 Peng Wu1,3,4
Peng Wu1,3,4 Yong Liu1,2,3,4*
Yong Liu1,2,3,4* Chunhou Li1,2,3,4*Yayuan Xiao1,3,4Teng Wang1,2,3,4Lin Lin1,3,4Yufang Xie1,3,4
Chunhou Li1,2,3,4*Yayuan Xiao1,3,4Teng Wang1,2,3,4Lin Lin1,3,4Yufang Xie1,3,4- 1Key Laboratory of South China Sea Fishery Resources Exploitation and Utilization, Ministry of Agriculture and Rural Affairs, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China
- 2Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 3Scientific Observation and Research Field Station of Pearl River Estuary Ecosystem, Guangzhou, China
- 4Guangdong Provincial Key Laboratory of Fishery Ecology and Environment, Guangzhou, China
By their nature and geographical location, estuaries shape different marine habitats via freshwater and seawater interactions. Thus, fish intestinal microbiota, as mediated by estuary habitat fluctuations, are fundamentally important but rarely studied. Similarly, it is unclear how, and to what extent, water microbiota influences fish intestinal microbiota in different estuary habitats. In this study, the euryhaline fish species, Collichthys lucidus from three different habitats in the Pearl River estuary (PRE) was investigated to determine the influence of habitat fluctuation on intestinal microbiota. The three water environments selected for sample collection were very different, particularly for chlorophyll-a, suspended solid, and nutrient constituents. Using high-throughput sequencing of 16S rRNA gene amplicons, we observed that dominant microbial genera in surrounding estuary waters or fish intestines were seldom shared. The most dominant genera in water samples were Candidatus Actinomarina and HIMB11, while Bifidobacterium, Stenotrophomonas, Escherichia-Shigella and Rhodopseudomonas were more abundant in fish intestines. Fish hosts can shape fish intestinal microbiota. However, microbial exchange was also found between fish intestines and water samples. The frequency of microbial exchange between fish intestines and water samples was increased from upstream to downstream estuary points, and was influenced by changes in seawater salinity in the estuary. Finally, core intestinal microbiota from C. lucidus was analyzed, and showed that Bifidobacterium, Rhodopseudomonas, Escherichia-Shigella, Acinetobacter, and Stenotrophomonas were highly abundant. These microbiota were theoretically implicated in immune responses, nutrient metabolism, probiotics, and potential pathogen behaviors. Overall, these data highlighted the composition of C. lucidus intestinal microbiota in different habitats across the PRE.
Introduction
Estuaries are transition zones between the land and the sea, and represent a dynamic system where freshwater meets seawater. These interactions make estuaries unique habitats in terms of habitat diversity and species productivity (Mitra, 2015; Kamrani et al., 2016; Zhou et al., 2019b). It was previously shown that protective estuary environments may facilitate the generation of over half of all marine fish (Mitra, 2015). Moreover, spatial differences in fish assemblages in estuaries were primarily attributed to unique salinity, water temperature, primary productivity, turbidity, and water nutrient conditions (Eick and Thiel, 2014; Zhou et al., 2019b). However, insights on how fish adapt to variable physico-chemical features in these environments requires further investigation (Molina et al., 2020).
The intestinal microbiota is considered an “extra organ”, and plays a key role in fish adaption to the environment, mediating nutrient metabolism, immune responses, and gut homeostasis (Li et al., 2015; Egerton et al., 2018; Butt and Volkoff, 2019). Fish intestinal microbiota originate from the eggs, the first feed, and the surrounding waters, and develop a complex, habitat-driven composition (Dehler et al., 2017a; Egerton et al., 2018). Generally, factors affecting intestinal microbiota are categorized as: 1) environmental factors, 2) diet, and 3) host-associated factors (Talwar et al., 2018). Previous studies have focused on fish intestinal microbiota in aquaculture, its relationship with the environment, and demonstrated intestinal microbiota roles for host health and welfare (Dehler et al., 2017a; Egerton et al., 2018; Sun et al., 2019). However, the intestinal microbiota from wild fish requires more investigation (Egerton et al., 2018).
In estuaries, tidal action mixes inland freshwater with seawater, generating spatial variations in salinity, nutrients, oxygen, turbidity, and organic pollutants (Fu et al., 2003; Mitra, 2015; Wu et al., 2017). However, little is known how fish intestinal microbiota respond to these physico-chemical variations. Also, spatial distributions of water microbial communities in estuaries are different, e.g., Proteobacteria classes vary significantly between freshwater and saltwater environments, whereas Actinobacteria are more abundant in freshwater areas (Kirchman et al., 2005; Zhang et al., 2006; Feng et al., 2009). Furthermore, the main sources of microbes in fish come from surrounding waters and their diet (Wu et al., 2012; Dehler et al., 2017a; Egerton et al., 2018). However, limited attention has been given to the exchange of microbial communities between fish intestines and estuary waters. Previous studies have suggested that microbial taxa/operational taxonomic units (OTUs), recognized as core microbiota, are constant in fish, regardless of fish populations or geographical locations (Ghanbari et al., 2015; Givens et al., 2015; Kokou et al., 2019).
The Pearl River estuary (PRE) is a subtropical estuary, located on the south coast of China, with an annual rainfall of 1600–2300 mm (Huang et al., 2003). The PRE is divided into different regions, comprising the Shiziyang Channel, Lingding Bay, and the northern South China Sea (Wu et al., 2014). The Pearl River is the second largest river in China, with an annual average river discharge of 10,524 m3 s−1 (Zhao, 1990; Huang et al., 2003). Approximately 20% of the total flow appears in the dry season from October to March, with 80% in the wet season, from April to September (Zhao, 1990; Huang et al., 2003). In the dry season, seawater covers most of the estuary (Ying, 1994).
Collichthys lucidus is an economically important fish species, widely distributed across the PRE. C. lucidus is a small-sized species and often lives in the benthic zones of coastal waters (Liu et al., 2015). This fish is short-lived with a life span of about 3 years and becomes first sexually mature when it is about 80 cm (Zhuang, 2018). C. lucidus usually feeds on zoobenthos, small fishes and mysidacea (Zhuang, 2018). In 1986, catch levels in the PRE were reported at 4,000 tons/year, accounting for 26.0% of the total fish biomass by bottom trawling (He and Li, 1988). However, C. lucidus populations and biomass have decreased rapidly in recent years (Huang et al., 2018). In this study, we collected C. lucidus samples from three different habitats along the PRE, to investigate intestinal microbiota composition, following variations in the water environment. Furthermore, we also investigated relationships between fish intestinal microbiota and water microbiota from individual PRE sites. These data provided key insights on C. lucidus intestinal microbiota interactions with estuary water.
Materials and Methods
Sample Collection and Measurements
Fish (intestines) and water samples were collected in triplicate at three sampling sites along the PRE, in the dry season (December 2019) (Supplementary Figure S1). Site A1 was situated near the Shiziyang Channel, while sites A2 and A3 were located in Lingding Bay and the northern South China Sea, respectively, (Supplementary Figure S1). C. lucidus was collected from each site using bottom trawling. Three adult fish (average body length; 9.7–10.3 cm) from each site were chosen for intestinal sampling. The whole intestine was aseptically dissected in situ using sterile scissors. Then, the contents were squeezed out, collected into sterile plastic cryotubes, and stored in liquid nitrogen for DNA extraction. Water samples were taken from the surface and bottom layers of each individual site, and mixed. Mixed water from each site (1 L) was filtered in situ through a 0.2 μm pore-size membrane (Millipore, United States), and the membrane immediately stored in liquid nitrogen for DNA extraction. The remaining water was used for physicochemical analysis following previously published protocols [“The specification for marine monitoring” GB 17378.4 (2007), China]; and Wu et al. (2017). Salinity, temperature, pH and dissolved oxygen (DO) were determined in situ using the YSI Pro Plus meter (YSI Inc. Yellow Springs, United States). Chlorophyll a (Chl a) was extracted in 10 ml 90% acetone in the dark for 24 h in a refrigerator and measured using spectrophotometer (Shimadzu, Japan). Oil from the seawater was extracted with n-hexane and measured using UV-spectrophotometer according to the standard curve. The suspended solids from 0.5 L seawater were measured gravimetrically on a pre-weighted Whatman GF/C filter. NO2-N, NO3-N, NH4-N, and PO4-P were analyzed using a continuous flow injection analyzer (AA3, Seal Analytical, UK). Water environmental parameters are shown (Supplementary Table S1), and indicated differences in chlorophyll a (Chl a) concentrations, suspended solids (SS), and nutrients (NO2-N, NO3-N, NH4-N, and PO4-P) along the PRE.
DNA Extraction and High-Throughput Sequencing
Total genomic DNA from filter membranes and intestinal contents (0.2 g) were extracted using the E. Z.N.A® Water DNA Kit (Omega, United States) and QIAamp® Fast DNA Stool Mini Kit (Qiagen, United States), respectively, according to manufacturer’s instructions. PCR was performed on DNA by targeting the V3–V4 region of the microbial 16S rRNA gene. The following primers were used; 341F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTATCTAAT-3′) which were synthesized by Sangon Biotech (Shanghai, China). A final 50 μL PCR reaction volume consisted of; 5 μL 10 × KOD buffer, 1.5 μL each primer (5 μM), 5 μL 2.5 mM dNTPs, 1 μL KOD polymerase, and 20 ng DNA template. PCR amplifications (95°C for 2 min, followed by 27 cycles at 98°C for 10 s, 62°C for 30 s, and 68°C for 30 s and a final extension at 68°C for 10 min) were conducted in triplicate. Amplified PCR products were then extracted and purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States). These purified amplicons were pooled in equimolar amounts and sequenced on an Illumina HiSeq 2500 platform, using a 2 × 250 base pair (bp) paired-end strategy (Gene Denovo Co., Guangzhou, China). Raw reads from intestine and water samples were deposited into the Sequence Read Archive (SRA) database of the NCBI, under accession numbers; PRJNA647310 and PRJNA647485, respectively.
Sequence Analysis
After sequencing, reads containing >10% unknown nucleotides and <50% bases with quality Q-values > 20, were removed to generate clean reads. Paired-end clean reads were then merged as raw tags using FLASH (V1.2.11), with a minimum overlap of 10 bp, and mismatch error rates of 2% (Chen et al., 2018). Raw tags were then received after merging paired-end clean reads using FLASH (V1.2.11) (Magoc and Salzberg, 2011). Raw tags were processed with QIIME (V1.9.1) software to generate clean tags under specific filtering conditions (Caporaso et al., 2010). Subsequently, UCHIME algorithms were used to filter clean tags, remove chimeric tags, and derive effective reads (Edgar et al., 2011). Finally, these effective reads were clustered into operational taxonomic units (OTUs) using UPARSE (V9.2.64), with 97% sequence similarity (Edgar, 2013). A dominant sequence was chosen to represent each OTU, and taxonomic assignments analyzed using the RDP classifier (V2.2) Wang et al. (2007) in the SILVA database (V128) (Pruesse et al., 2007). QIIME (V1.9.1) was used to calculate α-diversity indices, including Shannon, Simpson, and Chao1 indices (Caporaso et al., 2010).
Statistical Analysis
Principal coordinates analysis (PCoA) was conducted based on the Bray-Curtis distance of microbial phylogeny, using R (V4.0.0) software. The relative abundance of dominant bacterial phyla/classes (top 20) was performed using Microsoft Excel. A heat map was constructed using STAMP (V2.1.3) software Parks et al. (2014), where samples were analyzed using the relative abundance of dominant bacterial communities at genus levels (top 40). Significant dominant microbial community differences between fish intestines and water samples at the phylum/class (top 20) and genus level (top 40) were compared and analyzed using a two-sided Welch’s t-test (p < 0.05) by STAMP (V2.1.3) software (Parks et al., 2014). Analysis of similarities (ANOSIM) was used to test for differences in microbial communities between intestine and water samples, based on the Bray-Curtis distance matrix (p < 0.05). The R value from ANOSIM was used to determine any overlaps in microbial communities (Buttigieg and Ramette, 2014). Mantel tests and Pearson’s correlation analyses were used to examine correlations between microbial communities and environmental factors, using the ggcor R package (V3.6.1).
Results
High-Throughput Sequencing Data and Operational Taxonomic Units
The Illumina HiSeq sequencing platform produced a total of 1,698,194 raw tags from intestine and water samples. In total, 3,321 OTUs (intestine: 1,217, water: 2,349) were identified at 97% sequence similarity. OTUs in water samples were much higher than intestine samples at the same site. Specifically, average OTU numbers in water samples ranged between 1,822 and 2,797 across the three sites, while this range was 841 and 1,047 for intestine samples. The α-diversity (Shannon and Chao1) index of microbial communities in water samples was also higher. In addition, for individual intestine or water samples, OTU numbers and the Shannon index from site A2 were the highest, but no significant differences were observed between sites (p > 0.05). The bacterial coverage of each sample was >98.0%, indicating most bacteria in samples were represented and identified (Supplementary Table S2). PCoA based on Bray-Curtis distances of OTU abundance of each sample, indicated that intestine and water samples were separated from each other (Figure 1). However, intestine samples from different sites were closely clustered together, with a high similarity. Water samples from the three sites were clustered into another group, but exhibited low similarity between sites. Our ANOSIM data further confirmed that microbial communities between intestine and water samples from the same site were well separated (R > 0.9). However, microbial community distribution between intestine samples were barely separated (Supplementary Table S3).
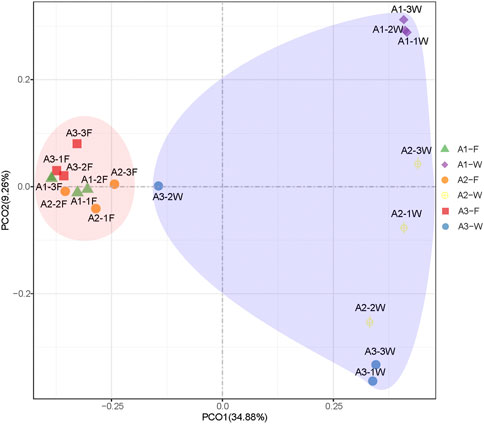
FIGURE 1. PCoA of microbial communities in intestine (F) and water (W) samples from the Pearl River estuary, based on Bray-Curtis distance.
Microbial Composition Between Intestine and Water Samples
The microbial composition of the top 20 phyla in each sample is shown (Figure 2A). The relative abundance of these phyla from individual samples accounted for >95% of all sequences. Proteobacteria was the most abundant taxa in all samples. In addition, Actinobacteria, Cyanobacteria, Firmicutes, Bacteroidetes, and Planctomycetes were also dominant in intestine and water samples. Within Proteobacteria, Gammaproteobacteria was the most dominant class, followed by Alphaproteobacteria and Deltaproteobacteria in all samples. However, the dominant phyla and proteobacterial classes between fish intestine and water samples (Figure 3A) were analyzed to test whether there were significant differences (p < 0.05) in the two groups. Actinobacteria in water samples (mean; 32.5%) were more abundant than intestine samples (mean; 12.8%), while, Alphaproteobacteria, Marinimicrobia SAR406, and Armatimonadetes were more abundant in water samples, with means of 14.0, 0.4, and 0.02%, respectively. When compared with water samples, Firmicutes, Acidobacteria, Gemmatimonadetes, and Chloroflexi displayed a significantly higher abundance in intestine samples (p < 0.05).
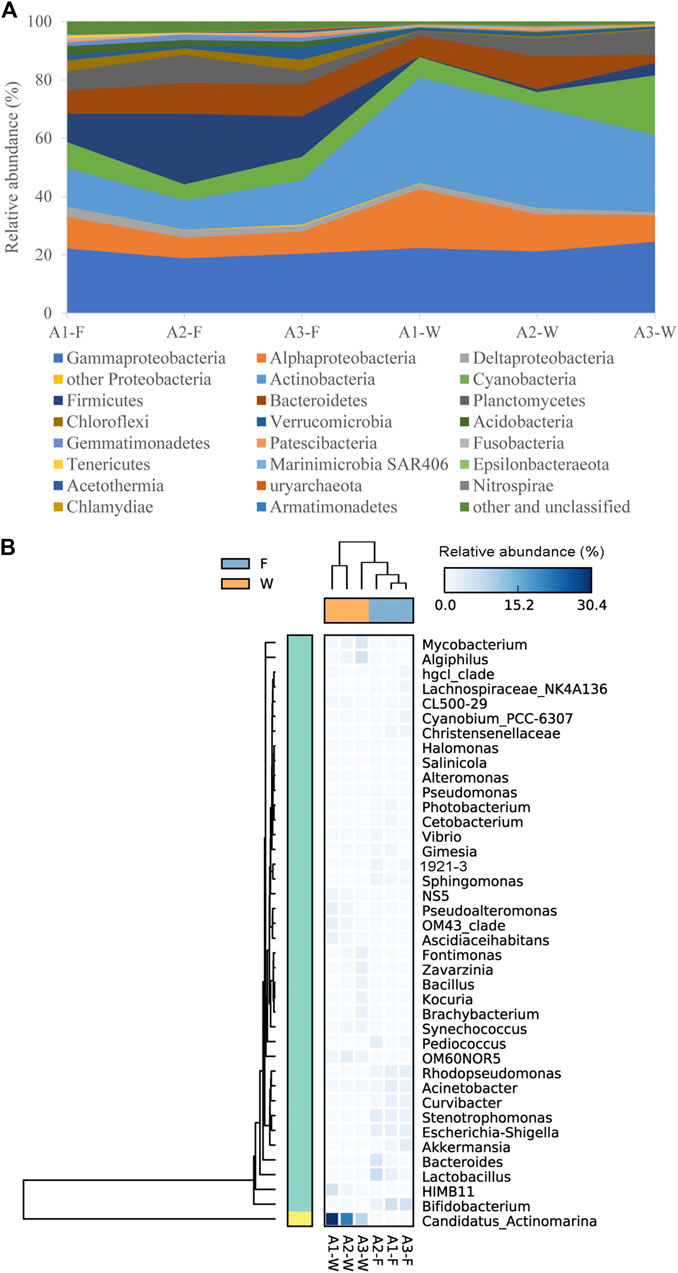
FIGURE 2. Relative abundance of dominant microbial communities in intestine (F) and water (W) samples from different sites along the Pearl River estuary, at phylum/class (A) and genus (B) levels.
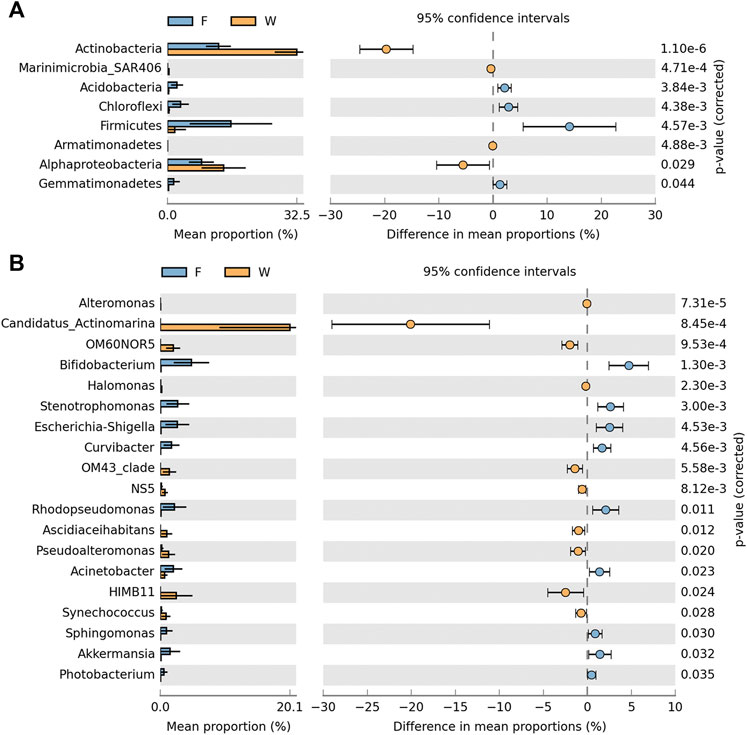
FIGURE 3. Dominant microbial community significant differences between intestine (F) and water (W) samples from the Pearl River estuary at phylum/class (A) and genus (B) levels (Welch’s t-test, p < 0.05).
Using a heat map, the top 40 genera were selected to show the microbial composition of intestine and water samples (Figure 2B). Candidatus Actinomarina, HIMB11, Algiphilus, and Mycobacterium were the dominant genera in water samples (>2%), especially Candidatus Actinomarina which belonged to the Actinobacteria phylum, and showed the highest abundance (20.1%). However, the most dominant genera in intestine samples (>2%) included Bifidobacterium, Lactobacillus, Stenotrophomonas, Escherichia-Shigella, Rhodopseudomonas, and Acinetobacter. Moreover, 19 of the top 40 genera exhibited significant differences between intestine and water samples (p < 0.05) (Figure 3B). The nine genera; Bifidobacterium, Stenotrophomonas, Escherichia-Shigella, Curvibacter, Rhodopseudomonas, Acinetobacter, Sphingomonas, Akkermansia, and Photobacterium were significantly higher in intestine samples (p < 0.05), while the remaining 10, including Candidatus Actinomarina, HIMB11, OM60NOR5, OM43 clade, Pseudoalteromonas, etc., were much higher in water samples (p < 0.05).
Microbial Community Relationships Between Intestine and Water Samples
Microbial exchange between intestine and water samples from the same site was analyzed (Figure 4). The number of shared OTUs at sites A1, A2, and A3 were 152, 200, and 176, respectively. These OTUs accounted for 17.0–20.9% of total OTUs in intestinal samples from the three sites, while this was 7.2–8.7% in water samples (Figures 4 A–C). However, shared OTUs represented a higher proportion of total sequences in intestinal samples (39.4–47.0%) (Table 1). In water samples, shared OTUs at sites A1, A2, and A3 corresponded to 13.8, 33.4, and 44.9% of total sequences (Table 1). Therefore, the exchange of microbial communities between intestine and water samples was more pronounced at downstream PRE sampling sites.
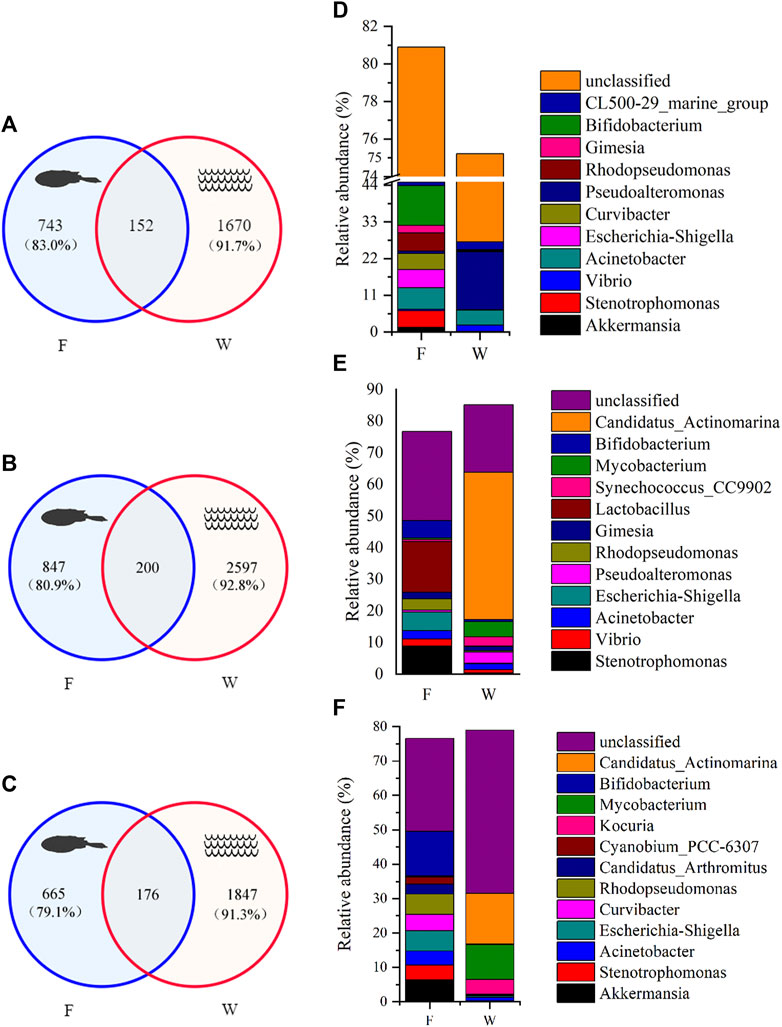
FIGURE 4. Microbial composition between fish intestine (F) and water (W) samples from the Pearl River estuary. (A–C) Venn diagrams of OTU composition in intestine and water samples from individual sites, A1, A2, and A3 (correspond to A–C). The percentage in parentheses indicates the contribution of unique OTUs to total OTUs in each sample; (D–F) The relative abundance of dominant microbial genera based on shared OTU analysis for the three sites.
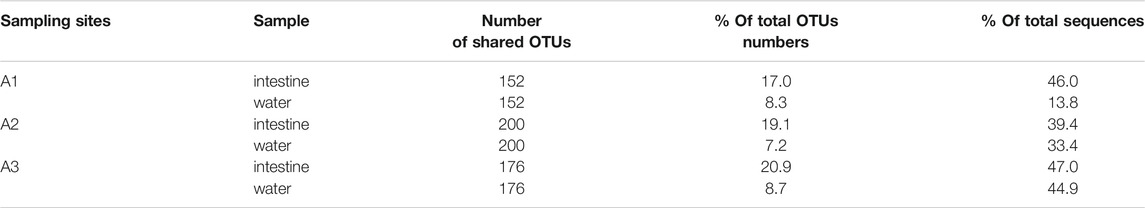
TABLE 1. Composition of shared OTUs in intestine and water samples from different Pearl River estuary sites (A1, A2, and A3).
At each site, the taxonomic assignment of shared OTUs was also analyzed, and dominant genera taxa identified (Figures 4 D–F). We observed differences in the relative abundance of dominant genera between intestine and water samples at the same site, due to the high abundance of special genus (Pseudoalteromonas, Candidatus Actinomarina, Mycobacterium) in water samples, but not in intestine samples. For example, Pseudoalteromonas had a very high abundance in water samples at site A1, accounting for 17.3% of total shared sequences. Candidatus Actinomarina (46.6%) was the most dominant genus in water samples at site A2. Similarly, Candidatus Actinomarina (14.8%) and Mycobacterium (10.1%) were more abundant at site A3. Nevertheless, microbial exchange between intestine and water samples was also observed. Acinetobacter and CL500–29 marine group were the dominant groups in both intestine and water samples at site A1, while Vibrio, Acinetobacter and Gimesia were exchanged frequently between samples at site A2. Intestine and water samples at site A3 shared the dominant genus, Acinetobacter. In general, this genus was shared by both intestine and water samples, and showed a constant dominance at all sites. Moreover, the relationship between microbial communities and environmental factors, using Mantel tests (Figure 5), revealed that intestinal microbiota exhibited a good correlation with oil, whereas water microbiota were correlated with suspended solids (SS), salinity, NO2-N, and NO3-N.
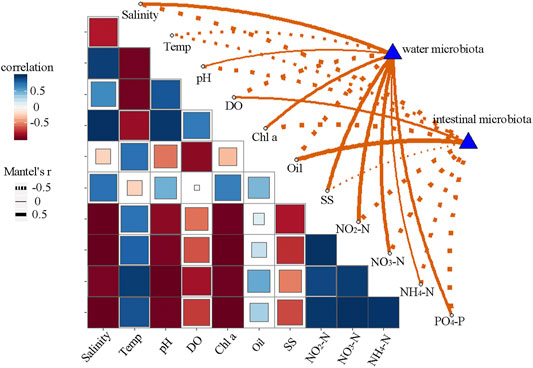
FIGURE 5. Relationship between microbial communities (Bray-Curtis distance) and environmental factors, using Mantel tests. Temp, temperature; DO, dissolved oxygen; Chl a, chlorophyll a; SS, suspended solids.
The Intestinal Microbiota of C. lucidus
The microbial composition of C. lucidus intestine samples from all sites was also analyzed (Figure 6). The shared OTUs of intestine samples from all sites were 412, and were identified as core intestinal microbiota. These OTUs at sites A1, A2, and A3 corresponded to 46.0, 39.4, and 49.0% of the total OTUs in each sample. However, shared OTUs accounted for 76.7, 72.6, and 80.6% of total sequences in intestine samples from sites A1, A2, and, A3, respectively, (Figure 6A). Furthermore, microbial composition was demonstrated based on the analysis of shared OTUs (Figure 6B). These were mainly assigned to 14 dominant genera, which represented 30.2–32.9% of total shared sequences from the three individual intestine samples. Also, 5 of the 14 genera showed low variability levels between each sample, including Bifidobacterium, Rhodopseudomonas, Escherichia-Shigella, Acinetobacter, and Stenotrophomonas. In addition, a high proportion of sequences belonging to unclassified taxa were found in shared sequences (30.1–43.9%).
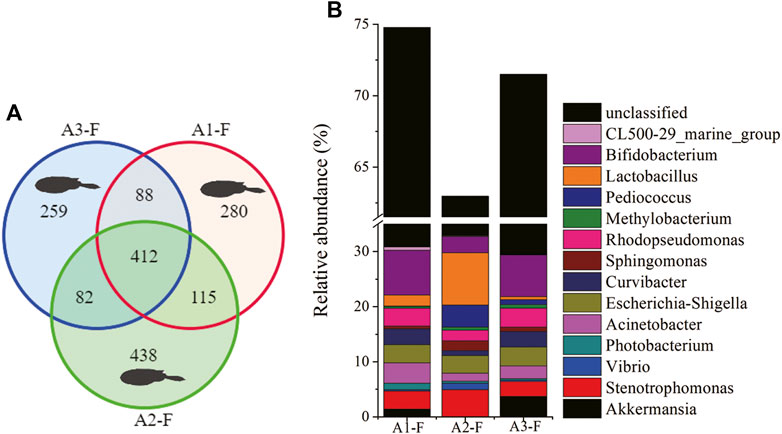
FIGURE 6. Composition of microbial communities in fish intestine samples from different Pearl River estuary sites (A1, A2, and A3). (A) Venn diagrams show OTU composition (fish: F); (B) The relative abundance of dominant microbial genera based on shared OTU analysis.
Discussion
By investigating fish intestinal microbiota and its association with estuary waters, we can improve our understanding of how fish adapt to dynamic estuary environments. In this study, C. lucidus, which was widely distributed in the PRE, was selected to investigate changes in intestinal microbiota across estuary transitions, using Illumina high-throughput sequencing. Furthermore, microbial exchange between intestine and water samples along the PRE was also analyzed.
We observed that Proteobacteria, Actinobacteria, Cyanobacteria, Firmicutes, Bacteroidetes, and Planctomycetes were highly abundant in C. lucidus intestines. These phyla also dominate other fish species (Givens et al., 2015; Gao et al., 2020). We also observed differences between water microbiota and C. lucidus intestinal microbiota from the PRE, while no significant differences were observed in microbial composition in intestines at different habitats in the PRE. Candidatus Actinomarina and HIMB11 genera were most dominant in water samples, but seldom discovered in intestines. In contrast, Bifidobacterium, Stenotrophomonas, Escherichia-Shigella, and Rhodopseudomonas genera were abundant in intestine samples, but rare in water samples. Previous studies similarly reported that dominant microbial groups in surrounding waters were not observed in the intestines of habitant fish (Schmidt et al., 2015; Yan et al., 2016). A possible reason could be that fish intestinal microbiota are substantially shaped by the host, and that host effects decrease microbial interactions with surrounding environments (Schmidt et al., 2015; Yan et al., 2016). Importantly, this is the first report characterizing such host effects in C. lucidus in the PRE. In addition, host effect was also identified during the migration of the Salmo salar (Schmidt et al., 2015; Llewellyn et al., 2016; Dehler et al., 2017b; Rudi et al., 2018). However, host effects may be weakened by selective variations, such as host immunity, physiology, and development (Bolnick et al., 2014; Schmidt et al., 2015; Yan et al., 2016).
Surrounding waters are important sources of intestinal microbiota for fish (Wu et al., 2012; Dehler et al., 2017a; Egerton et al., 2018). However, it is unclear how, and to what extent, water microbiota influences fish intestinal microbiota in different estuary habitats. The OTUs of water samples shared with intestinal samples at sites A1, A2, and A3, accounted for 13.8, 33.4, and 44.9% of total sequences, respectively, showing an increasing microbial exchange tendency from PRE upstream to downstream points. This observation may be related to changes in salinity. The aforementioned S. salar study indicated that fish drink continuously to compensate for water loss during freshwater to seawater transitions, thus microbial exchange between intestinal and water samples were increased when S. salar was transferred to a seawater environment (Dehler et al., 2017b). Meanwhile, the dominant taxa no matter in fish intestinal samples or in water samples showed a low frequency of exchange from each site of the whole estuary (Figure 4). Similar results for dominant taxa in hosts did not share preferences with these in rearing environment was observed in aquaculture environment (Sun et al., 2019; Xiong et al., 2019). However, Acinetobacter constantly appeared in both intestine and water samples along the estuary. Acinetobacter is a dominant taxa in marine fish intestines (Wang et al., 2018; Givens et al., 2015; Gomez and Balcazar, 2008). Some Acinetobacter species play important roles in fish digestive processes Ray et al. (2012), Ringo et al. (2016), while other species are opportunistic pathogens and induce fish intestinal inflammation (Zhou et al., 2019a). In addition, we observed a large proportion of unclassified taxa which had colonized fish intestines from the surrounding water, but the function and relevance of this group requires further investigation. Moreover, the relationship between microbial communities and environmental variables indicated that oil pollutants affected fish intestinal microbiota (Figure 5). It was suggested that pollutants may act as environmental stressors to weaken host immune systems, thereby influencing intestinal microbiota (Hansen and Olafsen, 1999; Zeglin, 2015). However, water microbiota was associated with suspended solids (SS), salinity, NO2-N, and NO3-N levels (Figure 5). These common constituents influence the water microbiota in aquaculture (Vasemagi et al., 2017; Sun et al., 2019).
When compared with water samples, shared OTUs accounted more for proportion of either the number of total OTUs or the total sequences in the intestinal samples (Table 1), indicating the importance of core intestinal microbiota for C. lucidus. Several highly abundant genera were consistently observed in the core intestinal microbiota, including Bifidobacterium, Rhodopseudomonas, Escherichia-Shigella, Acinetobacter, and Stenotrophomonas (Figure 6). Bifidobacterium is a Gram-positive obligate anaerobe which prevents pathogenic bacterial invasion into the intestinal environment of humans and animals (Sekirov et al., 2010; Fukuda et al., 2011). However, it is not common in marine fish, whereas Bifidobacterium is more common in some freshwater fish (Kopecny et al., 2010; Vlkova et al., 2012). Rhodopseudomonas is a photosynthetic bacteria, often recognized as a probiotic species, and associated with growth promoters or immune responses in fish, e.g., R. palustris (Zhou et al., 2010; Wang, 2011; Feckaninova et al., 2017). Escherichia-Shigella is frequently identified in fish intestinal samples, and is known as a potential pathogen (Sun et al., 2019; Zheng et al., 2019; Gao et al., 2020). Acinetobacter is common in fish intestines, the functions of which have been outlined above. Most of the Stenotrophomonas species have key roles in nitrogen and sulfur cycles Ryan et al. (2009), but S. maltophilia is often identified as an opportunistic pathogen in aquaculture (Geng et al., 2010; Abraham et al., 2016). The presence of such opportunistic pathogens in fish intestines suggests the intestinal tract is a potential pathogen access route (Roeselers et al., 2011; Li et al., 2015). Core intestinal microbiota components were also observed in Salmo salarDehler et al. (2017a), Rudi et al. (2018), zebra fish Roeselers et al. (2011), European sea-bass (Kokou et al., 2019), carps (grass carp, crucian carp and bighead carp) Wu et al. (2012), Li et al. (2015), rainbow trout Wong et al. (2013), and several other marine fish (Givens et al., 2015; Gao et al., 2020). Based on our data, the core intestinal microbiota of C. lucidus harbored bacteria associated with immune response, nutrient metabolism, probiotic actions, and potential pathogen behaviors.
Conclusion
Interactions between the intestinal microbiota of an euryhaline fish species and water environments along an estuary were investigated in this study. Dominant genera in intestine samples or water samples were seldom exchanged. While differences existed between the water microbiota and intestinal microbiota, some microbial taxa, e.g., Acinetobacter were constantly exchanged. Furthermore, these microbial exchanges were increased from upstream to downstream estuary points. Salinity changes may have influenced these microbial exchanges between samples (Figure 7). In conclusion, we analyzed the core intestinal microbiota of C. lucidus across different estuary points, and identified potential microbiota functions, incorporating immune responses, nutrient metabolism, probiotic actions, and potential pathogen behaviors.
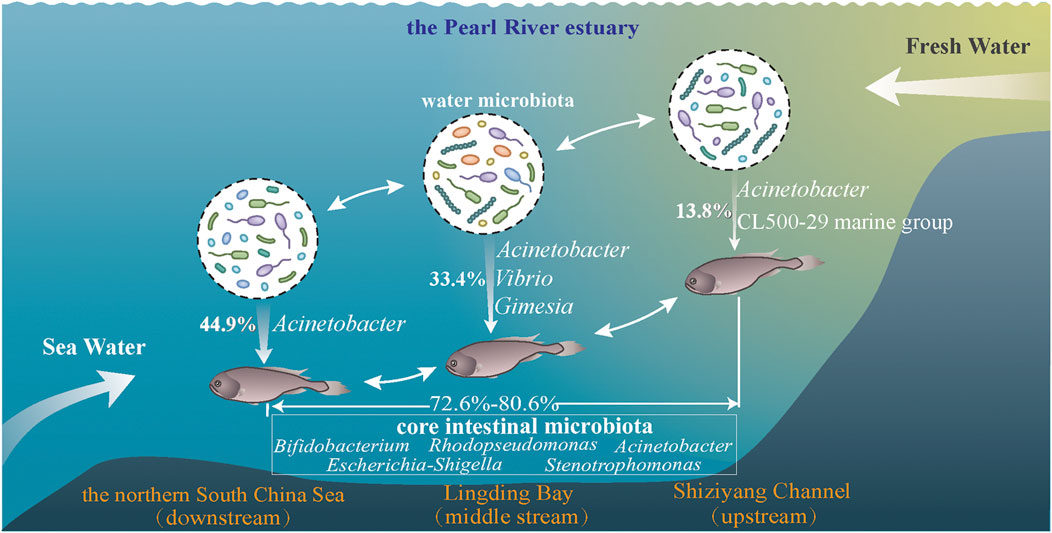
FIGURE 7. Schematic diagram showing the composition and interactions of intestinal microbiota from C. lucidus with surrounding study sites (A1, A2, and A3) along the Pearl River estuary. The fresh water movement is represented from right to left.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by Committee on Laboratory Animal Welfare and Ethics of South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences (nhdf2020-03).
Author Contributions
Conceptualization, PW, YL, and CL; investigation, PW, YX, LL, and YX; methodology, PW, YX, and YX; formal analysis, PW; writing original draft, PW and YX; writing-review and editing, PW, YL, CL, and TW; data curation, YLand LL; funding acquisition, CL and PW.
Funding
This research was supported by National Key R&D Program of China (2019YFD0901204, 2019YFD0901201), Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0605), Guangdong Basic and Applied Basic Research Foundation (2019B1515120065), Science and Technology Planning Project of Guangdong Province (2019B121201001), Fundamental and Applied Fundamental Research Major Program of Guangdong Province (2019B030302004–05), Central Public-interest Scientific Institution Basal Research Fund, CAFS (No. 2020TD16), Central Public-interest Scientific Institution Basal Research Fund, SCSFRI, CAFS (No. 2021SD04, NO. 2019TS28).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the academic editor and reviewers for their insightful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2021.675856/full#supplementary-material.
References
Abraham, T. J., Paul, P., Adikesavalu, H., Patra, A., and Banerjee, S. (2016). Stenotrophomonas Maltophilia as an Opportunistic Pathogen in Cultured African Catfish Clarias gariepinus (Burchell, 1822). Aquaculture 450, 168–172. doi:10.1016/j.aquaculture.2015.07.015
Bolnick, D. I., Snowberg, L. K., Hirsch, P. E., Lauber, C. L., Knight, R., Caporaso, J. G., et al. (2014). Individuals' Diet Diversity Influences Gut Microbial Diversity in Two Freshwater Fish (Threespine Stickleback and Eurasian Perch). Ecol. Lett. 17, 979–987. doi:10.1111/ele.12301
Butt, R. L., and Volkoff, H. (2019). Gut Microbiota and Energy Homeostasis in Fish. Front. Endocrinol. 10. 9, doi:10.3389/fendo.2019.00009
Buttigieg, P. L., and Ramette, A. (2014). A Guide to Statistical Analysis in Microbial Ecology: a Community-Focused, Living Review of Multivariate Data Analyses. Fems Microbiol. Ecol. 90, 543–550. doi:10.1111/1574-6941.12437
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 7, 335–336. doi:10.1038/nmeth.f.303
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34, i884–i890. doi:10.1093/bioinformatics/bty560
Dehler, C. E., Secombes, C. J., and Martin, S. A. M. (2017a). Environmental and Physiological Factors Shape the Gut Microbiota of Atlantic Salmon Parr (Salmo salar L.). Aquaculture 467, 149–157. doi:10.1016/j.aquaculture.2016.07.017
Dehler, C. E., Secombes, C. J., and Martin, S. A. M. (2017b). Seawater Transfer Alters the Intestinal Microbiota Profiles of Atlantic Salmon (Salmo salar L.). Sci. Rep. 7. 13877, doi:10.1038/s41598-017-13249-8
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 27, 2194–2200. doi:10.1093/bioinformatics/btr381
Edgar, R. C. (2013). UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 10, 996–998. doi:10.1038/NMETH.2604
Egerton, S., Culloty, S., Whooley, J., Stanton, C., and Ross, R. P. (2018). The Gut Microbiota of Marine Fish. Front. Microbiol. 9. 873, doi:10.3389/fmicb.2018.00873
Eick, D., and Thiel, R. (2014). Fish Assemblage Patterns in the Elbe Estuary: Guild Composition, Spatial and Temporal Structure, and Influence of Environmental Factors. Mar. Biodiv 44, 559–580. doi:10.1007/s12526-014-0225-4
Feckaninova, A., Koscova, J., Mudronova, D., Popelka, P., and Toropilova, J. (2017). The Use of Probiotic Bacteria against Aeromonas Infections in Salmonid Aquaculture. Aquaculture 469, 1–8. doi:10.1016/j.aquaculture.2016.11.042
Feng, B.-W., Li, X.-R., Wang, J.-H., Hu, Z.-Y., Meng, H., Xiang, L.-Y., et al. (2009). Bacterial Diversity of Water and Sediment in the Changjiang Estuary and Coastal Area of the East China Sea. Fems Microbiol. Ecol. 70, 236–248. doi:10.1111/j.1574-6941.2009.00772.x
Fu, J., Mai, B., Sheng, G., Zhang, G., Wang, X., Peng, P. a., et al. (2003). Persistent Organic Pollutants in Environment of the Pearl River Delta, China: an Overview. Chemosphere 52, 1411–1422. doi:10.1016/S0045-6535(03)00477-6
Fukuda, S., Toh, H., Hase, K., Oshima, K., Nakanishi, Y., Yoshimura, K., et al. (2011). Bifidobacteria Can Protect from Enteropathogenic Infection through Production of Acetate. Nature 469, 543–547. doi:10.1038/nature09646
Gao, Y.-M., Zou, K.-S., Zhou, L., Huang, X.-D., Li, Y.-Y., Gao, X.-Y., et al. (2020). Deep Insights into Gut Microbiota in Four Carnivorous Coral Reef Fishes from the South China Sea. Microorganisms 8, 426. doi:10.3390/microorganisms8030426
Gb 17378.4, (2007). The Specification for Marine Monitoring Part 4: Seawater Analysis. Beijing, China: Standards Press. in Chinese.
Geng, Y., Wang, K., Chen, D., Huang, X., He, M., and Yin, Z. (2010). Stenotrophomonas Maltophilia, an Emerging Opportunist Pathogen for Cultured Channel Catfish, Ictalurus punctatus, in China. Aquaculture 308, 132–135. doi:10.1016/j.aquaculture.2010.08.032
Ghanbari, M., Kneifel, W., and Domig, K. J. (2015). A New View of the Fish Gut Microbiome: Advances from Next-Generation Sequencing. Aquaculture 448, 464–475. doi:10.1016/j.aquaculture.2015.06.033
Givens, C., Ransom, B., Bano, N., and Hollibaugh, J. (2015). Comparison of the Gut Microbiomes of 12 Bony Fish and 3 Shark Species. Mar. Ecol. Prog. Ser. 518, 209–223. doi:10.3354/meps11034
Gómez, G. D., and Balcázar, J. L. (2008). A Review on the Interactions between Gut Microbiota and Innate Immunity of Fish: Table 1. FEMS Immunol. Med. Microbiol. 52, 145–154. doi:10.1111/j.1574-695X.2007.00343.x
Hansen, G. H., and Olafsen, J. A. (1999). Bacterial Interactions in Early Life Stages of Marine Cold Water Fish. Microb. Ecol. 38, 1–26. doi:10.1007/s002489900158
He, B. Q., and Li, H. Q. (1988). Stock Assessment of Collichthys Lucidus in Pearl River Estuary. J. Fish. China 12 (2), 125–134. in Chinese with English abstract.
Huang, J. W., Sun, D. R., Liu, Y., Liu, S. N., Shan, B. B., Yang, C. P., et al. (2018). Diversity of Fish Community in Sousa Chinensis Nature Reserve of Pearl River Estuary. J. South. Agric. 49 (5), 1000–1007. in Chinese with English abstract.
Huang, X. P., Huang, L. M., and Yue, W. Z. (2003). The Characteristics of Nutrients and Eutrophication in the Pearl River Estuary, South China. Mar. Pollut. Bull. 47, 30–36. doi:10.1016/S0025-326X(02)00474-5
Kamrani, E., Sharifinia, M., and Hashemi, S. H. (2016). Analyses of Fish Community Structure Changes in Three Subtropical Estuaries from the Iranian Coastal Waters. Mar. Biodiv 46, 561–577. doi:10.1007/s12526-015-0398-5
Kirchman, D. L., Dittel, A. I., Malmstrom, R. R., and Cottrell, M. T. (2005). Biogeography of Major Bacterial Groups in the Delaware Estuary. Limnol. Oceanogr. 50, 1697–1706. doi:10.4319/lo.2005.50.5.1697
Kokou, F., Sasson, G., Friedman, J., Eyal, S., Ovadia, O., Harpaz, S., et al. (2019). Core Gut Microbial Communities Are Maintained by Beneficial Interactions and Strain Variability in Fish. Nat. Microbiol. 4, 2456–2465. doi:10.1038/s41564-019-0560-0
Kopecny, J., Mrazek, J., and Killer, J. (2010). The Presence of Bifidobacteria in Social Insects, Fish and Reptiles. Folia Microbiol. (Praha) 55, 336–339. doi:10.1007/s12223-010-0053-2
Li, T., Long, M., Gatesoupe, F.-J., Zhang, Q., Li, A., and Gong, X. (2015). Comparative Analysis of the Intestinal Bacterial Communities in Different Species of Carp by Pyrosequencing. Microb. Ecol. 69, 25–36. doi:10.1007/s00248-014-0480-8
Liu, H., Jiang, T., Huang, H., Shen, X., Zhu, J., and Yang, J. (2015). Estuarine Dependency in Collichthys Lucidus of the Yangtze River Estuary as Revealed by the Environmental Signature of Otolith Strontium and Calcium. Environ. Biol. Fish. 98 (1), 165–172. doi:10.1007/s10641-014-0246-7
Llewellyn, M. S., McGinnity, P., Dionne, M., Letourneau, J., Thonier, F., Carvalho, G. R., et al. (2016). The Biogeography of the Atlantic Salmon (Salmo salar) Gut Microbiome. ISME J. 10, 1280–1284. doi:10.1038/ismej.2015.189
Magoc, T., and Salzberg, S. L. (2011). FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 27, 2957–2963. doi:10.1093/bioinformatics/btr507
Molina, A., Duque, G., and Cogua, P. (2020). Influences of Environmental Conditions in the Fish Assemblage Structure of a Tropical Estuary. Mar. Biodivers. 50, 5. doi:10.1007/s12526-019-01023-0
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 30, 3123–3124. doi:10.1093/bioinformatics/btu494
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a Comprehensive Online Resource for Quality Checked and Aligned Ribosomal RNA Sequence Data Compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi:10.1093/nar/gkm864
Ray, A. K., Ghosh, K., and Ringø, E. (2012). Enzyme-producing Bacteria Isolated from Fish Gut: a Review. Aquacult Nutr. 18, 465–492. doi:10.1111/j.1365-2095.2012.00943.x
Ringø, E., Zhou, Z., Vecino, J. L. G., Wadsworth, S., Romero, J., Krogdahl, Å., et al. (2016). Effect of Dietary Components on the Gut Microbiota of Aquatic Animals. A Never‐ending Story? Aquacult Nutr. 22, 219–282. doi:10.1111/anu.12346
Roeselers, G., Mittge, E. K., Stephens, W. Z., Parichy, D. M., Cavanaugh, C. M., Guillemin, K., et al. (2011). Evidence for a Core Gut Microbiota in the Zebrafish. ISME J. 5, 1595–1608. doi:10.1038/ismej.2011.38
Rudi, K., Angell, I. L., Pope, P. B., Vik, J. O., Sandve, S. R., and Snipen, L.-G. (2018). Stable Core Gut Microbiota across the Freshwater-To-Saltwater Transition for Farmed Atlantic Salmon. Appl. Environ. Microbiol. 84. e01974. doi:10.1128/AEM.01974-17
Ryan, R. P., Monchy, S., Cardinale, M., Taghavi, S., Crossman, L., Avison, M. B., et al. (2009). The Versatility and Adaptation of Bacteria from the Genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525. doi:10.1038/nrmicro2163
Schmidt, V. T., Smith, K. F., Melvin, D. W., and Amaral-Zettler, L. A. (2015). Community Assembly of a Euryhaline Fish Microbiome during Salinity Acclimation. Mol. Ecol. 24, 2537–2550. doi:10.1111/mec.13177
Sekirov, I., Russell, S. L., Antunes, L. C. M., and Finlay, B. B. (2010). Gut Microbiota in Health and Disease. Physiol. Rev. 90, 859–904. doi:10.1152/physrev.00045.2009
Sun, F., Wang, Y., Wang, C., Zhang, L., Tu, K., and Zheng, Z. (2019). Insights into the Intestinal Microbiota of Several Aquatic Organisms and Association with the Surrounding Environment. Aquaculture 507, 196–202. doi:10.1016/j.aquaculture.2019.04.026
Talwar, C., Nagar, S., Lal, R., and Negi, R. K. (2018). Fish Gut Microbiome: Current Approaches and Future Perspectives. Indian J. Microbiol. 58, 397–414. doi:10.1007/s12088-018-0760-y
Vasemägi, A., Visse, M., and Kisand, V. (2017). Effect of Environmental Factors and an Emerging Parasitic Disease on Gut Microbiome of Wild Salmonid Fish. Msphere 2. e00418, doi:10.1128/mSphere.00418-17
Vlková, E., Kalous, L., Bunešová, V., Rylková, K., Světlíková, R., and Rada, V. (2012). Occurrence of Bifidobacteria and Lactobacilli in Digestive Tract of Some Freshwater Fishes. Biologia 67, 411–416. doi:10.2478/s11756-012-0017-x
Wang, A. R., Ran, C., Ringø, E., and Zhou, Z. G. (2018). Progress in Fish Gastrointestinal Microbiota Research. Rev. Aquacult 10, 626–640. doi:10.1111/raq.12191
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Aem 73, 5261–5267. doi:10.1128/AEM.00062-07
Wang, Y. (2011). Use of Probiotics Bacillus Coagulans, Rhodopseudomonas Palustris and Lactobacillus Acidophilus as Growth Promoters in Grass Carp (Ctenopharyngodon Idella) Fingerlings. Aquacult Nutr. 17, E372–E378. doi:10.1111/j.1365-2095.2010.00771.x
Wong, S., Waldrop, T., Summerfelt, S., Davidson, J., Barrows, F., Kenney, P. B., et al. (2013). Aquacultured Rainbow Trout (Oncorhynchus mykiss) Possess a Large Core Intestinal Microbiota that Is Resistant to Variation in Diet and Rearing Density. Appl. Environ. Microbiol. 79, 4974–4984. doi:10.1128/AEM.00924-13
Wu, M., Wang, Y., Dong, J., Sun, F., Wang, Y., and Hong, Y. (2017). Spatial Assessment of Water Quality Using Chemometrics in the Pearl River Estuary, China. Front. Earth Sci. 11, 114–126. doi:10.1007/s11707-016-0585-0
Wu, P., Wang, Y.-S., Sun, F.-L., Wu, M.-L., and Peng, Y.-l. (2014). Bacterial Polycyclic Aromatic Hydrocarbon Ring-Hydroxylating Dioxygenases in the Sediments from the Pearl River Estuary, China. Appl. Microbiol. Biotechnol. 98, 875–884. doi:10.1007/s00253-013-4854-5
Wu, S., Wang, G., Angert, E. R., Wang, W., Li, W., and Zou, H. (2012). Composition, Diversity, and Origin of the Bacterial Community in Grass Carp Intestine. Plos One 7 (2), e30440. doi:10.1371/journal.pone.0030440
Xiong, J., Xuan, L., Yu, W., Zhu, J., Qiu, Q., and Chen, J. (2019). Spatiotemporal Successions of Shrimp Gut Microbial Colonization: High Consistency Despite Distinct Species Pool. Environ. Microbiol. 21, 1383–1394. doi:10.1111/1462-2920.14578
Yan, Q., Li, J., Yu, Y., Wang, J., He, Z., Van Nostrand, J. D., et al. (2016). Environmental Filtering Decreases with Fish Development for the Assembly of Gut Microbiota. Environ. Microbiol. 18, 4739–4754. doi:10.1111/1462-2920.13365
Ying, Z. F. (1994). The Frontal Classification and its Influence on Sedimentation in Lingdingyang of Pearl River Estuary. Tropic Oceanology 13 (2), 25–31. in Chinese with English abstract.
Zeglin, L. H. (2015). Stream Microbial Diversity in Response to Environmental Changes: Review and Synthesis of Existing Research. Front. Microbiol. 6. doi:10.3389/fmicb.2015.00454
Zhang, Y., Jiao, N., Cottrell, M., and Kirchman, D. (2006). Contribution of Major Bacterial Groups to Bacterial Biomass Production along a Salinity Gradient in the South China Sea. Aquat. Microb. Ecol. 43, 233–241. doi:10.3354/ame043233
Zheng, X., Yang, R., Hu, J., Lin, S., Gu, Z., and Ma, Z. (2019). The Gut Microbiota Community and Antioxidant Enzymes Activity of Barramundi Reared at Seawater and Freshwater. Fish Shellfish Immunol. 89, 127–131. doi:10.1016/j.fsi.2019.03.054
Zhou, L., Lin, K.-t., Gan, L., Sun, J.-j., Guo, C.-j., Liu, L., et al. (2019a). Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study. Microorganisms 7, 465. doi:10.3390/microorganisms7100465
Zhou, L., Wang, G., Kuang, T., Guo, D., and Li, G. (2019b). Fish Assemblage in the Pearl River Estuary: Spatial‐seasonal Variation, Environmental Influence and Trends over the Past Three Decades. J. Appl. Ichthyol. 35, 884–895. doi:10.1111/jai.13912
Keywords: microbial exchange, water environment, host effects, water microbiota, habitat transition
Citation: Wu P, Liu Y, Li C, Xiao Y, Wang T, Lin L and Xie Y (2021) The Composition of Intestinal Microbiota From Collichthys lucidus and Its Interaction With Microbiota From Waters Along the Pearl River Estuary in China. Front. Environ. Sci. 9:675856. doi: 10.3389/fenvs.2021.675856
Received: 04 March 2021; Accepted: 14 April 2021;
Published: 29 April 2021.
Edited by:
Xiaoping Huang, South China Sea Institute of Oceanology, ChinaReviewed by:
Yehui Tan, South China Sea Institute of Oceanology, ChinaYiguo Hong, Guangzhou University, China
Copyright © 2021 Wu, Liu, Li, Xiao, Wang, Lin and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Liu, bGl1eW9uZ0BzY3NmcmkuYWMuY24=; Chunhou Li, c2NzbGNoQHZpcC4xNjMuY29t