Caldilinea had significant spatial variability at different sample sites. For the metabolic pathways ko00564 and ko00190, the different sampling sites had a high similarity in species composition and the same dominant genera, such as Thiobacillus and unclassified Gammaproteobacteria; however, the variability in the spatial distribution according to sampling sites of the ko00190 pathway as a whole was more significant compared to the ko00440 pathway. In contrast, the ko00440 metabolic pathway was represented by low levels of different genera of bacteria at each sampling site and therefore generally did not contribute to the dominant bacteria. Overall, the most dominant genus in the entire phosphorus metabolic pathway was Thiobacillus, and unclassified Deltaproteobacteria, Caldilinea, and unclassified Anaerolineae also influenced the phosphorus metabolic process.
Phosphorus metabolic processes are mainly divided into two parts: phosphate metabolism and cellular material synthesis (Figure 4). Functional genes in the ko00030, ko00562, and ko00440 pathways involved in phosphate metabolism encode proteins involved in carbohydrate metabolism and amino acid metabolism, while the ko00190 and ko00564 pathways involved in cellular substance synthesis belong to the functional classification of energy metabolism and lipid metabolism. Among these, energy metabolism is central to microbial metabolism and therefore the ko00190 pathway played an important role in phosphorus metabolism. Studies have shown that there are multiple enzymes and genes involved in phosphorus metabolism. The functional genes predicted in the metagenome analysis were compared with the KEGG database, and the functional genes that were more closely involved in the process of phosphorus cycling in the Daihai sediments are shown in Table 2. Combined with Figure 3D, the abundance of functional genes related to phosphorus metabolism in the Daihai sediments, its functional genes control and synthesize phytase; alkaline phosphatase and acid phosphatase; polyphosphate kinase and exopolyphosphatase, which are involved in phytate hydrolysis; phosphate hydrolysis; polyphosphate synthesis; and exopolyphosphatase (Zhou et al., 2020).

TABLE 2. Functional genes related to phosphorus metabolism detected in Daihai sediments.
Some responses between physicochemical indicators and dominant bacterial communities existed in the surface sediments of the Daihai basin (Figure 5), but the effects were not consistent across metabolic pathways and sample sites. With the help of Monte Carlo tests (Table 3), results demonstrate that EC was the main factor affecting bacterial activity in the related pathways of phosphorus metabolism, and related studies have shown that EC was also an important index to measure salinity in sediments in previous research. Therefore, under the strong influence of EC, we speculate that salinity affects the composition and structure of the phosphorus metabolic microbial community (McKinney et al., 2019; Joo H W et al., 2021; de Santana et al., 2022). Although salinity did not directly show the strong influence degree in the data results, salinity, as an important environmental factor index, is still a hot issue worthy of discussion under the positive influence of EC. Similarly, N/P ratio and pH also play different roles in the different metabolic pathways, and the contribution of pH is slightly greater than that of N/P ratio (Luo et al., 2020; Zhu et al., 2021; Zhang et al., 2022; Jackson et al., 2009). The results of the analysis showed that the content of the microbial community at the different sample sites showed spatial and temporal variability, and there was no clear similarity between the sites. Overall, EC, pH, salinity, and the N/P ratio at different sites had an influence on the phosphorus metabolic processes, but the ko00030 and ko00562 pathways had more significant effects based on their relative abundance. In the major pathways ko00030, ko00562, and ko00190, salinity all showed a positive correlation with Bacteroidetes, but negatively with Euryarchaea, where the ko00190 pathway also showed a positive correlation with Actinobacteria, Chloroflexi, and Spirochaetes. Under the influence of EC, the effect of pH and N/P ratio on each dominant phylum was less than that of salinity. And pH showed a positive correlation with Bacteroidetes in each pathway, while N/P ratio showed a negative correlation only in the two pathways ko00030 and ko00562 with the highest contribution. In each pathway, salinity was negatively correlated with N/P ratio uniformity, but positively correlated with pH in the ko00190 pathway. In the secondary pathways ko00564 and ko00440, salinity and pH were positively correlated with Bacteroidetes, and only N/P ratio was negatively correlated with Bacteroidetes in the ko00564 pathway. The effect of salinity on microbial community was greater than that of pH and N/P ratio. At this time, salinity was negatively correlated with N/P ratio and positively correlated with pH. According to the relative abundance figures of each pathway species, the metabolic function of ko00440 had the lowest response to the whole phosphorus metabolic process.
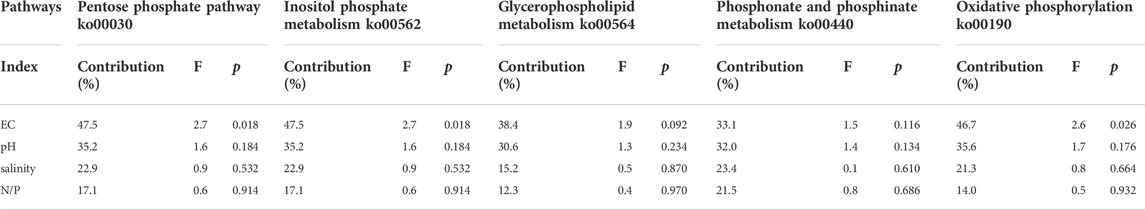
TABLE 3. Redundancy analysis (RDA) and Monte Carlo test results.
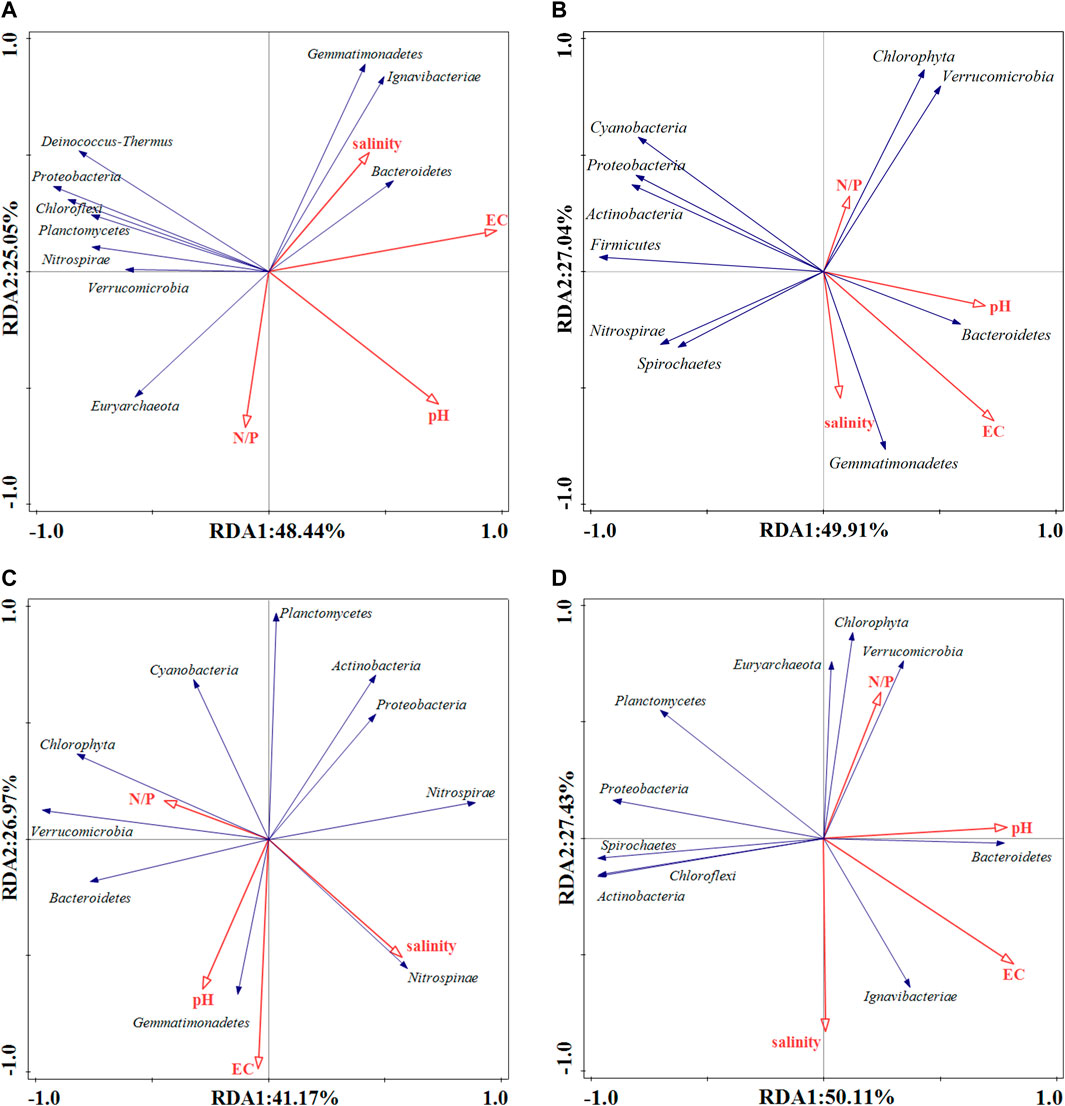
FIGURE 5. Three-sequence diagram of the redundancy analysis of phosphorus metabolism-related pathways in Daihai sediments at the phylum level for samples, species, and environmental factors. Analysis results for pathways (A) ko00030 and ko00562, (B) ko00564, (C) ko00440, and (D) ko00190.
As an important part of the ecological environment, the structural characteristics of the dominant bacterial flora and the changes in physicochemical indicators are very closely related (Jackson et al., 2009; Yu et al., 2022). Figure 6 reveals the relationship between species and environmental factors during phosphorus metabolism in Daihai sediments at the genus level. In the ko00030 and ko00562 pathways, Thioalkalivibrio, Desulfococcus, and unclassified Dehalococcoidia were significantly correlated with salinity (p < 0.05), and unclassified Actinobacteria showed a significant negative correlation with the N/P. In the ko00190 pathway, N/P ratio had a significantly negative correlation with Candidatus Actinomarina and Ardenticatena, and had significantly positive correlation with Desulfococcus (p < 0.01), and in ko00564 pathway, Desulfococcus had significantly positive correlation with N/P ratio. At the same time, EC showed a significant negative correlation with Anaerolinea. The unclassified Actinobacteria in the two pathways were significantly negatively correlated with N/P ratio, and only Thioalkalivibrio was significantly negatively correlated with pH in the ko00190 pathway. In the ko00440 pathway with the lowest response to phosphorus metabolism, salinity played the greatest role, which was significantly negatively correlated with Pelobacter, while Hydrogenophaga and Pelobacter were significantly positively correlated with salinity and N/P ratio, respectively. Desulfococcus was significantly negatively correlated with salinity. Due to the significant effects of N/P ratio and salinity on genus level bacteria during the whole phosphorus metabolism process, it can be speculated that the two have an irreplaceable role in the lake ecosystem. In the related pathways of phosphorus metabolism, N/P ratio and salinity had obvious effects on genus level bacteria, and only pH was observed in the ko00190 pathway.
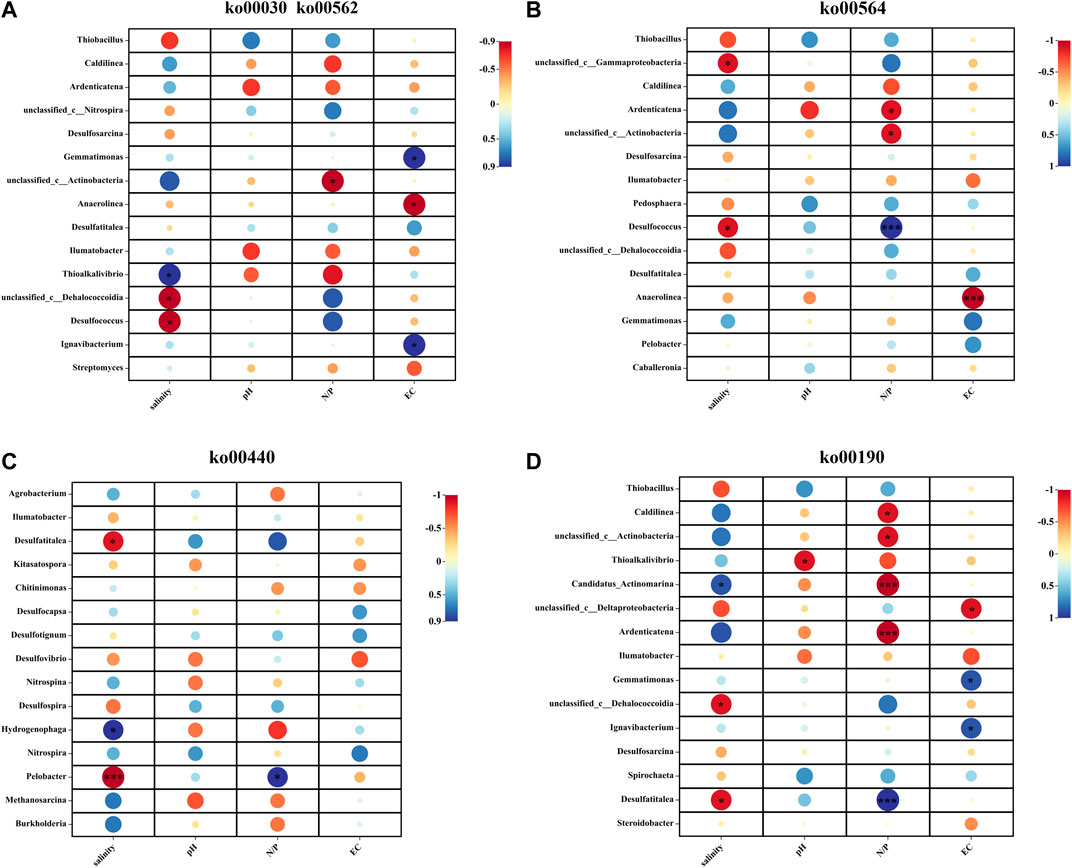
FIGURE 6. Heat map of correlations between samples, species, and environmental factors at the genus level for phosphorus metabolism-related pathways in Daihai sediments. Analysis results for pathways (A) ko00030 and ko00562, (B) ko00564, (C) ko00440, and (D) ko00190.
Therefore, the magnitude of the influence of environmental factors on the diversity of the bacterial community characteristics during phosphorus metabolism in Daihai sediments was ranked as salinity > N/P ratio > pH. Numerous studies have shown that salinity is the main factor controlling microbial communities in saline lakes, as the energy metabolic processes of microorganisms in saline environments are limited by the osmotic pressure of salinity (Xie 2012; Yang 2015; Yang et al., 2020; Yu et al., 2022). In Daihai sediments, salinity had a positive correlation with most bacterial communities, and changes in salinity led to changes in the microbial community composition as well as a loss of biodiversity, which affected the biochemical cycling of carbon, nitrogen, and phosphorus in the ecosystem, leading to an imbalance in the water ecological structure of Daihai Lake.
It is notable that functional genes annotated in sediment phosphorus metabolism may play an important role in the survival of microorganisms in lake bottom sediments (Zhou et al., 2020). For example, phytic acid is an organophosphate compound widely found in plant roots. Plant decay in the water column enters the sediment, and therefore the sediment also contains phytic acid, which is a “reservoir” of phosphorus. However, the phosphorus cannot be used directly by algae and instead must be converted into orthophosphate through a series of hydrolyses, thus generating an increase in bioavailable phosphorus in the water column (Feng et al., 2016; Feng et al., 2018; Zhou et al., 2020). The metagenome annotations results indicated the low levels of phytate hydrolases in the Daihai sediments, and reflected the serious ecological degradation in the Daihai basin. These problems are mainly due to the frequent occurrence of diseases that have led to fish species decline or even disappearance, and the continuous reduction of benthic organisms and submerged plants, resulting in an imbalance in the lake water ecosystem, which intrinsically operates at a low level year round. It is speculated that this may be related to the granulation of phosphorus and the morphological characteristics of phosphorus, which to a certain extent limits the N/P ratio of the lake and thus directly affects the survival of algae in Daihai Lake.
The PHO, aphA, phoN, phoA, phoB, and phoD genes encoding phosphate hydrolases were predicted in the samples, but the relative abundances of genes encoding for phosphate hydrolysis functions were higher for alkaline phosphatases than for acid phosphatases in the Daihai sediments. The ppk gene encoding polyphosphate kinase and the PPX1 and ppx genes encoding epithelial polyphosphatase were predicted in all five sediment samples. The relative abundance of the ppk gene was higher than that of ppx, and the activity of polyphosphate kinase was slightly higher than that of epithelial polyphosphatase at different sampling sites. ppk and ppx encode for key enzymes in the respective synthesis and decomposition of polyphosphate, and their activities are directly related to the ability to perform phosphorus removal, anaerobic phosphorus release, and aerobic phosphate uptake (Peng et al., 2021; Xiao et al., 2016; Zhao et al., 2011; Zheng et al., 2013). According to the results of the KEGG database, the functional potential of genes for phosphorus cycling in different lake areas differed slightly, and reference to previous studies revealed that elemental nitrogen and phosphate were closely related to the taxonomic and functional changes of microorganisms (Luo et al., 2020; Nelson et al., 2016; Wu et al., 2022; Song et al., 2021). In the entire phosphorus metabolic process, only two pathways, ko00030 and ko00562, could be annotated nitrogen metabolism-relevant genes (Figure 3D). There were 18 types of genes common to both pathways, and the represented enzymes were all oxidoreductases. The ko00562 pathway had seven nitrogen metabolism genes specific to its pathway, also annotated as oxidoreductases. This further demonstrates that the biogeochemical cycling of sediment phosphorus is closely related to the nitrogen cycle. The results showed that the key functional genes of the nitrogen cycling process, including the nif genes of the nitrogen fixation pathway (nif D/H/K), the amo A/B/C genes of the nitrification pathway, the nir and nor genes of the denitrification pathway, and the nrfA gene of the anaerobic ammonia oxidation process, all showed significant spatial and temporal abundance differences in the Daihai sediments. Studies have shown that narG, nirS, and norB are the main functional genes closely associated with the sediment nitrogen cycle (Nelson et al., 2016; Kuypers et al., 2018; Wu et al., 2022; Song et al., 2021). narG encodes a nitrate reductase [EC: 1.7.5.1 1.7.99.4] that catalyzes the reduction of NO3− to NO2− by denitrification; nirS encodes a cytochrome-containing cd1 nitrite reductase [EC: 1.7.2.1 1.7.99.1] that catalyzes the reduction of NO2− to NO through denitrification; and the NO reductase [EC: 1.7.2.5] encoded by norB also catalyzes the reduction of NO to N2O through denitrification. Thus, functional genes for denitrification in microorganisms play an important role in phosphorus metabolism. Similarly, among the nitrogen metabolism genes specific to the ko00562 pathway, only the nasA gene was present in high abundance, while the remaining nirA, pmoC-amoC, pmoA-amoA, pmoB-amoB, NR, and narB genes had very low abundance, further concluding that these six specific genes have minimal influence on the process of phosphorus metabolism.
Based on the main functional genes of nitrogen metabolism, narG, nirS, and norB, a Spearman correlation analysis was conducted between the three genes and related functional genes of phosphorus metabolism, including the KEGG functional orthologs K01083, K01093, K01078, K01077, K01113, K00937, K01514, and K01524, and the relationships between these were analyzed (Table 4). There was a very significant (p < 0.01) positive correlation between K01093 and norB and between K01113 and nirS, while there was a significant (p < 0.05) positive correlation between K01524, K01078, and K00937 and norB. The denitrifying genes nirS and norB had a strong influence on phosphorus metabolism, and norB had a specific influence on K01083, a phytic acid hydrolase. Additionally, norB played a role in the hydrolysis of phosphoric acid via an acid phosphatase gene (K01078) and the synthesis and decomposition of polyphosphate by K01524 and K00937, while nirS had a significant influence on the promotion of phosphoric acid hydrolysis via an alkaline phosphatase gene (K01113).

TABLE 4. Spearman correlation analysis.
(1) Surface sediments were collected from Daihai Lake during the summer at sites with high salinity variability, and the bacterial taxa in the phosphorus metabolic process in the Daihai sediments were identified mainly as Proteobacteria, Chloroflexi, Firmicutes, Actinobacteria, and Bacteroidetes. The dominant genus was Thiobacillus.
(2) The pentose phosphate pathway (ko00030) and inositol phosphate metabolism pathway (ko00562) had major roles in the process of phosphorus metabolism, and different bacterial compositions selected for different metabolic pathways, which were related to bacterial functional genes and sediment physical and chemical properties. Nitrogen metabolism was linked to phosphorus metabolism, and there was interaction between norB and nirS genes with the denitrification and functional genes of phosphorus metabolism, which once again verified the conjecture that effective control of lake eutrophication requires simultaneous control of nitrogen and phosphorus nutrients.
(3) In major metabolic pathways (ko00030, ko00562, ko00190), salinity had positive correlations with Bacteroidetes and negatively correlated with Euryarchaeota. The ko00190 pathway also showed positive correlation with Actinobacteria, Chloroflexi, and Spirochaetes. In the secondary pathway (ko00564 and ko00440), salinity and pH were positively correlated with Bacteroidetes, only the N/P ratio was negatively correlated with Bacteroidetes in the ko00564 pathway, and environmental factors influenced the diversity of the bacterial community characteristics in the order of salinity > N/P ratio > pH.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA877385).
WY: conceptualization, methodology, and Resources. JM: data curation, validation, and writing—original draft. YZ: visualization. WL: supervision, and project administration. ZY: investigation. WF: writing—review, and editing.
This work was supported by the National Natural Science Foundation of China (Grant no. 42167018) and the Inner Mongolia Autonomous Region Science and Technology Plan Project (Grant no. 2021GG0410).
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Banerjee, S., Schlaeppi, K., and van der Heijden, M. G. A. (2018). Keystone taxa as drivers of microbiome structure and functioning. J. Nat. Rev. Microbiol. 16 (9), 567–576. doi:10.1038/s41579-018-0024-1
Buchfink, B., Xie, C., and Huson, D. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12 (1), 59-60. doi:10.1038/nmeth.3176
Campos, M., Rilling, J. I., Acuña, J. J., Valenzuela, T., Larama, G., Peña-Cortés, F., et al. (2021). Spatiotemporal variations and relationships of phosphorus, phosphomonoesterases, and bacterial communities in sediments from two Chilean rivers. J. Sci. Total Environ. 776, 145782. doi:10.1016/j.scitotenv.2021.145782
Cao, H., Chen, R., Wang, L., Jiang, L., Yang, F., Zheng, S., et al. (2016). Soil pH, total phosphorus, climate and distance are the major factors influencing microbial activity at a regional spatial scale. J. Sci. Rep. 6 (1), 1–10. doi:10.1038/srep25815
de Santana, C. O., Spealman, P., Azulai, D., Reid, M., Dueker, E. M., and Perron, G. G. (2022). Bacteria communities and water quality parameters in riverine water and sediments near wastewater discharges. Sci. Data 9 (1), 1–8. doi:10.1038/s41597-022-01686-8
Ding, S., Zhang, Q., Dong, J., Chen, Z., and Chen, S. (2018). Microbial community structure and its relationship to heavy metals in Shenzhen and Hong Kong mangrove sediments. J. Chin. J. Ecol. 37 (10), 3018–3030. doi:10.13292/j.1000-4890.201810.009
Duarte, B., Freitas, J., and Cacador, I. (2012). Sediment microbial activities and physic-chemistry as progress indicators of salt marsh restoration processes. J. Ecol. Indic. 19, 231–239. doi:10.1016/j.ecolind.2011.07.014
Feng, W., Wu, F., He, Z., Song, F., Zhu, Y., Giesy, J. P., et al. (2018). Simulated bioavailability of phosphorus from aquatic macrophytes and phytoplankton by aqueous suspension and incubation with alkaline phosphatase. J. Sci. Total Environ. 616, 1431–1439. doi:10.1016/j.scitotenv.2017.10.172
Feng, W., Yang, F., Zhang, C., Liu, J., Song, F., Chen, H., et al. (2020). Composition characterization and biotransformation of dissolved, particulate and algae organic phosphorus in eutrophic lakes. J. Environ. Pollut. 265, 114838. doi:10.1016/j.envpol.2020.114838
Feng, W., Zhu, Y., Wu, F., Meng, W., Giesy, J. P., He, Z., et al. (2016). Characterization of phosphorus forms in lake macrophytes and algae by solution P-31 nuclear magnetic resonance spectroscopy. J. Environ. Sci. Pollut. Res. 23 (8), 7288–7297. doi:10.1007/s11356-015-5913-5
Fraser, M. W., Gleeson, D. B., Grierson, P. F., Laverock, B., and Kendrick, G. A. (2018). Metagenomic evidence of microbial community responsiveness to phosphorus and salinity gradients in seagrass sediments. J. Front. Microbiol. 9, 1703. doi:10.3389/fmicb.2018.01703
He, Q., Wang, H., Chen, L., Gao, S., Zhang, W., and Song, J. (2020). Elevated salinity deteriorated enhanced biological phosphorus removal in an aerobic granular sludge sequencing batch reactor performing simultaneous nitrification, denitrification and phosphorus removal. J. Hazard. Mater. 390, 121782. doi:10.1016/j.jhazmat.2019.121782
Hollister, E. B., Engledow, A. S., Hammett, A. J. M., Provin, T. L., Wilkinson, H. H., and Gentry, T. J. (2010). Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 4 (6), 829–838. doi:10.1038/ismej.2010.3
Hongxia, M., Jingfeng, F., Jiwen, L., Jie, S., Zhiyi, W., and Yantao, W. (2021). Full-length 16S rRNA gene sequencing reveals spatiotemporal dynamics of bacterial community in a heavily polluted estuary, China. Environ. Pollut. 275, 116567. doi:10.1016/j.envpol.2021.116567
Hu, M., Le, Y., Sardans, J., Yan, R., Zhong, Y., and Sun, D. (2022). Moderate salinity improves the availability of soil P by regulating P-cycling microbial communities in coastal wetlands. Glob. Change Biol. doi:10.1111/gcb.16465
Huang, J. (2021). The influence of salinity on the microbial community structure and functional stability in Qinghai-Tibetan lakes. D. China University of Geosciences. Wuhan, China. doi:10.27492/d.cnki.gzdzu.2021.000284
Huang, L., Du, S., Fan, L., Lin, X., Wang, H., and Zhang, Y. (2011). Microbial activity facilitates phosphorus adsorption to shallow lake sediment. J. J. Soils Sediments 11 (1), 185–193. doi:10.1007/s11368-010-0305-4
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., and Frischkorn, K. R. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1 (1), 1–17. doi:10.1186/2049-2618-1-22
Hyatt, D., Chen, G. L., Locascio, P. F., Lang, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. J. BMC Bioinforma. 11 (1), 119. doi:10.1186/1471-2105-11-119
Jackson, C. R., and Vallaire, S. C. (2009). Effects of salinity and nutrients on microbial assemblages in Louisiana wetland sediments. J. Wetl. 29 (1), 277–287. doi:10.1672/08-86.1
Jiang, H., Deng, S., Huang, Q., Dong, H., and Yu, B. (2010). Response of aerobic anoxygenic phototrophic bacterial diversity to environment conditions in saline lakes and Daotang River on the Tibetan Plateau, NW China. J. Geomicrobiol. J. 27 (5), 400–408. doi:10.1080/01490450903480269
Joo, H. W., Kwon, T. H., Lee, S. R., and Yuxin, W. (2021). Relaxation behavior in low-frequency complex conductivity of sands caused by bacterial growth and biofilm formation by Shewanella oneidensis under a high-salinity condition[J]. Geophysics 86 (6), B389–B400. doi:10.1190/geo2020-0213.1
Kinsman-Costello, L. E., Sheik, C. S., Sheldon, N. D., Burton, G. A., Costello, D. M., Marcus, D., et al. (2017). Groundwater shapes sediment biogeochemistry and microbial diversity in a submerged Great Lake sinkhole. J. Geobiol. 15 (2), 225–239. doi:10.1111/gbi.12215
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. J. Nat. Rev. Microbiol. 16 (5), 263–276. doi:10.1038/nrmicro.2018.9
Li, D., Liu, C., Luo, R., Sadakane, K., and Lam, T. W. (2015). Megahit: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31 (10), 1674-1676. doi:10.1093/bioinformatics/btv033
Li, P., An, Q., Wang, X., Sun, S., Li, X., and Zheng, N. (2022). Analysis on diversity and structure of microbial community in river sediment of Siping section of Liaohe River. J/OL. Environ. Sci. 43 (5), 2586–2594. doi:10.13227/j.hjkx.202107032
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity[J]. Proc. Natl. Acad. Sci. U. S. A. 104 (27). doi:10.1073/pnas.0611525104
Luo, G., Xue, C., Jiang, Q., Xiao, Y., Zhang, F., Guo, S., et al. (2020). Soil carbon, nitrogen, and phosphorus cycling microbial populations and their resistance to global change depend on soil C: N: P stoichiometry. J. Msystems. 5 (3), 001622–e220. doi:10.1128/mSystems.00162-20
McKinney, R., Hanson, A., Johnson, R., and Charpentier, M. (2019). Seasonal variation in apparent conductivity and soil salinity at two Narragansett Bay, RI salt marshes. PeerJ 7, e8074. doi:10.7717/peerj.8074
Ming, H., Fan, J., Liu, J., Su, J., Wan, Z., Wang, Y., et al. (2021). Full-length 16S rRNA gene sequencing reveals spatiotemporal dynamics of bacterial community in a heavily polluted estuary, China. J. Environ. Pollut. 275, 116567. doi:10.1016/j.envpol.2021.116567
Nelson, M. B., Martiny, A. C., and Martiny, J. B. H. (2016). Global biogeography of microbial nitrogen-cycling traits in soil. J. Proc. Natl. Acad. Sci. 113 (29), 8033–8040. doi:10.1073/pnas.1601070113
Noguchi, H., Park, J., and Takagi, T. (2006). MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34 (19), 5623 5630. doi:10.1093/nar/gkl723
Ouyang, L., Chen, H., Liu, X., Wong, M., Xu, F., Yang, X., et al. (2020). Characteristics of spatial and seasonal bacterial community structures in a river under anthropogenic disturbances. J. Environ. Pollut. 264, 114818. doi:10.1016/j.envpol.2020.114818
Pang, X., Liu, H., Liu, X., Yu, X., Kou, X., Xu, Z., et al. (2021). Analysis of lake area and water lever dynamic and its driving forces of Daihai Lake in recent 30 years. J. J. Inn. Mong. Univ. Nat. Sci. Ed. 52 (03), 311–321. doi:10.13484/j.nmgdxxbzk.20210313
Peng, K., Dong, Z., Di, Y., and Guo, X. (2021). Contrasting analysis of microbial community composition in the water and sediments of the North Canal based on 16 rRNA high-throughput sequencing. J. Environ. Sci. 42 (11), 5424–5432. doi:10.13227/j.hjkx.202104122
Qin, B., Zhou, J., Elser, J. J., Gardner, W. S., Deng, J., and Brookes, J. D. (2020). Water depth underpins the relative roles and fates of nitrogen and phosphorus in lakes. J. Environ. Sci. Technol. 54 (6), 3191–3198. doi:10.1021/acs.est.9b05858
Reinhard, C. T., Planavsky, N. J., Gill, B. C., Ozaki, K., Robbins, L. J., and Lyons, T. M. (2017). Evolution of the global phosphorus cycle[J]. Nature 541 (7637), 386–389. doi:10.1038/nature20772
Ren, C., Chen, J., Deng, J., Zhao, F., Han, X., Yang, G., et al. (2017). Response of microbial diversity to C: N: P stoichiometry in fine root and microbial biomass following afforestation. J. Biol. Fertil. Soils 53 (4), 457–468. doi:10.1007/s00374-017-1197-x
Song, J., Ming, H., Shi, T., Su, J., Chen, Q., Jin, Y., et al. (2021). Analysis of microbial functional genes involved in major biogeochmical cycles in sediments of Liaohe River estuary. J. Oceanol. Limnologia Sinica. 52 (04), 904–916. doi:10.11693/hyhz20201200341
Tao, Y., Du, S., Li, Z., Liu, S., Zhu, M., Liu, X., et al. (2021). Successional dynamics of molecular ecological network of nammox microbial communities under elevated salinity. Water Res. 188A116540. ISSN 0043-1354. doi:10.1016/j.watres.2020.114540
Wang, B. (2019). Investigation on structure and function of carbon-fixing microbial communities in sediments of northern Qinghai-Tibetan Plateau lakes. Wuhan, China. D. China University of Geosciences. doi:10.27492/d.cnki.gzdzu.2019.000110
Wang, H. (2019). Research on the interaction mechanism of microbial community structure and nitrogen and phosphorus in lake sediment. Beijing, China: D. North China Electric Power University Bei Jing. doi:10.27140/d.cnki.ghbbu.2019.001010
Wu, H., Ruan, C., Wan, W., Li, S., Pei, D., Han, M., et al. (2022). Progress of functional genes related to soil nitrogen cycling Based on knowledge mapping . J/OL. Acta Pedol. Sin 60 (1). doi:10.11766/trxb202110270580
Wu, L. (2019). Response of microbial community structure in eutrophic Taihu sediments to environmental factors. J. Appl. Environ. Biol. 25 (06), 1470–1476. doi:10.19675/j.cnki.1006-687x.2019.02007
Xiao, Y., Liu, X., Liang, Y., Niu, J., Zhang, X., Ma, Li., et al. (2016). Insights into functional genes and taxonomical/phylogenetic diversity of microbial communities in biological heap leaching system and their correlation with functions. J. Appl. Microbiol. Biotechnol. 100 (22), 9745–9756. doi:10.1007/s00253-016-7819-7
Xie, G. (2012). Spatial heterogeneity of bacteria community compositions and the environmental factors in Lake Bosten. Anhui, China: D. Anhui Agricultural University.
Xu, W., Gao, Q., He, C., Shi, Q., Zheng, Q., and Hua, Z. (2020). Using ESI FT-ICR MS to characterize dissolved organic matter in salt lakes with different salinity. J. Environ. Sci. Technol. 54 (20), 12929–12937. doi:10.1021/acs.est.0c01681
Yang, H. (2020). Analysis of water ecological environment problems and control measures in Daihai Lake. J. Water Conservancy Inn. Mong. 2020 (9), 33–34.
Yang, J., Chen, Y., She, W., Xial, H., Wang, Z., Wang, H., et al. (2020a). Deciphering linkages between microbial communities and priming effects in lake sediments with different salinity. J. J. Geophys. Res. Biogeosciences. 125 (11), e2019JG005611. doi:10.1029/2019jg005611
Yang, J., Jiang, H., Dong, H., and Liu, Y. (2019). A comprehensive census of lake microbial diversity on a global scale. Sci. China Life Sci 62, 1320–1331. doi:10.1007/s11427-018-9525-9
Yang, J. (2015). Microbial response to environmental changes in Qinghai-Tibetan Lakes and its environmental implications. Wuhan, China. D. China University of Geosciences.
Yang, W., Wu, F., Shen, H., Li, W., and Zhang, S. (2020). Structure of bacteria community in the Nanhai Lake at frozen. J. J. Irrigation Drainage 39 (10), 75–81. doi:10.13522/j.cnki.ggps.2019414
Yu, H., Zhong, Q., Peng, Y., Zheng, X., Xiao, F., Wu, B., et al. (2022). Environmental filtering by pH and salinity jointly drives prokaryotic community assembly in coastal wetland sediments. J. Front. Mar. Sci 8, 792294. doi:10.3389/fmars.2021.792294
Zhang, H., Huo, S., Xiao, Z., He, Z., Yang, Z., Yegaer, K. M., et al. (2021b). Climate and nutrient-driven regime shifts of cyanobacterial communities in low-latitude plateau lakes. J. Environ. Sci. Technol. 55 (5), 3408–3418. doi:10.1021/acs.est.0c05234
Zhang, L., Yu, C., Chen, K., and Lv, X. (2021a). Evaluation and analysis of water environmental quality of Daihai in 2020. J. Water Conservancy Inn. Mong. 2021 (12), 7–9.
Zhang, P., Xie, X., Li, Q., Gan, Z., Hu, T., Yang, J., et al. (2022). Microbial community structure and its response to environment in mangrove sediments of Dongzhai Port. J. J. Earth Sci. 47 (3), 1122–1135. doi:10.3799/dqkx.2022.025
Zhang, Z. (2021). Bacteria taxonomic diversities and functional profiles in estuarine wetland. Shanghai, China: D. East China Normal University. doi:10.27149/d.cnki.ghdsu.2021.000526
Zhao, Y., Hui, Z., Chao, X., Nie, E., Li, H., He, J., et al. (2011). Efficiency of two-stage combinations of subsurface vertical down-flow and up-flow constructed wetland systems for treating variation in influent C/N ratios of domestic wastewater. J. Ecol. Eng. 37 (10), 1546–1554. doi:10.1016/j.ecoleng.2011.06.005
Zheng, X., Sun, P., Lou, J., Fang, Z., Guo, M., Song, Y., et al. (2013). The long-term effect of nitrite on the granule-based enhanced biological phosphorus removal system and the reversibility. J. Bioresour. Technol. 132, 333–341. doi:10.1016/j.biortech.2013.01.042
Zhou, J., Fu, Y., Zhang, M., Ding, S., Pan, S., Liu, Y., et al. (2020). Effect of modified coal gangue on Phosphorus in overlying water and key genes of phosphorus metabolism in sediment. J. China Environ. Sci. 40 (11), 4998–5009. doi:10.19674/j.cnki.issn1000-6923.2020.0553
Keywords: plateau, lake sediment, salinity, phosphorus metabolism, microorganisms, community composition
Citation: Yang W, Ma J, Zhen Y, Li W, Yao Z and Feng W (2022) Community characteristics and functional gene response analysis of phosphorus-metabolizing bacteria in plateau saline lake sediments. Front. Environ. Sci. 10:994104. doi: 10.3389/fenvs.2022.994104
Received: 14 July 2022; Accepted: 07 November 2022;
Published: 22 November 2022.
Edited by:
Christopher J. Martyniuk, University of Florida, United StatesReviewed by:
Hongchen Jiang, China University of Geosciences Wuhan, ChinaCopyright © 2022 Yang, Ma, Zhen, Li, Yao and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiying Feng, ZmVuZ3dlaXlpbmdAYnVhYS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.