 Philippa L. Ascough1*
Philippa L. Ascough1* Fiona Brock2
Fiona Brock2 Margaret E. Collinson3
Margaret E. Collinson3 Jon D. Painter2
Jon D. Painter2 David W. Lane2
David W. Lane2 Michael I. Bird4,5
Michael I. Bird4,5- 1NERC Radiocarbon Facility, Scottish Universities Environmental Research Centre, East Kilbride, United Kingdom
- 2Cranfield Forensic Institute, Cranfield University, Defence Academy of the United Kingdom, Shrivenham, United Kingdom
- 3Department of Earth Sciences, Royal Holloway University of London, Egham, United Kingdom
- 4Centre for Tropical Environmental and Sustainability, College of Science and Engineering, James Cook University, Cairns, QLD, Australia
- 5ARC Centre of Excellence for Australian Biodiversity and Heritage, College of Science and Engineering, James Cook University, Cairns, QLD, Australia
Pyrogenic Carbon (PyC) is ubiquitous in global environments, and is now known to form a significant, and dynamic component of the global carbon cycle, with at least some forms of PyC persisting in their depositional environment for many millennia. Despite this, the factors that determine the turnover of PyC remain poorly understood, as do the physical and chemical changes that this material undergoes when exposed to the environment over tens of thousands of years. Here, we present the results of an investigation to address these knowledge gaps through chemical and physical analysis of a suite of wood PyC samples exposed to the environment for varying time periods, to a maximum of >90,000 years. This includes an assessment of the quantity of resistant carbon, known as Stable Polyaromatic Carbon (SPAC) vs. more chemically labile carbon in the samples. We find that, although production temperature is likely to determine the initial “degradation potential” of PyC, an extended exposure to environmental conditions does not necessarily mean that remaining PyC always progresses to a “SPAC-dominant” state. Instead, some ancient PyC can be composed largely of chemical components typically thought of as environmentally labile, and it is likely that the depositional environment drives the trajectory of preservation vs. loss of PyC over time. This has important implications for the size of global PyC stocks, which may have been underestimated, and also for the potential loss of previously stored PyC, when its depositional environment alters through environmental or climatic changes.
Introduction
Pyrogenic Carbon (PyC) is a term for biomass that has undergone chemical alteration by exposure to high temperatures and low oxygen availability (i.e., pyrolysis) and comprises a continuum from lightly charred substances to highly condensed soot (e.g., Preston and Schmidt, 2006; Bird and Ascough, 2012). This paper focuses upon the macro-charcoal component of this continuum, covering material produced in both natural and anthropogenically-mediated events, including where the purpose of the latter was not the production of charcoal itself per se (for example, domestic fires). PyC is an important source of archaeological and palaeoenvironmental information, e.g., as a proxy for fire frequency, or the type, abundance, and stable isotopic composition of vegetation burnt at a site (Aleman et al., 2018; Hawthorne et al., 2018). Charcoal is also frequently used for radiocarbon age measurement (Bird, 2007). PyC is ideal for these purposes as its significant production rate in fires and environmental persistence means it is both abundant and ubiquitous in the natural environment (e.g., Bird et al., 2015; Coppola and Druffel, 2016; Cotrufo et al., 2016; Reisser et al., 2016; Santin et al., 2016). In suitable depositional environments in terrestrial, freshwater, and marine settings, PyC has been shown to persist with half-lives of the order of >1,000 years (Preston and Schmidt, 2006), and is present in the geological record (e.g., Scott, 2010).
The high carbon content and long environmental lifetime of PyC has led to the concept of “Biochar” for carbon sequestration; this is pyrogenic carbon produced intentionally by controlled pyrolysis and “intended for use as a soil application or broader for environmental management” (Lehmann and Joseph, 2015). The environmental persistence of PyC is predominantly attributed to a chemical structure containing abundant carbon aromatic rings, which require high energy input to break down, compared to non-polyaromatic structures such as cellulose. These ring structures grow in size with increasing production temperature and/or pyrolysis duration (Ascough et al., 2008). The importance of PyC is also increasingly apparent to the modern global carbon cycle, both in terms of its size as a global carbon stock, and its environmental dynamics (Bird et al., 2015; Santín et al., 2015).
Despite the importance of PyC in a range of environmental research questions (e.g., global carbon cycling and soil interactions), the role of PyC in these areas, and the interactions it undergoes with its environment, remain poorly understood. It is now quite clear that although its aromatic structure confers the potential for long-term preservation, PyC is not an environmentally inert substance, but can be degraded to CO2 (e.g., Bird et al., 2017), can be chemically altered (Knicker, 2011), and can mediate processes in soils, such as priming effects during processing of non-PyC organic matter (e.g., Tilston et al., 2016). Important knowledge gaps include the specific ways in which PyC interacts with its environment, the timescales over which these interactions occur, and the drivers of these processes. Previous work has indicated there is a time-dependent “maturation” of PyC in the environment, in which its various chemical sub-components are progressively degraded (e.g., Ascough et al., 2011a). These sub-components are defined by their chemical resistance to degradation, and broadly consist of labile, semi-labile, and Stable Polycyclic Aromatic Carbon (SPAC) (c.f. Ascough et al., 2011a; Bird et al., 2015). This concept implies that there should be a relationship between the abundance of these components in a particular PyC sample, and the time over which it has been exposed to environmental conditions. The ultimate state would be a near-dominance of SPAC, potentially followed by complete chemical breakdown and loss of the PyC, if the SPAC itself is eventually exposed to degradation in either particularly aggressive chemical environments, or by exposure to chemical attack (e.g., oxidation) over very extended time periods (Ascough et al., 2011a). This may not be directly linear, as the rate of alteration should be a function of the intensity of degradation processes, both abiotic (e.g., photooxidation, Wang et al., 2017), and biotic (Wang et al., 2016; Woo et al., 2016). These clearly could vary through time if environmental conditions (e.g., soil pH, moisture regime, etc.) were to fluctuate. However, in the above paradigm, there would be an overall progressive shift toward the SPAC-only state, before potential total loss.
In the above concept, the time period over which PyC remained “intact” in any given context would be dictated firstly by production conditions, predominantly temperature. As production temperature increases, so does aromaticity, shifting the initial structure of the PyC to a “SPAC-dominant” state (Ascough et al., 2008; McBeath et al., 2015). In identical environments therefore, the PyC produced at higher temperature would be preserved over a longer period than material produced at lower temperatures. Secondly, depositional conditions would dictate the fate of PyC, with identical PyC subjected to conditions of high and low “degradation potential” being altered at different rates. This concept is appealing, as, if correct, it could provide a means to model and/or predict PyC behavior (e.g., C release from PyC degradation) on large geographic and temporal scales.
To date, systematic chemical investigation of these processes, the timescales over which they operate, and how closely PyC in the environment follows the “maturation” concept, has largely used laboratory experiments, or field studies of <10 years duration. These studies provide important insights but would be usefully consolidated by studies of PyC that have been exposed to the environment for the extended half-lives of PyC that have been proposed (e.g., >1,000 years; Preston and Schmidt, 2006). This is desirable as there is evidence that the residence times of PyC predicted by laboratory incubations can be shorter than those measured for “real world” conditions (see Knicker, 2011), and that the initial chemistry (likely vital in determining the trajectory of PyC change) of laboratory-produced PyC often used in experiments, including field trials, differs significantly from that produced in “natural” events such as wildfires (Santín et al., 2017). This paper therefore provides a detailed chemical analysis of a suite of PyC of varying antiquity, recovered from palaeoenvironmental and archaeological deposits.
Analysis of PyC of demonstrably older age is important as a counterpart to studies that are, justifiably, highlighting the potential for rapid alteration and degradation of PyC on short (<centennial) timescales (e.g., Czimczik et al., 2003; Ohlson et al., 2009; Singh et al., 2012). This is because a significant proportion of PyC existing in our Earth system is dated (e.g., by radiometric dating), to periods greatly in excess of these short timeframes. This paper aims to contribute to our knowledge of what enables such contrasts in the timescales of degradation, specifically: (i) whether this is a clear function of initial chemistry (i.e., via high production temperature), (ii) whether the hypothesis that more ancient material should be progressively dominated by SPAC is correct, and (iii) what the impact of depositional conditions may be upon PyC chemistry/status after these extended timescales. The latter is most difficult to ascertain, as records of conditions over such extended timescales are often problematic. However, these questions are insufficiently addressed by analysis of material from short-term trials or laboratory treatments without requiring significant assumptions surrounding the equivalence of processes observed in these settings to those in operation over millennia in the environment.
Methodology
Samples
PyC used in this study comprised six different materials (Table 1). Samples were selected for inclusion in the study from archaeological and palaeoenvironmental samples on the basis of known depositional environment, and demonstrable resistance to chemical changes in pH and laboratory oxidation from prior experiments. The size of the samples (individual fragments of PyC) ranged from c. 3 cm in length to c. 10 cm in length, and were hence all macroscopic in scale. The aim was that these would form end members of production types and depositional environment (see Table 1). As a contrast, a sample of macro-charcoal was produced in an open fire (simulating archaeological hearth conditions) for this research, but which not exposed to environmental deposition. This was achieved by heating wood of Pinus sylvestris (Caledonian Pine) on an open fire for 60 min. Oxygen availability was restricted during production of these samples by wrapping the wood in aluminum foil during heating. Previous work (Ascough et al., 2011a) found a marked difference between PyC that was exposed to the environment, and freshly-produced material at a range of temperatures. We therefore included this sample as a “control” example representing what the ancient material included in the study likely resembled prior to deposition, using an open fire, rather than laboratory pyrolysis (e.g., in a muffle furnace) or industrial process. We acknowledge this is a generalization, due to the small sample size, but the aim was to identify any consistent differences that were apparent across the “environmental exposure” material vs. the freshly-produced material. We also note that none of our materials were produced in wildfires. Instead, for our samples, the heating duration during production would most likely have exceeded the minimum 60 min that Guo and Bustin (1998) found that further heating at a constant temperature did not result in significant further changes in PyC chemistry. This increases the likelihood that our freshly-produced PyC represents a reasonable indicator of general PyC chemistry prior to deposition.
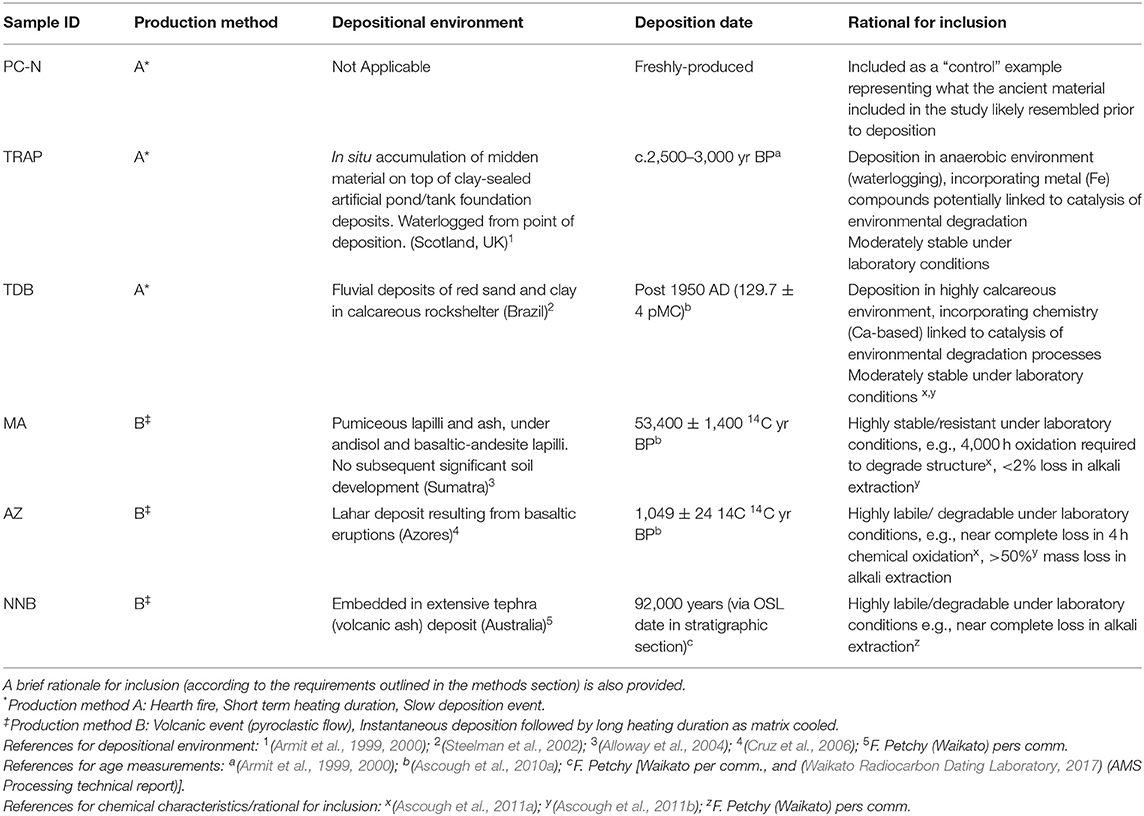
Table 1. Details of sample material used in this study, including production method, depositional environment and age.
The age of the samples was previously obtained via either radiocarbon (14C) measurement, or stratigraphic grounds. One sample dated to <50 years (129.7 ± 0.4 pMC) in the environment, while the remainder had been exposed to environmental conditions for 1,000 to >90,000 years. Material was obtained from previously excavated palaeoenvironmental or archaeological sites with permission from the excavators. Where material was small in size or embedded in a soil matrix, material was isolated from the sedimentary matrix by conventional water floatation, which removed visible soil contaminants. Samples were visually inspected in the lab and further soil material was removed by sonication in deionized (Milli-QTM) water in the lab for 2 h, followed by air-drying at 40°C.
Reflectance of PyC: Temperature of Formation, Anatomical Structure, and Mineral Content
Mean Random Reflectance (Romean) is a robust measure from which to assess formation temperature of PyC even after an extended period exposed to environmental conditions, and independent of wood species feedstock (see Scott and Glasspool, 2005; Ascough et al., 2010b). In the present study, we used Romean as a measure of the key production condition (temperature) that dictates the chemical structure of PyC prior to entering the environment. Typical production temperatures mediated by human activity span 300–800°C (though reaching >1,100°C in metal smelting and casting; McParland et al., 2009), and are broadly paralleled by natural PyC production temperatures in non-anthropogenic fires (Stinson and Wright, 1969; Stronach and McNaughton, 1989); Romean operates well-across this full range of temperatures. Assessing formation temperature is important, as temperature is directly correlated with the size and extent of aromatic domains in PyC, which is a direct predictor of its resistance to chemical degradation (e.g., by oxidation). This also dictates the proportions of PyC that fall into the “labile,” “semi-labile,” and “SPAC” categories before exposure to environmental conditions (Bird et al., 2015).
Samples were embedded in resin and polished (using 60-, 240-, 400-, and 600- grit SiC papers followed by 0.3 micron alumina on Buehler Texmet paper and 0.05 micron alumina on silk) to provide a surface for study in either the transverse or longitudinal plane of the material. Polished blocks were viewed in reflected light under immersion oil (Cargille type A, density 0.923 g/cc at 23°C, RI of 1.514) using a Leica reflected light microscope and either ×20 or ×50 oil immersion objectives. Reflectance data, Romean, are based on measurement of 100 points per specimen. The microscope was calibrated using one of two sets of standards depending on visual indication of the likely reflectance (i) low reflectance standard set [YAG, Spinel, Silica glass] (ii) Medium-High reflectance standard set [Cubic Zironium, GGG. YAG]. Where the transverse section was polished (specimens AZ and PC-N, Figures 1A,C) the corner junctions of thicker cell walls were used to provide a large enough area to measure reliably. For polished longitudinal sections (specimens TRAP and MA Figures 1B,D) all areas of the cell walls were available for measurement. Each measurement was from a different cell and a transect across the specimen was used. Specimen NNB presented a very different appearance in the polished block (Figure 2). In this case no cell walls could be measured (either not polished or too narrow) and the measurements presented are from the inferred organic cell infills (Figure 2). For all samples it was ensured that the area for measurement was in focus and care was taken to avoid scratches on the blocks where possible. Values of Romean and standard deviation are provided in Table 2.

Figure 1. Reflectance images, from polished blocks under oil, (A) AZ showing very low reflectance of conifer cell walls either side of a growth ring seen in transverse section; (B) TRAP Low reflectance of wood seen in longitudinal section, perforation plate at bottom left corner suggests angiosperm wood; (C) PC-N, anatomy as in A but high reflectance; (D) MA High reflectance of wood seen in longitudinal section, bordered pits at top right suggest gymnosperm.

Figure 2. Reflectance image, from polished block under oil, of Sample NNB showing very low reflectance of cell infills (inferred organic) in wood rays seen in tangential longitudinal section. Cell walls either not polished or too thin to measure reflectance. Bright silvery speckly areas within some cell lumina are inferred to be mineral, based on brightness and straight edges (e.g., right of center near base of image). Orange material, some of which may be mineral, is within the block i.e., unpolished.
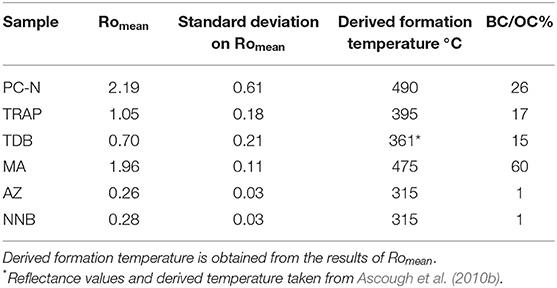
Table 2. Results of Reflectance Spectroscopy and Hydropyrolysis (HyPy) investigations [Black Carbon (BC) as a proportion of Organic Carbon (OC)] performed upon the sample materials listed in Table 1.
Anatomical data seen in the polished blocks, including cell and tissue structure and were recorded. Reflectance differing from that of the recogniseable plant cells was noted and is inferred to be mineral (e.g., very brightly reflecting tiny specks and some silvery gray cell infills).
All data for specimen TDB are taken directly from Ascough et al. (2010b) sample there named Env-5.
Values of Romean were converted to production temperatures using the equation experimentally determined for PyC reflectance in Ascough et al. (2010b):
(where y = Romean and x = temperature).
Hydropyrolysis
Hydropyrolysis (HyPy) was used to assess the content of polycyclic aromatic carbon structures of ≥7 rings (also known as Black Carbon as defined by HyPy: BCHyPy) in the samples on a secure chemical basis (Ascough et al., 2009; Meredith et al., 2012). Immediately after formation, the proportion of these (resistant) chemical structures is a function of production temperature (Ascough et al., 2008). The process separates the larger polycyclic aromatic domains from labile chemical structures in the sample, and therefore provides a quantitative assessment of the proportion of the sample that exists in either the polycyclic aromatic or non-aromatic (or small aromatic structural) chemical forms.
Samples were crushed in a pestle and mortar to <1 mm. 100–300 mg of sample was loaded with a catalyst using an aqueous/methanol (1:1) solution of ammonium dioxydithiomolybdate [(NH4)2MoO2S2]. As these were individual PyC samples, the catalyst was added at 10% by weight (nominal Mo loading of 0.5%). Samples were gently agitated to ensure full coating with catalyst, before drying under air. Catalyst-loaded samples were pyrolysed at 550°C under a hydrogen pressure of 150 bar and sweep gas flow of 5 L min−1 (ATP). The proportion of sample composed of ≥7 aromatic rings (also known as Black Carbon: BC) was obtained by comparison of the organic carbon (OC) content of the catalyst loaded samples with their HyPy residues (defined as BC/OC%). The OC content of samples before and after HyPy was obtained by elemental analysis on a Costech elemental analyzer (EA). The BCHyPy content of the sample was then calculated via:
Raman Spectroscopy
Raman spectroscopy provides an analysis of the underlying chemical structure of the material analyzed. For PyC, two major peaks relating to carbon chemistry are of particular interest: the G (or graphitic) peak indicative of highly stable, ordered carbon atoms at ca. 1,575 cm−1 and the D (or disordered) peak at ca. 1,300–1,350 cm−1 caused by the presence of defects and discontinuities in crystallites (e.g., Alon et al., 2002; McDonald-Wharry et al., 2013). The G peak therefore relates to highly polycyclic aromatic domains (e.g., those comprising SPAC), that are chemically resistant, and the D peak to more disordered carbon chemical structures, encompassing the “labile” and “semi-labile” fractions of PyC. Raman spectroscopy was included in this study to complement the information provided by hydropyrolysis, as this provides a direct qualitative assessment of the chemistry of the sample; while HyPy provides a robust quantitative measure, it does not directly observe the carbon chemistry during the process.
Raman measurements were made in air at room temperature using a Horiba Jobin Yvon Raman microscope employing a ×50 brightfield objective (NA 0.75, working distance ~ 0.5 mm). Polarization state was not defined. Continuous excitation at 532 nm (green) was produced by a frequency-doubled Nd:YAG laser, with power at sample estimated to be 5–10 mW, focused into a spot estimated to be 2–3 μm diameter. Laser power was set using a combination of variable laser drive current and fixed-transmission neutral density filters on the microscope, arranged to avoid visible signs of sample pyrolysis, as judged by observation images obtained in reflected light using white light illumination and captured by the microscope's onboard color video camera. Each measurement consisted of six separate scans, each of 60 s exposure on the CCD detector, with the spectrograph set to capture the range 1,000–2,000 cm−1. The first dataset was discarded (as it often resulted in the loss of loose surface powder material) and the remaining 5 averaged. Spectral resolution was estimated as ~5 cm−1.
Fourier Transform Infrared Spectroscopy (FTIR)
Fourier Transform Infrared Spectroscopy (FTIR) uses the mid-infrared region of the electromagnetic spectrum (~4,000–400 cm−1) to reveal qualitative detail regarding the nature of chemical bonds in a sample (e.g., Guo and Bustin, 1998; Nishimiya et al., 1998; Moore and Owen, 2001). This has been used for PyC to identify a range of bonds relating to aromatic and aliphatic structures, and highlight the presence of material such as cellulose, which can be attributed to the precursor biomass, or non-PyC contaminants from the environment (e.g., Ascough et al., 2011b). In addition, bonds characteristic of mineral components (e.g., silica-based rock-derived material), can also be detected in the spectra. We applied FTIR in this study to be able to attribute any non-polycyclic aromatic material in the samples to specific groups of chemical compounds or structures that are present in the environment, including those that can be derived from incomplete pyrolysis of biomass (i.e., the original PyC precursor), and from in situ degradation of PyC itself (e.g., material resembling substances traditionally classed as humic; c.f. Trompowsky et al., 2005; Ascough et al., 2011b).
Several small scrapings of each sample were taken with a clean scalpel for analysis using a Bruker Alpha mid-IR with a diamond crystal and Platinum ATR (Attentuated Total Reflectance). Each sample was placed directly onto the crystal and scanned 16 times in absorbance mode between 4,000 and 400 cm−1 wavenumbers, with 4 cm−1 resolution. The background was subtracted and individual peaks were identified using OpusTM software. The chemical bonds represented by these peaks were identified through reference to the existing literature (Guo and Bustin, 1998; Bustin and Guo, 1999; Benites et al., 2005; Trompowsky et al., 2005).
X-ray Fluorescence (XRF)
X-ray Fluorescence (XRF) was used to identify the mineral elements present in the samples (detectable elements between sodium and uranium). XRF has a greater sensitivity than SEM-EDX for identifying the mineral infill elements, but does not have the spatial resolution of the SEM.
The elemental compositions of samples were assessed by using a Seiko Instruments SEA6000VX XRF spectrometer. Measurements were taken in a helium atmosphere to enhance sensitivity to lighter elements, and over a 3 × 3 mm area at the instrument's normal focusing distance. The spectrometer's rhodium target X-ray tube was operated at 50 kV, with automatic beam current and with additional filtration to reduce background noise in the vicinity of the characteristic X-ray lines for the elements of interest. Each spectrum was collected for 60 s and a semi-quantitative calculation of concentrations of the elements was obtained by using the instrument's standard fundamental parameters program with the balance of the sample assumed to be carbon.
Two different surfaces of PyC sample TDB were analyzed, due to differing surface coloration (lighter material and darker surface), presumably reflecting residual depositional contamination on an external surface.
Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy
Scanning Electron Microsopy (SEM) was used to image the preservation state of the original precursor plant structure in the macro-PyC and, where relevant, mineral infills. Chemical degradation or alteration of the PyC is often accompanied by physical alteration, for example, physical fragmentation, cracking, splitting (Spokas et al., 2014), and fungal or microbiota ingress (Bird et al., 2014). We therefore investigated evidence for these processes using a qualitative approach. Energy dispersive X-ray spectroscopy (EDX) was used in parallel to analyse the elemental composition of any mineral content within the PyC structure. Soil minerals have been linked to catalysis of alteration and degradation of PyC structure during environmental deposition, with evidence that severely degraded PyC is often chemically or physically incorporating mineral substances in some way (Hockaday et al., 2007; Bird et al., 2014, 2017).
For SEM analysis, where possible, pieces of charcoal were cut with a clean scalpel so that only freshly exposed surfaces were imaged, but surface data was interpreted with caution to ensure that only in situ particulate material was analyzed rather than any external contamination. The PyC pieces were mounted on sticky conductive carbon tabs on aluminum stubs prior to placing in the SEM. Analysis was conducted using a Hitachi SU5000 in variable pressure mode, with an attached EDAX TEAM microanalysis system. The conditions used were; a 20 kV accelerating voltage, a chamber pressure of 60 Pa and a nominal working distance of 10 mm (optimal for elemental analysis). Backscattered electron imaging was used throughout the analysis, with the detector set to compositional contrast. The TEAM software enabled elemental spectra to be acquired from selected area(s) of interest on each image, with an acquisition time of 100 s for each EDX spectrum. Elemental maps were similarly acquired, but the acquisition was left to run (summing scan frames) until the map was sufficiently clear to enable visualization of topographical and compositional variations within the maps. Map collection times therefore varied between samples but were a minimum of 1,000 s.
Results
Reflectance of PyC
Temperature of Formation
Results of Romean for all samples are presented in Table 2. Two samples have fairly high reflectance Romean 2.91 (PC-N) and 1.96 (MA), Figures 1C,D); two low reflectance values Romean 1.05 (TRAP, Figure 1B) and 0.70 (TDB, Ascough et al., 2010b) and two very low reflectance near the limits of measurable values with the system used here (AZ and NNB; Figures 1A, 2).
The freshly-produced PyC (PC-N) reflectance indicates production around 490°C. The SD of 0.61 shows variable temperature distribution over even a relatively small sample during production in the Recent hearth fire. The two PyC samples produced in ancient hearth fires returned lower Romean values (with much lower SD), indicating production at around 395°C (TRAP) and 361°C (TDB).
Sample MA, recovered from a pyroclastic deposit associated with an eruption dating to beyond 50 ka BP (Alloway et al., 2004), gave high Romean values (and low SD), translating to production temperatures of around 475°C similar to sample PC-N from the Recent hearth fire. Samples AZ and NNB were also formed during pyroclastic events and both have very low Romean with very low SD corresponding to formation at around 315°C. Different depositional environments (Table 1) may explain these different temperatures of formation. It should be noted that reflectance for sample NNB was measured on what are inferred to be organic cell infills of former cell lumina and not on cell walls. The value is therefore not directly comparable with that for other samples. No higher reflecting organic material was present anywhere in the sample.
Plant Anatomy and Mineral Content
Samples PC-N and AZ both represent gymnosperm (probably conifer; known to be the conifer genus Pinus for PC-N) wood with transverse sections of tracheids seen either side of a growth ring in transverse section (Figures 1A,C). Samples TRAP and MA are also wood but are seen in longitudinal section. Bordered pits in MA indicate gymnosperm wood (Figure 1D), whereas a complex perforation plate in TRAP (bottom left of Figure 1B) is indicative of angiosperm wood. Sample NNB shows uniseriate and biseriate (to multiseriate) rays of wood seen in tangential longitudinal section with material, inferred to be organic, infilling many of the cell lumina (Figure 2). No data are available for TDB which was studied previously (Ascough et al., 2010b sample Env-5).
The few, very small, bright flecks in samples AZ and PC-N (Figures 1A,C) are likely to be mineral. However, those in cell lumina could be derived from dust during handling rather than original presence in the sample. TRAP (Figure 1B) clearly contains abundant mineral infill (speckly grayish to gray-orange material) in the cell lumina. This is also seen to a much lesser extent in MA. Although sample NNB is hard to interpret the bright speckly silvery material is interpreted to be mineral, both based on reflectance brightness and some straight edges in places (Figure 2). Some of the orange speckly material may also be mineral but unpolished. No data are available for TDB which was studied previously (Ascough et al., 2010b sample Env-5).
Hydropyrolysis
Results of BC/OC% derived from hydropyrolysis for all samples are presented in Table 2. Of the three samples (PC-N, TRAP, and TDB) produced on hearth/open fires (361–490°C), the freshly-produced material pyrolyzed at the highest temperature (PC-N) contained the largest proportion of highly polycyclic aromatic material. Approximately one quarter of the carbon in this sample was composed of material comprising seven or more aromatic rings. The carbon in the two ancient hearth samples (TRAP and TDB) contained 17 and 15% of highly polycyclic aromatic material, respectively. The 34°C difference in production temperature between these two samples did not correspond to a substantial difference in polycyclic aromatic carbon, although the PyC produced at the higher temperature (TRAP) appeared slightly more polycyclic aromatic.
Material produced in pyroclastic events differed widely in terms of polycyclic aromatic content. The material produced at the highest temperatures (475°C), emplaced in a non-organic deposit (MA) contained a very high quantity of polycyclic aromatic material (60% BC/OC). This sample is hence likely to be very resistant to chemical or biological attack, with many aromatic bonds that would require substantial energy input to break. In contrast, AZ and NNB, both produced at low temperatures (c. 315°C), contain very little polycyclic aromatic material (1% of total organic carbon is of this chemical form). This means that, although these materials are likely to contain more polycyclic aromatic material in total than 1% of OC, it is hosted mainly in chemical domains that are smaller than seven aromatic rings in extent. It is also likely that a high proportion of these samples is non-aromatic in chemical composition.
Raman Spectroscopy
The Raman spectra of all samples are shown in Figure 3. The modern sample PC-N shows distinct peaks corresponding to the Raman shift (cm−1) for both the G and D bands (centered on 1,575 and 1,350 cm−1, respectively). Increasing extent and size of polycyclic aromatic domains in PyC is usually represented by narrower G bands, positioned closer to 1,575 cm−1 (Tuinstra and Koenig, 1969; Knight and White, 1989). The results support the interpretation of PC-N as containing a high proportion of crystalline (graphite-like) material, relative to other samples, with the exception of MA. The MA sample is highly crystalline, also with a very well-defined G peak.
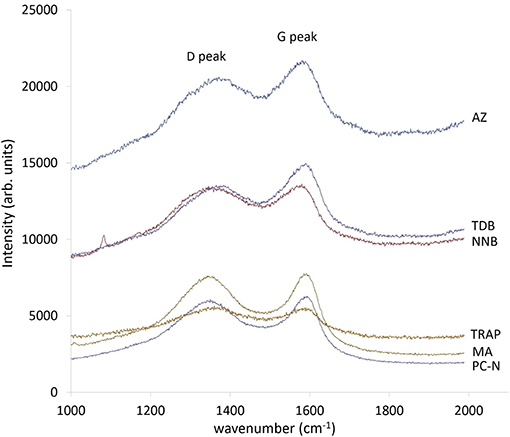
Figure 3. Raman spectra of all samples analyzed in this study. For convenience, spectra with broad similarities are grouped on the figure, although all material analyzed displayed some form of both G and D peaks; this, and the similarities and contrasts between material, is fully discussed in the text.
Humic substances are known to fluoresce strongly (Yang and Wang, 1997), and the relative intensity of Raman spectra of archaeological charcoal has been shown to reflect the amount of humic substances present (Alon et al., 2002). The PC-N and MA spectra both have low intensities relative to the other samples, suggesting low levels of humic substances, which correlates to the high quantities of polycyclic aromatic material observed by HyPy.
The Raman spectra of samples AZ and NNB (low temperature pyroclastic formation, low polycyclic aromatic content as assessed by HyPy), appear similar to each other, although AZ has a much higher relative intensity. These both contain G and D peaks, but the relative height of the D peak is lower than the G peak, and the peaks themselves broader, when compared to the more crystalline PC-N and MA. The G peaks of AZ and NNB are also at slightly lower wavenumbers. This indicates a greater proportion of disorganized carbon within the structure of these samples.
Sample TDB also has very broad peaks shifted to lower wavenumbers than those of PC-N and MA, and appears to be somewhat more crystalline than AZ and NNB. Samples AZ, NNB, and TDB all have much higher intensities relative to MA and PC-N, potentially reflecting higher humic content than the latter samples. However, there is not a direct correlation between low OC values as measured by HyPy and relative Raman intensity.
Sample TRAP differs from all other samples, as distinct peaks in the region for carbon (either organized or disorganized) are very muted, likely due to the lower proportion of carbon within the sample itself relative to the mineral content. Because of this, the G and D peaks are difficult to distinguish from the remainder of the signal, but appear to be of broadly equivalent intensity. The relative intensity of the TRAP spectrum is similar to that of both MA and PC-N, but should be interpreted with caution due to the contribution of the mineral content.
Fourier Transform Infrared Spectroscopy (FTIR)
The FTIR spectra for all samples are shown in Figure 4. Peak assignments are detailed in Table 3. The majority of signal for all spectra was concentrated in the “fingerprint” region of 600–2,000 cm−1, hence discussion below focusses on this region. For the two most chemically aromatic samples (PC-N and MA), there is strong signal for aromatic C=C ring structures at 1,570–1,580 cm−1, and a well-defined series of peaks for aromatic CH deformation at 700–900 cm−1. Both spectra also contain a broad and indistinct signal centered at c. 1,180 cm−1 (PC-N), and 1,106 cm−1 (MA). In MA this region contains more structure, indicating a wider range of distinct different bonding environments. The region has been assigned to the C-O stretch of esters, phenols and ethers (Marshall et al., 2005). Both MA and PC-N also contain small amounts of signal at c. 1,690 cm−1. This likely relates to carboxylic acids (COOH), although the latter are more frequently assigned at 1,700 cm−1. This interpretation is supported by the presence of structure at 1,250–1,270 cm−1 within the broad region mentioned above.
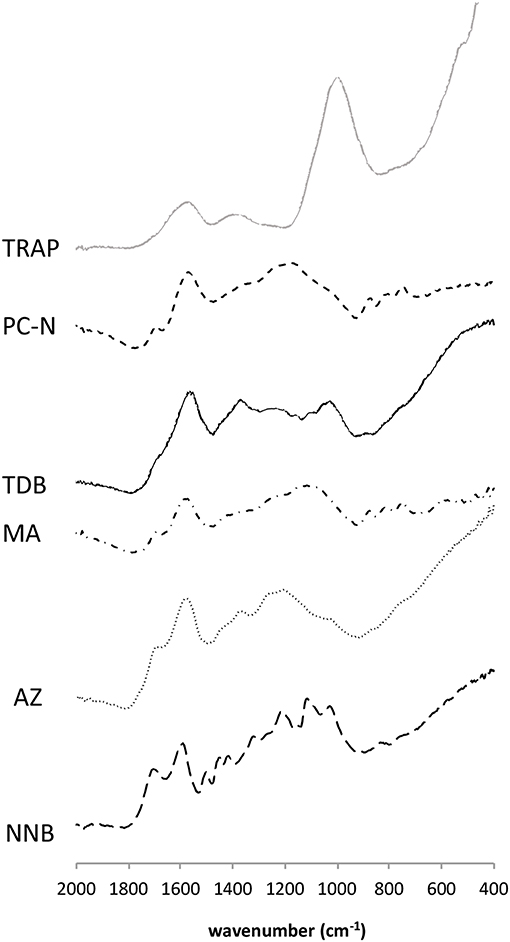
Figure 4. FTIR spectra of all samples. As discussed in the text, the figure focuses upon the “fingerprint” region between 400–2,000 cm−1.
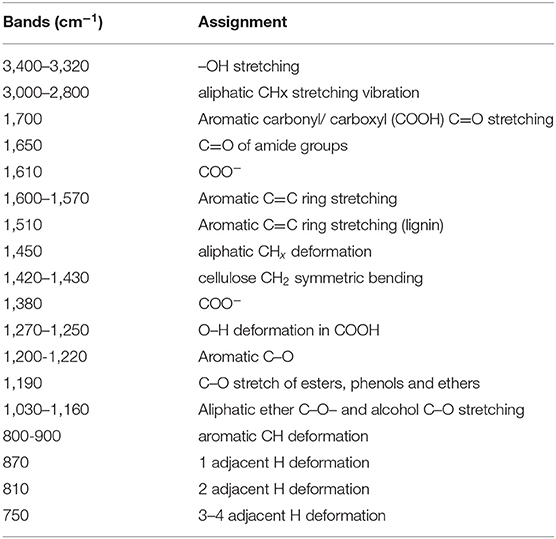
Table 3. Assignment of Infra-Red peaks for analysis of FTIR spectra (From Ascough et al., 2011b, assignments after Guo and Bustin, 1998; Bustin and Guo, 1999; Benites et al., 2005; Trompowsky et al., 2005).
Samples TDB, NNB, and AZ also contain prominent signal for C=C ring structures. In NNB this centers on 1,590 cm−1, and is shifted to slightly lower wavenumbers in TDB and AZ. TDB, and NNB also contain identifiable peaks for aromatic C-H in the region 700–900 cm−1, but these are much less distinct than for material such as PC-N and MA. In AZ this signal is not clear in the spectrum. In TDB peaks at c. 1,380 cm−1 are likely to relate to COOH (COO−) groups, and signal at c. 1,245 cm−1 to O–H deformation in COOH. In NNB bands for COOH are evident in the spectrum, particularly at 1,700 cm−1. Carboxylic groups are represented in AZ by very minor peaks around 1,700 cm−1, and greater signal in the c. 1,250 cm−1 region. A peak at c. 1,380 cm−1 is also present. All three of these samples contain signal for oxygenated functional groups of cellulose, including aliphatic ether and alcohol bonding (C-O-C and alcohol OH groups) at 1,030–1,160 cm−1 (Guo and Bustin, 1998; Colom and Carrillo, 2002). NNB also contains signal relating to lignin aromatic stretching at c. 1500 cm−1, and aliphatic CHx deformation at 1,450 cm−1 (Owen and Thomas, 1989).
Sample TRAP contains little distinct signal compared to the other PyC materials. This relates to a clear peak for C=C ring structures at 1,570 cm−1, and for COOH (COO−) groups at c. 1,390 cm−1. The most prominent peak is centered on 1,000 cm−1. This is lower than the usual range for aliphatic ether linkages. It is possible this relates to some aliphatic structures, or aromatic linkages in alkenes, however it seems most likely that this represents a strong signal for iron phosphates, derived from the depositional environment (c.f. Palacios et al., 2012).
X-ray Fluorescence (XRF)
Results of XRF analysis are shown in Table 4. Samples were examined for 12 elements which were observed to be present in one or more sample. Note PC-N had not been exposed to environmental depositional conditions, and therefore serves as an example of elemental abundances levels that can be present naturally in PyC immediately following production, although we acknowledge that the specific quantity of these will vary somewhat depending on feedstock. This therefore serves as an indicator of minimum values. This sample contained low levels of tested elements across all elements analyzed.
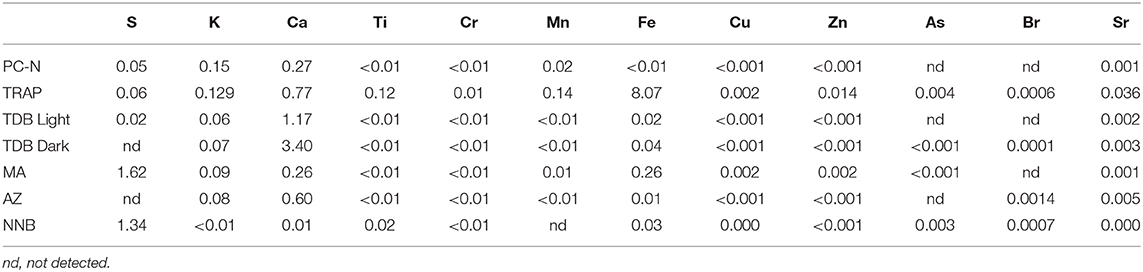
Table 4. Concentrations (weight %) of the elements observed in the PyC samples as determined by XRF (balance C).
For the remaining PyC samples, AZ, TDB, and TRAP contained Ca at abundances higher than other samples. Calcium was particularly high in TDB, with Ca content more than twice as high as all other PyC samples. This is consistent with deposition in a calcareous environment, highlighting that minerals from the environment had ingressed, and at least partially infilled, the PyC through openings in the cell lumina structure. Sulfur was present in much higher quantities in MA and NNB than all other PyC, consistent with a pyroclastic formation mechanism, where sulfur minerals are commonly present (i.e., iron sulfide). MA also contained quantities of Fe. In sample TRAP however, the abundance of elements measured by XRF was higher in the categories of Fe, Zn, and Sr, than in all other samples. Fe in particular was anomalously high in this sample, consistent with its depositional environment in a waterlogged, anaerobic setting high in soluble iron minerals, again showing that elements from the environment freely ingress to PyC during deposition.
Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy
SEM micrographs for the samples are shown in Figures 5A–F. The purpose of SEM examination was to investigate the physical structure of the samples, identifying whether significant physical degradation (e.g., cracking) was present, and also examining for the presence of mineral infills. EDX was used to identify the composition of mineral material present on or in the samples.

Figure 5. Scanning Electron Microscopy images for all samples analyzed, showing areas that are representative of the overall structure of the specimen. Images are ordered: (A) PC-N; (B) AZ; (C) MA; (D) NNB; (E) TRAP; (F) TDB. PC-N and AZ show bordered pits in longitudinal fracture of conifer wood; MA shows radial longitudinal fracture of conifer wood with rays (e.g., right of image); NNB shows tangential longitudinal fracture of conifer wood with uniseriate and biseriate rays; TRAP and TDB show little plant anatomy (see text) due to presence of mineral.
The freshly produced PC-N had the expected structure of a modern PyC showing very clear plant anatomy of bordered pits on cell walls, characteristic of gymnosperm (and known to be the conifer Pinus) wood seen in longitudinal section, with some cracking (Figure 5A). Compositionally the inorganic or mineral content, present at minor or trace levels, was K, Ca, Mg, P, S, and Cl, as expected for an uncontaminated wood (Herzog et al., 2006). The natural mineral inclusions are within the cell walls and so are not observable in the SEM micrograph. AZ (Figure 5B) shows the same plant anatomy as PC-N. MA (Figure 5C) is wood seen in radial longitudinal section (rays running slightly obliquely from top to bottom evident at near right and left of image); similar width in most cells running left to right in image (probably tracheids) suggests gymnosperm wood. Both the AZ and MA specimens were similarly free from particulate contamination, with the visible debris being fragments of the PyC likely to be generated during sample preparation. Visually, there was no direct evidence of infill in the SEM (Figures 5B,C) to explain the XRF results, although the EDX spectrum for AZ did have a minor Ca peak, whereas MA produced a minor S peak. This suggests that the infill does not penetrate throughout the entirety of the samples as the SEM and XRF analyses were on different regions of each PyC sample. We also note that this does not preclude the deposition/precipitation of Ca-rich minerals on internal pores or surfaces on a scale that may only be visible at higher magnification, or at the molecular level via the strong affinity of Ca2+ to carboxylic groups (Hardy et al., 2017). Specimen NNB (Figure 5D) shows wood in tangential longitudinal section with mainly uniseriate rays and one biseriate ray near base left of image. Other cells may be fibers or tracheids. Specimen NNB had a relatively uniform, if minor, mineral infill forming small particles which were iron rich with phosphorus (the latter not detectable by XRF due to the low energies involved).
The greatest abundance of mineral was observed in the TRAP and TDB specimens. Most plant anatomy is obscured in the SEM images (Figures 5E,F) though small patches with many small round holes in TDB (Figure 5F) might be the pitted walls of vessel elements indicative of angiosperm wood and lineation (top left to bottom right) in TRAP (Figure 5E) suggests a longitudinal section through wood. The mineral infill is packed throughout the sample indicating the ingress throughout the material in this area. However, the particulate appearance and composition is notably different between the two highlighting the environmental source differences; TRAP contained an iron-rich mineral with a minor phosphorus component; TDB contains what is likely a clay mineral (aluminum silicate) with additional cation Ca and K accommodation (the Al, Si, and O not detected by XRF).
Discussion
Although observing specific processes operating on the PyC samples prior to our study has not been possible in the present study, we have used samples about which there is broad information on depositional environment. Samples were selected to cover a range of environments for which literature evidence suggests there can be an impact upon PyC alteration/degradation rates. For example, including material from waterlogged/anaerobic vs. aerated environments (Table 1) is useful, as evidence suggests strongly reduced PyC degradation under the former conditions (Nguyen and Lehmann, 2009; Foereid et al., 2011; Knicker, 2011). We have also used material for which we have information on the nature of the production event (e.g., hearth fire vs. pyroclastic event). For example, for MA, the depositional environment is very low in organic matter (Alloway et al., 2004), suggesting the original pyroclastic material (which provided the formation medium) was predominantly mineral in nature (e.g., pumice, lava). In contrast, AZ was also formed during a pyroclastic event, but the depositional environment suggests that the material forming the formation medium was more organic than for MA, likely to have been associated with a lahar event (Cruz et al., 2006). NNB was also formed during a pyroclastic event, during burial by a large volume of hot volcanic ash. For all three samples (MA, AZ, and NNB), it is likely that the duration of exposure to elevated temperatures (during subsequent cooling of the pyroclastic deposit) was long, compared to that of PC-N, TRAP, and TDB.
Although observing specific processes operating on the PyC samples in detail is not practical in the present study, we have been able to obtain material about which there is broad information on depositional environment. Samples were selected to cover a range of environments for which literature evidence suggests there is an impact upon PyC alteration/degradation rates. For example, including material from waterlogged/anaerobic vs. aerated environments is useful, as evidence suggests strongly reduced PyC degradation under the former conditions (Nguyen and Lehmann, 2009; Foereid et al., 2011; Knicker, 2011). We have also been able to obtain material for which we have information on the type and temperature of production. This enables us to investigate the range of chemical composition of ancient PyC (up to >90 Ka BP). These are complemented by material freshly-produced and not subjected to environmental deposition, and material subjected to environmental conditions for decadal timescales. One key feature of the research presented here is a consideration of whether (ancient and modern) PyC that is “intact” when in situ may not retain this stability when the depositional environment changes, for example through climatic changes, or exposure to the atmosphere/excavation, even when stored in the lab for a period of weeks, months, years, prior to analysis. There is evidence that carbon compounds resulting from the alteration of initial PyC structure can remain at the site of alteration (i.e., associated with the remaining PyC structure); this process seems to be mediated by soil chemistry, particularly the presence of calcium ions (Bird et al., 2017; Munksgaard et al., 2018).
The results presented provide corroboration of the hypothesis that the chemical characteristics of long-lived PyC in the environment are highly variable. It has become apparent over recent years that PyC is neither a homogeneous material, nor environmentally inert (e.g., Ascough et al., 2011a; Bird et al., 2015). It is also apparent that while carbon dominates in the chemical composition of PyC, a wide range of other elements are also integral to its structure and are present in significant quantities (e.g., Ascough et al., 2008; Bird et al., 2017). These elements also appear to play a role in modulating the fate of PyC in the environment. Aromaticity is a key feature of PyC, being fundamental to the chemical structure of freshly-produced PyC. Bond energies of individual aromatic rings are ~193.71 Kcal mol−1, and increase proportionally to the size of polyaromatic structures (Maitland, 2005), in comparison to much lower bond energies in non-aromatic components, such as C-C (80.711 Kcal mol−1) and C-H (92.241 Kcal mol−1). As aromatic bonds require substantial energy input to break down, compared to non-aromatic material (e.g., C-C bonds in cellulose), it is logical that the aromaticity of PyC is an underlying driver of PyC long-term preservation in the environment. However, the research presented here shows that formation temperature (and hence aromaticity) is not the sole determinant of PyC preservation in the environment over tens of thousands of years, and neither is the length of time to which PyC has been exposed to the environment per se.
It is important to note that a key distinction in the following discussion is between degradation (that involves the original PyC components physically and/or chemically being lost from the in situ environment) and alteration in situ (whereby the original PyC chemistry is changed, but the products of this change remain in situ). Degradation can occur either via mineralisation of the PyC (or its alteration products) directly to CO2 at the deposition site, or via processes that lead to physical or chemical transport of the PyC (or its alteration products) away from the in situ environment. These processes vary in their nature, but the net effect (loss of the PyC components from the material initially deposited) is the same.
Our results indicate that very long-lived PyC in the environment can be composed nearly entirely of chemically labile components. Both AZ and NNB on visual inspection appear to be intact PyC fragments, and plant anatomical structure is visible in them under both reflectance microscopy and SEM. However, both samples have a very low content of polycyclic aromatic structures [seven aromatic rings or greater (1%)], and significant quantities of non-aromatic bonding, as observed by FTIR. The latter is consistent with incompletely thermally altered material, and/or a lower order of aromatisation during production at temperatures c. 315°C (as derived from Romean). It is possible that the current chemical composition of these materials to some extent reflects post-depositional alteration processes, however we note that low-order aromaticity and incomplete thermal alteration is quite consistent with the chemical structure of PyC produced at these lower temperatures (e.g., Ascough et al., 2008). We acknowledge the sample size here is small, but this suggests that production temperature is not necessarily a determinant of whether a significant proportion of PyC remains in situ in an unmineralized state (and hence retaining carbon in the global sedimentary, not atmospheric sink) for millennia. The implication of this is that the PyC global sink (as defined on a chemical basis, i.e., aromaticity) could be larger than hypothesized, if material of low aromaticity (whether the result of low-temperature production or post-depositional alteration) is retained in a specific location for very extended timescales. The results of the present study imply that the depositional setting for this is important; both AZ and NNB were subject to rapid burial in relatively extensive (in three dimensions) deposits. This is consistent with findings from other workers (e.g., Marin-Spiotta et al., 2014), who have identified large stocks of PyC preserved on timescales of the order of 10,000 years, where rapid burial and subsequent lack of disturbance to the depositional environment appears to have been a key process that leads to the preservation of PyC of low aromaticity. It is important to note that the condition of the plant material prior to transformation to PyC is not known for any of the samples included in this study (with the exception of PC-N), in particular which were still living (especially at the time of a volcanic event) and which were already dead and had begun to degrade.
Conversely, the low-SPAC samples (AZ and NNB) are very chemically labile when exposed to oxidation or changes in pH in a laboratory environment. A separate sub-sample of AZ was assessed by Ascough et al. (2011a), who exposed this material to oxidation by potassium dichromate. This resulted in complete loss of the material extremely rapidly, when compared to a range of other PyC, both freshly-produced and of various ages and from natural depositional environments. Combined with the present study, this raises the point that PyC that is long-lived in a particular depositional environment will not necessarily remain stable if that environment changes. Examples of change would be exposure to oxidation via excavation (Marin-Spiotta et al., 2014), and changes in soil pH via climatic changes or alteration in land-use practice (c.f. Hardy et al., 2017). Although ancient PyC does not necessarily contain ever-increasing levels of SPAC as a precondition for its preservation, chemical degradability (at least in laboratory conditions) does appear strongly linked to the amount of SPAC in the sample, in an inverse proportional relationship (Ascough et al., 2010a, 2011a).
The results also confirm that production at higher temperatures does directly result in more highly aromatic PyC. The range of production temperatures inferred for the six samples by Romean corresponds well to the proportion of polycyclic aromatic carbon making up the sample as measured via hydropyrolysis, and also broadly to the relative prominence of the G band in Raman spectroscopy. If production temperature apparently remains a reliable (at least in semi-quantitative terms) indicator of SPAC content in PyC even after millennia in the environment, this does support the existing interpretation that, under constant environmental conditions, the degradability of the PyC is primarily driven by initial production temperature. However, the results of the present study also emphasize that it is appropriate to consider PyC as having an initial degradation potential following production (c.f. Ascough et al., 2011a; Bird et al., 2015). The pathway that a particular PyC sample then follows is heavily modulated by the conditions of deposition (e.g., fast vs. slow burial, shallow vs. deep burial), and the chemistry of the depositional environment (e.g., oxic vs. anoxic, pH) through time (Braadbaart et al., 2009; Knoblauch et al., 2011; Zimmermann et al., 2012; Marin-Spiotta et al., 2014). A key point is that structures of seven aromatic rings or greater (as isolated by Hypy) is a feature of all samples analyzed, and that even material produced at the low end of conditions conventionally understood to result in PyC (c. 315°C) still contains at least a proportion of SPAC. This is observed in the results of Raman spectroscopy, where G band peaks are evident throughout the samples, and in the proportion of highly polycyclic aromatic carbon in samples identified via hydropyrolysis.
This study has shown that PyC samples can appear physically similar but simultaneously exhibit fundamental chemical differences. In particular, the similarity in visible wood cellular structure observed by both reflectance and SEM in PC-N and AZ (Figures 1A,C, 5A,B) belie the significant difference in polycyclic aromatic carbon content between the two samples. The differences in inorganic content of individual samples measured by XRF and SEM-EDX are partly due to the greater depth penetration of XRF and the wider range of elements detectable by SEM-EDX, but also reflect sample heterogeneity. The SEM-EDX results show that, even in a small suite of sample PyC, mineral material from the depositional environment can become an integral part of the gross structure of the PyC sample, and that this is a feature of both oxic and anoxic environments. Consideration of both the XRF and SEM-EDX data indicate that the infill is localized within the PyC samples, consistent with mineral laden water permeating through fissures in the material. In the case of the TRAP samples in particular, the authigenic mineral lines and infills individual cell lumina without causing obvious disruption to the cellular structure, which may potentially protect the remaining organic matter from alteration and degradation. Information on the mineral chemistry of the depositional environment is available for TRAP (Armit et al., 1999, 2000) and TDB (Steelman et al., 2002), which is consistent with the information on PyC mineral content analyzed in the present study (see Table 1). This is further confirmation that compounds potentially linked to catalysis of environmental degradation can migrate from the depositional environment to become incorporated into the PyC structure, for example in micropores and coating interior surfaces.
Conclusion
The work presented here uses wood PyC to address the factors identified in the introduction, which lead to the contrasts in the timescales of PyC turnover that have been observed globally, namely: (i) is this mainly a function of initial chemistry (i.e., linked to production temperature), (ii) is more ancient material progressively dominated by SPAC, and (iii) what impact do depositional conditions have upon PyC chemistry/status over tens of thousands of years. We show, firstly, that, within the sample set investigated, although initial production temperature is a driver of the “degradation potential” of PyC immediately after production, the trajectory of PyC in the environment (in terms of degradation or alteration vs. preservation) is not only driven by its initial chemistry, but is strongly modulated by depositional environment, and possibly, features of the initial depositional event. The latter may include depth and rapidity of deposition, and the chemistry of the matrix in which the PyC was first emplaced. Secondly, the composition of ancient PyC is not necessarily a function of its age, even after millennia exposed to the environment. Instead, even PyC thousands of years old can be composed of material that is chemically labile, and/or is of low aromaticity. The implications of this are two-fold; that the long-term PyC sink, usually identified as the most highly aromatic chemical component of PyC, such as SPAC, is likely to be larger than previously thought, because of potential for long-term preservation of the labile/semi-labile fraction in specific depositional environments. However, the second implication is that when the environmental context of PyC alters, this situation of long-term preservation may not necessarily continue. At present, the size of PyC budgets in depositional environments is largely unknown, and further work should focus on refining these estimates, particularly as hotspots' of long-term PyC preservation may be vulnerable to land-use and climatic or environmental change. Finally, the specific impact of depositional conditions upon PyC chemistry/status remains unclear. This is a difficult problem to address, however we show that the chemistry of the depositional environment is reflected in the elements associated with PyC, some of which, particularly calcium and iron, have been linked with catalysis of PyC alteration, and mediation of environmental interactions. To extend this knowledge, it is clear that further investigations of PyC degradation vs. preservation, and the carbon sequestration status of this material, would greatly benefit from multi-technique approaches, which integrate physical investigation of structural features with a range of chemical characterization methods, in order to obtain as full a picture as possible of this material in the environment.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
PA led drafting of the manuscript. PA, FB, and JP jointly prepared overall structuring, tables, and figures. All authors had intellectual input to the study and contributed to the preparation and revision of the manuscript. MB informed study design and rationale. MC performed measurement of Romean with reflectance photography and their interpretations and provided botanical interpretation. PA performed Hydropyrolysis experiments. FB undertook FTIR, and Raman analysis. JP undertook SEM-EDX analysis, including sample preparation and interpretation of data. DL undertook analysis, preparation, and interpretation for XRF measurements.
Funding
Overall financial support was provided by NERC (NE/F017456/1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This publication represents a contribution from the Scottish Alliance for Geoscience, Environment, and Society (SAGES). Samples were kindly provided by Brent Alloway (MA), Mike Church (TRAP), Neide Guidon (TDB), Fiona Petchey (NNB). FTIR was undertaken in the Center for Defense Chemistry, Cranfield University. We thank Adrian Mustey (Cranfield University) for preparation of resin blocks for reflectance analysis. At Royal Holloway MEC would like to thank Sharon Gibbons for technical support and Neil Holloway for re-polishing of resin blocks when necessary.
References
Aleman, J., Hennebelle, A., Vannière, B., Blarquez, O., and Global Paleofire Working Group. (2018). Sparking new opportunities for charcoal-based fire history reconstructions. Fire 1:7. doi: 10.3390/fire1010007
Alloway, B. V., Pribadi, A., Westgate, J. A., Bird, M., Fifield, L. K., Hogg, A., et al. (2004). Correspondence between glass-FT and AMS 14 C ages of silicic pyroclastic density current (PDC) deposits sourced from Maninjau caldera, west-central Sumatra. Earth Planet. Sci. Lett. 227, 121–133. doi: 10.1016/j.epsl.2004.08.014
Alon, D., Mintz, G., Cohen, I., Weiner, S., and Boaretto, E. (2002). The use of Raman spectroscopy to monitor the removal of humic substances from charcoal: quality control for 14C dating of charcoal. Radiocarbon 44, 1–11. doi: 10.1017/S0033822200064638
Armit, I., Dunwell, A., and Hunter, F. (1999). Traprain Law Summit Project. East Lothian. Unpublished Data Structure Report
Armit, I., Dunwell, A., and Hunter, F. (2000). Traprain Law Summit Project. East Lothian. Unpublished Data Structure Report
Ascough, P., Bird, M. I., Scott, A. E., Collinson, M. E., Cohen-Ofri, I., Snape, C. E., et al. (2010b). Charcoal reflectance measurements: implications for structural characterization and assessment of diagenetic alteration. J. Archaeol. Sci. 37, 1590–1599. doi: 10.1016/j.jas.2010.01.020
Ascough, P., Bird, M. I., Wormald, P., Snape, C. E., and Apperley, D. (2008). Influence of pyrolysis variables and starting material on charcoal stable isotopic and molecular characteristics. Geochim. Cosmochim. Acta 72, 6090–6102. doi: 10.1016/j.gca.2008.10.009
Ascough, P. L., Bird, M. I., Brock, F., Higham, T. F. G., Meredith, W., Snape, C., et al. (2009). Hydropyrolysis as a new tool for radiocarbon pretreatment and the quantification of black carbon. Quat. Geochronol. 4, 140–147. doi: 10.1016/j.quageo.2008.11.001
Ascough, P. L., Bird, M. I., Francis, S. M., and Lebl, T. (2011b). Alkali extraction of archaeological and geological charcoal: evidence for diagenetic degradation and formation of humic acids. J. Archaeol. Sci. 38, 69–78. doi: 10.1016/j.jas.2010.08.011
Ascough, P. L., Bird, M. I., Francis, S. M., Thornton, B., Midwood, A., Scott, A. C., et al. (2011a). Variability in oxidative degradation of charcoal: influence of production variables and environmental exposure. Geochim. Cosmochim. Acta 75, 2361–2378. doi: 10.1016/j.gca.2011.02.002
Ascough, P. L., Bird, M. I., Meredith, W., Wood, R. E., Snape, C. E., Brock, F., et al. (2010a). Hydropyrolysis: implications for radiocarbon pre-treatment and characterization of black carbon. Radiocarbon 52, 1336–1350. doi: 10.1017/S0033822200046427
Benites, V. M., Mendonca, E. S., Schaefer, C. E. G. R., Novotny, E. H., Reis, E. L., and Ker, J. C. (2005). Properties of black soil humic acids from high altitude rocky complexes in Brazil. Geoderma 127, 104–113. doi: 10.1016/j.geoderma.2004.11.020
Bird, M. I. (2007). “Radiocarbon dating: charcoal” in Encyclopedia of Quaternary Science, ed S. Elias, (Elsevier) 2950–2958. doi: 10.1016/B0-44-452747-8/00046-6
Bird, M. I., and Ascough, P. L. (2012). Isotopes in pyrogenic carbon: a review. Org. Geochem. 42, 1529–1539. doi: 10.1016/j.orggeochem.2010.09.005
Bird, M. I., Levchenko, V., Ascough, P., Meredith, W., Wurster, C. M., Williams, A., et al. (2014). The efficiency of charcoal decontamination for radiocarbon dating by three pre-treatments – ABOX, ABA and hypy. Quat. Geochronol. 22, 25–32 doi: 10.1016/j.quageo.2014.02.003
Bird, M. I., McBeath, A. V., Ascough, P. L., Levchenko, V. A., Wurster, C. M., Munksgaard, N. C., et al. (2017). Loss and gain of carbon during char degradation. Soil Biol. Biochem. 106, 80–89. doi: 10.1016/j.soilbio.2016.12.012
Bird, M. I., Wynn, J. G., Saiz, G., Wurster, C. M., and McBeath, A. (2015). The pyrogenic carbon cycle. Ann. Rev. Earth Planet. Sci. 43, 273–298. doi: 10.1146/annurev-earth-060614-105038
Braadbaart, F., Poole, I., and Van Brussel, A. A. (2009). Preservation potential of charcoal in alkaline environments: an experimental approach and implications for the archaeological record. J. Archaeol. Sci. 36, 1672–1679. doi: 10.1016/j.jas.2009.03.006
Bustin, R. M., and Guo, Y. (1999). Abrupt changes (jumps) in reflectance values and chemical compositions of artificial charcoals and inertinite in coals. Int. J. Coal Geol. 38, 237–260. doi: 10.1016/S0166-5162(98)00025-1
Colom, X., and Carrillo, F. (2002). Crystallinity changes in lyocell and viscose-type fibres by caustic treatment. Euro. Polymer J. 38, 2225–2230. doi: 10.1016/S0014-3057(02)00132-5
Coppola, A. I., and Druffel, E. R. (2016). Cycling of black carbon in the ocean. Geophys. Res. Lett. 43, 4477–4482. doi: 10.1002/2016GL068574
Cotrufo, M. F., Boot, C. M., Kampf, S., Nelson, P. A., Brogan, D. J., Covino, T., et al. (2016). Redistribution of pyrogenic carbon from hillslopes to stream corridors following a large montane wildfire. Glob. Biogeochem. Cycles 30, 1348–1355. doi: 10.1002/2016GB005467
Cruz, J. V., Antunes, P., Amaral, C., Franca, Z., and Nunes, J. C. (2006). Volcanic lakes of the Azores archipelago (Portugal): Geological setting and geochemical characterization. J. Volcanol. Geotherm. Res. 156, 135–157. doi: 10.1016/j.jvolgeores.2006.03.008
Czimczik, C. I., Preston, C. M., Schmidt, M. W., and Schulze, E. D. (2003). How surface fire in Siberian Scots pine forests affects soil organic carbon in the forest floor: stocks, molecular structure, and conversion to black carbon (charcoal). Glob. Biogeoch. Cycles 17. doi: 10.1029/2002GB001956
Foereid, B., Lehmann, J., and Major, J. (2011). Modeling black carbon degradation and movement in soil. Plant Soil 345, 223–236. doi: 10.1007/s11104-011-0773-3
Guo, Y., and Bustin, R. M. (1998). FTIR spectroscopy and reflectance of modern charcoals and fungal decayed woods: implications for studies of inertinite in coals. Int. J. Coal Geol. 37, 29–53. doi: 10.1016/S0166-5162(98)00019-6
Hardy, B., Leifeld, J., Knicker, H., Dufey, J. E., Deforce, K., and Cornélis, J. T. (2017). Long term change in chemical properties of preindustrial charcoal particles aged in forest and agricultural temperate soil. Org. Geochem. 107, 33–45. doi: 10.1016/j.orggeochem.2017.02.008
Hawthorne, D., Mustaphi, C. J. C., Aleman, J. C., Blarquez, O., Colombaroli, D., Daniau, A. L., et al. (2018). Global Modern Charcoal Dataset (GMCD): a tool for exploring proxy-fire linkages and spatial patterns of biomass burning. Quat. Int. 488, 3–17. doi: 10.1016/j.quaint.2017.03.046
Herzog, A., Reznik, B., Chen, T., Graule, T., and Vogt, U. (2006). Structural changes in activated wood-based carbons: correlation between specific surface area and localization of molecular-sized pores. Holzforschung 60, 85–92. doi: 10.1515/HF.2006.015
Hockaday, W. C., Grannas, A. M., Kim, S., and Hatcher, P. G. (2007). The transformation and mobility of charcoal in a fire-impacted watershed. Geochim. Cosmochim. Acta 71, 3432–3445. doi: 10.1016/j.gca.2007.02.023
Knicker, H. (2011). Pyrogenic organic matter in soil: Its origin and occurrence, its chemistry and survival in soil environments. Quat. Int. 243, 251–263. doi: 10.1016/j.quaint.2011.02.037
Knight, D. S., and White, W. B. (1989). Characterization of diamond films by Raman spectroscopy. J. Mater. Res. 4, 385–393. doi: 10.1557/JMR.1989.0385
Knoblauch, C., Maarifat, A. A., Pfeiffer, E. M., and Haefele, S. M. (2011). Degradability of black carbon and its impact on trace gas fluxes and carbon turnover in paddy soils. Soil Biol. Biochem. 43, 1768–1778. doi: 10.1016/j.soilbio.2010.07.012
Lehmann, J., and Joseph, S. (2015). “Biochar for environmental management: an introduction,” in Biochar for Environmental Management (Routledge), 33–46.
Marin-Spiotta, E., Chaopricha, N. T., Plante, A. F., Diefendorf, A. F., Mueller, C. W., Grandy, A. S., et al. (2014). Long-term stabilization of deep soil carbon by fire and burial during early Holocene climate change. Nat. Geosci. 7:428. doi: 10.1038/ngeo2169
Marshall, C. P., Kamali Kannangara, G. S., Alvarez, R., and Wilson, M. A. (2005). Characterisation of insoluble charcoal in Weipa bauxite. Carbon 43, 1279–1285. doi: 10.1016/j.carbon.2004.12.024
McBeath, A. V., Wurster, C. M., and Bird, M. I. (2015). Influence of feedstock properties and pyrolysis conditions on biochar carbon stability as determined by hydrogen pyrolysis. Biomass Bioenergy 73, 155–173. doi: 10.1016/j.biombioe.2014.12.022
McDonald-Wharry, J., Manley-Harris, M., and Pickering, K. (2013). Carbonisation of biomass-derived chars and the thermal reduction of a graphene oxide sample studied using Raman spectroscopy. Carbon 59, 383–405. doi: 10.1016/j.carbon.2013.03.033
McParland, L. C., Scott, A. C., Collinson, M. E., and Campbell, G. (2009). The use of reflectance values for the interpretation of natural and anthropogenic charcoal assemblages. Archaeol. Anthropol. Sci. 1, 249–261. doi: 10.1007/s12520-009-0018-z
Meredith, W., Ascough, P. L., Bird, M. I., Large, D. J., Snape, C. E., Sun, Y., et al. (2012). Assessment of hydropyrolysis as a method for the quantification of black carbon using standard reference materials. Geochim. Cosmochim. Acta 97, 131–147. doi: 10.1016/j.gca.2012.08.037
Moore, A. K., and Owen, N. L. (2001). Infrared spectroscopic studies of solid wood. Appl. Spectrosc. Rev. 36, 65–86. doi: 10.1081/ASR-100103090
Munksgaard, N. C., McBeath, A. V., Ascough, P. L., Levchenko, V. A., Williams, A., and Bird, M. I. (2018). Partitioning of microbially respired CO2 between indigenous and exogenous carbon sources during biochar degradation using radiocarbon and stable carbon isotopes. Radiocarbon 61, 573–586. doi: 10.1017/RDC.2018.128
Nguyen, B. T., and Lehmann, J. (2009). Black carbon decomposition under varying water regimes. Org. Geochem. 40, 846–853. doi: 10.1016/j.orggeochem.2009.05.004
Nishimiya, K., Hata, T., Imamura, Y., and Ishihara, S. (1998). Analysis of chemical structure of wood charcoal by X-ray photoelectron spectroscopy. J. Wood Sci. 44, 56–61. doi: 10.1007/BF00521875
Ohlson, M., Dahlberg, B., Økland, T., Brown, K. J., and Halvorsen, R. (2009). The charcoal carbon pool in boreal forest soils. Nat. Geosci. 2, 692. doi: 10.1038/ngeo617
Owen, N. L., and Thomas, D. W. (1989). Infrared studies of “hard” and “soft” woods. Appl. Spectr. 43, 451–455. doi: 10.1366/0003702894202760
Palacios, E., Leret, P., Fernández, J. F., De Aza, A. H., and Rodríguez, M. A. (2012). Synthesis of amorphous acid iron phosphate nanoparticles. J. Nanoparticle Res. 14:1131. doi: 10.1007/s11051-012-1131-y
Preston, C. M., and Schmidt, M. W. I. (2006). Black (pyrogenic) carbon in boreal forests: a synthesis of current knowledge and uncertainties. Biogeosci. Disc. 3, 211–271. doi: 10.5194/bgd-3-211-2006
Reisser, M., Purves, R. S., Schmidt, M. W., and Abiven, S. (2016). Pyrogenic carbon in soils: a literature-based inventory and a global estimation of its content in soil organic carbon and stocks. Front. Earth Sci. 4:80. doi: 10.3389/feart.2016.00080
Santin, C., Doerr, S. H., Kane, E. S., Masiello, C. A., Ohlson, M., de la Rosa, J. M., et al. (2016). Towards a global assessment of pyrogenic carbon from vegetation fires. Glob. Change Biol. 22, 76–91. doi: 10.1111/gcb.12985
Santín, C., Doerr, S. H., Merino, A., Bucheli, T. D., Bryant, R., Ascough, P., et al. (2017). Carbon sequestration potential and physicochemical properties differ between wildfire charcoals and slow-pyrolysis biochars. Sci. Rep. 7:11233. doi: 10.1038/s41598-017-10455-2
Santín, C., Doerr, S. H., Preston, C. M., and González-Rodríguez, G. (2015). Pyrogenic organic matter production from wildfires: a missing sink in the global carbon cycle. Glob. Change Biol. 21, 1621–1633. doi: 10.1111/gcb.12800
Scott, A. C. (2010). Charcoal recognition, taphonomy and uses in palaeoenvironmental analysis. Palaeogeogr. Palaeoclimatol. Palaeoecol. 291, 11–39. doi: 10.1016/j.palaeo.2009.12.012
Scott, A. C., and Glasspool, I. J. (2005). Charcoal reflectance as a proxy for the emplacement temperature of pyroclastic flow deposits. Geology 33, 589–592. doi: 10.1130/G21474.1
Singh, N., Abiven, S., Torn, M. S., and Schmidt, M. W. (2012). Fire-derived organic carbon in soil turns over on a centennial scale. Biogeosciences 9, 2847–2857. doi: 10.5194/bg-9-2847-2012
Spokas, K. A., Novak, J. M., Masiello, C. A., Johnson, M. G., Colosky, E. C., Ippolito, J. A., et al. (2014). Physical disintegration of biochar: an overlooked process. Environ. Sci. Technol. Lett. 1, 326–332. doi: 10.1021/ez500199t
Steelman, K. L., Rickman, R., Rowe, M. W., Boutton, T. W., Russ, J., and Guidon, N. (2002). “Accelerator mass spectrometric radiocarbon ages of an oxalate accretion and rock paintings at Toca do Serrote da Bastiana, Brazil,” in Archaeological Chemistry VI: Materials, Methods, and Meaning, ed K. A. Jakes (Washington, DC: American Chemical Society), 22–35. doi: 10.1021/bk-2002-0831.ch003
Stinson, K. J., and Wright, H. A. (1969). Temperatures of headfires in the southern mixed prairie of Texas. J. Range Manag. 22, 169–174. doi: 10.2307/3896335
Stronach, N. R. H., and McNaughton, S. J. (1989). Grassland fire dynamics in the Serengeti ecosystem, and a potential method of retrospectively estimating fire energy. J. Appl. Ecol. 26, 1025–1033. doi: 10.2307/2403709
Tilston, E. L., Ascough, P. L., Garnett, M. H., and Bird, M. I. (2016). Quantifying charcoal degradation and negative priming of soil organic matter with a radiocarbon-dead tracer. Radiocarbon 58, 905–919. doi: 10.1017/RDC.2016.45
Trompowsky, P. M., Benites, V. M., Madari, B. E., Pimenta, A. S., Hockaday, W. C., and Hatcher, P. G. (2005). Characterization of humic like substances obtained by chemical oxidation of eucalyptus charcoal. Org. Geochem. 36,1480–1489. doi: 10.1016/j.orggeochem.2005.08.001
Tuinstra, F., and Koenig, J. L. (1969). Characterization of graphite fiber surfaces with Raman spectroscopy. J. Chem. Phys. 53, 1126–1130. doi: 10.1063/1.1674108
Waikato Radiocarbon Dating Laboratory (2017). AMS Processing Technical Report. Available online at: https://radiocarbondating.com/__data/assets/pdf_file/0010/387712/Waikato-Radiocarbon-Dating-Laboratory-AMS-Processing-Technical-Report-2017.pdf (accessed September 9, 2019).
Wang, J., Xiong, Z., and Kuzyakov, Y. (2016). Biochar stability in soil: meta-analysis of decomposition and priming effects. GCB Bioenergy 8, 512–523. doi: 10.1111/gcbb.12266
Wang, R., Gibson, C. D., Berry, T. D., Jiang, Y., Bird, J. A., and Filley, T. R. (2017). Photooxidation of pyrogenic organic matter reduces its reactive, labile C pool and the apparent soil oxidative microbial enzyme response. Geoderma 293, 10–18. doi: 10.1016/j.geoderma.2017.01.011
Woo, S. H., Enders, A., and Lehmann, J. (2016). Microbial mineralization of pyrogenic organic matter in different mineral systems. Org. Geochem. 98, 18–26. doi: 10.1016/j.orggeochem.2016.05.006
Yang, Y.-H., and Wang, T. (1997). Fourier transform Raman spectroscopic characterization of humic substances. Vib. Spectr. 14, 105–112. doi: 10.1016/S0924-2031(96)00044-6
Keywords: charcoal, preservation, aromaticity, wood, pyrogenic carbon
Citation: Ascough PL, Brock F, Collinson ME, Painter JD, Lane DW and Bird MI (2020) Chemical Characteristics of Macroscopic Pyrogenic Carbon Following Millennial-Scale Environmental Exposure. Front. Environ. Sci. 7:203. doi: 10.3389/fenvs.2019.00203
Received: 22 September 2019; Accepted: 17 December 2019;
Published: 21 January 2020.
Edited by:
Jean-Thomas Cornelis, Gembloux Agro-Bio Tech, University of Liege, BelgiumReviewed by:
Giovanni Mastrolonardo, University of Florence, ItalyBrieuc Hardy, Walloon Agricultural Research Centre, Belgium
Copyright © 2020 Ascough, Brock, Collinson, Painter, Lane and Bird. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippa L. Ascough, cGhpbGlwcGEuYXNjb3VnaEBnbGEuYWMudWs=