 Dinesh Kumar
Dinesh Kumar Zarna Patel
Zarna Patel Priti Pandit
Priti Pandit Ramesh Pandit
Ramesh Pandit Amrutlal Patel
Amrutlal Patel Madhvi Joshi
Madhvi Joshi Chaitanya Joshi
Chaitanya Joshi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci. , 02 September 2021
Sec. Water and Wastewater Management
Volume 9 - 2021 | https://doi.org/10.3389/fenvs.2021.720707
This article is part of the Research Topic Efficient Treatment of Industrial Wastewater with Microbiome and Synthetic Biology View all 5 articles
Textile industries play an important role in uplifting the national economies worldwide. Nevertheless, they generate a huge amount of intensive colored effluent, which is a serious threat to the environment. The microbial communities present in these highly polluted environmental sites help in remediating pollutants naturally. However, little is known about their genes and enzymes in the textile wastewater systems. In this study, we explored the microbial community structure and their functional capability in three different wastewater systems, i.e., industry sites, effluent treatment plant (ETP), and common effluent treatment plant (CETP). Our findings based on shotgun metagenomics highlight the varied bacterial diversity at the three industry sites. Overall, the major dominant phyla in the industry site and CETP samples were Proteobacteria and Bacteroidetes, while in the ETP site, Firmicutes, Cyanobacteria, and Proteobacteria were predominant. The final discharge sample site was having a higher proportion of the Proteobacteria and Bacteroidetes. Aeromonas caviae, Desulfovibrio desulfuricans, Klebsiella pneumoniae, Pseudomonas stutzeri, Shewanella decolorationis, Shewanella oneidensis, Shewanella putrefaciens, and Vibrio cholera were the abundant species across the three sites. Furthermore, this research study identified the key microbial genes encoding enzymes having a known role in textile dye and aromatic compound degradation. Functional annotation of the shotgun metagenome samples indicates the presence of reductase, azoreductase, nitrate/nitrite reductase, and oxidoreductase enzyme encoding genes. Our findings provide the shotgun metagenomics-based approach for mining the textile dye degrading genes and genomic insights into the bioremediation of textile industrial effluent.
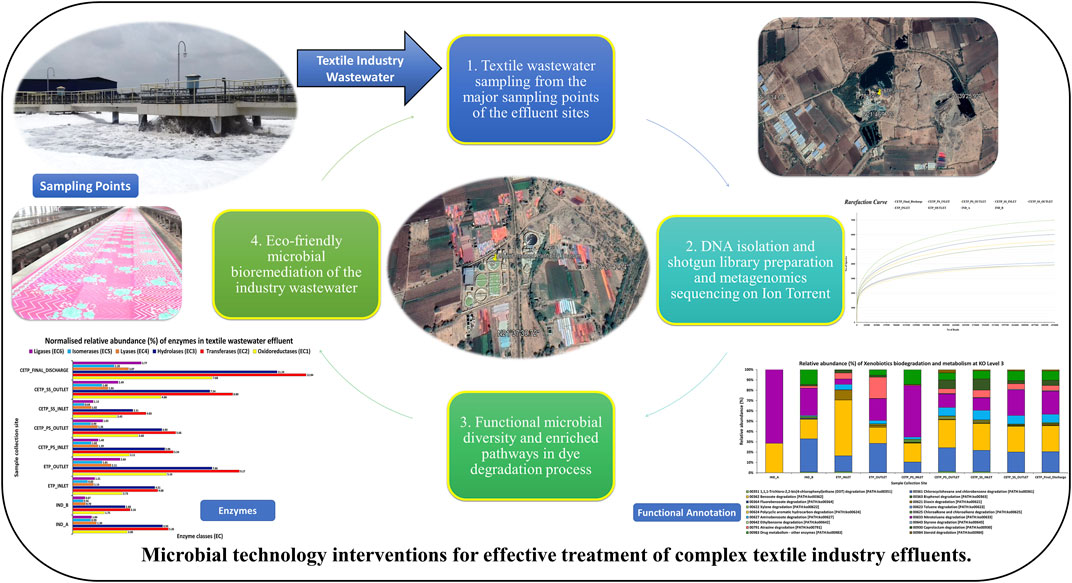
GRAPHICAL ABSTRACT
The environmental pollution and toxic substances produced by the industrial manufacturing process are major challenges faced worldwide. Textile industries heavily rely on water during pre-processing of raw material and dyeing and printing of fabric and simultaneously consume a huge amount of synthetic chemicals and pigments. The pollutants generated from several steps involved in the textile industries create various ecological problems due to the generation of a large amount of wastewater containing pollutants. Textile industry wastewater contains a variety of persistent coloring pollutants and synthetic dyes, phenols, formaldehyde, phthalates, surfactants, chlorophenol and aromatic compounds and heavy metals such as lead (Pb), cadmium (Cd), arsenic (As), chromium (Cr), zinc (Zn), and nickel (Ni), chlorides, and sulfates (Hossain et al., 2018; Kishor et al., 2021). Synthetic chemicals, pigments, and dyes add to the complexity of the wastewater effluents and sludge, causing damage to the environment (Chen et al., 2021; Manickam and Vijay, 2021; Methneni et al., 2021).
Subsequently, the usage of synthetic dye has been increased in the textile and dyeing industry due to their cost-effectiveness, high stability to light, temperature, and detergents, and resistance to microbial attack as compared to natural dye (El-Kassas and Mohamed, 2014; Anantharaman et al., 2016; Kumar et al., 2019). Almost 10,000 different synthetic dyes are currently manufactured, mainly azo dyes, and are frequently used by the textile and various other industries for dyeing because they are more economically stable than natural dyes (Xu et al., 2007; He et al., 2016; Garg and Tripathi, 2017). Azo dyes are the largest class of synthetic dyes, which are characterized by the presence of azo (-N=N-) groups in their structure (Verma et al., 2012; Yaseen and Scholz, 2019). These chemical dyes are toxic and highly persistent in the environment and their metabolic products are mutagenic and carcinogenic in nature (Xu et al., 2007). Therefore, understanding azo dye degrading enzymes and functional genes is crucial for elucidating their molecular mechanisms for the development of efficient bioremediation technology (Dixit and Garg, 2018). Further, investigating the complex dynamics of microbial diversity and community structure of the textile wastewater treatment sites can provide crucial information regarding novel molecular pathways, biosynthetic gene clusters (BGCs), and textile dye degrading genes encoding enzymes.
Microorganisms play an important role in the degradation of various pollutants through their adapted metabolic and enzymatic activity. However, the potential relationship between bacterial taxa and the niches occupied by specific microorganisms in textile wastewater and sludge is largely unknown. Therefore, the characterization and identification of microbes and their genes mediating textile dye degradation can be an ecofriendly way to treat such a complex industry effluent. Therefore, this study was undertaken to explore the microbial community and their potential genes, which may have a role in the bioremediation of textile industry effluent. Therefore, for this study, we collected textile industry effluent samples from the ETP and CETP located in Jetpur, Gujarat, India, and carried out whole-genome shotgun sequencing.
The textile industry wastewater samples were collected from Jetpur, Gujarat, India (21°37′36.42″N, 70°37′33.03″E), which is one of the largest textile industry hubs in India. The geotagged images of sampling site locations are depicted in Figure 1, while Supplementary Table S1 provides the details of the sampling site with approximate operational members and wastewater treatment capacity. The textile wastewater samples were collected in the sterile 50 ml falcon tubes from a total of nine sampling points representing three different sites, namely, textile industry site, effluent treatment plant (ETP), and common effluent treatment plant (CETP) site. The samples were stored at 4°C for downstream processing. The samples from industry sites (IND_A and IND_B) were collected from the print houses of the textile industry sites, while wastewater samples were collected from the common effluent treatment plant (CETP) and effluent treatment plant (ETP) sites.

FIGURE 1. Wastewater effluent samples were collected from the Jetpur textile industry cluster from Gujarat, India, which is a major hub of textile-based industries. The sampling sites included were effluent treatment plant (ETP, 1) (A) and common effluent treatment plant (CETP, 1) (B), where the final discharge samples were treated in the lagoon by the phytoremediation process with the nearby plant species and wash area from the dyeing and print house. The reddish/orange color indicates the washing area around the effluent treatment plant.
The total genomic DNA was extracted using the DNeasy PowerWater DNA isolation kit (QIAGEN) as per the manufacturer’s protocol. The quality of total genomic DNA was checked on 0.8% agarose gel visualized on a gel documentation system (Bio-Rad). The quantity and purity of the DNA were assessed using the QIAxpert System (QIAGEN) and Qubit 4 Fluorometer (Invitrogen). Approximately 250 ng of genomic DNA was used for the shotgun library preparation using the Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific) according to the manufacture’s protocol. For the library size selection, we used E-Gel SizeSelect II, 2% agarose gel electrophoresis (Invitrogen), and Agencourt AMPure XP kit (Beckman Coulter) was used for purification. Shotgun metagenome sequencing was carried out using the Ion torrent S5 platform with 400 bp chemistry and Ion 530 chip.
Shotgun metagenome sequencing raw data files were uploaded on the MG-RAST version 4.0.3 (Aziz et al., 2008) server for taxonomic and functional analysis. After quality control and filtering (dereplication, removing the artificial sequences, and ambiguous bases), sequences with the “Best Hit” criteria were used for the taxonomical analysis with default parameter (i.e., E-value of 1 × 10−5, the minimum identity of 60%, and a minimum length of 15 amino acids). For the functional annotation, each of the metagenomic reads was annotated against the NCBI reference sequence (RefSeq) database in MG-RAST with default parameters (i.e., E-value of 1 × 10−5 with 60% identity).
For functional annotation, reads were further processed using the SqueezeMeta bioinformatics analysis pipeline (Tamames and Puente-Sánchez, 2019). The co-assembly mode was used for the annotation of genes in each metagenome sample. MEGAHIT v1.2.9 (Li D. et al., 2015) and Prodigal (Hyatt et al., 2010) were used for the assembly and gene prediction, respectively. The predicted genes were searched for homology against the KEGG database for the functional annotation (Kanehisa et al., 2016) of the genes in the contigs. The genes encoding for the enzymes involved in the textile dye degradation were identified from the nine different sampling points representing three different sites, and the biosynthetic gene clusters were reconstructed based on the enzyme encoding genes (Supplementary Table S2). For the identification of specific functional gene prediction, KEGG IDs were manually filtered based on the criteria of their role in involvement in the gene clusters known for textile degradation with predicted function based on Enzyme Commission (EC) number.
Estimation of parametric and non-parametric diversity indices was processed using the PAST v4.0.3 software (PAST Hammer et al., 2001). Further, based on the KEGG and COG functional annotation, the metagenome samples were clustered following the principal component analysis (PCA). PCA plot and sample clustering dendrogram were done using the Microbiome Analyst tool (Dhariwal et al., 2017), and the heatmap of the xenobiotic compound degrading genes was constructed using Clustvis (Metsalu and Vilo, 2015).
The sequencing output generated from shotgun sequencing is tabulated in Supplementary Table S3. The rarefaction curve depicts the species richness and sequencing depth in the shotgun metagenome samples. Parametric and non-parametric diversity indices of the microbial community in the textile industry effluent, ETP, and CETP are shown in Table 1. The maximum number of microbial taxa (161) was assigned to the final discharge sample of the common effluent treatment plant (CETP_Final_Discharge), while the minimum (11) was present in the textile industry sample IND_A. Bacterial richness (Shannon’s H index) was highest for CETP_Final_Discharge, while lower microbial communities were observed in the textile industry sample (IND_B). The non-parametric diversity index, i.e., Chao1, was found maximum in the CETP_Final_Discharge and lowest in the textile industry sample (IND_A). The Operational Taxonomic Units (OTUs) table (Supplementary Table S4) provides details of the taxonomic abundance of the different sampling points, and details provided in Supplementary Table S5 highlight the metagenome assembly statistics. The details of the taxonomy assigned to the metagenome-assembled contigs are shown in Supplementary Table S6.
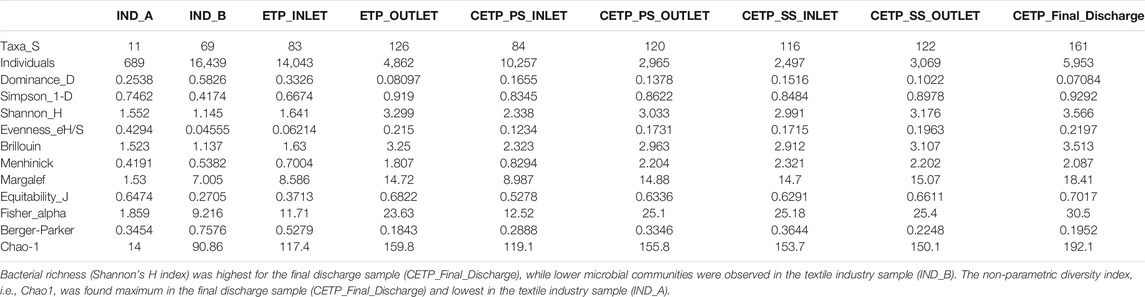
TABLE 1. Diversity indices calculated at the family level from different sampling points highlight the taxonomic richness and dominance.
Microbial diversity of the textile industry wastewater samples from three different sample collection sites indicated the dominance of the well-known synthetic dye degrading microbial population among all the samples. At the phylum level, Proteobacteria was abundant in IND_A (48.77%) followed by CETP_SS_INLET (37.08%), CETP_PS_INLET (32.94%), INLET (21.22%), ETP_OUTLET (19.68%), CETP_SS_OUTLET (16.85%), and IND_B (9.02%). Bacteroidetes were found to be abundant in the samples from textile industry sites (IND_B, 82.55%), where the dyeing and printing processes were carried out, followed by CETP_PS_INLET (34.23%) and CETP_SS_OUTLET (32.71%), while the Firmicutes were dominant in samples CETP_Final_Discharge (27.03%), ETP_INLET (24.95%), and CETP_SS_OUTLET (21.34%), as highlighted in Figure 2. It is interesting to note that the dominance of Bacteroidetes in the industry effluent sites (IND_A and IND_B) is also evident from the literature where the dominant taxonomic abundance was possibly involved in the cleavage of azo dye compounds under anaerobic circumstances (Li C. et al., 2015; Waktole et al., 2020; Samuchiwal et al., 2021), highlighting the bioremediation potential of the taxonomically dominant microbial communities in the textile industry effluents. The inlet sample from the effluent treatment plant contains a mixture of compounds used during the textile printing and processing and provides better opportunities for the microbial communities to thrive on a multitude of compounds. Unclassified sequences (derived from Bacteria) were abundant in ETP_INLET (52.79%) which is the effluent treatment plant inlet site, followed by CETP_PS_OUTLET (33.46%) at the primary stage outlet site, CETP_SS_INLET (36.44%) at the secondary stage inlet site (IND_A) (34.54%), textile industry site (CETP_PS_INLET) (24.55%), primary stage inlet (CETP_SS_OUTLET) (19.97%) secondary stage outlet, CETP_Final_Discharge (19.52%) at the final outlet site of the common effluent treatment plant site, and ETP_OUTLET (17.56%) and IND_B (7.24%) sample at the industry site. It shows that as the treatment of wastewater processes, the proportion of uncultured bacteria is reduced. This may be because very few uncultured bacteria from the initial load have adapted to the conditions in the treatment plant and may play a crucial role in the degradation of waste from the textile industries. However, the role of the unclassified sequences needs to be explored further as microbial activity and adaptability in the textile industry effluents provide a biological mechanism for the reduction of the azo group of compounds. Bacterial enzymes, especially oxidoreductases, play an important role in the degradation of synthetic dyes and the dominant microbial community structure at the family level of classification reveals key species known for adaptability under highly toxic wastewater. At the family level, Shewanellaceae (48.95%), Bacteroidaceae (29.86%), and Campylobacteraceae (6.16%) were found in textile industry site A (IND_A) sample. On the other hand, in the textile industry site B (IND_B) sample, Bacteroidaceae (92.67%), Shewanellaceae (3.21%), and Aeromonadeaceae (1.48%) were dominant. Further exploration at the family level of taxonomic dominance highlighted the possible role of catalytic enzymes encoded by the genes present in these microbial communities and significantly highlighted the structural and functional performance to adapt and degrade the textile dyes, complex aromatic and benzene ring compounds, and synthetic pigments. Shewanellaceae and Bacteroidaceae are well-known taxonomic families that are characterized from the industrial sludge and wastewater treatment plants, process industries, and fermenting and tannery waste treatment sites that contain highly toxic and complex wastewater generated during the processing steps (Xu et al., 2007; Meerbergen et al., 2017). Therefore, we anticipate the potential role and decipher the metagenomics features identified through dominant taxonomic abundance in the textile industry wastewater samples collected from the industry sites, common effluent treatment plant (CETP), and effluent treatment plant (ETP). Similarly, in the effluent treatment plant (ETP) inlet sample (ETP_INLET), Rhodocyclaceae (51.65%) and Ectothiorhodospiraceae (48.34%) were dominant, while in the sample ETP_OUTLET, Ectothiorhodospiraceae (34.14%), Oceanospirillaceae (15.89%), and Piscirickettsiaceae (15.46%) were dominant. Simultaneously, the dominant microbial populations were analyzed, which are associated with different treatment processes of the effluent treatment plant (ETP), common effluent treatment plant (CETP), and dyeing and printing wash area of the local textile print houses as revealed by the taxonomic abundance. Furthermore, the relative abundance of Bacteroidaceae was higher in the CETP_PS_INLET (66.94%) primary stage inlet of the CETP than that of the CETP_PS_OUTLET (23.38%) primary stage outlet of the common effluent treatment plant. Moreover, the Desulfovibrionaceae (9.41%) and Enterobacteriaceae (7.05%) were also present in CETP_PS_INLET, whereas 30.18% remained unassigned. It was observed that in the CETP_SS_INLET and CETP_SS_OUTLET samples, Pseudomonaceae and Flavobacteriaceae were 1.20%. Desulfovibrionaceae and Pseudomonaceae have a relative abundance >1%. However, CETP_SS_OUTLET showed the predominance of Bacteroidaceace (71.52%) among all the metagenome datasets, while the dominant bacterial family in CETP_Final_Discharge was represented by Bacteroidaceae (53.07%), Enterobacteriaceae (10.47%), and Desulfovibrionaceae (8.63%), as mentioned in Figure 3. Furthermore, the potential role of the abundant taxonomic families is also interesting to understand in nutrient recycling and biogeochemical cycling processes during the treatment process. The results indicated that multiple deterministic processes, such as effluent load, biogeochemical cycling process, nutrient requirement, adaptive capacity, and biological competition, possibly established the microbial community profiles in textile industry sites, CETP, and ETP. Moreover, based on microbial diversity assessment, purposeful bacteria such as sulfur and nitrogen cycling-related bacteria were widely distributed in the textile wastewater process showing strong biological associations among them (Watsuntorn et al., 2019). At the genus level, the top three genera in textile industry site A (Ind_A) metagenome sample were Shewanella (53.43%), Bacteroides (17.30%), and Denitrovibrio (6.13%); in textile industry site B (IND_B), they were Bacteroides (76.33%), Shewanella (7.62%), and Pseudomonas (2.26%). In the effluent treatment plant (ETP) sites, Bacillus (30.10%), Polaromonas (7.09%), and Acidovorax (6.81%) were dominant at the ETP inlet site (ETP_INLET), while Cyanothece (9.39%), Oscillatoria (8.56%), and Microcoleus (8.46%) were dominant in ETP_OUTLET. Bacteroides (41.43%), Desulfovibrio (17.13%), Sulfurospirillum (12.58%), Aeromonas (5.16%), and Shewanella (3.79%) were dominant in CETP_PS_INLET, whereas Leadbetterella (11.62%), Hyphomonas (9.72%), and Pseudomonas (7.63%) were dominant in CETP_PS_OUTLET. Similarly, Pseudomonas (12.29%) and Hyphomonas (10.95%) were dominant in the CETP_SS_INLET, while Bacteroides (36.70%), Desulfovibrio (8.72%), Clostridium (6.32%), Pseudomonas (4.45%), and Denitrovibrio (2.77%) were abundant in the common effluent treatment plant secondary stage outlet site (CETP_SS_OUTLET). Lastly, in the final wastewater discharge site (CETP_Final_Discharge), the relative abundance of the genus Bacteroides (24.52%) decreases, whereas genera Desulfovibrio (9.78%), Pseudomonas (8.38%), and Clostridium (7.02%) increased as compared to the textile industry effluent sites (IND_A and IND_B) sampling points, as shown in Figure 4. The major species present in the sample textile industry (IND_A) were identified as Shewanella xiamenensis (46.88%), Bacteroides graminisolvens (29.86%), and Sulfurospirillum sp. MES (6.16%), whereas Bacteroides graminisolvens (92.67%), Shewanella xiamenensis (2.54%), and Aeromonascaviae (1.48%) were predominant in textile industry site (IND_B), and effluent treatment plant (ETP) has an abundance of the species Thauera sp. (51.65%), Ectothiorhodospira sp. PHS-1 (48.34%), Ectothiorhodospira sp. PHS-1 (34.14%), Thioalkalimicrobium aerophilum (12.27%), and Nitrincola lacisaponensis (12.11%). Novel strains of Gram-negative, non-motile, non-spore-forming rods Bacteroides graminisolvens are reported to be xylanolytic under anaerobe isolated from methanogenic bioreactors at an optimal pH of 7.2 and the optimum growth temperature was 30–35°C (Hatamoto et al., 2014). Moreover, Bacteroides graminisolvens (65.56%), Klebsiella sp. (6.88%), and Desulfovibrio desulfuricans (5.81%), Bacteroides graminisolvens (23.38%), and Flavobacterium sasangense (16.14%) were dominant in the primary stage inlet site (CETP_PS_INLET). On the other hand, Pseudomonas stutzeri (14.06%) and Flavobacterium sasangense (12.01%) were dominant in the secondary stage inlet (CETP_SS_INLET). At the species level, in the secondary stage outlet (CETP_SS_OUTLET), Bacteroides graminisolvens (71.58%), Desulfovibrio desulfuricans (5.49%), and Pseudomonas stutzeri (5.18%) were dominant. In the CETP_Final_Discharge sample, the dominance of Bacteroides graminisolvens (53.07%) and Klebsiella sp. (10.47%) was observed. The roles of identified catalytic enzyme encoding genes from the sample collection site and the dominant bacterial communities, as reflected in the relative abundance (%) of the determined biosynthetic gene clusters, have to be studied and understood because they might play a significant role in the complete breakdown of the complex chemical dyes during the treatment process.
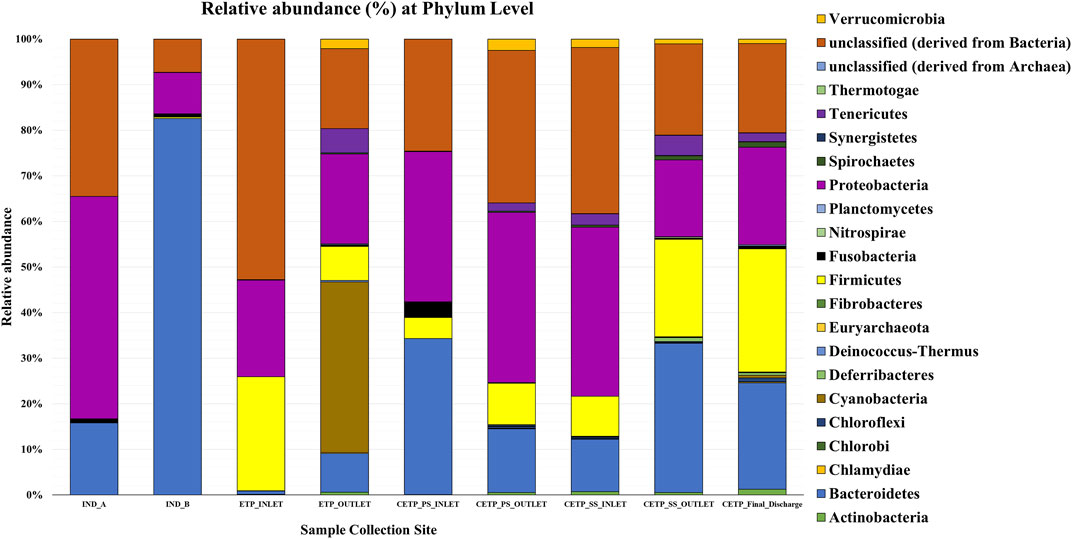
FIGURE 2. Relative abundance (%) of the phylum-level taxonomic diversity in different metagenome samples collected from the textile industry wastewater site, common effluent treatment plant (CETP), and effluent treatment plant (ETP).
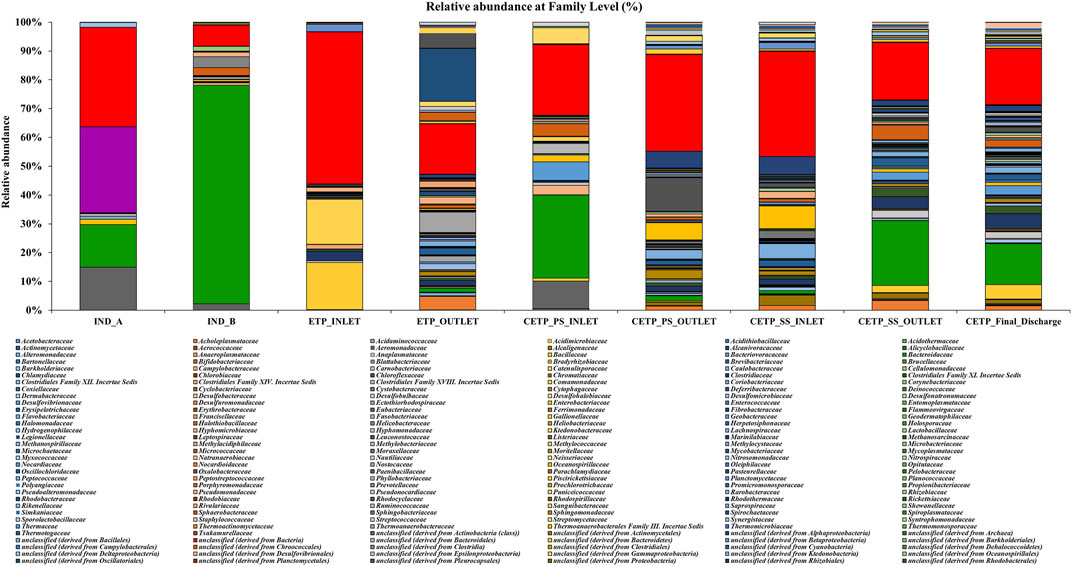
FIGURE 3. Relative abundance (%) of the microbial diversity at the family level of the different textile industry effluent metagenome samples highlighting the dominance of the Shewanellaceae, Enterobacteriaceae, Bacteroidaceae, Aeromonadaceae, and Pseudomonadaceae in the industry samples (IND_A and IND_B). Comamonadaceae, Bacillaceae, Nostocaceae, and Burkholderiaceae were dominant in ETP inlet samples. Bacteroidaceae, Desulfovibrionaceae, Aeromonadaceae, Enterobacteriaceae, Campylobacteraceae, Vibrionaceae, Flavobacteriaceae, and Alcaligenaceae were dominant in the CETP primary stage treatment sites. In contrast, Alcaligenaceae, Pseudomonadaceae, Flavobacteriaceae, Burkholderiaceae, Bacteroidaceae, and Clostridiaceae were families higher in abundance at CETP secondary stage sites. The dominant families at the final stage (CETP_Final_Discharge) sites were Bacteroidaceae, Alcaligenaceae, Clostridiaceae, Desulfovibrionaceae, Enterobacteriaceae, Pseudomonadaceae, and Bacillaceae.
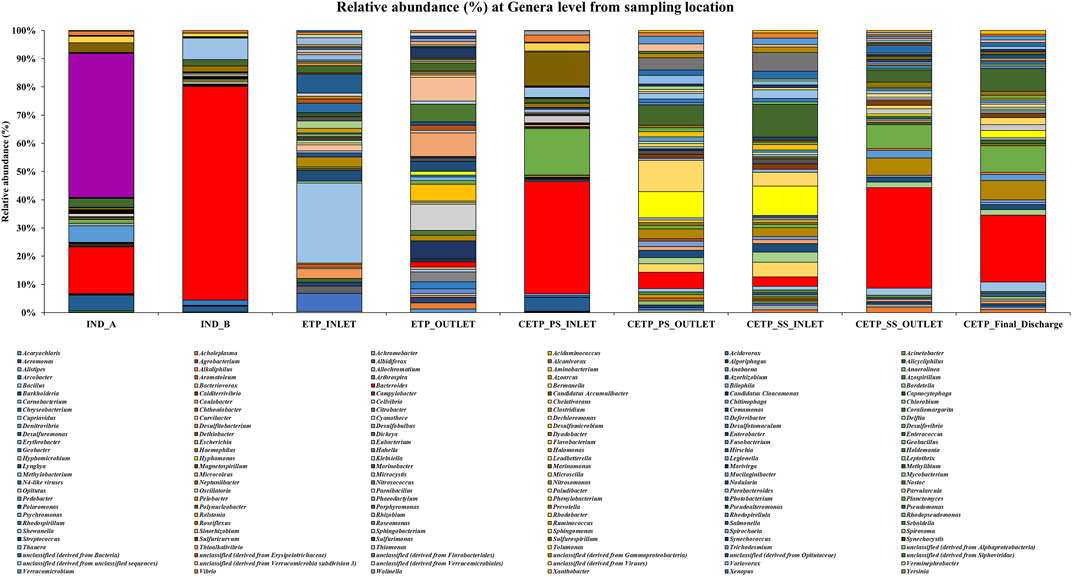
FIGURE 4. Relative abundance (%) of the genera level in different metagenome samples collected from the textile industry wastewater site, common effluent treatment plant (CETP), and effluent treatment plant (ETP).
Furthermore, based on the enriched metabolic pathway analysis and enzymatic abundance features, potential candidate gene families indicate xenobiotic degradation, aromatic compound, and synthetic pigment degradation, as highlighted in Figure 5. Furthermore, understanding complex microbial interactions at the genomic level could help develop suitable microbial technologies for the removal of recalcitrant compounds (Dai et al., 2020). Enzymatic degradation of azo dyes, polyaromatic compounds, and phenolic complexes by nitroreductase and azoreductase has been widely studied and reported to help in the process of remediating textile industry-based wastewater and treatment process (Khan and Malik, 2018; Yesilada et al., 2018; Mishra et al., 2020). These observations demonstrated that oxidoreductase, nitroreductase, and azoreductase enzymes from microorganisms were effectively helpful in the decolorization and degradation of synthetic textile dyes. The dominant microbial communities further highlight the significant role in the textile dye degradation process and remain the possible source of the dye degrading genes encoding the catalytic enzymes during the treatment process. Further, enzymatic reduction of azo dyes requires the addition of cofactors as electron donors for the reductive cleavage resulting in the production of aromatic amines, which are more potentially toxic health and environmental hazards. Similarly, an engineering approach and photocatalytic-based integrated design for optimal activity and efficient detoxification of wastewater containing harmful substances can be a helpful strategy at highly variable environmental conditions (Mate and Alcalde, 2015; Cardoso et al., 2016).
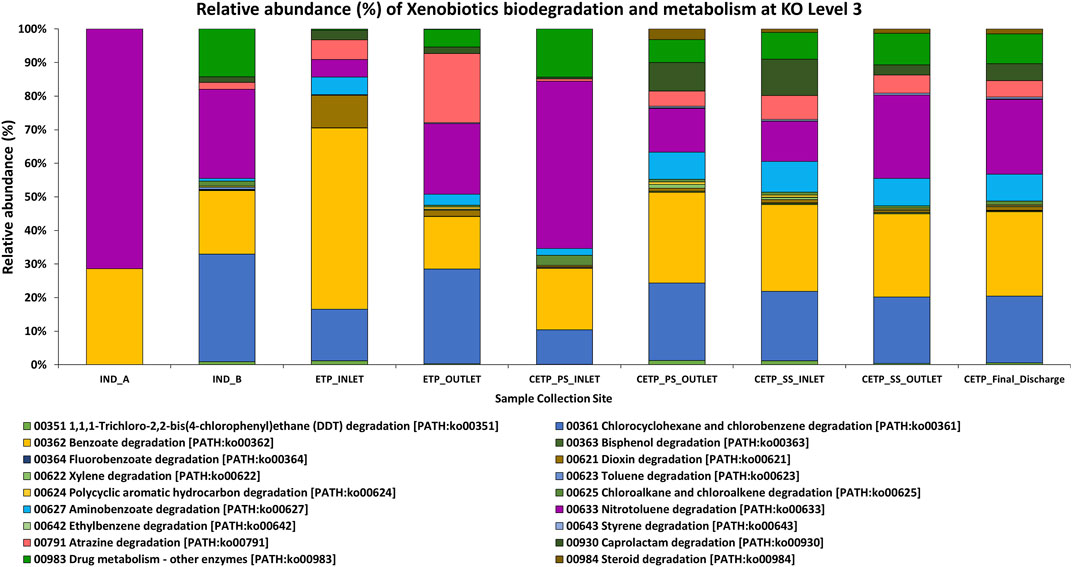
FIGURE 5. Relative abundance of the xenobiotic degradation and metabolism at KO Level 3 (KO-3), highlighting the metabolic functions of the key pathways involved in the synthetic chemical degradation in the textile wastewater and industry effluent treatment process. Samples from the industry effluent sites (IND_A and IND_B) were dominated by nitrotoluene degradation and benzoate degradation. In the ETP sites, chlorocyclohexane and chlorobenzene degradation was found, while the aminobenzoate degradation pathways were dominant in the secondary treatment and final discharge site.
The microbial diversity of the textile industry effluents harbors the dye degrading genes and remains completely unexplored in the context of the uncultured microbial community dynamics. This research study findings indicate that the identified dominant bacterial communities were known for the bioremediation potential for the heavy metals, aromatic compounds, synthetic dyes and pigments, chemicals, and xenobiotic compounds from the highly polluted sites (Dixit and Garg, 2019; Redfern et al., 2019). Bacterial isolates help mediate the treatment process since they are capable of utilizing degraded byproducts of compounds as electron acceptors, including oxygen, iron, manganese, sulfate, nitrate, and nitrite. Microbial enzymes play an important role in degrading various xenobiotic compounds (Shah and Jha, 2013; Mate and Alcalde, 2015). Moreover, enzymes including oxidoreductases, monooxygenases, dioxygenases, peroxidases, and laccases have an important role in the bioremediation of various xenobiotic compounds. The details of the predicted and annotated features are shown in Supplementary Table S7 from the shotgun metagenome assembly, while Figure 6 highlights the read normalized relative abundance of the enzyme classes gene families identified from the different metagenome samples from textile industry effluent sites, common effluent treatment plant (CETP), and effluent treatment plant (ETP) sites.
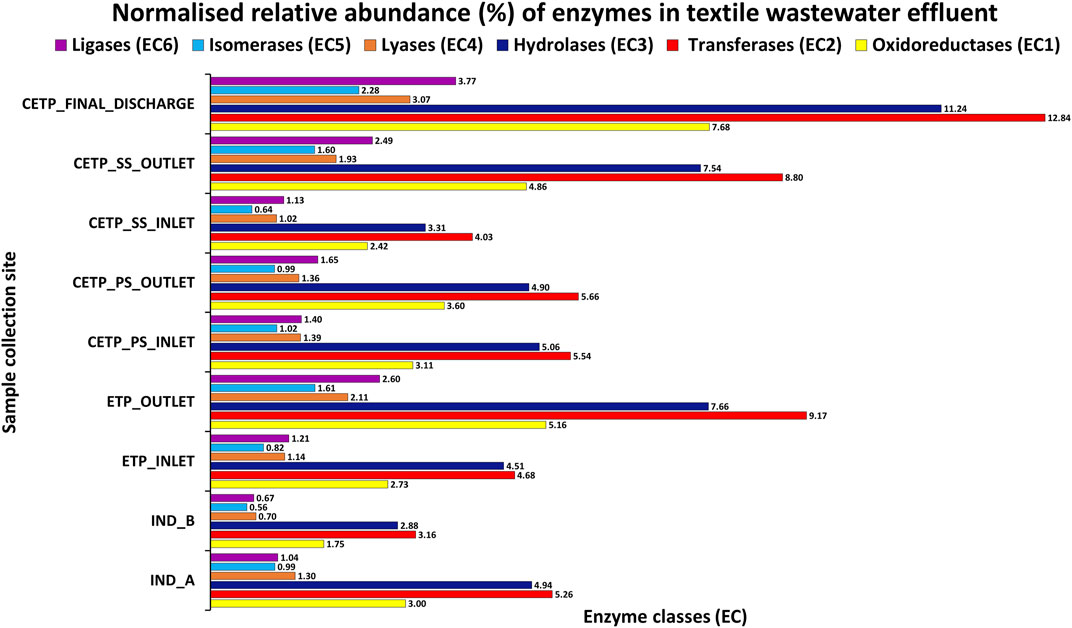
FIGURE 6. Reads of the normalized relative abundance (%) of the enzyme classes gene families of the different metagenome samples from textile industry effluent sites, common effluent treatment plant (CETP), and effluent treatment plant (ETP).
Microbial oxidoreductases can mediate the degradation of synthetic and harmful pollutants, reduce the toxicity caused by xenobiotic chemical compounds, and reduce heavy metals present in the textile industry wastewater through their oxidation and reduction potential. Similarly, monooxygenases and dioxygenases can play a key role in the degradation and detoxification of aromatic amines, phenols, and benzoate compounds through hydroxylation and aromatic ring cleavage, while peroxidases help in bioremediation processes due to their structural stability and capacity to oxidize a wide range of chemical substrates (Forgacs et al., 2004). Similarly, Laccases can act on different kinds of polluting substances, such as aromatic hydrocarbons, paints, polymers, synthetic dyes, chemicals, and pesticide substances, via oxidation reactions, decarboxylation, and demethylation, which further can oxidize phenols and polyphenols, and oxidation and reduction of heavy metals, polyamines, azo group compounds, and aryl diamines groups present in the textile industry wastewater treatment process (Liu et al., 2011; Mate and Alcalde, 2015; Gaur et al., 2018; Yesilada et al., 2018). Similarly, NADH: quinone oxidoreductase (complex I) is a multi-subunit integral membrane enzyme found in both bacteria and eukaryotic cell organelles. Even though little is known about the unconventional role of these enzymes in the textile wastewater treatment process, they play a critical role in bacterial adaptation to the diverse energetic lifestyles in biogeochemical cycling processes under harsh environmental conditions at polluted sites, including aerobic and anaerobic conditions. Comparative genomic studies suggest a wide role for NADH: quinone oxidoreductase in re-oxidizing NADH produced from various biological and catabolic reactions, including the tricarboxylic acid (TCA) cycle and fatty acid beta-oxidation. Together, these findings highlight the importance of enzymes and their encoding genes in shaping diverse physiological functions across the diverse microbial communities and domains mediating the bioremediation process in the industrial wastewater treatment process of the industry effluents (Kaur et al., 2018; Yang et al., 2020). Furthermore, the presence of the nitro group (-NO2) compounds in the textile wastewater process highlights the importance of the nitrate/nitrite oxidoreductase class of enzymes. Synthetic pigments and chemical compounds are part of the textile dye industry and printing houses. Therefore, aromatic and benzene ring nitro-compounds pollution has been a serious threat in environmental sites and water bodies, particularly due to the industrial wastewater being released into freshwater ecosystems. Bacterial heterotrophic nitrifier and aerobic denitrifier such as Klebsiella pneumoniae were isolated and reported from domestic wastewater and were also identified in the analysis of the metagenome samples with higher abundance in various samples at species level taxonomic identification suggesting a much broader role in bioremediation process at harsh environmental conditions (Dixit and Garg, 2018). The overall abundance of the assigned enzyme classes to the assembled contigs of the assembled shotgun metagenome samples. The oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases were present in different sampling sites collected from Jetpur, Gujarat, as mentioned in Supplementary Table S8. The bioremediation process by bacterial species may further mitigate changes to the nitrogen and sulfur cycling process, thus helping maintain the survival fitness and nutrient cycling balance of the prokaryotic community with adapted functions in the wastewater treatment process. The key genes encoding for nitrate/nitrite reductase were and were nirA, nirB, nirD, nirI, nirK, nirS, NIT-6, nrfA, nrfG, nrfH, narG, narZ, nxrA, narH, narY, and nxrB, and their abundance was quantified from the metagenome samples. These findings highlight the significance and the important role of nitrite reductases as important enzymes with better bioremediation potential for the textile industry effluents. Similarly, azoreductases act on the azo group of compounds and help in the degradation and bioremediation process of the toxic compounds present in the textile industry effluent and wastewater in a non-toxic and ecofriendly approach for the effective treatment of the hazardous chemical substances present in the effluent sites. This research study reveals that the important genes encoding azoreductase enzymes were acpD and azoR encoding FMN-dependent NADH-azoreductase known for the bioremediation potential for the industry effluent sites. The part of the textile dyes used for dying fabrics does not bind to the fabric, and as a result, it is released in the wastewater effluent (Robinson et al., 2001). These dyes can be harmful and carcinogenic in nature (Alves de Lima et al., 2007); therefore, their removal from the industry effluent is essential to reduce the ecological threats. All these serious concerns have prompted scientific research into both mechanical and electrochemical reduction of azo dyes and the use of both aerobic and anaerobic bacteria for bioremediation as integrated technology for efficient process development. Apart from the synthetic dyes and pigments, textile industries also utilize other intermediate chemicals, such as sodium hydroxide (NaOH), oxalic acid, hydrogen peroxide, and acetic acid, during the pre-processing of textile fabrics before the dyeing and printing processes, which may react with the chemical dyes and form recalcitrant compounds that can hinder the treatment process and influence the functional potential of the microbial diversity of the effective microbial population (Chen et al.; Stolz, 2001; Xu et al., 2007; Wijetunga et al., 2010; Ghodake et al., 2011; Yang et al., 2020). Therefore, metagenome studies can better highlight and aid the selection criteria for the microbial consortium development for the effective treatment process of the wastewater synthetic dyes, pigments, and chemicals. The detection and identification of the aldehyde oxidoreductase and 2-oxoacid ferredoxin oxidoreductase enzymes also highlight the role of the electron acceptor components essential in the functional potential of the specific enzyme activity in the wastewater treatment process, thus effective in the bioremediation processes under complex intermediates and harmful compounds of the hazardous in nature. Similarly, the microbial genes encoding these catalytic enzymes were detected in different metagenome samples across all the sampling points under investigation of this study. The important genes encoding catalytic enzymes were detected and represented by korA, oorA, oforA, korB, oorB, and oforB. The relative abundance (%) as represented in Figure 6 of the xenobiotic degradation and metabolism at KO Level 3 (KO-3) highlights the metabolic functions of the key pathways involved in the synthetic chemical degradation during the textile wastewater and industry effluent treatment process. The samples from the textile industry effluent sites (IND_A and IND_B) were dominated by nitrotoluene degradation and benzoate degradation. In contrast, chlorocyclohexane and chlorobenzene degradation was evident in the effluent treatment plant (ETP) and common effluent treatment plant (CETP) sites, while amino-benzoate degradation pathways were dominant in the secondary treatment and final discharge site, which might be the intermediate degradation products of the synthetic dyes and toxic chemical compounds used in the dyeing and printing process of the fabrics.
Metagenome analysis from the textile wastewater samples indicates that it is crucial for the microorganisms in dye-contaminated sites to adapt and perform survival functions under highly polluted environmental conditions. Furthermore, the genes encoding the transporter functions are ABC transporters, amino acid substrate-binding proteins, peptidases, permeases, and transferases. The relative abundance (%) of the key oxidoreductase enzymes found in the textile industry effluent sites, common effluent treatment plant (CETP), and effluent treatment plant (ETP) is given in Figure 7. The dominant catalytic enzymes identified in the industry effluent sites (IND_A and IND_B) were NADH-quinone oxidoreductase, NADH oxidoreductase, and pyruvate-ferredoxin oxidoreductase. The dominant microbial enzymes in the ETP sites were represented by NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase. Similarly, the CETP dataset showed the dominance of NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase. In contrast, the sample collected from the final discharge site (CETP_Final_Discharge) showed the dominance of the uncharacterized oxidoreductase apart from the NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase. These enzymes perform basic metabolic functions and maintain osmoregulatory processes for bacterial adaptation and survival in polluted sites where xenobiotic compounds are abundant. Based on the gene mining and combining the tricarboxylic acid (TCA) cycle, β-oxidation and electron transport chain processes of the microbial communities were deciphered from the taxonomic abundance; the majority of the research studies were explained from the perspective of the important textile dye degrading enzymes in the metabolic pathway at the individual gene level. However, only a few literature reports have discussed the degrading enzymes, their encoding functional genes along with the source of the bacterial species, mechanism of action in each step of the degradation pathway, and pilot-scale end-of-pipe removal of volatile aromatic compounds from textile dyeing wastewater treating plant (Freedman et al., 2005; Chen B. et al., 2009; Chen B.-Y. et al., 2009; Yaseen and Scholz, 2019). Therefore, these proposed gene clusters further provide a deeper insight into the molecular degradation mechanism of textile dyes during the industrial wastewater treatment process.
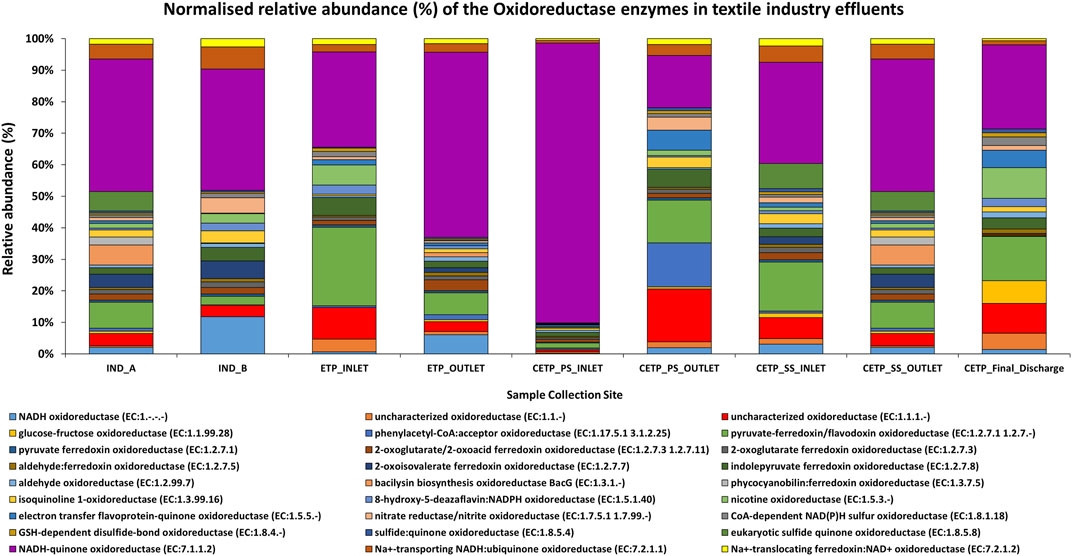
FIGURE 7. Normalized relative abundance (%) of the key oxidoreductase enzymes found in different samples of the textile industry effluent sites. The dominant enzymes identified in the industry effluent sites (IND_A and IND_B) were NADH-quinone oxidoreductase, NADH oxidoreductase, and pyruvate-ferredoxin oxidoreductase enzymes. The dominant enzymes present in the ETP sites were NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase. Similarly, CETP sites were found with the dominance of NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase. The uncharacterized oxidoreductase, apart from the NADH-quinone oxidoreductase and pyruvate-ferredoxin/flavodoxin oxidoreductase, was dominant in the final discharge samples.
Multiple copies of known genes encoding enzymes recognized for dye degradation were detected from the textile industry wastewater samples using the shotgun metagenomics approach, which further points towards the key role of integrated biosynthetic gene clusters (BGCs) for the synthetic pigments and chemical dye degradation during the wastewater treatment process. The results of the metagenome dataset collected from the industrial sites were dominated by the presence of oxidoreductase and reductase classes of enzymes. Therefore, sequence-based screening of shotgun metagenomics combined with the database annotations such as Kyoto Encyclopedia of Genes and Genomes (KEGG) and Cluster of Orthologous Groups of Proteins (COG) provides a robust approach for identification and characterization of the abundance of the enriched genes encoding for aromatic compound catabolism. Furthermore, wastewater microbial communities are often harbored by mobile genetic elements, such as plasmids, transposons, and integrative and conjugative elements that effectively disseminate the catabolic traits to phylogenetically distinct and diverse bacterial species (Ying et al., 2002; Jain et al., 2020). Previous research studies have shown that functional genes are a powerful biomarker for determining the potential of indigenous microbial communities to degrade xenobiotic compounds (More et al., 2014; Miao et al., 2015; Zhang et al., 2019; An et al., 2020; Li et al., 2020) during the wastewater and industrial effluent treatment process. Figure 8 shows the genes involved in proposed biosynthetic gene clusters (BGCs) for the textile dye degradation in the textile industry effluents. BCG-A indicates the involvement of 14 genes encoding different enzymes for the bioremediation potential in the wastewater treatment process. The higher abundance of the methyl-accepting chemotaxis proteins (MCPs) encoding genes significantly points towards the role of adaptation of the bacterial communities in the polluted environmental conditions, which further act as chemotaxis signal transducer protein sensing oxygen and were indicative of redox potential of the industry effluent sites (Engström and Hazelbauer, 1980). According to metagenome studies, textile dye degrading pathways are rich and abundant, with higher copy numbers of dye degrading enzyme encoding genes than the whole genome sequences. Therefore, we further speculate that these research findings shall considerably enhance the known gene pool diversity of the textile dye degrading genes and their encoded enzymes from the shotgun metagenomics approach.
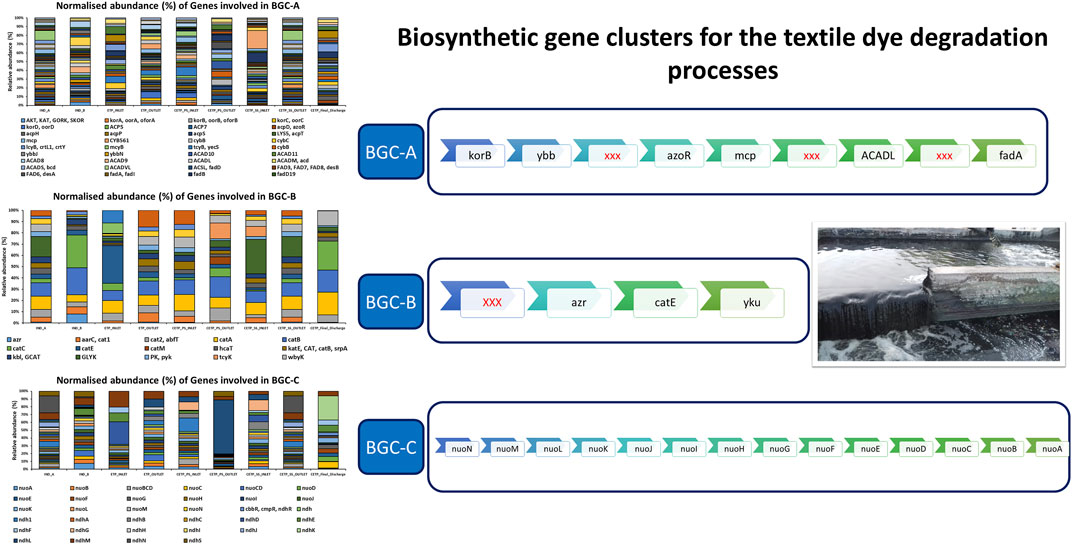
FIGURE 8. Relative abundance (%) of the gene involved in the biosynthetic gene clusters (BGCs) involved in the degradation of the aromatic compounds, synthetic dyes, and pigments.
Thus, the results of this research study suggest that textile industry effluents are probably influencing the occurrence and abundance of synthetic and xenobiotic compound degrading genes across the sampling points. At the same time, the functional potential of the enzyme encoding needs to be further examined for critical gaps in the preliminary experiment in the design of the treatment strategies with scope to improve the efficiency of the textile industry wastewater treatments plants. Furthermore, validation of the polyaromatic hydrocarbon (PAH) and textile dye degradation functional genes in treatment sites can be a useful approach for biomonitoring the bioremediation process at the industry effluent sites (Zhang et al., 2019). Additionally, combined with the metatranscriptomic approach, it can be used to understand the significant role of the functional genes contributing to microbial communities for textile degradation during active metabolic processes. Also, systematic studies on the effect of azo dye gradients on azo dye-related degradation genes are required to assess the role of azo dyes on the proliferation and prevalence of azo dye-related degradation genes in environmental sites (Sho et al., 2004; Suenaga et al., 2009; Zhuang et al., 2020; Srinivasan and Sadasivam, 2021; Wang et al., 2021). Furthermore, in the continuum of the recent research studies and the context of the textile industry-based shotgun metagenomics approach, we have tried to address the functional potential of the microbial community structure dominant in the industrial wastewater, ETP, and CETP sites. Thus, leveraging the genomic attributes defining the microbial population and genes encoding the catalytic enzymes could help in the ecological and efficient treatment process.
This research study highlights the overall dominance of the Shewanella decolorationis, Pseudomonas stutzeri, Aeromonas caviae, Desulfovibrio desulfuricans, and Klebsiella pneumoniae at the species level in all metagenome samples in all industrial and ETP sites. The hidden diversity is an untapped source of the biotechnological intervention based on the enzymatic degradation of the recovery of the useful products through recycling and process optimization specifically suited to the industrial wastewater treatment process. This further makes them highly suited for bioremediation of contaminated sites and waste removal process, and hundreds of uncharacterized cultivable species remain unexplored from these highly polluted environmental sites. This study reports the shotgun metagenome-based approach to decipher the microbial community from textile industry effluent treatment process and industry sites revealing the relative abundance of the significantly important genes encoding catalytic enzymes useful for the bioremediation of wastewater. Further, it highlights the integrated genomics approach by exploring the textile dye degrading genes and microbial community structure from the entire metagenome gene pool of the cultured and uncultivated microorganisms by shotgun sequencing. These research findings will help in designing efficient biological methods for the treatment of textile industry wastewaters.
The raw reads generated via sequencing of the samples from the textile industry have been submitted to the National Centre for Biotechnology Information (NCBI) under the Sequence Read Archive (SRA) under BioProject PRJNA655112.
DK was responsible for sample collection, performing the experiments, data analysis, and manuscript writing; ZP contributed to sample collection, performing the experiments, and data analysis; PP was responsible for the scientific discussion and writing of the manuscript; RP was responsible for data analysis and writing and editing of the manuscript; AP contributed to the scientific discussion, editing, and critical review; MJ was responsible for the scientific discussion, editing, and critical review; CJ was responsible for the conceptualization, experiment design, supervision, scientific discussion, editing, and critical review.
This work was supported by the Department of Science and Technology (DST), Government of Gujarat, Gandhinagar, India.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge the Department of Science and Technology (DST), the Government of Gujarat, and the Gujarat State Biotechnology Mission (GSBTM) for the financial assistance and infrastructure support for the research work. The authors also would like to acknowledge and highly appreciate the support of the staff members involved in the sample collections and their assistance in visiting the dyeing and printing industry sites, which has significantly helped in experimental analysis and data interpretation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2021.720707/full#supplementary-material
TCA, tricarboxylic acid cycle; ETP, effluent treatment plant; CETP, common effluent treatment plant; PCA, Principal Component Analysis; OT2, Ion One Touch 2; ES, Ion Enrichment System; KEGG, Kyoto Encyclopedia of Genes and Genomes; COG, Cluster of Orthologous Groups of Proteins; MG-RAST, Metagenome Rapid Annotation using Subsystem Technology; PAH, polyaromatic hydrocarbon; MCPs, methyl-accepting chemotaxis proteins.
Alves de Lima, R. O., Bazo, A. P., Salvadori, D. M. F., Rech, C. M., de Palma Oliveira, D., and de Aragão Umbuzeiro, G. (2007). Mutagenic and Carcinogenic Potential of a Textile Azo Dye Processing Plant Effluent that Impacts a Drinking Water Source. Mutat. Res./Genetic Toxicol. Environ. Mutagen. 626, 53–60. doi:10.1016/j.mrgentox.2006.08.002
An, X., Chen, Y., Chen, G., Feng, L., and Zhang, Q. (2020). Integrated Metagenomic and Metaproteomic Analyses Reveal Potential Degradation Mechanism of Azo Dye-Direct Black G by Thermophilic Microflora. Ecotoxicol. Environ. Saf. 196, 110557. doi:10.1016/j.ecoenv.2020.110557
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of Microbial Genomes Shed Light on Interconnected Biogeochemical Processes in an Aquifer System. Nat. Commun. 7, 13219. doi:10.1038/ncomms13219
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genomics 9, 75. doi:10.1186/1471-2164-9-75
Cardoso, J. C., Bessegato, G. G., and Boldrin Zanoni, M. V. (2016). Efficiency Comparison of Ozonation, Photolysis, Photocatalysis and Photoelectrocatalysis Methods in Real Textile Wastewater Decolorization. Water Res. 98, 39–46. doi:10.1016/j.watres.2016.04.004
Chen, B., Lin, K., and Wang, Y. (2009a). Undefined Revealing Interactive Toxicity of Aromatic Amines to Azo Dye Decolorizer Aeromonas Hydrophila. Elsevier. Available at: https://www.sciencedirect.com/science/article/pii/S0304389408016683 (Accessed July 21, 2020).
Chen, B.-Y., Lin, K.-W., Wang, Y.-M., and Yen, C.-Y. (2009b). Revealing Interactive Toxicity of Aromatic Amines to Azo Dye Decolorizer Aeromonas Hydrophila. J. Hazard. Mater. 166, 187–194. doi:10.1016/j.jhazmat.2008.11.030
Chen, X., Ning, X.-a., Lai, X., Wang, Y., Zhang, Y., and He, Y. (2021). Chlorophenols in Textile Dyeing Sludge: Pollution Characteristics and Environmental Risk Control. J. Hazard. Mater. 416, 125721. doi:10.1016/j.jhazmat.2021.125721
Dai, Q., Zhang, S., Liu, H., Huang, J., and Li, L. (2020). Sulfide-Mediated Azo Dye Degradation and Microbial Community Analysis in a Single-Chamber Air Cathode Microbial Fuel Cell. Bioelectrochemistry 131. doi:10.1016/j.bioelechem.2019.107349
Dhariwal, A., Chong, J., Habib, S., King, I. L., Agellon, L. B., and Xia, J. (2017). MicrobiomeAnalyst: a Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. academic.oup.com. Available at: https://academic.oup.com/nar/article-abstract/45/W1/W180/3760191 (Accessed March 29, 2021).
Dixit, S., and Garg, S. (2018). Biodegradation of Environmentally Hazardous Azo Dyes and Aromatic Amines Using Klebsiella pneumoniae. J. Environ. Eng. (United States 144. doi:10.1061/(ASCE)EE.1943-7870.0001353
Dixit, S., and Garg, S. (2019). Development of an Efficient Recombinant Bacterium and its Application in the Degradation of Environmentally Hazardous Azo Dyes. Int. J. Environ. Sci. Technol. 16, 7137–7146. doi:10.1007/s13762-018-2054-7
El-Kassas, H. Y., and Mohamed, L. A. (2014). Bioremediation of the Textile Waste Effluent by Chlorella Vulgaris. Egypt. J. Aquat. Res. 40, 301–308. doi:10.1016/j.ejar.2014.08.003
Engström, P., and Hazelbauer, G. L. (1980). Multiple Methylation of Methyl-Accepting Chemotaxis Proteins During Adaptation of E. coli to Chemical Stimuli. Cell 20, 165–171. doi:10.1016/0092-8674(80)90244-5
Forgacs, E., Cserháti, T., and Oros, G. (2004). Removal of Synthetic Dyes from Wastewaters: A Review. Environ. Int. 30, 953–971. doi:10.1016/j.envint.2004.02.001
Freedman, D. L., Payauys, A. M., and Karanfil, T. (2005). The Effect of Nutrient Deficiency on Removal of Organic Solvents from Textile Manufacturing Wastewater during Activated Sludge Treatment. Environ. Technol. 26, 179–188. doi:10.1080/09593332608618570
Garg, S. K., and Tripathi, M. (2017). Microbial Strategies for Discoloration and Detoxification of Azo Dyes from Textile Effluents. Res. J. Microbiol. 12, 1–19. doi:10.3923/jm.2017.1.19
Gaur, N., Narasimhulu, K., and Y, P. (2018). Recent Advances in the Bio-Remediation of Persistent Organic Pollutants and its Effect on Environment. J. Clean. Prod. 198, 1602–1631. doi:10.1016/j.jclepro.2018.07.076
Ghodake, G., Jadhav, U., Tamboli, D., Kagalkar, A., and Govindwar, S. (2011). Decolorization of Textile Dyes and Degradation of Mono-Azo Dye Amaranth by Acinetobacter Calcoaceticus NCIM 2890. Indian J. Microbiol. 51, 501–508. doi:10.1007/s12088-011-0131-4
Hammer, Ø., Harper, D. A. T., and Ryan, P.D. (2001). Past: Paleontological Statistics Software Package for Education and Data Analysis. Available at: https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=PAST+Hammer+et+al.%2C+2001&btnG= (Accessed March 29, 2021).
Hatamoto, M., Kaneshige, M., Nakamura, A., and Yamaguchi, T. (2014). Bacteroides Luti Sp. nov., an Anaerobic, Cellulolytic and Xylanolytic Bacterium Isolated from Methanogenic Sludge. Int. J. Syst. Evol. Microbiol. 64, 1770–1774. doi:10.1099/ijs.0.056630-0
He, S., Sun, W., Wang, J., Chen, L., Zhang, Y., and Yu, J. (2016). Enhancement of Biodegradability of Real Textile and Dyeing Wastewater by Electron Beam Irradiation. Radiat. Phys. Chem. 124, 203–207. doi:10.1016/j.radphyschem.2015.11.033
Hossain, L., Sarker, S. K., and Khan, M. S. (2018). Evaluation of Present and Future Wastewater Impacts of Textile Dyeing Industries in Bangladesh. Environ. Dev. 26, 23–33. doi:10.1016/j.envdev.2018.03.005
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinf. 11. doi:10.1186/1471-2105-11-119
Jain, K., Desai, C., Tiwari, O., and Madamwar, D. (2020). “Dyes: Effect on the Environment and Biosphere and Their Remediation Constraints,” in Microbial Bioremediation & Biodegradation (Springer Singapore), 73–94. doi:10.1007/978-981-15-1812-6_3
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 44, D457–D462. doi:10.1093/nar/gkv1070
Kaur, P., Kushwaha, J. P., and Sangal, V. K. (2018). Electrocatalytic Oxidative Treatment of Real Textile Wastewater in Continuous Reactor: Degradation Pathway and Disposability Study. J. Hazard. Mater. 346, 242–252. doi:10.1016/j.jhazmat.2017.12.044
Khan, S., and Malik, A. (2018). Toxicity Evaluation of Textile Effluents and Role of Native Soil Bacterium in Biodegradation of a Textile Dye. Environ. Sci. Pollut. Res. 25, 4446–4458. doi:10.1007/s11356-017-0783-7
Kishor, R., Purchase, D., Saratale, G. D., Saratale, R. G., Ferreira, L. F. R., Bilal, M., et al. (2021). Ecotoxicological and Health Concerns of Persistent Coloring Pollutants of Textile Industry Wastewater and Treatment Approaches for Environmental Safety. J. Environ. Chem. Eng. 9, 105012. doi:10.1016/j.jece.2020.105012
Kumar, D., Pandit, P. D., Patel, Z., Bhairappanavar, S. B., and Das, J. (2019). “Perspectives, Scope, Advancements, and Challenges of Microbial Technologies Treating Textile Industry Effluents,” in Microbial Wastewater Treatment (Elsevier), 237–260. doi:10.1016/b978-0-12-816809-7.00011-7
Li, C., Ren, H., Yin, E., Tang, S., Li, Y., and Cao, J. (2015a). Pilot-scale Study on Nitrogen and Aromatic Compounds Removal in Printing and Dyeing Wastewater by Reinforced Hydrolysis-Denitrification Coupling Process and its Microbial Community Analysis. Environ. Sci. Pollut. Res. 22, 9483–9493. doi:10.1007/s11356-015-4124-4
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015b). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi:10.1093/bioinformatics/btv033
Li, J., Liang, Y., Miao, Y., Wang, D., Jia, S., and Liu, C.-H. (2020). Metagenomic Insights into Aniline Effects on Microbial Community and Biological Sulfate Reduction Pathways during Anaerobic Treatment of High-Sulfate Wastewater. Sci. Total Environ. 742, 140537. doi:10.1016/j.scitotenv.2020.140537
Liu, Y. H., Ye, M., Lu, Y., Zhang, X., and Li, G. (2011). Improving the Decolorization for Textile Dyes of a Metagenome-Derived Alkaline Laccase by Directed Evolution. Appl. Microbiol. Biotechnol. 91, 667–675. doi:10.1007/s00253-011-3292-5
Manickam, P., and Vijay, D. (2021). “Chemical Hazards in Textiles,” in Chemical Management in Textiles and Fashion (Elsevier), 19–52. doi:10.1016/b978-0-12-820494-8.00002-2
Mate, D. M., and Alcalde, M. (2015). Digital CSIC Laccase Engineering: from Rational Design to Directed Evolution. Available at: https://www.sciencedirect.com/science/article/pii/S0734975014001943 (Accessed March 29, 2021).
Meerbergen, K., Van Geel, M., Waud, M., Willems, K. A., Dewil, R., Van Impe, J., et al. (2017). Assessing the Composition of Microbial Communities in Textile Wastewater Treatment Plants in Comparison with Municipal Wastewater Treatment Plants. MicrobiologyOpen 6, e00413–13. doi:10.1002/mbo3.413
Methneni, N., Morales-González, J. A., Jaziri, A., Mansour, H. B., and Fernandez-Serrano, M. (2021). Persistent Organic and Inorganic Pollutants in the Effluents from the Textile Dyeing Industries: Ecotoxicology Appraisal via a Battery of Biotests. Environ. Res. 196, 110956. doi:10.1016/j.envres.2021.110956
Metsalu, T., and Vilo, J. (2015). ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res., 43, W566–W570. online 43. doi:10.1093/nar/gkv468
Miao, Y., Liao, R., Zhang, X.-X., Wang, Y., Wang, Z., Shi, P., et al. (2015). Metagenomic Insights into Cr(VI) Effect on Microbial Communities and Functional Genes of an Expanded Granular Sludge Bed Reactor Treating High-Nitrate Wastewater. Water Res. 76, 43–52. doi:10.1016/j.watres.2015.02.042
Mishra, S., Nayak, J. K., and Maiti, A. (2020). Bacteria-mediated Bio-Degradation of Reactive Azo Dyes Coupled with Bio-Energy Generation from Model Wastewater. Clean. Techn Environ. Pol. 22, 651–667. doi:10.1007/s10098-020-01809-y
More, R. P., Mitra, S., Raju, S. C., Kapley, A., and Purohit, H. J. (2014). Mining and Assessment of Catabolic Pathways in the Metagenome of a Common Effluent Treatment Plant to Induce the Degradative Capacity of Biomass. Bioresour. Technol. 153, 137–146. doi:10.1016/j.biortech.2013.11.065
Redfern, L. K., Gardner, C. M., Hodzic, E., Ferguson, P. L., Hsu-Kim, H., and Gunsch, C. K. (2019). A New Framework for Approaching Precision Bioremediation of PAH Contaminated Soils. J. Hazard. Mater. 378, 120859. doi:10.1016/j.jhazmat.2019.120859
Robinson, T., McMullan, G., Marchant, R., and Nigam, P. (2001). Remediation of Dyes in Textile Effluent: A Critical Review on Current Treatment Technologies with a Proposed Alternative. Bioresour. Technol. 77, 247–255. doi:10.1016/S0960-8524(00)00080-8
Samuchiwal, S., Gola, D., and Malik, A. (2021). Decolourization of Textile Effluent Using Native Microbial Consortium Enriched from Textile Industry Effluent. J. Hazard. Mater. 402, 123835. doi:10.1016/j.jhazmat.2020.123835
Shah, R., and Jha, S. (2013). Alishewanellasp. Strain GIDC-5, Arsenite Hyper-Tolerant Bacteria Isolated from Industrial Effluent of South Gujarat, India. Chem. Ecol. 29, 427–436. doi:10.1080/02757540.2013.774379
Sho, M., Hamel, C., and Greer, C. W. (2004). Two Distinct Gene Clusters Encode Pyrene Degradation in Mycobacterium Sp. Strain S65. FEMS Microbiol. Ecol. 48, 209–220. doi:10.1016/j.femsec.2004.01.011
Srinivasan, S., and Sadasivam, S. K. (2021). Biodegradation of Textile Azo Dyes by Textile Effluent Non-adapted and Adapted Aeromonas Hydrophila. Environ. Res. 194, 110643. doi:10.1016/j.envres.2020.110643
Stolz, A. (2001). Basic and Applied Aspects in the Microbial Degradation of Azo Dyes. Appl. Microbiol. Biotechnol. 56, 69–80. doi:10.1007/s002530100686
Suenaga, H., Koyama, Y., Miyakoshi, M., Miyazaki, R., Yano, H., Sota, M., et al. (2009). Novel Organization of Aromatic Degradation Pathway Genes in a Microbial Community as Revealed by Metagenomic Analysis. ISME J. 3, 1335–1348. doi:10.1038/ismej.2009.76
Tamames, J., and Puente-Sánchez, F. (2019). SqueezeMeta, a Highly Portable, Fully Automatic Metagenomic Analysis Pipeline. Front. Microbiol. 9, 3349. doi:10.3389/fmicb.2018.03349
Verma, A. K., Dash, R. R., and Bhunia, P. (2012). A Review on Chemical Coagulation/flocculation Technologies for Removal of Colour from Textile Wastewaters. J. Environ. Manage. 93, 154–168. doi:10.1016/j.jenvman.2011.09.012
Berkessa, Y. W., Yan, B., Li, T., Jegatheesan, V., and Zhang, Y. (2020). Treatment of Anthraquinone Dye Textile Wastewater Using Anaerobic Dynamic Membrane Bioreactor: Performance and Microbial Dynamics. Chemosphere 238, 124539. doi:10.1016/j.chemosphere.2019.124539
Wang, X., Xia, R., Sun, M., and Hu, F. (2021). Metagenomic Sequencing Reveals Detoxifying and Tolerant Functional Genes in Predominant Bacteria Assist Metaphire Guillelmi Adapt to Soil Vanadium Exposure. J. Hazard. Mater. 415, 125666. doi:10.1016/j.jhazmat.2021.125666
Watsuntorn, W., Ruangchainikom, C., Rene, E. R., Lens, P. N. L., and Chulalaksananukul, W. (2019). Comparison of Sulphide and Nitrate Removal from Synthetic Wastewater by Pure and Mixed Cultures of Nitrate-Reducing, Sulphide-Oxidizing Bacteria. Bioresour. Technol. 272, 40–47. doi:10.1016/j.biortech.2018.09.125
Wijetunga, S., Li, X.-F., and Jian, C. (2010). Effect of Organic Load on Decolourization of Textile Wastewater Containing Acid Dyes in Upflow Anaerobic Sludge Blanket Reactor. J. Hazard. Mater. 177, 792–798. doi:10.1016/j.jhazmat.2009.12.103
Xu, M., Guo, J., and Sun, G. (2007). Biodegradation of Textile Azo Dye by Shewanella Decolorationis S12 under Microaerophilic Conditions. Appl. Microbiol. Biotechnol. 76, 719–726. doi:10.1007/s00253-007-1032-7
Yang, X., Chen, Y., Guo, F., Liu, X., Su, X., and He, Q. (2020). Metagenomic Analysis of the Biotoxicity of Titanium Dioxide Nanoparticles to Microbial Nitrogen Transformation in Constructed Wetlands. J. Hazard. Mater. 384, 121376. doi:10.1016/j.jhazmat.2019.121376
Yaseen, D. A., and Scholz, M. (2019). Textile Dye Wastewater Characteristics and Constituents of Synthetic Effluents: a Critical Review. Int. J. Environ. Sci. Technol. 16, 1193–1226. doi:10.1007/s13762-018-2130-z
Yesilada, O., Birhanli, E., and Geckil, H. (2018). “Bioremediation and Decolorization of Textile Dyes by White Rot Fungi and Laccase Enzymes. Fungal Biol. Mycoremediation Environ. Sustainability 2, 121–153. doi:10.1007/978-3-319-77386-5_5
Ying, G.-G., Williams, B., and Kookana, R. (2002). Environmental Fate of Alkylphenols and Alkylphenol Ethoxylates-A Review. Environ. Int. 28, 215–226. doi:10.1016/S0160-4120(02)00017-X
Zhang, S., Hu, Z., and Wang, H. (2019). Metagenomic Analysis Exhibited the Co-metabolism of Polycyclic Aromatic Hydrocarbons by Bacterial Community from Estuarine Sediment. Environ. Int. 129, 308–319. doi:10.1016/j.envint.2019.05.028
Keywords: metagenomics, biosynthetic gene clusters (BGCs), bioremediation, textile industry effluent, wastewater
Citation: Kumar D, Patel Z, Pandit P, Pandit R, Patel A, Joshi M and Joshi C (2021) Textile Industry Wastewaters From Jetpur, Gujarat, India, Are Dominated by Shewanellaceae, Bacteroidaceae, and Pseudomonadaceae Harboring Genes Encoding Catalytic Enzymes for Textile Dye Degradation. Front. Environ. Sci. 9:720707. doi: 10.3389/fenvs.2021.720707
Received: 04 June 2021; Accepted: 29 July 2021;
Published: 02 September 2021.
Edited by:
Shadi Rahimi, Chalmers University of Technology, SwedenReviewed by:
Ram Naresh Bharagava, Babasaheb Bhimrao Ambedkar University, IndiaCopyright © 2021 Kumar, Patel, Pandit, Pandit, Patel, Joshi and Joshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaitanya Joshi, ZGlyZWN0b3JAZ2JyYy5yZXMuaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.