 Nnanake-Abasi O. Offiong1*†‡
Nnanake-Abasi O. Offiong1*†‡ John B. Edet2†‡
John B. Edet2†‡ Solomon E. Shaibu3‡
Solomon E. Shaibu3‡ Nyaknno E. Akan4Edidiong O. Atakpa5‡
Nyaknno E. Akan4Edidiong O. Atakpa5‡ Edmond Sanganyado6*‡Imeh J. Okop7‡Nsikak U. Benson1,8‡
Edmond Sanganyado6*‡Imeh J. Okop7‡Nsikak U. Benson1,8‡ Anthony Okoh9
Anthony Okoh9- 1Department of Chemical Sciences, Topfaith University, Mkpatak, Nigeria
- 2Department of Chemical and Process Engineering, University of Strathclyde, Glasgow, United Kingdom
- 3Department of Chemistry, University of Uyo, Uyo, Nigeria
- 4Department of Chemistry, University of Ibadan, Ibadan, Nigeria
- 5Ocean College, Zhejiang University, Zhoushan, Zhejiang, China
- 6Department of Applied Sciences, Northumbria University, Newcastle-Upon-Tyne, United Kingdom
- 7Department of Chemistry, Akwa Ibom State University, Ikot Akpaden, Nigeria
- 8Institut des Sciences Analytiques, Université Claude Bernard Lyon 1, Villeurbanne, France
- 9SAMRC Microbial Water Quality Monitoring Centre, University of Fort Hare, Alice, South Africa
Metagenomics is the study of genetic information, including the sequences and genomes of microorganisms present in an environment. Since 1998, the full-scale application of this technique to environmental chemistry has brought significant advances in the characterization of the nature and chemical composition/distribution of contaminants present in environmental matrices of contaminated and/or remediated sites. This has been critical in the selection of microorganisms and has contributed significantly to the success of this biological treatment over the years. Metagenomics has gone through different phases of development, which ranges from initial sequencing strategies to next-generation sequencing (NGS), which is a recently developed technology to obtain more robust deoxyribonucleic acid (DNA) profile of microorganisms devoid of chimeric sequences which reduces the quality of metagenomic data. Therefore, the objective of this review is to evaluate the applications of metagenomics in the understanding of environmental dynamics of chemical contaminants during remediation studies. Also, this review presents the relationship between biological characteristics of microorganisms and chemical properties of chemical compounds, which forms the basis of bioremediation and could be useful in developing predictive models that could enhance remediation efficiency. In conclusion, metagenomic techniques have improved the characterisation of chemical contaminants in the environment and provides a correlation for useful prediction of the type of contaminant expected in various environmental matrices.
1 Introduction
The chemical composition of an environmental matrix has been useful in describing the nature of interaction, changes in the natural environment, the health of living organisms as well as certain human-induced phenomena such as global warming. Characterization of the chemical composition of the environment can either be qualitative, requiring the identification of the compounds present, or quantitative, involving the determination of the concentration of the analytes. The conventional method of determining the chemical composition of the environment involves collecting of samples, extracting the target contaminant from the matrix into solution, and measuring the concentration of the analyte of interest using appropriate equipment. It is important to know the type of contaminant and its concentration in the matrices, as the natural environment has become a sink to chemical compounds, which could be traced to industrial effluents, emissions, and other anthropogenic activities. This poses health risks to living organisms in the environment as the natural equilibrium is gradually altered, culminating into various diseases and other stressors. The implications of allowing chemical contaminants to accumulate in the environment are massive. This ranges from far-reaching consequences including the malfunction of sensitive organs in living organisms in the environment, interference with natural processes that depletes oxygen required by organisms for various biological processes, competition with essential nutrients required for growth, or interference with food production within an ecosystem, amongst others. These deleterious impacts are not only felt by plants and animals in the environment, but these are also extended to human beings either through direct ingestion of contaminated water, inhalation of contaminant-laden air, dermal contact with contaminated soil, and/or indirectly by consuming plants and animals that have assimilated these contaminants from the environment.
While knowing the environmental fate, transport and toxicity of chemical contaminants is essential in environmental risk assessment, it is also important to develop efficient methods of removing the contaminants from the environment without causing harm to organisms or the environment itself. The genetic information of microorganisms has been widely used as the fingerprint of specific chemical contaminants in the environment, which serves as a prerequisite for selection as a potential agent for bioremediation among the myriads of biota present in the environment. This may be attributed to their increased abundance and diversity, as well as their role in the biogeochemical cycling of chemical compounds (Hemme et al., 2010).
In this regard, metagenomics, which is a technical term for the assessment of genetic profile of microorganisms as a means of obtaining detailed information of the functions and sequence of microbial genomes present in an environment has offered a reliable means of characterizing different chemical contaminants for the purpose of remediation. It is worth noting that the levels of chemical contaminants in the environment is not fixed, therefore it is expected that there will be fluctuation in the true value of these contaminants with time due to varying degrees of anthropogenic activities and some natural processes that influences the concentration of these contaminants in the environment. Moreover, metagenomic analysis offers a means of assessing the response of ecosystems to these environmental changes by providing detailed adaptive features of microorganisms in the matrix (Hemme et al., 2010). Interestingly, the abundance of certain microorganisms in a contaminated environment has been used to predict specific chemical compounds expected to be the dominant contaminant. This is because certain microbes have been found to consistently congregate within locations where specific chemical contaminants predominate. For instance, a positive correlation has been established between the relative abundance of acidobacteria and concentration of heavy metals such as arsenic, Mercury and copper in the environment (Xu et al., 2022).
Although the distribution of certain microorganisms increases in the presence of some compounds, there are cases where depletion is observed in the presence of other chemical contaminants. A typical example is the negative trend reported to exist between the amount of acidobacteria, planctomycetes, proteobacteria and the level of chromium compounds (Sun et al., 2019). This correlation between the abundance of microorganisms and the concentration of chemical contaminants suggests that the presence of these organisms may have an influence on the properties, composition, and distribution of such contaminants in the ecosystem. The scientific explanation of this relationship is still unclear; therefore, it would be interesting to investigate the correlation existing between the abundance of microorganisms and the concentration of chemical contaminants in the environment. This will be discussed in detail in subsequent sections. Recent studies have indicated that metagenomics has been used to explore the bioremediation of diesel contaminated soil in Canada (Yergeau et al., 2012), degradation of polycyclic aromatic hydrocarbon in sediments in India (Gosai et al., 2022), improving bioremediation via isotope probing (Uhlig et al., 2013) and the use of culturable and non-culturable microbial species (Wani et al., 2022), among other studies as documented in different review articles (Bell et al., 2012; Techtmann and Hazen, 2016; Datta et al., 2020).
In order to provide an overview of this trend as reported in literature, Table 1 summarises the relationship between concentration of chemical contaminants and the number of microorganisms, while Table 2 presents selected metagenomic analyses associated with environmental chemical components. The purpose of these analyses, which range from simple biological functions or microbial distribution to prediction of biochemical mechanisms, is mainly used to explain complex procedures such as bioremediation. For bioremediation to be effective, the environmental conditions must be favourable for the biochemical process and interaction between microorganisms, contaminants, nutrients, and electron acceptors/donors (Sturman et al., 1995). This is because in situ biodegradation can be limited by factors such as bioavailability of the contaminant, the contact time between microorganisms and contaminants as well as bioaccessibility (Gill et al., 2014). This means that biodegradation processes may likely occur at the subsurface of the contaminated environment, but not at the rate that is expected to mitigate risks at the site (Gill et al., 2014).
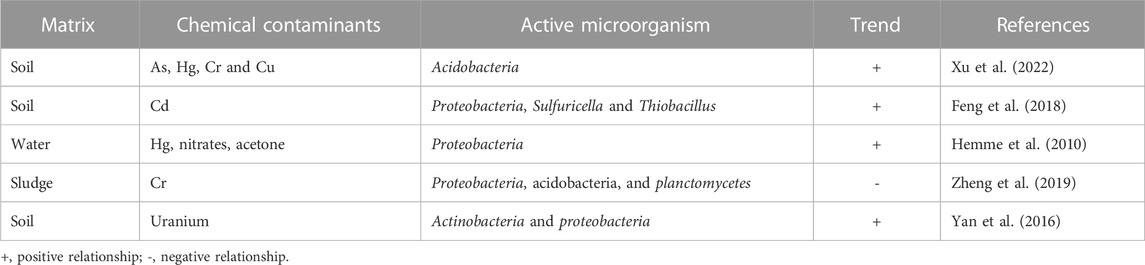
TABLE 1. Microorganisms found in different contaminated environments.
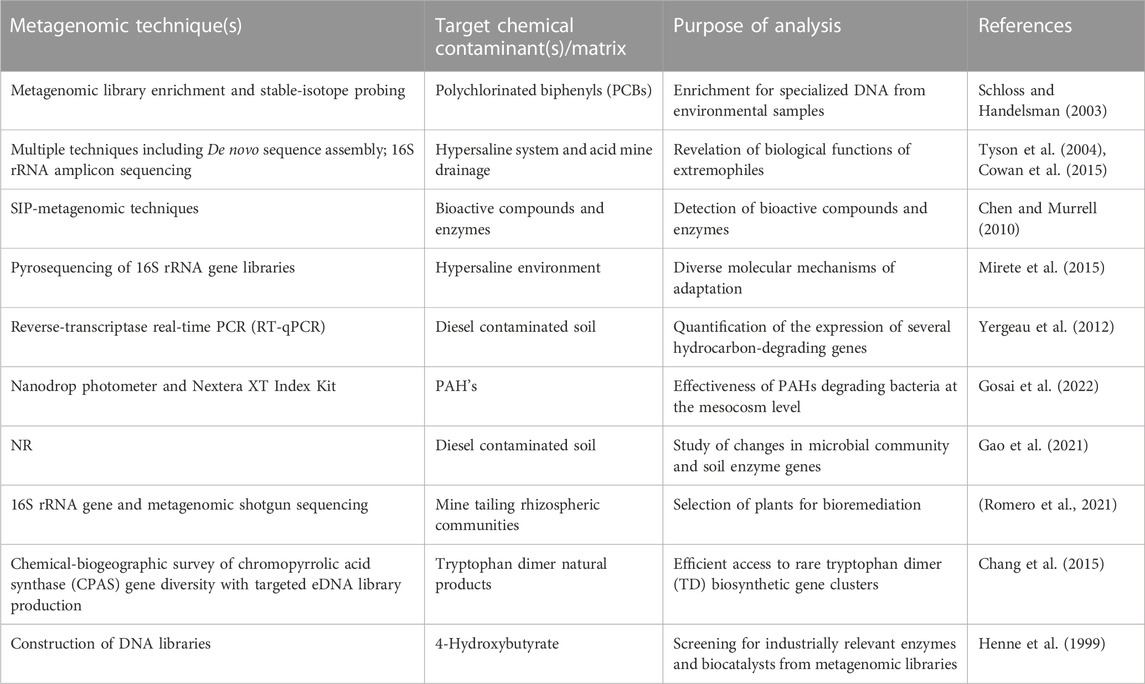
TABLE 2. Selected metagenomic analyses associated with environmental chemical components.
Therefore, the curiosity surrounding this review include the following questions: (I) Why does certain microorganism display high tolerance with increasing concentration of some chemical compounds but are depleted in the presence of other compounds? (II) How does microorganism break down chemical compounds in the environment and how does it affect their metabolic activities? (III) What adaptive features do microorganisms possess that is responsible for increased tolerance to high levels of chemical contaminants in the environment? (IV) Can the genetic sequence of microorganisms be used to predict the presence or degree of tolerance to a particular chemical contaminant in the environment? The answers to these questions could potentially establish a trend between the genetic information of microorganisms and chemical contaminants in the environment, identify structural and metabolic changes occurring during adaptation to increasing levels of chemical contaminants as well as establish a relationship between genetic information of microorganisms and tolerance level to certain chemical compounds which could be used to develop theoretical models to predict remediation efficiency.
2 Synopsis of metagenomics as a predictive tool for environmental chemists
Despite the importance of metagenomic technique to the study of bioremediation of chemical contaminants, the genetic information of microorganisms does not provide a direct quantitative measurement of the level of chemical contaminant in the environment. Instead, it measures the number of microbial communities in effluents or contaminated matrices (Sharma et al., 2021b). This means that the development of a deoxyribonucleic acid (DNA) library for a contaminated environment will require both identification of the type of contaminant present and the concentration using suitable standard chemical procedures recommended for the type of contaminant present or suspected to be present. In this review, we place emphasis on the application of metagenomic techniques and the development of DNA library from different environmental matrices to aid in possible prediction of the likely distribution or presence of chemical contaminants. In addition, the microorganisms selected as agents of bioremediation are known to be tolerant against high concentrations of the target contaminants and can break down toxic organic compounds to less toxic components. Speculations surrounding this behaviour as presented by evolutionary analysis have suggested that lateral gene transfer (LGT) could play a significant role (Hemme et al., 2010), which may be due to the presence of mobile genetic elements such as plasmids, integrons and sequences of insertion (Luo et al., 2017). These provide the basis of utilizing microorganisms for remediation of chemical contaminants from the environment. In addition, studies have indicated that these adaptive features of microorganisms in different media is a necessary requirement for selecting the type of microorganism for bioremediation (Dell’Anno et al., 2023; Gosai et al., 2022; Sharma et al., 2021a).
In order to provide a well-defined perspective for this review and account for uniqueness of information, a survey of review articles related to metagenomic analysis of chemical contaminants in the environment were scouted in addition to research articles published within the past 2 decades. Specific attention was paid to the areas that are connected to bioremediation of the chemical contaminants in the environment. Several review articles published nearly every year from 2004 to 2021 on the subject area were found (Figure 1). This search was necessary to enable identification of the areas that have not been covered in these reviews as well as linking the different perspectives of metagenomic analysis of environmental samples discussed in the review articles. This is to provide a holistic view of application of the technique in environmental analysis. In Figure 1, it can be observed that the number of review articles sampled from a population of 71 publications in the field was observed to increase in recent years (2015–2021) compared to 2004–2008, which is an indication of increasing interest in this area of research. The publications were obtained through searches conducted via Google Scholar and Scopus databases using key words such as “metagenomics”; “environmental remediation”, “environmental analysis”, and “environmental samples”. An overview of the microorganisms found in different contaminated environments are presented in Table 1 (trends indicated are based on results reported and not on statistical significance). However, detailed relationship between the genetic information of microorganisms and levels of chemical compounds in the environment are still lacking. The review articles presented in Figure 1 consists of different areas of metagenomics, though there are few overlapping concepts. However, each review article may be regarded as a subset of the entire field of metagenomics which requires connection to estimate the depth of research that has been carried out in this field as well as assessing the gaps that still needs further study, which is the focus of this review.
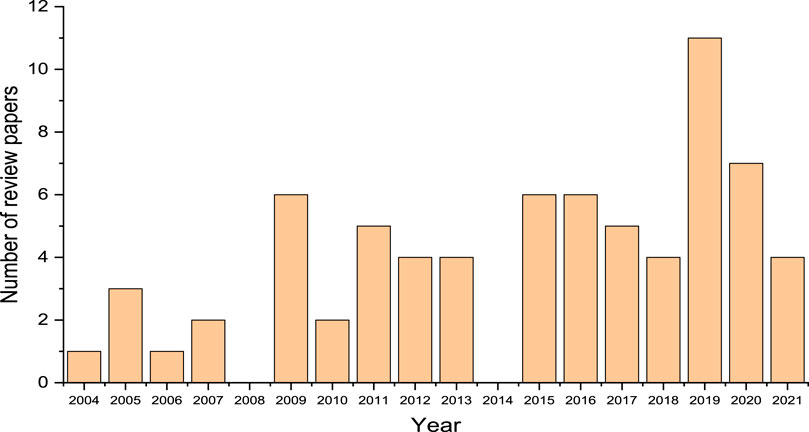
FIGURE 1. Plot of number of review papers on metagenomic analysis of environmental samples versus year of publication (Total number of review papers sampled is 71).
Therefore, the main aim of this work is to assess the progress in developing DNA library for different chemical contaminants for the past 2 decades and identify the key challenges that affect research in this field as well as areas that still require in-depth study. This will help to establish the correlation that exist between microorganisms and chemical compounds which account for their use in bioremediation, track the progress in different matrices for the past 2 decades, as well as the challenges and research needs that limits scientific advancement in this area of research. This could be a positive step towards optimization of the process of bioremediation as well as useful for researchers who might be interested in developing predictive models that could enhance remediation of chemical contaminants from the environment.
3 State of the art
The changes and developments in the collection, storage and metagenomic analysis of environmental samples are presented and discussed in this section. The reliability and accuracy of the results obtained from analysis of environmental samples depend significantly on the sampling plan, storage conditions, the time between collection of samples and analysis, and the analytical method used. If any of these steps are compromised for any reason, the accuracy of the result may likely be affected resulting in erroneous conclusions. Therefore, a survey of the sampling plan, storage and analysis for different environmental matrices are presented and discussed.
3.1 Environmental sample collection and storage for metagenomic analysis
Sample collection is an important step in environmental analyses as it forms the bedrock on which further analysis is based and it is the foundation that determines the reliability of the outcome. Therefore, it is important to obtain samples representative of the location under study. A definite sampling design tailored to meet the requirements for the environmental matrix is expected to be in place prior to visiting the site. For soil matrix, the collection of representative samples for metagenomic analysis has been identified to be quite challenging compared to other matrices due to diversity of species present as well as the size of microbial communities (Daniel, 2005). This may be categorized into functional, taxonomic and phylogenetic diversities, which complicates the biological and physicochemical properties of soil environments leading to spatial heterogeneity of the matrix. Variation in the number of soil microorganisms depend on type of soil, porosity, pH, and moisture content. In order to successfully determine the degree of microbial diversity as well as the genetic variation in soil samples, a large soil surveys is recommended (Daniel, 2005). This is important in constructing reliable DNA libraries characteristic of the sample location. The first step towards constructing a DNA library is to collect samples from the study location. In most studies, researchers prefer collecting a small amount of composite soil samples of about 500 g or less from a depth of 10–20 cm over an extended area due to large microbial populations (Henne et al., 1999; Daniel, 2005). This is used to evaluate the composite genetic pool of the sample location which provides information on the overall DNA composition of the site.
Furthermore, it is highly advisable to carry out microbial analysis immediately after collection from source. Also, the transport conditions from the source to the place of analysis should be properly assessed to prevent external interference or change in properties and composition. Although the ideal case would be immediate analysis after sample collection, there may be situations where this may not be possible due to location of the sampling site or other logistics. In such situations, it is important that the time between sample collection and analysis be kept to a minimum and the storage conditions should prevent contamination or proliferation of microorganisms. This is because the microbial load of samples stored over a period may not be representative of the samples in the field. Besides, it is necessary that physicochemical as well as biological characteristics of samples be determined as this influences the diversity of microorganisms as well as its adherence to soil. Storage of samples under unconducive conditions could lead to a change in some of these biological and chemical properties of the samples. The storage conditions of samples collected for metagenomic analysis as reported in the literature are summarized in Table 3, while the factors that affect the distribution of microorganisms in different matrices are shown in Figure 2. Different sample containers have been used to collect samples from different matrices; for example, plastic containers are mostly used for soils and sediments (Feng et al., 2018), filters for air (Aalismail et al., 2019) and sterile Nalgene carboy for water (Brumfield et al., 2021). Although these containers only serve as holders to aid in transporting the samples from the source to the place of analysis, nevertheless, they are expected to be free from contaminants and must be sterilized prior to use. In general, the parameters selected during sample collection for metagenomic analysis usually depend on the experimental purpose and objective of the study (Table 3).

TABLE 3. Key parameters during sample collection for metagenomic analysis.
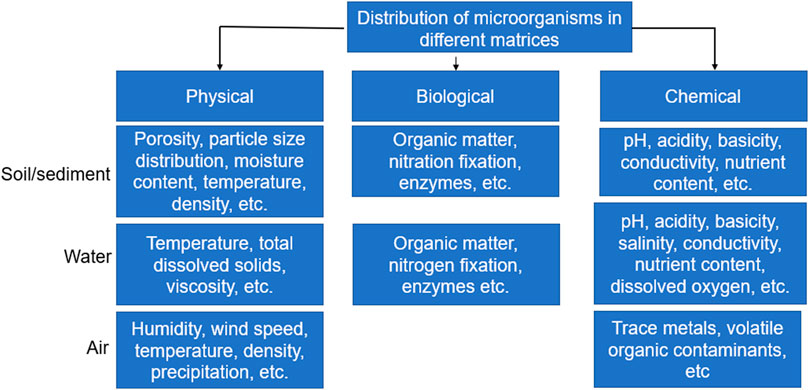
FIGURE 2. Factors affecting the distribution of microorganisms in different environments.
Composition of microorganisms in air samples depend on location, time as well as animal and anthropogenic activities. Unlike microorganisms in soil and water matrices which have limited mobility, microorganisms in air can spread more rapidly covering a larger distance within a short period. Some studies have indicated the possibility of microorganisms in air spreading between different continents (Be et al., 2015; Aalismail et al., 2019), which makes monitoring of air quality a priority. Besides, due to the unrestricted mobility of microorganisms in air, it may be difficult to prove that outdoor air samples collected at a particular sample point are strictly representative of that location. At best, an outdoor sampling plan can only provide an estimate of the microorganisms in the air over a particular geographical area. Therefore, while sampling air for metagenomic analysis, it is preferable (recommended) to split large sample areas into components with reasonable distance between each sample area. Then, sterile air samplers are used to collect samples over the entire area of interest.
In collecting air samples, some researchers prefer using specialized air samplers containing different compartments each playing specific roles (Yooseph et al., 2013). In another study, impingers connected to aspirators were used to collect outdoor air samples for metagenomic analysis (Cha et al., 2017). Collection of indoor air samples may not be as demanding and complicated as outdoor samples as this may be collected after closing all outlets. However, some researchers suggested that it might be reasonable to obtain large volume of samples from air handling units of some modern ventilation systems instead of using special air samplers for collection of indoor samples (Tringe et al., 2008). This strategy depends on the objective of the research. If the aim is to determine the indoor air quality exclusively, then it might not be advisable to sample air from ventilation systems as there is constant exchange of indoor and outdoor air in most ventilation systems. Besides, it is likely that the ventilation system itself might serve as host for microorganisms which might be different from the microbial load of the target matrix.
3.2 Metagenomic analysis of environmental samples
The steps involved in obtaining DNA information from environmental samples include extraction, isolation, purification, and sequencing. This is then used to construct DNA libraries of the environmental matrix. Although most publications on metagenomic analysis of environmental samples have not really provided detailed information on how analyses were carried out. Therefore, the information provided in this review is an overview of the standard procedures available in the literature and few modifications that have been reported by researchers working in the field of metagenomics.
3.2.1 Extraction, isolation, and purification of DNA from environmental samples
Direct extraction and isolation of DNA from environmental samples has taken precedence over the traditional culturing techniques as a means of overcoming the limitation posed by some microorganisms that has been tagged as “unculturable”; though this term has been considered inappropriate as it is possible that no suitable culturing technique has been discovered for such organisms yet (Lewis et al., 2010). Conventional methods of obtaining bacteria from contaminated soils such as polymerase chain reaction and denaturing gradient gel electrophoresis techniques were initially recognized as reliable means of detecting gene sequences and genetically engineered microorganisms in the environment (Porteous et al., 1994). However, recent studies have reported that these techniques are insufficient to provide detailed information of soil microorganisms (Yan et al., 2016). Therefore, new sequencing methods (next-generation sequencing) have been developed to obtain more detailed information of microorganisms in contaminated matrices. This breakthrough obviously comes with reduction in the cost of DNA sequence analysis (Luo et al., 2023). This also involves decoding the functional role of microbial communities in the detoxification, degradation, and transformation of toxic chemical compounds (Sharma et al., 2021a). The steps involved in creating a DNA library from environmental samples are shown in Figure 3.
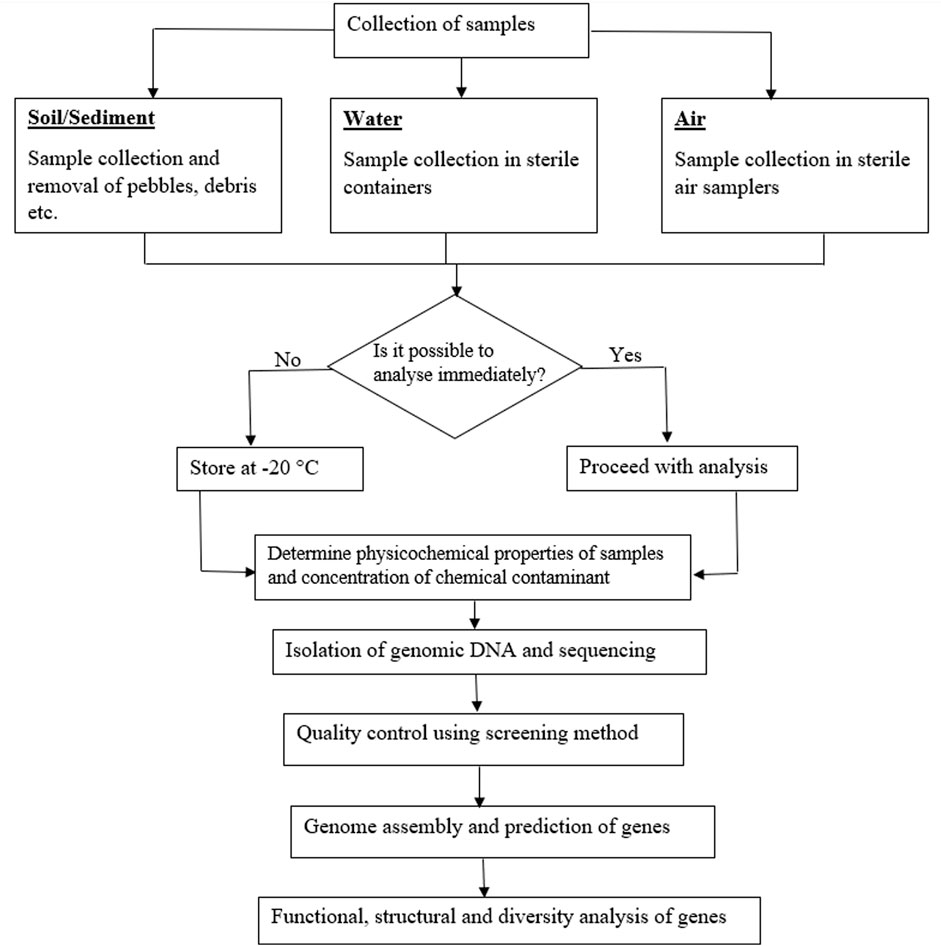
FIGURE 3. Flow chart showing the steps involved in metagenomic analysis of environmental samples.
Genetic information of microorganisms in contaminated environment can either be obtained from analysis of 16S rRNA amplified PCR fragments or all DNA extracted from the environmental sample (Sharma et al., 2021b). The extraction process requires the use of microbial extraction genomic kits after initial high-speed vortex at about 10,000 rpm to release the microbial cells from the bulk matrix, and further treatment of the residue with lysozyme and proteinase K (Ding et al., 2019). Polymerase chain reaction (PCR) may be used to amplify the DNA prior to creating a library for the matrix. Analysis of the 16S rRNA as well as quality control may be carried out using special software packages such as QIIME (Quantitative Insights Into Microbial Ecology), USEARCH and quality control toolkits (Ding et al., 2019).
Analysis of genomic data can be quite challenging despite significant improvements in technology. This is due to significant diversity of microbial species within the environment, which has negative implication on accurate representation of reads, or even greater complicacies associated with analysis of low-quality reads. Some researchers prefer analysing a broader spectrum of contiguous genomic fragments usually contracted to contigs. However, this increases the heterogeneity, which may culminate in generation of chimeric contigs. Therefore, there is need to provide an explicit workflow that takes into consideration the diversity of species as well as conditions that suggest consistency between information obtained from reads and contigs. Information obtained from samples are usually compared to a reference database to trace the origin of each sequence. In the literature, MEGAN, a microbiome analysis software that uses Last Common Ancestor (LCA) algorithm have been employed to assign sequences to a specific taxon (Haro-Moreno et al., 2018). This is then subjected to further analysis to evaluate the quality of the taxa as well as statistical significance of the results.
4 Application of metagenomic techniques for remediation of organic and trace metals contaminants
The remediation of different chemical contaminants in the environment will be discussed separately in two sections. This will consist of bioremediation of trace metals and organic compounds (polycyclic aromatic hydrocarbons, emerging organic contaminants, and persistent organic pollutants). This is aimed at providing insight to the mechanism of remediation as the pathways for these group of contaminants is likely to be different as well as the genetic information of microorganisms responsible for this activity. Metagenomic analysis allows a powerful elucidation of biotransformation and interactions of chemical components in environmental matrices (Handelsmanl et al., 1998; Offiong et al., 2022). The microbial community analysis and biological functions enables highly precise and efficient optimization of protocols to achieve remediation goals.
4.1 Bioremediation of inorganic and organometallic compounds
Inorganic compounds, especially trace metals are highly persistent in the environment and can be solubilized under acidic conditions which enhances rapid uptake of chemical contaminants by living organisms. As most inorganic compounds are non-biodegradable, the most viable means of remediating these contaminants from the environment involves change in oxidation state of the metal from a more toxic and soluble form to a less soluble and less toxic state. For instance, chromium exists predominantly in the hexavalent (Cr6+) and trivalent states (Cr3+). The hexavalent form is highly toxic, soluble, carcinogenic, it is predominantly found in natural aquifers and has been noted by USEPA to be one of the chemicals with the highest threat to human beings, while the trivalent form is less soluble and less toxic (Marsh and McInerney, 2001; Cheung and Gu, 2007). A summary of selected research projects on remediation of metallic and organometallic contaminated sites with aspects of metagenomics are presented in Table 4.
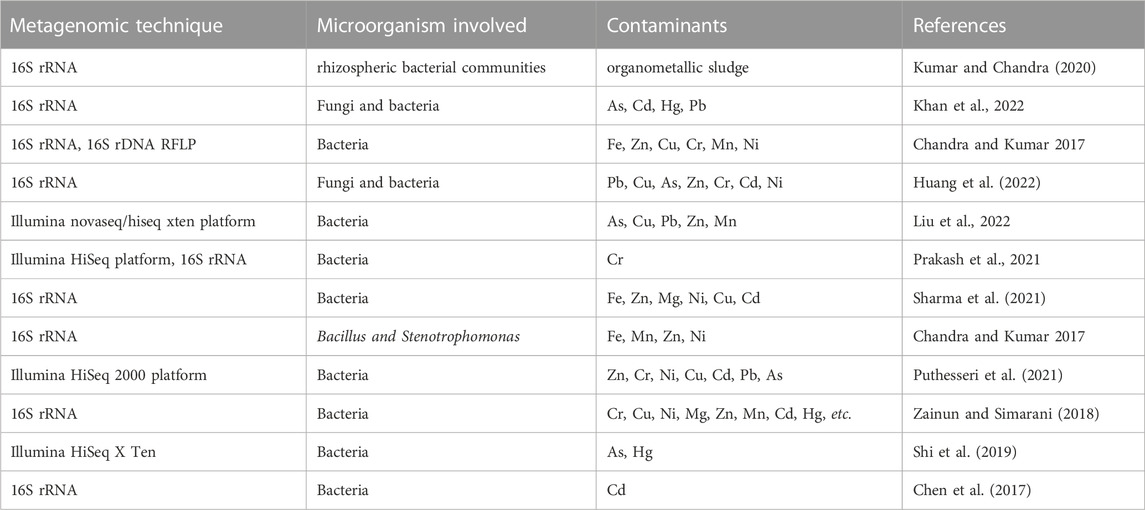
TABLE 4. Metagenomic study of heavy metals and organometallic contaminated sites.
Although some metal ions are useful for growth and metabolism of microorganisms, excess metal ions beyond a certain threshold are detrimental to microorganisms. Therefore, these organisms have an in-built biological system that triggers certain response when there is a change in the concentration of these contaminants in the environment. One of such response is the activation of metalloregulatory proteins, which serves as a metal sensing agent that controls metal homeostasis in microorganisms. These proteins serve as ligands that bind to the metal, thus, initiating the regulatory process. The efficacy as well as tendency of this binding process is different across metals as shown by the Irving-William series, Cu (I) > Zn (II) > Ni (II) > Co (II) > Fe (II) > Mn (II) > Mg (II) > Ca (II) (Barnett et al., 2012). Therefore, it is suspected that the reason why certain microorganisms thrive under increased concentration of some chemical contaminants but are decreased or extinct in the presence of other contaminants may likely be due to the differences in the binding potential of these ligands to different metals. This is because it has been reported that failure to trap these excess metal ions will culminate in termination of growth and ultimate death of microorganisms under these conditions (Chandrangsu et al., 2017).
Cheung and Gu (2007) have documented the conversion of hexavalent chromium to trivalent chromium in both aerated and anoxic conditions by microorganisms as described in Figure 4 (Cheung and Gu, 2007). A summary of this report is that under aerobic conditions, the resistance to hexavalent chromium ions by Pseudomonas aeruginosa results in a decreased uptake and enhanced efflux from the cell membrane of the microorganism, this results in formation of unstable intermediates before reaching the stable trivalent state, while anaerobic pathway involves direct transition from hexavalent to trivalent state (Cheung and Gu, 2007). Interestingly, some microorganisms especially the acidophiles such as pyrococcus, acidianus, sulfurisphaera among others have adaptive features such as the release of ATP synthase, chaperones, DNA repair proteins, etc., that helps in maintaining internal chemical balance especially changes in pH which would interrupt its metabolic activities (Tripathi et al., 2021).
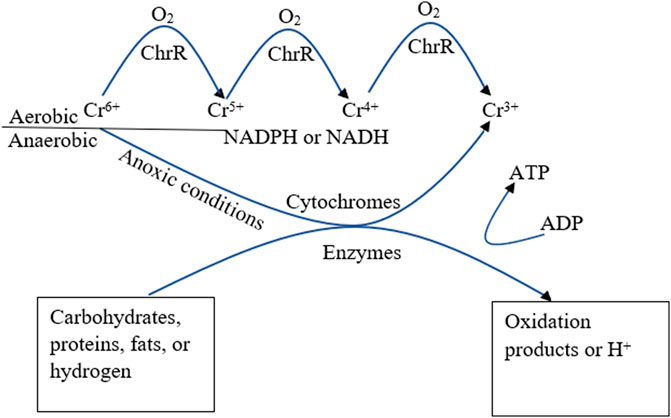
FIGURE 4. Mechanism of conversion from Cr6+ to Cr3+ under aerated and anoxic conditions and enzymatic role (Cheung and Gu, 2007).
The mechanism of remediation of heavy metals using bacteria and fungi are shown in Figures 5, 6 respectively. Three important steps have been reported in the bioremediation of heavy metals using bacteria. This includes: Physical biosorption based on weak attractive forces (Van der Waal’s forces), the formation of complex at the cell surface following the interaction between heavy metal ion and active groups of the bacteria as well as ion exchange of chemical components of the cell wall with polysaccharides (de Alernca et al., 2017). This results in extracellular sequestration and efflux of the peptide bound ions from the bacterial cells. This transformation of heavy metal and subsequent efflux from the cell has been reported to be the reason for increased resistance of certain bacteria against toxic heavy metal at different contaminated sites (Verma and Kuila, 2019).
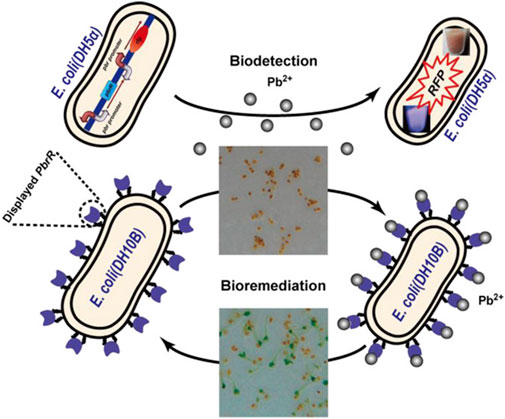
FIGURE 5. Mechanism of remediation of heavy metals using bacteria (Wei et al., 2014).
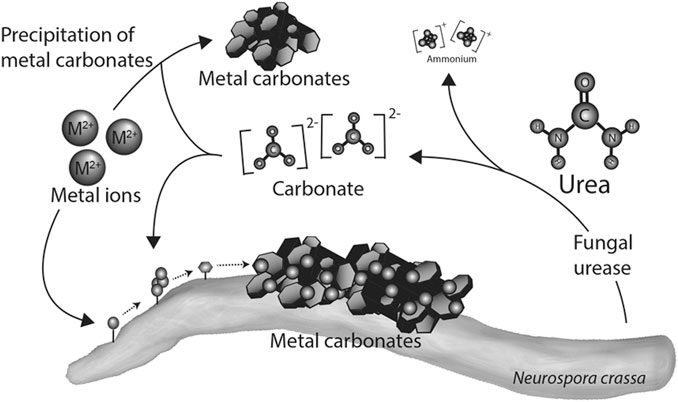
FIGURE 6. Mechanism of heavy metal removal using fungi (Li et al., 2014).
For heavy metal remediation using fungi, the adsorption of the metal ion to the cell wall of the fungi has been reported as the first step in the remediation process (Riaz et al., 2021). This is facilitated by the presence of functional groups such as carboxyl, hydroxyl, carbonyl, nitrile groups among others on the surface of the fungus which adsorbs with heavy metal (Chen et al., 2022). This might result in a change in morphology of the mycelial surface which restricts further penetration of metal ions (Chen et al., 2022). It is also possible that there could be intracellular bioaccumulation of metal ions rather than adsorption to the bacterial cell wall as reported by Sharma et al. (2021).
4.2 Bioremediation of organic compounds
Organic contaminants such as polycyclic aromatic hydrocarbons (PAHs), persistent organic pollutants (POPs) and emerging organic contaminants (EOCs) are the main groups of organic contaminants that has been the focus of environmental scientists over the years. The ability of microorganisms to metabolize aliphatic and aromatic organic compounds for the purpose of accessing carbon or obtaining energy, thereby converting these contaminants to carbon dioxide, water and biomass has been the basis of bioremediation of organic compounds (Bouwer and Alexander, 1993). This process has been proven to be effective and reliable, however, the heterogeneity of environmental matrices like soil implies that changes in physical and chemical conditions can make the matrix unsuitable for biodegradation of these contaminants. Therefore, pretreatment and conditioning measures are sometimes employed to improve remediation efficiency. This include bio-stimulation, where nutrients or fertilizer are added to the soil as well as bioaugmentation, which involves the addition of microorganism-laden materials such as sludge or compost to boost the remediation process (Vogel, 1996). The electrokinetics of these supplementary remediation processes has been well revised elsewhere (Gill et al., 2014). Furthermore, it has been reported that 16S rRNA gene analysis of soil contaminated with diesel and other fuels, which has been conditioned prior to remediation confirmed the presence of alphaproteobacterial, actinobacteria, firmicutes, betaproteobacteria and deltaproteobacteria, which significantly improves remediation of organic contaminants from the environment.
In a recent study, biostimulation was used for the remediation of a crude oil contaminated site via biodegradation of total petroleum hydrocarbons (TPHs) and PAHs by a consortium of bacteria observed using metagenomic analysis (Rahmeh et al., 2021). The diversity investigation revealed a group of microbes belonging to a diverse genus could be employed for remediation/restoration purposes especially in temperate regions. Similarly, rhizoremediation was also reviewed for the degradation of PAH and closely monitored by different omic techniques (metagenomics, metatranscriptomics, metaproteomics and genomics) to assess the role played by the microbiome in the remediation process (Kotoky et al., 2018). Proposition and description of a probable genetic modification of the rhizosphere microbiome for enhanced degradation of PAH has been highlighted in the literature using these advanced genetic technologies (Kotoky et al., 2018). At different environmental components and various stages of an oil supply chain, monitoring the degradation of PAHs is critical due to anthropogenic and natural interferences.
Therefore, it is on this premise that the genetic, metabolic pathways and identity of biodegraders of PAHs were studied to gain insights into the different optimization techniques for bioremediation as well as prevention of crude oil deterioration by (Hidalgo et al., 2020) using metagenomics. However, some biotic and abiotic factors were also opined to affect the efficiency of the bioremediation process (Hidalgo et al., 2020). Elsewhere, Actinomycetales and Rhizobiales were found to be present in a crude oil laden soil, but the former was the dominant microorganism while Actinobacteria known for their potent degradation potentials of PAH’s were similarly identified (Scow and Hicks, 2005). Though not often reported, Weathered diesel oil (WDO) with poor biodegradation and volatility features is a pollution threat when released inadvertently into the environment and enhanced bioremediation of WDO via biopile processes was investigated with metagenomic tools. High TPH removal of more than 70% was removed while heavy constituents in the WDO could be removed by Sphingomonas but Pseudomonas was responsible for lower fractions (Liu et al., 2021). The valuable information revealed during the bioremediation of organic compounds via metagenomic studies is vital and indispensable elements for optimization and efficient ecosystem restoration purposes.
The bioremediation of target organic compounds from aquifers and other aquatic sources also depends on the biological, physical and chemical properties of the aquatic matrix. In fact, a strong correlation between the abundance of microorganisms and these properties have been reported (Scow and Hicks, 2005; Santini et al., 2015). Several researchers have agreed that using multivariate statistics or artificial neural networks to link geochemical properties of an environmental matrix with microbial community data could be very vital in enhancing bioremediation of organic and other chemical contaminants from the environment (Feris et al., 2004; Palumbo et al., 2004; Scow and Hicks, 2005). In addition, the need to provide evidence for the remediation of these contaminants using microorganisms have resulted in evolution of techniques to study the effect of cell properties such as electrophoretic mobility, hydrophobic and electrostatic interactions on rates of attachment to particles as well as tracking inoculated populations following injection into aquifers (Scow and Hicks, 2005). A compendium of metagenomic-assisted bioremediation studies is presented in Table 5. As an example, for biodegradation of benzene, toluene, ethylbenzene and xylene (BTEX) in soil contaminated with petroleum hydrocarbons, it was demonstrated that during the central metabolism, the bacterial consortium used contained Coding DNA sequences (CDSs) required for ortho-cleavage of catechol such as catechol 1, 2-dioxygenase (catA), muconate cycloisomerase (catB), muconolactone D-isomerase (catC), and 3-oxoadipate enol-lactonases (pcaD and pcaL) as shown in Figure 7.
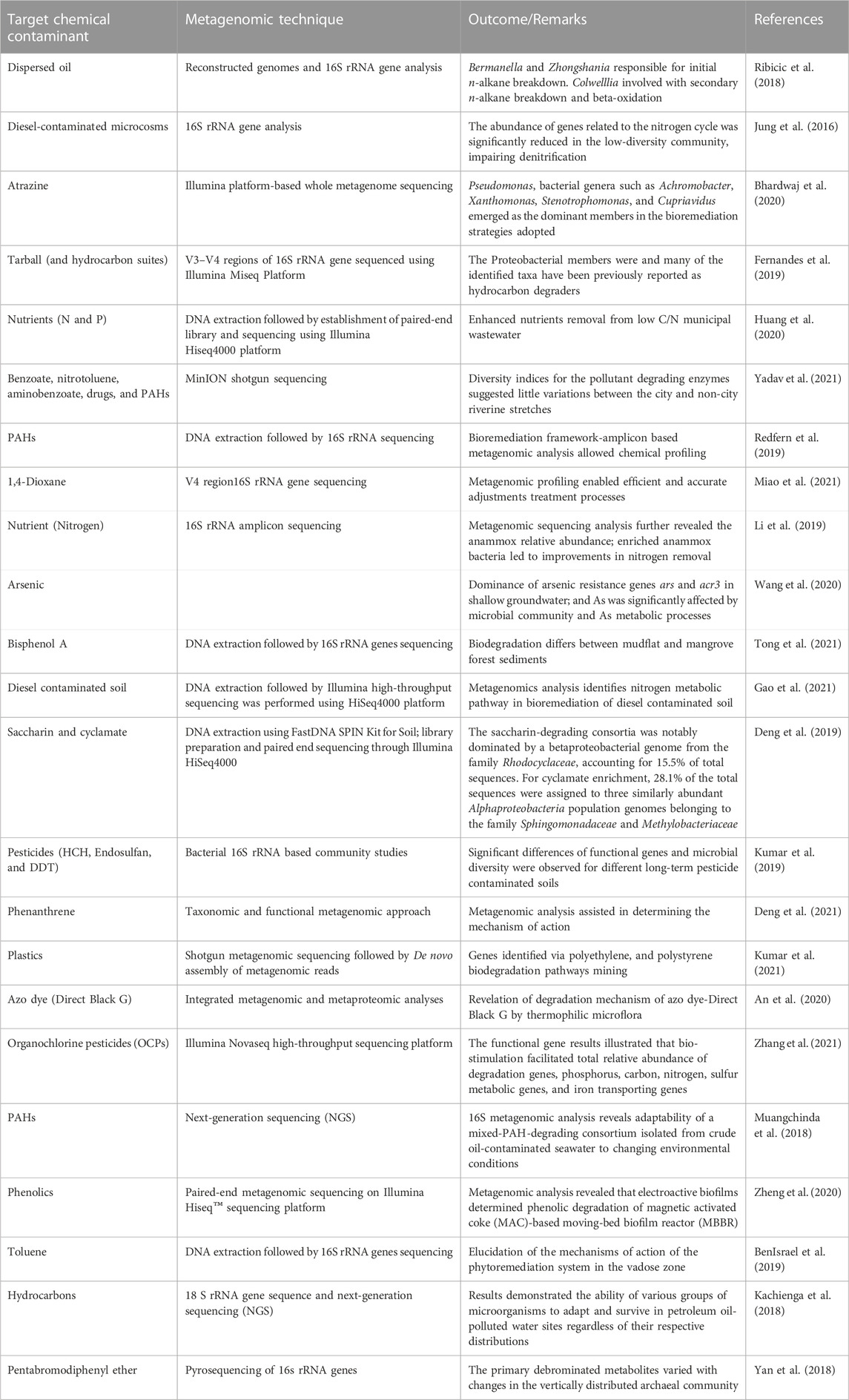
TABLE 5. Metagenomic-assisted bioremediation studies.
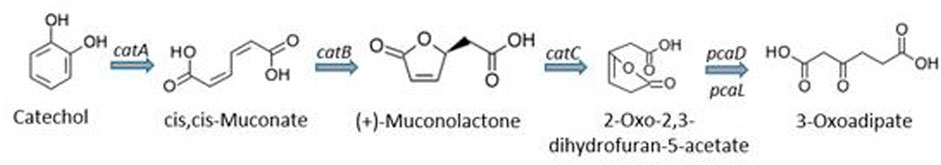
FIGURE 7. Ortho-cleavage of catechol and subsequent degradation reactions (Eze, 2021).
5 Research challenges and way forward
Despite improvement in technology in recent years, there are still challenges that need to be addressed in order to improve the quality of data obtained from metagenomic analysis of environmental samples. One important challenge is how to obtain representative samples of microbial communities, this is because of increased diversity of environmental samples particularly soil, sediments, and aquatic matrices. One recommendation to solve this problem is to introduce push-pull tests to the analysis of microbial populations (Scow and Hicks, 2005). This involves injection of a controlled test solution into an aquifer, then the test solution is subsequently extracted and analysed for chemical, physical and biological characteristics of the matrix. Even though this will give information on the properties of the target matrix, which may serve as a guide to sample collection, it may not be useful for large sample areas with increased diversity of species and variability in properties. Therefore, it might be better to split large sample areas into different components and obtain homogenous samples within areas that have similar properties.
Another major challenge is the collection of samples in aquatic and sediment matrices without interfering with the composition of both matrices. Even though the sediment bed is beneath the water table; the properties of these matrices are likely to be different. It is expected that the sediment should have large variation in microbial community compared to the water above it. Care must be taken when collecting sediment samples to avoid interference with the overhead water matrix. Some researchers have suggested suspension of a solid phase support matrices such as bio-sep beads that could help in collecting actively growing surface associated bacteria in aquatic matrices (Scow and Hicks, 2005). If this device should be used to collect sediment samples, then, it should be adequately shielded from the water layer till it reaches the sediment bed.
Furthermore, technical challenges arising during analysis include variable contact of microorganisms with the target contaminant as well as bioavailability of the chemical compound of interest. This may arise due to certain factors like transport phenomena, porosity of the matrix and level of interaction of the contaminant with organic matter or other components in the matrix. To address this issue, the incorporation of electrokinetics to the study of biodegradation of chemical contaminants in order to monitor transport phenomena, porosity, bioavailability of contaminants and microbial communities which may provide useful information during sample collection were suggested (Gill et al., 2014). Besides, gene assembly and prediction are another major challenge especially during modelling. Also, it was stressed that the reconstruction phase of the whole genome from the sequence reads using fragment assemblers is almost impossible to achieve for multiple microbial environments due to complexity of the target species and short length of sequencing reads (Wooley and Ye, 2010), which explains why metagenomic sequences are usually analysed as a collection of short reads.
Furthermore, the presence of chimera during 16S rRNA analysis could affect estimation of species diversity if not adequately filtered. Chimeras are mainly formed from two different parent sequences and are commonly found to appear during 16S rRNA analysis. Development of improved protocols such as emulsion polymerase chain reaction (ePCR), which allows amplification of DNA in samples has been useful in preventing the formation of chimeras and certain artifacts between similar DNA sequences. Also, computational tools such as Ballerophone, Pintail and Mallard have played a significant role in removing chimeric data, though improvement in the function of these tools have been suggested to allow efficient functioning with large 16S rRNA datasets derived from barcoded pyrosequencing (Wooley and Ye, 2010).
6 Conclusion
The collection of genetic information from microorganisms domiciled in a contaminated environment has been useful in predicting the type of contaminant expected in the matrix, which is usually validated by carrying out a more detailed chemical analysis. The diversity of species across different environmental matrices has necessitated a robust sampling plan and analysis to ensure high quality data representative of the sample location under study. Metagenomic analyses discussed in this review are targeted at assessing the process of bioremediation, which employs the functions of microorganisms in remediating chemical contaminants from the environment. This has been found to provide useful information that correlates the biological characteristics of microorganisms to environmental chemical properties. In addition, it has been observed that the interactions of enzymes at the cell membrane of microorganisms with the chemical contaminant in the environment is one of the determining factors of the efficacy of bioremediation, in addition to accessibility of the microbial cell to the target contaminant, which is determined by solubility of the contaminant, and the porosity of the matrix. Also, variation in the binding potential of the proteins in microbial cells with the chemical contaminant has been identified as the reason for the differences in the abundance of microorganism in the presence of different chemical species.
The collection of representative samples within a contaminated environment, the choice of sample containers and storage conditions to prevent proliferation of microorganisms, and the time of analysis have all been identified as key factors that affect the quality of metagenomic data at the initial stages. Also, the choice of sample container as well as transport conditions of the collected samples from the field to the point of analysis should be taken into consideration as well. This is done to ensure that the samples collected for metagenomic analysis in the laboratory reflect the true condition of the field. The choice of sequencing technique also has an influence on the quality of DNA information contained in each matrix’s library. Even though analysis of metagenomic sequences using 16S rRNA has been useful in providing in-depth DNA information of environmental samples, the high possibility of generating chimeras, which affects the quality of metagenomic data, has been identified as a key challenge, inspiring the development of more sophisticated computational tools that filter the data as a scientific response to ensure improved results. Despite improvement in technology, some challenges have been identified, including collecting representative samples, especially in a matrix that is characterized by increased diversity of species, and transporting of target chemical species within the matrix. This calls for a definite sampling plan tailored to meet the needs of the matrix under investigation. Also, incorporating electrokinetics into the entire metagenomics process has been suggested to improve the quality of samples collected from the environment.
Furthermore, the efficacy of the remediation process depends on the chemistry of the contaminants as well as the biological characteristics of the microorganisms. Since this is influenced by the interaction between the proteins in the cell membrane of microorganisms, which serves as ligands that bind to the contaminant, a detailed study of these parameters could provide useful information that can ensure control over the entire remediation process. Furthermore, microorganisms could be cultured or even modified in the laboratory and introduced to certain contaminated matrices that may not naturally have microorganisms that could break down these contaminants or convert it to less toxic component. Also, the chemical properties of these contaminants and the cell properties of microorganisms may likely be useful in building biochemical models that could predict remediation efficiency of different microorganisms as initial assessment prior to deployment of these organisms to the field.
Author contributions
Conceptualization, N-AO and NB; methodology, N-AO, NB, and JE; investigation, N-AO, JE, and SS; resources, N-AO; data interpretation, N-AO, JE, SS, NA, EA, ES, and IO; writing—original draft preparation, N-AO and JE; writing—review and editing, N-AO, JE, SS, NA, EA, ES, IO, and NB; supervision, N-AO, NB, ES, and AO. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
The authors are grateful to the South Africa Medical Research Council for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aalismail, N. A., Ngugi, D. K., Díaz-Rúa, R., Alam, I., Cusack, M., and Duarte, C. M. (2019). Functional metagenomic analysis of dust-associated microbiomes above the Red Sea. Sci. Rep. 9, 1–12. doi:10.1038/s41598-019-50194-0
Acharya, K., Blackburn, A., Mohammed, J., Haile, A. T., Hiruy, A. M., and Werner, D. (2020). Metagenomic water quality monitoring with a portable laboratory. Water Res. 184, 116112. doi:10.1016/j.watres.2020.116112
An, X., Chen, Y., Chen, G., Feng, L., and Zhang, Q. (2020). Integrated metagenomic and metaproteomic analyses reveal potential degradation mechanism of azo dye-Direct Black G by thermophilic microflora. Ecotoxicol. Environ. Saf. 196, 110557. doi:10.1016/j.ecoenv.2020.110557
Barnett, J. P., Scanlan, D. J., and Blindauer, C. A. (2012). Protein fractionation and detection for metalloproteomics: Challenges and approaches. Anal. Bioanal. Chem. 402, 3311–3322. doi:10.1007/s00216-012-5743-y
Be, N. A., Thissen, J. B., Fofanov, V. Y., Allen, J. E., Rojas, M., Golovko, G., et al. (2015). Metagenomic analysis of the airborne environment in urban spaces. Microb. Ecol. 69, 346–355. doi:10.1007/s00248-014-0517-z
Bell, T. H., Greer, C. W., and Yergeau, E. (2012). “Metagenomics potential for bioremediation,” in Encyclopedia of metagenomics (Berlin, Germany: Springer), 1–11.
BenIsrael, M., Wanner, P., Aravena, R., Parker, B. L., Haack, E. A., Tsao, D. T., et al. (2019). Toluene biodegradation in the vadose zone of a poplar phytoremediation system identified using metagenomics and toluene-specific stable carbon isotope analysis. Int. J. Phytoremediation 21, 60–69. doi:10.1080/15226514.2018.1523873
Bhardwaj, P., Singh, K. R., Jadeja, N. B., Phale, P. S., and Kapley, A. (2020). Atrazine bioremediation and its influence on soil microbial diversity by metagenomics analysis. Indian J. Microbiol. 60, 388–391. doi:10.1007/s12088-020-00877-4
Bouwer, E. J., and Alexander, J. B. Z. (1993). Bioremediation of organic compounds ? Putting microbial metabolism to work. Trends Biotechnol. 11, 360–367. doi:10.1016/0167-7799(93)90159-7
Brumfield, K. D., Cotruvo, J. A., Shanks, O. C., Sivaganesan, M., Hey, J., Hasan, N. A., et al. (2021). Metagenomic sequencing and quantitative real-time PCR for fecal pollution assessment in an urban watershed. Front. Water 3, 626849. doi:10.3389/frwa.2021.626849
Cha, S., Srinivasan, S., Jang, J. H., Lee, D., Lim, S., Kim, K. S., et al. (2017). Metagenomic analysis of airborne bacterial community and diversity in seoul, Korea, during December 2014, Asian dust event. PLoS One 12, 01706933–e170712. doi:10.1371/journal.pone.0170693
Chandra, R., and Kumar, V. (2017b). Detection of androgenic-mutagenic compounds and potential autochthonous bacterial communities during in situ bioremediation of post-methanated distillery sludge. Front. Microbiol. 8, 887. doi:10.3389/fmicb.2017.00887
Chandra, R., and Kumar, V. (2017a). Detection of Bacillus and Stenotrophomonas species growing in an organic acid and endocrine-disrupting chemical-rich environment of distillery spent wash and its phytotoxicity. Environ. Monit. Assess. 189, 26. doi:10.1007/s10661-016-5746-9
Chandrangsu, P., Rensing, C., and Helmann, J. D. (2017). Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 15, 338–350. doi:10.1038/nrmicro.2017.15
Chang, F.-Y., Ternei, M. A., Calle, P. Y., and Brady, S. F. (2015). Targeted metagenomics: Finding rare tryptophan dimer natural products in the environment. J. Am. Chem. Soc. 137, 6044–6052. doi:10.1021/jacs.5b01968
Chen, L., Zhang, X., Zhang, M., Zhu, Y., and Zhuo, R. (2022). Removal of heavy-metal pollutants by white rot fungi: Mechanisms, achievements, and perspectives. J. Clean. Prod. 354, 131681. doi:10.1016/j.jclepro.2022.131681
Chen, Y., and Murrell, J. C. (2010). When metagenomics meets stable-isotope probing: Progress and perspectives. Trends Microbiol. 18, 157–163. doi:10.1016/j.tim.2010.02.002
Chen, Z., Zheng, Y., Ding, C., Ren, X., Yuan, J., Sun, F., et al. (2017). Integrated metagenomics and molecular ecological network analysis of bacterial community composition during the phytoremediation of cadmium-contaminated soils by bioenergy crops. Ecotoxicol. Environ. Saf. 145, 111–118. doi:10.1016/j.ecoenv.2017.07.019
Cheung, K. H., and Gu, J. (2007). Mechanism of hexavalent chromium detoxification by microorganisms and bioremediation application potential: A review. Int. Biodeterior. Biodegrad. 59, 8–15. doi:10.1016/j.ibiod.2006.05.002
Cowan, D., Ramond, J.-B., Makhalanyane, T., and De Maayer, P. (2015). Metagenomics of extreme environments. Curr. Opin. Microbiol. 25, 97–102. doi:10.1016/j.mib.2015.05.005
Daniel, R. (2005). The metagenomics of soil. Nat. Rev. Microbiol. 3, 470–478. doi:10.1038/nrmicro1160
Datta, S., Rajnish, K. N., Samuel, M. S., Pugazlendhi, A., and Selvarajan, E. (2020). Metagenomic applications in microbial diversity, bioremediation, pollution monitoring, enzyme and drug discovery. A review. A Rev. Env. Chem. Lett. 18, 1229–1241. doi:10.1007/s10311-020-01010-z
de Alernca, F. L. S., Navoni, J. A., and Amaral, V. S. (2017). The use of bacterial bioremediation of metals in aquatic environments in the twenty-first century: A systematic review. Environ. Sci. Pollut. Res. 24, 16545–16559. doi:10.1007/s11356-017-9129-8
Dell’Anno, F., Joaquim van Zyl, L., Trindade, M., Buschi, E., Cannavacciuolo, A., Pepi, M., et al. (2023). Microbiome enrichment from contaminated marine sediments unveils novel bacterial strains for petroleum hydrocarbon and heavy metal bioremediation. Environ. Pollut. 317, 120772. doi:10.1016/j.envpol.2022.120772
Deng, F., Dou, R., Sun, J., Li, J., and Dang, Z. (2021). Phenanthrene degradation in soil using biochar hybrid modi fi ed bio-microcapsules: Determining the mechanism of action via comparative metagenomic analysis. Sci. Total Environ. 775, 145798. doi:10.1016/j.scitotenv.2021.145798
Deng, Y., Wang, Y., Xia, Y., Ni, A., Zhao, Y., and Zhang, T. (2019). Genomic resolution of bacterial populations in saccharin and cyclamate degradation. Sci. Total Environ. 658, 357–366. doi:10.1016/j.scitotenv.2018.12.162
Ding, W., Zhang, W., Alikunhi, N. M., Batang, Z., Pei, B., Wang, R., et al. (2019). Metagenomic analysis of zinc surface–associated marine biofilms. Microb. Ecol. 77, 406–416. doi:10.1007/s00248-018-01313-3
Djikeng, A., Kuzmickas, R., Anderson, N. G., and Spiro, D. J. (2009). Metagenomic analysis of RNA viruses in a fresh water lake. PLoS One 4, e7264. doi:10.1371/journal.pone.0007264
Eze, M. O. (2021). Metagenome analysis of a hydrocarbon-degrading bacterial consortium reveals the specific roles of BTEX biodegraders. Genes (Basel). 12, 98. doi:10.3390/genes12010098
Feng, G., Xie, T., Wang, X., Bai, J., Tang, L., Zhao, H., et al. (2018). Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 18, 11–13. doi:10.1186/s12866-018-1152-5
Feris, K. P., Hristova, K., Gebreyesus, B., Mackay, D., and Scow, K. M. (2004). A shallow BTEX and MTBE contaminated aquifer supports a diverse microbial community. Microb. Ecol. 48, 589–600. doi:10.1007/s00248-004-0001-2
Fernandes, C., Kankonkar, H., Meena, R. M., Menezes, G., Shenoy, B. D., and Khandeparker, R. (2019). Metagenomic analysis of tarball-associated bacteria from Goa, India. Mar. Pollut. Bull. 141, 398–403. doi:10.1016/j.marpolbul.2019.02.040
Gao, Y., Du, J., Bahar, M., Wang, H., Subashchandrabose, S., Duan, L., et al. (2021). Metagenomics analysis identifies nitrogen metabolic pathway in bioremediation of diesel contaminated soil. Chemosphere 271, 129566. doi:10.1016/j.chemosphere.2021.129566
Gill, R. T., Harbottle, M. J., Smith, J. W. N., and Thornton, S. F. (2014). Electrokinetic-enhanced bioremediation of organic contaminants: A review of processes and environmental applications. Chemosphere 107, 31–42. doi:10.1016/j.chemosphere.2014.03.019
Gosai, H. B., Panseriya, H. Z., Patel, P. G., Patel, A. C., Shankar, A., Varjani, S., et al. (2022). Exploring bacterial communities through metagenomics during bioremediation of polycyclic aromatic hydrocarbons from contaminated sediments. Sci. Total Environ. 842, 156794. doi:10.1016/j.scitotenv.2022.156794
Handelsmanl, J., Rondon, M. R., Goodman, R. M., Brady, S. F., and Clardy, J. (1998). Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 5, R245–R249. doi:10.1016/S1074-5521(98)90108-9
Haro-Moreno, J. M., López-Pérez, M., de la Torre, J. R., Picazo, A., Camacho, A., and Rodriguez-Valera, F. (2018). Fine metagenomic profile of the Mediterranean stratified and mixed water columns revealed by assembly and recruitment. Microbiome 6, 128. doi:10.1186/s40168-018-0513-5
Hemme, C. L., Deng, Y., Gentry, T. J., Fields, M. W., Wu, L., Barua, S., et al. (2010). Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 4, 660–672. doi:10.1038/ismej.2009.154
Henne, A., Daniel, R., Schmitz, R. A., and Gottschalk, G. (1999). Construction of environmental DNA libraries in Escherichia coli and screening for the presence of genes conferring utilization of 4-hydroxybutyrate. Appl. Environ. Microbiol. 65, 3901–3907. doi:10.1128/AEM.65.9.3901-3907.1999
Hidalgo, K. J., Sierra-Garcia, I. N., Dellagnezze, B. M., and de Oliveira, V. M. (2020). Metagenomic insights into the mechanisms for biodegradation of polycyclic aromatic hydrocarbons in the oil supply chain. Front. Microbiol. 11, 561506. doi:10.3389/fmicb.2020.561506
Huang, W., Gong, B., Wang, Y., Lin, Z., He, L., Zhou, J., et al. (2020). Metagenomic analysis reveals enhanced nutrients removal from low C/N municipal wastewater in a pilot-scale modified AAO system coupling electrolysis. Water Res. 173, 115530. doi:10.1016/j.watres.2020.115530
Huang, W. L., Wu, P. C., and Chiang, T. Y. (2022). Metagenomics: Potential for bioremediation of soil contaminated with heavy metals. Ecol. Genet. Genomics 22, 100111. doi:10.1016/j.egg.2021.100111
Jung, J., Philippot, L., and Park, W. (2016). Metagenomic and functional analyses of the consequences of reduction of bacterial diversity on soil functions and bioremediation in diesel-contaminated microcosms. Sci. Rep. 6, 23012. doi:10.1038/srep23012
Kachienga, L., Jitendra, K., and Momba, M. (2018). Metagenomic profiling for assessing microbial diversity and microbial adaptation to degradation of hydrocarbons in two South African petroleum-contaminated water aquifers. Sci. Rep. 8, 7564. doi:10.1038/s41598-018-25961-0
Khan, D., Kabiraj, A., Roy, R. K., Let, M., Mahji, K., and Bandopadhay, R. (2022). Bioremediation of heavy metals by metagenomic approaches, omics insight in environmental bioremediation. Singapore: Springer Nature.
Kotoky, R., Rajkumari, J., and Pandey, P. (2018). The rhizosphere microbiome: Signi fi cance in rhizoremediation of polyaromatic hydrocarbon contaminated soil. J. Environ. Manage. 217, 858–870. doi:10.1016/j.jenvman.2018.04.022
Kumar, R., Kumar, V., Bajaj, A., Tambat, S., and Manickam, N. (2019). Comparative microbiome analysis of two different long-term pesticide contaminated soils revealed the anthropogenic in fl uence on functional potential of microbial communities. Sci. Total Environ. 681, 413–423. doi:10.1016/j.scitotenv.2019.05.090
Kumar, R., Pandit, P., Kumar, D., Patel, Z., Pandya, L., Kumar, M., et al. (2021). Land fill microbiome harbour plastic degrading genes: A metagenomic study of solid waste dumping site of Gujarat, India. Sci. Total Environ. 779, 146184. doi:10.1016/j.scitotenv.2021.146184
Kumar, V., and Chandra, R. (2020). Metagenomics analysis of rhizospheric bacterial communities of Saccharum arundinaceum growing on organometallic sludge of sugarcane molasses-based distillery. Biotech 10, 316. doi:10.1007/s13205-020-02310-5
Lewis, D. J., Atwill, E. R., Lennox, M. S., Pereira, M. D. G., Miller, W. A., Conrad, P. A., et al. (2010). Management of microbial contamination in storm runoff from California coastal dairy pastures. J. Environ. Qual. 39, 1782–1789. doi:10.2134/jeq2009.0464
Li, J., Peng, Y., Zhang, L., Liu, J., Wang, X., Gao, R., et al. (2019). Quantify the contribution of anammox for enhanced nitrogen removal through metagenomic analysis and mass balance in an anoxic moving bed biofilm reactor. Water Res. 160, 178–187. doi:10.1016/j.watres.2019.05.070
Li, Q., Csetenyi, L., and Gadd, G. M. (2014). Biomineralization of metal carbonates by Neurospora crassa. Environ. Sci. Technol. 48 (24), 14409–14416. doi:10.1021/es5042546
Liu, C., Li, B., Chen, X., Dong, Y., and Lin, H. (2022). Insight into soilless revegetation of oligotrophic and heavy metal contaminated gold tailing pond by metagenomic analysis. J. Hazard. Mater. 435, 128881. doi:10.1016/j.jhazmat.2022.128881
Liu, P., Zhang, Y., Tang, Q., and Shi, S. (2021). Bioremediation of metal-contaminated soils by microbially-induced carbonate precipitation and its effects on ecotoxicity and long-term stability. Biochem. Eng. J. 166, 107856. doi:10.1016/j.bej.2020.107856
Luo, G., Li, B., Li, L. G., Zhang, T., and Angelidaki, I. (2017). Antibiotic resistance genes and correlations with microbial community and metal resistance genes in full-scale biogas reactors as revealed by metagenomic analysis. Environ. Sci. Technol. 51, 4069–4080. doi:10.1021/acs.est.6b05100
Luo, M., Ji, Y., Warton, D., and Yu, D. W. (2023). Extracting abundance information from DNA-based data. Mol. Ecol. Resour. 23, 174–189. doi:10.1111/1755-0998.13703
Marsh, T. L., and McInerney, M. J. (2001). Relationship of hydrogen bioavailability to chromate reduction in aquifer sediments. Appl. Environ. Microbiol. 67, 1517–1521. doi:10.1128/AEM.67.4.1517-1521.2001
Miao, Y., Heintz, M. B., Bell, C. H., Johnson, N. W., Polasko, A. L., Favero, D., et al. (2021). Profiling microbial community structures and functions in bioremediation strategies for treating 1,4-dioxane-contaminated groundwater. J. Hazard. Mat. 408, 124457. doi:10.1016/j.jhazmat.2020.124457
Mirete, S., Mora-Ruiz, M. R., Lamprecht-Grandío, M., de Figueras, C. G., Rosselló-Móra, R., and González-Pastor, J. E. (2015). Salt resistance genes revealed by functional metagenomics from brines and moderate-salinity rhizosphere within a hypersaline environment. Front. Microbiol. 6, 01121. doi:10.3389/fmicb.2015.01121
Muangchinda, C., Rungsihiranrut, A., Prombutara, P., Soonglerdsongpha, S., and Pinyakong, O. (2018). 16S metagenomic analysis reveals adaptability of a mixed-PAH-degrading consortium isolated from crude oil-contaminated seawater to changing environmental conditions. J. Hazard. Mat. 357, 119–127. doi:10.1016/j.jhazmat.2018.05.062
Offiong, N. O., Inam, E. J., Fatunla, O. K., Ofon, U. A., Abraham, N. A., and Essien, J. P. (2022). Metagenomic signature and total petroleum hydrocarbons distribution in a crude oil contaminated ultisol remediated with biochar–humus sediment slurry. Remediat. J. 32, 299–308. doi:10.1002/rem.21734
Palumbo, A. V., McCarthy, J. F., Amonette, J. E., Fisher, L. S., Wullschleger, S. D., and Lee Daniels, W. (2004). Prospects for enhancing carbon sequestration and reclamation of degraded lands with fossil-fuel combustion by-products. Adv. Environ. Res. 8, 425–438. doi:10.1016/S1093-0191(02)00124-7
Porteous, L. A., Armstrong, J. L., Seidler, R. J., and Watrud, L. S. (1994). An effective method to extract DNA from environmental samples for polymerase chain reaction amplification and DNA fingerprint analysis. Curr. Microbiol. 29, 301–307. doi:10.1007/BF01577445
Prakash, A. A., Rajasekar, A., Sarankumar, R. K., AlSahli, M. S., Devanesan, S., Aljaafreh, M. J., et al. (2021). Metagenomic analysis of microbial community and its role in bioelectrokinetic remediation of tannery contaminated soil. J. Hazard. Mater. 412, 125133. doi:10.1016/j.jhazmat.2021.125133
Puthesseri, R. M., Nair, H. P., Johny, T. K., and Bhat, S. G. (2021). Insights into the response of mangrove sediment microbiomes to heavy metal pollution: Ecological risk assessment and metagenomics perspectives. J. Environ. Manag. 298, 113492. doi:10.1016/j.jenvman.2021.113492
Rahmeh, R., Akbar, A., Kumar, V., Al-Mansour, H., Kishk, M., Ahmed, N., et al. (2021). Insights into bacterial community involved in bioremediation of aged oil-contaminated soil in arid environment. Evol. Bioinforma. 17, 117693432110168. doi:10.1177/11769343211016887
Redfern, L. K., Gardner, C. M., Hodzic, E., Ferguson, P. L., Hsu-Kim, H., and Gunsch, C. K. (2019). A new framework for approaching precision bioremediation of PAH contaminated soils. J. Hazard. Mat. 378, 120859. doi:10.1016/j.jhazmat.2019.120859
Riaz, M., Kamran, M., Fang, Y., Wang, Q., Cao, H., Yang, G., et al. (2021). Arbuscular mycorrhizal fungi-induced mitigation of heavy metal phytotoxicity in metal contaminated soils: A critical review. J. Hazard. Mater. 402, 123919. doi:10.1016/j.jhazmat.2020.123919
Ribicic, D., Netzer, R., Hazen, T. C., Techtmann, S. M., Drabløs, F., and Brakstad, O. G. (2018). Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 129, 370–378. doi:10.1016/j.marpolbul.2018.02.034
Romero, M. F., Gallego, D., Lechuga-Jimenez, A., Martinez, J. F., Barajas, H. R., Hayano-Kanashiro, C., et al. (2021). Metagenomics of mine tailing rhizospheric communities and its selection for plant establishment towards bioremediation. Microb. Res. 247, 126732. doi:10.1016/j.micres.2021.126732
Sangwan, N., Lata, P., Dwivedi, V., Singh, A., Niharika, N., Kaur, J., et al. (2012). Comparative metagenomic analysis of soil microbial communities across three hexachlorocyclohexane contamination levels. PLoS One 7, 462199–e46312. doi:10.1371/journal.pone.0046219
Santini, T. C., Kerr, J. L., and Warren, L. A. (2015). Microbially-driven strategies for bioremediation of bauxite residue. J. Hazard. Mat. 293, 131–157. doi:10.1016/j.jhazmat.2015.03.024
Schloss, P. D., and Handelsman, J. (2003). Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 14, 303–310. doi:10.1016/S0958-1669(03)00067-3
Scow, K. M., and Hicks, K. A. (2005). Natural attenuation and enhanced bioremediation of organic contaminants in groundwater. Curr. Opin. Biotechnol. 16, 246–253. doi:10.1016/j.copbio.2005.03.009
Sharma, P., Kumar, S., and Pandey, A. (2021a). Bioremediated techniques for remediation of metal pollutants using metagenomics approaches: A review. J. Environ. Chem. Eng. 9, 105684. doi:10.1016/j.jece.2021.105684
Sharma, P., Pandey, A. K., Kim, S. H., Singh, S. P., Chaturvedi, P., and Varjani, S. (2021). Critical review on microbial community during in-situ bioremediation of heavy metals from industrial wastewater. Environ. Technol. Innovation 24, 101826. doi:10.1016/j.eti.2021.101826
Sharma, P., Tripathi, S., and Chandra, R. (2021b). Metagenomic analysis for profiling of microbial communities and tolerance in metal-polluted pulp and paper industry wastewater. Bioresour. Technol. 324, 124681. doi:10.1016/j.biortech.2021.124681
Shi, L. D., Chen, Y. S., Du, J. J., Hu, Y. Q., Shapleigh, J. P., and Zhao, H. P. (2019). Metagenomic evidence for a methylocystis species capable of bioremediation of diverse heavy metals. Front. Microbiol. 9, 3297. doi:10.3389/fmicb.2018.03297
Sturman, P. J., Stewart, P. S., Cunningham, A. B., Bouwer, E. J., and Wolfram, J. H. (1995). Engineering scale-up of in situ bioremediation processes: A review. J. Contam. Hydrol. 19, 171–203. doi:10.1016/0169-7722(95)00017-P
Sun, F. L., Fan, L. L., Wang, Y. S., and Huang, L. Y. (2019). Metagenomic analysis of the inhibitory effect of chromium on microbial communities and removal efficiency in A2O sludge. J. Hazard. Mat. 368, 523–529. doi:10.1016/j.jhazmat.2019.01.076
Techtmann, S. M., and Hazen, T. C. (2016). Metagenomic applications in environmental monitoring and bioremediation. J. Ind. Microbiol. Biotechnol. 43, 1345–1354. doi:10.1007/s10295-016-1809-8
Tong, T., Li, R., Chen, J., Ke, Y., and Xie, S. (2021). Bisphenol A biodegradation differs between mud fl at and mangrove forest sediments. Chemosphere 270, 128664. doi:10.1016/j.chemosphere.2020.128664
Tringe, S. G., Zhang, T., Liu, X., Yu, Y., Lee, W. H., Yap, J., et al. (2008). The airborne metagenome in an indoor urban environment. PLoS One 3, e1862. doi:10.1371/journal.pone.0001862
Tripathi, S., Singh, K., and Chandra, R. (2021). “Adaptation of bacterial communities and plant strategies for amelioration and eco-restoration of an organometallic industrial waste polluted site,” in Microbes in Land Use Change Management. Elsevier, 870, 45–90. doi:10.1016/B978-0-12-824448-7.00005-X
Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43. doi:10.1038/nature02340
Uhlig, O., Leewis, M. C., Strejcek, M., Musilova, L., Mackova, M., Leigh, M. B., et al. (2013). Stable isotope probing in the metagenomics era: A bridge towards improved bioremediation. Biotechnol. Adv. 31, 154–165. doi:10.1016/j.biotechadv.2012.09.003
Verma, S., and Kuila, A. (2019). Bioremediation of heavy metals by microbial process. Environ. Technol. Innovation 14, 100369. doi:10.1016/j.eti.2019.100369
Vogel, T. M. (1996). Bioaugmentation as a soil bioremediation approach. Curr. Opin. Biotechnol. 7, 311–316. doi:10.1016/S0958-1669(96)80036-X
Wang, L., Yin, Z., and Jing, C. (2020). Metagenomic insights into microbial arsenic metabolism in shallow groundwater of Datong basin, China. Chemosphere 245, 125603. doi:10.1016/j.chemosphere.2019.125603
Wani, A. K., Akhtar, N., Naqash, N., Chopra, C., Singh, R., Kumar, V., et al. (2022). Bioprospecting culturable and unculturable microbial consortia through metagenomics for bioremediation. Clean. Chem. Eng. 2, 100017. doi:10.1016/j.clce.2022.100017
Wei, W., Liu, X., Sun, P., Wang, X., Zhu, H., Hong, M., et al. (2014). Simple whole-cell biodetection and bioremediation of heavy metals based on an engineered lead-specific operon. Environ. Sci. Technol. 48 (6), 3363–3371. doi:10.1021/es4046567
Wooley, J. C., and Ye, Y. (2010). Metagenomics: Facts and artifacts, and computational challenges. J. Comput. Sci. Technol. 25, 71–81. doi:10.1007/s11390-010-9306-4
Xu, R., Sun, X., Häggblom, M. M., Dong, Y., Zhang, M., Yang, Z., et al. (2022). Metabolic potentials of members of the class Acidobacteriia in metal-contaminated soils revealed by metagenomic analysis. Environ. Microbiol. 24, 803–818. doi:10.1111/1462-2920.15612
Yadav, R., Rajput, V., and Dharne, M. (2021). Functional metagenomic landscape of polluted river reveals potential genes involved in degradation of xenobiotic pollutants. Environ. Res. 192, 110332. doi:10.1016/j.envres.2020.110332
Yan, X., Luo, X., and Zhao, M. (2016). Metagenomic analysis of microbial community in uranium-contaminated soil. Appl. Microbiol. Biotechnol. 100, 299–310. doi:10.1007/s00253-015-7003-5
Yan, Y., Ma, M., Liu, X., Ma, W., and Li, Y. (2018). Vertical distribution of archaeal communities associated with anaerobic degradation of pentabromodiphenyl ether (BDE-99) in river-based groundwater recharge with reclaimed water. Environ. Sci. Pollut. Res. 25, 5154–5163. doi:10.1007/s11356-017-0034-y
Yergeau, E., Snaschagrin, S., Beaumier, D., and Greer, C. W. (2012). Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high arctic soils. Plos One 7, e30058. doi:10.1371/journal.pone.0030058
Yooseph, S., Andrews-Pfannkoch, C., Tenney, A., McQuaid, J., Williamson, S., Thiagarajan, M., et al. (2013). A metagenomic framework for the study of airborne microbial communities. PLoS One 8, e81862. doi:10.1371/journal.pone.0081862
Zainun, M. Y., and Simarani, K. (2018). Metagenomics profiling for assessing microbial diversity in both active and closed landfills. Sci. Total Environ. 616-617, 269–278. doi:10.1016/j.scitotenv.2017.10.266
Zhang, L., Loh, K. C., Lim, J. W., and Zhang, J. (2019). Bioinformatics analysis of metagenomics data of biogas-producing microbial communities in anaerobic digesters: A review. Renew. Sustain. Energy Rev. 100, 110–126. doi:10.1016/j.rser.2018.10.021
Zhang, Z., Wan, J., Liu, L., Ye, M., and Jiang, X. (2021). Metagenomics reveals functional profiling of microbial communities in OCP contaminated sites with rapeseed oil and tartaric acid biostimulation. J. Environ. Manage. 289, 112515. doi:10.1016/j.jenvman.2021.112515
Zheng, M., Shi, J., Xu, C., Han, Y., Zhang, Z., and Han, H. (2020). Insights into electroactive biofilms for enhanced phenolic degradation of coal pyrolysis wastewater (CPW) by magnetic activated coke (MAC): Metagenomic analysis in attached biofilm and suspended sludge. J. Hazard. Mat. 395, 122688. doi:10.1016/j.jhazmat.2020.122688
Keywords: metagenomics, bioremediation, microorganisms, chemical fate, environmental contaminants
Citation: Offiong N-AO, Edet JB, Shaibu SE, Akan NE, Atakpa EO, Sanganyado E, Okop IJ, Benson NU and Okoh A (2023) Metagenomics: an emerging tool for the chemistry of environmental remediation. Front. Environ. Chem. 4:1052697. doi: 10.3389/fenvc.2023.1052697
Received: 24 September 2022; Accepted: 27 April 2023;
Published: 09 May 2023.
Edited by:
Naser A. Anjum, Aligarh Muslim University, IndiaReviewed by:
Vineet Kumar, Central University of Rajasthan, IndiaYutao Peng, Sun Yat-sen University, China
Copyright © 2023 Offiong, Edet, Shaibu, Akan, Atakpa, Sanganyado, Okop, Benson and Okoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nnanake-Abasi O. Offiong, bm8ub2ZmaW9uZ0B0b3BmYWl0aC5lZHUubmc=; Edmond Sanganyado, ZWRtb25kLnNhbmdhbnlhZG9Abm9ydGh1bWJyaWEuYWMudWs=
†These authors have contributed equally to this work
‡ORCID: Nnanake-Abasi O. Offiong, orcid.org/0000-0003-4692-7891; John B. Edet, orcid.org/0000-0001-6316-2988; Solomon E. Shaibu, orcid.org/0000-0002-5845-4238; Edidiong O. Atakpa, orcid.org/0000-0002-2259-0967; Edmond Sanganyado, orcid.org/0000-0001-6244-3059; Imeh J. Okop, orcid.org/0000-0002-1487-6711; Nsikak U. Benson, orcid.org/0000-0002-1285-579X