 Piotr M. Kowalski
Piotr M. Kowalski Zhengda He
Zhengda He Oskar Cheong
Oskar Cheong- 1Forschungszentrum Jülich GmbH, Institute of Energy and Climate Research - IEK-13: Theory and Computation of Energy Materials, Jülich, Germany
- 2Jülich Aachen Research Alliance, JARA Energy & Center for Simulation and Data Science (CSD), Jülich, Germany
- 3Chair of Theory and Computation of Energy Materials, Faculty of Georesources and Materials Engineering, RWTH Aachen University, Aachen, Germany
LixFePO4 orthophosphates and fluorite- and pyrochlore-type zirconate materials are widely considered as functional compounds in energy storage devices, either as electrode or solid state electrolyte. These ceramic materials show enhanced cation exchange and anion conductivity properties that makes them attractive for various energy applications. In this contribution we discuss thermodynamic properties of LixFePO4 and yttria-stabilized zirconia compounds, including formation enthalpies, stability, and solubility limits. We found that at ambient conditions LixFePO4 has a large miscibility gap, which is consistent with existing experimental evidence. We show that cubic zirconia becomes stabilized with Y content of ~8%, which is in line with experimental observations. The computed activation energy of 0.92eV and ionic conductivity for oxygen diffusion in yttria-stabilized zirconia are also in line with the measured data, which shows that atomistic modeling can be applied for accurate prediction of key materials properties. We discuss these results with the existing simulation-based data on these materials produced by our group over the last decade. Last, but not least, we discuss similarities of the considered compounds in considering them as materials for energy storage and radiation damage resistant matrices for immobilization of radionuclides.
1. Introduction
MPO4 orthophosphates, with M cations being transition metals, rare-earth elements or actinides, are ceramic materials of interest in various research fields, including geochronology (Williams et al., 2007), geothermometry (Andrehs and Heinrich, 1998; Mogilevsky, 2007), energy storage (Iyer et al., 2006; Yamada et al., 2006; Dunn et al., 2015; Dong et al., 2017; Cerdas et al., 2018; Li et al., 2018; Phan et al., 2019), and nuclear waste management (Ewing and Wang, 2002; Neumeier et al., 2017a; Schlenz et al., 2018), to name but a few. Most of the potential applications come from high durability (e.g., radiation damage resistance) of these materials (Neumeier et al., 2017a; Phan et al., 2019). There exist large varieties of phosphate-based ceramics of different crystalline structures (e.g., cheralite, apatites, olivine, kosnarite, see Iyer et al., 2006; Neumeier et al., 2017a; Phan et al., 2019). The orthophosphates of interest for energy storage, FePO4 and LiFePO4, have olivine-type structure of orthorombic space group symmetry of Pnma in which Fe exists in an octahedral environment (Iyer et al., 2006; Maxisch and Ceder, 2006, see Figure 1 for visualization of the structures). It consists of eight-fold coordinated Fe3+ cations, which become reduced to Fe2+ upon incorporation of Li. Besides, there exist less stable FePO4 monoclinic, orthorombic, and trigonal phases with Fe in tetrahedraly coordinated crystallographic positions (Iyer et al., 2006; Yamada et al., 2006; Dong et al., 2017). LiFePO4 materials are one of the best known candidates for energy storage electrodes (Dunn et al., 2015; Cerdas et al., 2018). These exhibit high theoretical energy density of 170mAh/g and voltage of 3.5V (Phan et al., 2019). Because of this, these materials are considered for large scale energy storage devices, including batteries for hybrid and electric automobiles (Prosini et al., 2002; Dunn et al., 2015; Cerdas et al., 2018). Intercalation of Li ions into FePO4 results in formation of a solid solution between Li cations and vacant sites (Phan et al., 2019). Depending on the sizes of mixing species, a solid solution compounds could form a thermodynamically stable solid solution or a compound with a mixture of two phases, each rich in one of the cations (Kowalski and Li, 2016; Ji et al., 2019a). The second case indicates formation of a temperature dependent miscibility gap. The two endmember phases may be of the same type, like in the case of LixFePO4, or different phases, like in the monazite-xenotime system (Mogilevsky, 2007; Ji et al., 2019a). The formation of miscibility gap is correlated with the maximum solubility (Ji et al., 2019a). Such a temperature-dependent maximum solubility offers opportunity to use such a system as geothermometer (Andrehs and Heinrich, 1998; Mogilevsky, 2007). It is known from experiment that LixFePO4 solid solution posses a wide miscibility gap at ambient temperature (Yamada et al., 2006; Meethong et al., 2007; Li et al., 2018; Phan et al., 2019). In particular, studies of Yamada et al. (2006) indicate relatively low Li solubility limits in LixFePO4 system at x = 0.05 and 0.89. The thermodynamics of such a system, however, although has been modeled by CALPHAD method (Phan et al., 2019), has not been modeled in details using ab initio atomistic modeling methods and the results of various studies differ substantially (Phan et al., 2019).
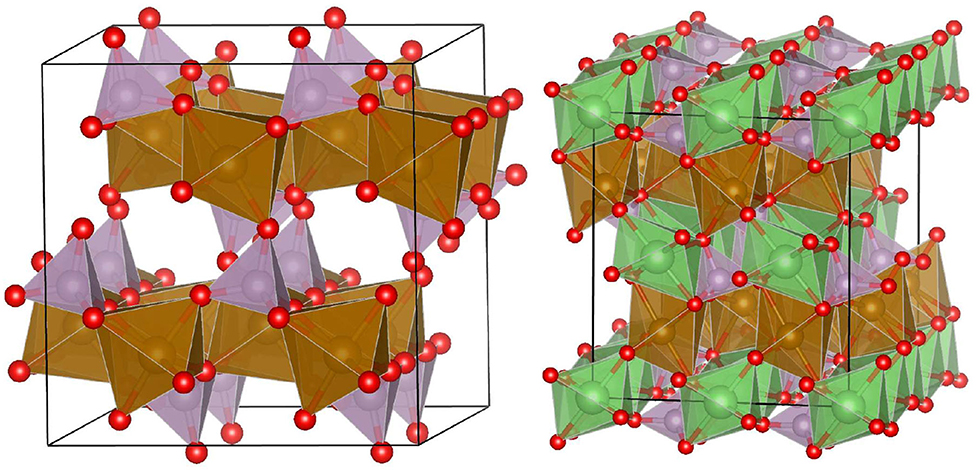
Figure 1. Structures of FePO4 (left) and LiFePO4 (right).
Zirconium-based ceramics have been shown to posses interesting ion-conduction characteristics (Diazguillen et al., 2008; Mandal et al., 2008; Xia et al., 2010; Anithakumari et al., 2016; Li and Kowalski, 2018), with yttria stabilized zirconia being one of the fastest ionic conductors (Kilo et al., 2003; Krishnamurthy et al., 2005b). It is thus used as solid electrolyte in various energy storage devices (Zakaria et al., 2020). The fast oxygen conduction properties are associated with formation and distribution of vacant sites (in fluorite and pyrochlore, Bukaemskiy et al., 2021) and interesting short- and long-range ordering phenomena (Wuensch, 2000; Yamamura, 2003; Anithakumari et al., 2016; Drey et al., 2020; Kowalski, 2020; Bukaemskiy et al., 2021). For instance, certain pyrochlore compounds exhibit high ionic conductivity (e.g., Eu2Zr2O7, Yamamura, 2003) and form a stable, cation-disordered solid phase (defect fluorite, Li et al., 2015). Such phases show interesting short- and long-range ordering (Shamblin et al., 2016; Drey et al., 2020; Kowalski, 2020; Bukaemskiy et al., 2021), with distribution of vacancies determining the amplitude of pre-exponential factor and efficiency of ionic conduction (Bukaemskiy et al., 2021). For instance, Bukaemskiy et al. (2021) demonstrated that the maximum of ionic conductivity in YO1.5 − ZrO2 system occurs at x = 0.146 due to vacancy avoidance phenomenon and related vacancy jump probability. This correlates well with the existing ionic conductivity data (Bukaemskiy et al., 2021). Cubic phase of zirconia (ZrO2) becomes stabilized upon doping with tri-valent elements, e.g., Y (Li et al., 1994; Kilo et al., 2003; Krishnamurthy et al., 2005b) at about 8% content of dopant. The formed solid solution shows very high ionic conductivity that increases significantly with dopant amount (Ioffe et al., 1978; Bukaemskiy et al., 2021). These compounds are thus considered as solid electrolyte for energy storage devices (Zakaria et al., 2020).
In the last two decades, atomistic modeling became a widely used research technique in various research fields, including energy materials (Chroneos et al., 2013; Jahn and Kowalski, 2014; Wu et al., 2019). We used it intensively over the past decade for computation of, for instance, various physical and chemical properties of orthophosphate- and zircon-based ceramics (e.g., Kowalski et al., 2015; Ji et al., 2019a). This is because steady advancements in high performance computing and computational software, especially in ab initio methods-based codes, allows nowadays for computation of complex systems containing hundreds of atoms from first principles (Jahn and Kowalski, 2014). Regarding ceramic compounds considered here, computational studies have been used to deliver information on: the structural (Rustad, 2012; Feng et al., 2013; Blanca-Romero et al., 2014; Beridze et al., 2016; Huittinen et al., 2017), the electronic structure (Tang and Holzwarth, 2003; Blanca-Romero et al., 2014; Kowalski et al., 2017a; Lee et al., 2017), the elastic (Wang et al., 2005; Feng et al., 2013; Ali et al., 2016; Kowalski and Li, 2016; Ji et al., 2017a; Kowalski et al., 2017b), the thermodynamic (Mogilevsky, 2007; Feng et al., 2013; Li et al., 2014; Kowalski et al., 2015, 2016; Ji et al., 2017b; Neumeier et al., 2017b; Eremin et al., 2019), the thermochemical (Rustad, 2012; Beridze et al., 2016; Kowalski, 2020), the electrochemical (Krishnamurthy et al., 2005b; Lee et al., 2017), and the radiation damage resistance (Kowalski et al., 2016; Li et al., 2016; Ji et al., 2017c; Jolley et al., 2017) parameters as well as materials at high-pressure (López-Solano et al., 2010; Stavrou et al., 2012; Ali et al., 2016; Shein and Shalaeva, 2016; Gomis et al., 2017). The relevant research activity increases steadily worldwide, with most of the papers published just recently. One important aspect is the correct calculations of compounds with d- and f- elements that contain strongly correlated electrons [e.g., Fe, Ni, lanthanides (Ln), actinides (An)]. In a series of papers we have shown that standard DFT approach often fails for such cases and these compounds must be carefully computed, including proper accounting for the correlation effects (Beridze and Kowalski, 2014; Blanca-Romero et al., 2014; Kowalski et al., 2015; Li and Kowalski, 2018). These simulations must be performed with methods beyond the standard DFT+U approach and include derivation of the Hubbard U parameter and careful choice of projectors for estimation of occupancy of d− and f− levels within the DFT+U scheme (Maxisch and Ceder, 2006; Kvashnina et al., 2018; Kick et al., 2019). In particular, we apply the linear response method (Cococcioni and de Gironcoli, 2005) with Wannier orbitals as representation of d or f states (Kvashnina et al., 2018) and here we will demonstrate impact of these procedures on the estimation of formation enthalpies and solubility limits of LixFePO4 compound.
In this contribution we provide an overview of the recent atomistic modeling activities on the orthophosphates and zirconates, focusing on the information that have been delivered by atomistic modeling activities at Forschungszentrum Jülich and that allowed on many occasions for better characterization of these materials, including long-term thermodynamic stability, thermochemical parameters, and thermal conductivity. Besides such overview, we present results of computation of thermochemical and thermodynamic parameters of LixFePO4 solid solution, with focus on the formation of miscibility gap in this system, as well as simulation of yttria-stabilized zirconia with focus on prediction the phase stability and ionic conductivity in this class of materials. We especially highlight a cross-linking, interdisciplinary character of our research, from which the general science community could highly benefit.
2. Computational Approach
In all ab initio1 calculations discussed here we used a density functional theory (DFT)-based quantum chemistry approach and calculations were performed with Quantum-ESPRESSO simulation package (Giannozzi et al., 2009). We applied the PBEsol exchange-correlation functional (Perdew et al., 2008), the ultrasoft pseudopotentials to represent the core electrons of the atoms (Vanderbilt, 1990) and the plane-wave energy cutoff of 50 Ryd. The PBEsol functional is specifically selected because it correctly reproduces slowly varying electron density limit and results in good structural parameters of solids (Perdew et al., 2008). This is important for consideration of, for instance, thermodynamics of solid solutions (Li et al., 2014; Kowalski and Li, 2016; Ji et al., 2019a). Following our broad experience on computation of lanthanide orthophosphates and zirconates (e.g., Blanca-Romero et al., 2014; Li et al., 2015; Beridze et al., 2016) we applied the self-consistent DFT+U approach. The DFT+U calculations were performed with the Hubbard U parameter values computed from first principles using the linear response method of Cococcioni and de Gironcoli (2005). In order to apply realistic projectors for occupations of d orbitals of Fe we used Poor Man Wannier scheme implemented in Quantum-ESPRESSO. This computational setup was extensively tested by us in several studies and, among others, proved to give very good results for orthophosphates and zirconates (Blanca-Romero et al., 2014; Beridze et al., 2016; Finkeldei et al., 2017). The activation barriers were computed using Nudged Elastic Band (NEB) method implemented in Quantum-ESPRESSO, with 10 images and climbing image procedure to compute the transition state. The exponential pre-factors were computed with the aid of transition state theory (Moynihan et al., 1982) and probability of vacancy migration derived by Bukaemskiy et al. (2021), so that the ionic conductivity, σ, is estimated using the modified here formula of Moynihan et al. (1982):
where Ze is the charge of the carrier, kb is the Boltzmann constant, Na is Avogadro number, d is the distance of the jump, Ea is the activation barrier, T is the temperature and ν0 is the attempt frequency. The first part in the square bracket reflects the vacancy migration probability contribution to the pre-exponential factor derived by Bukaemskiy et al. (2021). The attempt frequency ν0 was estimated from the computation of phonon spectra of the initial (IS) and transition states (TS), as
The computation of LixFePO4 phases were performed with 2x2x1 supercells (96 atoms for FePO4 and 112 atoms for LiFePO4 phases) using the 2x2x2 k-point grid. The oxides were computed as: Fe2O3 (Pnmm symmetry, with supercell containing 30 atoms and 4x6x2 k-point grid), hexagonal P2O5 as is in Blanca-Romero et al. (2014) and Y2O3 as cubic oxide. The magnetic arrangements in iron phases were computed using the models of Whittingham et al. (2005) and Lee et al. (2017).
The Hubbard U parameters computed with the linear response method are listed in Table 1. As in our previous studies (Beridze and Kowalski, 2014; Beridze et al., 2016; Kvashnina et al., 2018; Sun et al., 2020) we see strong dependence on the Fe redox state, with the U parameter for Fe(III) being ~0.5 eV larger than for Fe(II). This is well-consistent with previous studies of FePO4 and LiFePO4 phases by Maxisch and Ceder (2006), who obtained 4.9 and 3.7eV for both phases (taken as effective value, Ueff = U−J), respectively.

Table 1. The computed Hubbard U parameters for Fe in Fe2O3, FePO4 and LiFePO4 compounds.
3. Results and Discussion
3.1. Structural Data
The first test of a computational method is its ability to reproduce the measured lattice parameters of the computed crystalline solid. The lattice parameters of considered materials were measured by different studies and are well-known. These are collected in Table 2 and compared to the computed data.
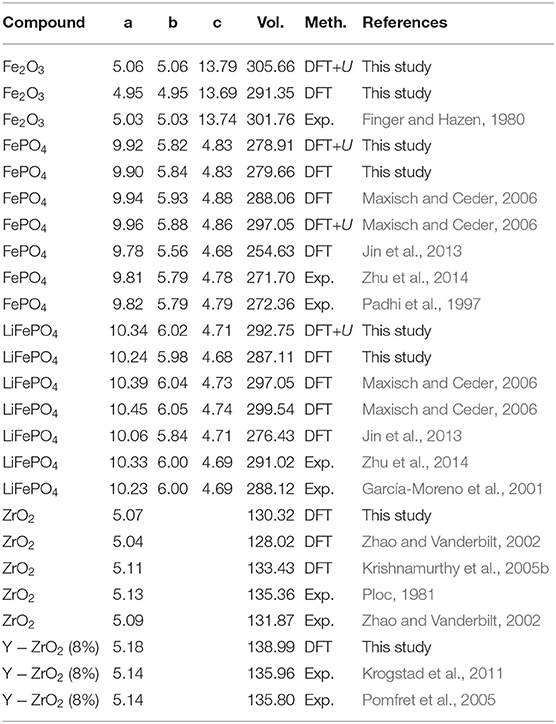
Table 2. The computed and measured lattice parameters of Fe2O3, FePO4, LiFePO4 and cubic zirconia compounds.
Our previous studies showed that the structural parameters of lanthanide-orthophosphates are very sensitive to the applied computational method, especially to the exchange-correlation functional (Blanca-Romero et al., 2014). A correct treatment of strongly correlated 4f electrons also plays an important role in those cases. Here, with our computational setup we got much better fit to the measured data than Maxisch and Ceder (2006), who applied the PBE exchange-correlation functional that tends to overestimate lattice parameters and volumes, which is also evident in the data collected in Table 2. Blanca-Romero et al. (2014) performed extensive tests of the capability of different DFT-based approaches to reproduce the measured lattice parameters and bond-distances of monazite-type lanthanide-orthophosphates. In that paper, we found that the standard DFT method with explicitly computed 4f electrons overestimates the lattice parameters and bond-lengths by up to 3%, which is consistent with previous studies (Wang et al., 2005; Rustad, 2012). A very good match to the experimental values of structural parameters was achieved applying the parameter free DFT+U method, with the PBEsol exchange-correlation functional (Perdew et al., 2008) and the Hubbard U parameters derived ab initio. An improved description of structures by the PBEsol exchange-correlation functional is an important property of that functional. It recovers the known solution for slowly varying electron densities and with that it improves the description of structural parameters over widely used GGA functionals such as PBE (see discussion by Perdew et al., 2008). It is evident also for zirconate-based materials. In our past studies of Nd2−xZr2+xO7+x/2 compound, with the PBEsol exchange-correlation functional we got perfect match to the lattice parameter in case of the pyrochlore phase (Finkeldei et al., 2017).
In the most recent contribution we computed the MUO4 compounds with M = Ni, Fe, Co, Cd, and found that only by using the correct projectors for estimation of occupation of d orbitals, e.g., Wannier functions, we could reproduce experimentally seen structural distortions (Murphy et al., 2021). The problem arises from the fact that with the standard DFT+U approach, when using atomic orbitals as projectors, the total occupancy of the d or f states of interest is much higher than the actual one (Kick et al., 2019; Murphy et al., 2021). As illustrated in Table 3, in our case it gives ~1.3 excess electrons for Fe(III) and ~0.5 for Fe(II). This is cured when using Wannier orbitals as projectors. Unfortunately, the forces and cell optimizations with this scheme are not yet implemented in Quantum-ESPRESSO or any equivalent codes, so we could not perform geometry optimization with such a more realistic approach.
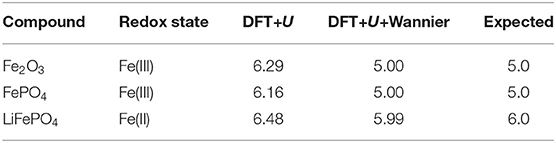
Table 3. The computed projected total number of d electrons per Fe atom in Fe2O3, FePO4 and LiFePO4 compounds using the DFT+U approach with atomic orbitals and Wannier functions as projectors.
3.2. Formation Enthalpies
The formation enthalpies from oxides for series of lanthanide orthophosphates have been measured by Ushakov et al. (2001) and of FePO4 and LiFePO4 by Iyer et al. (2006). Rustad (2012) noticed that there is a systematic offset between the computed and measured values for LnPO4 of ~40 kJ/mol, with the computed enthalpies being less exothermic. Blanca-Romero et al. (2014) have shown that this offset is present also in the DFT+U calculations and is to a large extent Ln-cation independent, which rules out the 4f electrons correlations as a contributing factor. They attributed this to the overestimation of P-O bond lengths, and thus volumes, of the LnPO4 and P2O5 compounds. Beridze et al. (2016) have found an identical offset for xenotime phase. When a constant shift of ~30 kJ/mol is applied to the computed formation enthalpies, the measured values are nicely reproduced. The computed formation enthalpies for FePO4 and LiFePO4 compounds are reported in Table 4. It is evident that DFT heavily underestimates the values (taking absolute values) by 40–100 kJ/mol. This is significantly improved with the DFT+U approach with an error of 20 kJ/mol. Best result, however, is obtained with the DFT+U method when Wanner functions are used as projectors of Fe d states occupations. In this scheme, the computed formation enthalpies are within 10 kJ/mol. Accurate prediction of formation enthalpies are crucial for correct estimate of thermodynamic parameters of solid solution, including solubilities (section 3.4).

Table 4. The computed here with different methods and measured (Iyer et al., 2006) formation enthalpies from oxides of FePO4 and LiFePO4 compounds.
The most stable phase of ZrO2 is monoclinic. However, upon doping with tri-valent elements it undergoes phase transition to cubic phase, with possible triclinic phase as an intermediate. The experimental evidence shows transition to that phase at ~8% of YO1.5 (Lee et al., 2003; Götsch et al., 2016; Ahamer et al., 2017). In Figure 2, we show the results of computation of formation enthalpies for the three phases of ZrO2 (monoclinic, tetragonal, and cubic). The results are plotted together with the experimental data of Lee et al. (2003). The computed values show clearly that at ~ 8% content of Y the enthalpy of cubic phase becomes the lowest and that phase most stable, which is well consistent with the aforementioned experimental data. Moreover, the computed formation enthalpy as a function of Y content is well consistent with the measured values. Also the computed enthalpy difference between monoclinic and cubic phases of ~14 kJ/mol is well consistent with the previous measurements and estimates (ranging from 6 to 22 kJ/mol, with the best measured value of 10 kJ/mol, Lee et al., 2003).
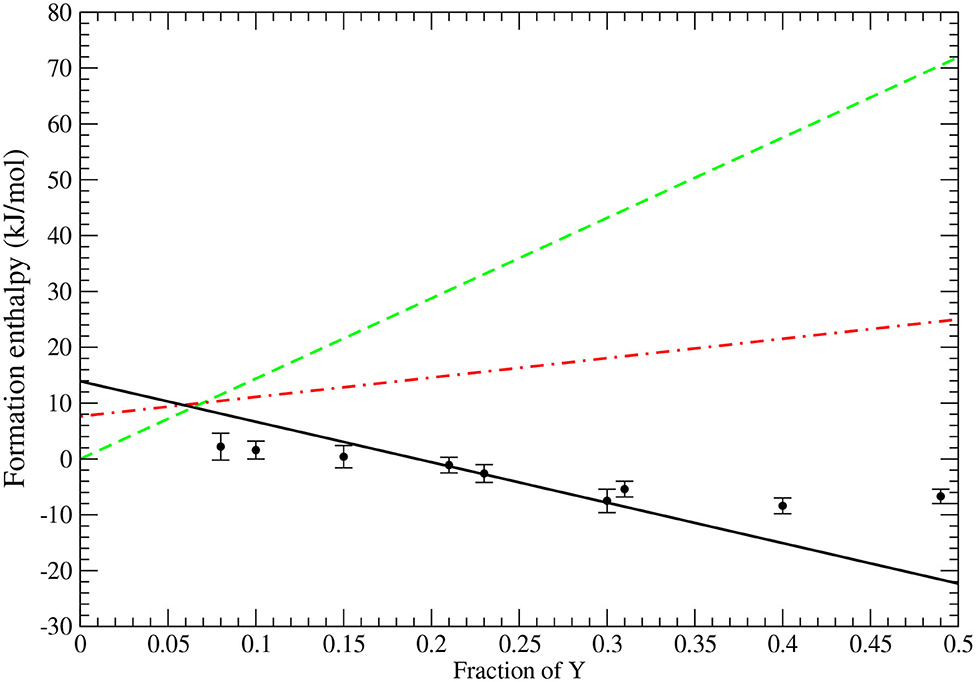
Figure 2. The computed formation enthalpies of YO1.5 − ZrO2 system. The lines represent results for monoclinic (dashed green), trigonal (dot-dashed red), and cubic (solid black) phases. The data comes from Lee et al. (2003).
3.3. Elastic, Thermodynamic, and Thermal Conductivity Parameters
Besides formation enthalpies, our previous studies show good ability of atomistic modeling to predict the elastic, thermodynamic, and thermal parameters of considered systems. Li et al. (2014), Kowalski and Li (2016), and Kowalski et al. (2017b) computed the elastic parameters of series of lanthanide orthophosphates and obtained good agreements with the available experimental data. With these computed data they provided good estimates of parameters that are key for modeling of solid solutions within these compounds. Maxisch and Ceder (2006) computed elastic parameters of FePO4 and LiFePO4 compounds, which we will use for estimates of thermodynamic parameters of solid solutions in LixFePO4 system (section 3.4). In the follow-up studies of lanthanide phosphate we computed heat capacities for series of these compounds and explained the quasirandom-like behavior of heat capacity along lanthanide series by the lanthanide cation-dependent contribution from thermal excitation of 4f electrons (Schottky effect, Kowalski et al., 2015, 2017b; Ji et al., 2017a). Ji et al. (2019b) computed thermal conductivity for series of lanthanide phosphates and were able to derive accurate information on phonon mean free path in these systems. These studies demonstrate that with appropriate computational setup, accurate information on various physical parameters can be delivered by atomistic simulations and materials effectively screened for desired physical characteristics. This has been used by us to deliver crucial information on orthophosphate-based ceramics for immobilization of actinides (Huittinen et al., 2017, 2018; Ji et al., 2019a).
Besides computing orthophosphate-type ceramics, we derived various parameters for fluorite- and pyrochlore-type compounds. Among those, with atomistic modeling we computed defect formation energies (Li et al., 2015; Li and Kowalski, 2018), barriers for oxygen diffusion (Li and Kowalski, 2018; Bukaemskiy et al., 2021), structural parameters (Finkeldei et al., 2017, 2020; Bukaemskiy et al., 2021), formation enthalpies (Finkeldei et al., 2017; Kowalski, 2020), and properties of doped systems (Finkeldei et al., 2017, 2020). Interestingly, we found that ability of a material to effectively conduct oxygen is also responsible for enhanced radiation damage resistance of selective pyrochlore compounds and stability of defect pyrochlore (defect fluorite) phase (Li et al., 2015; Li and Kowalski, 2018). In addition, Li and Kowalski (2018) have shown that the formation of split vacancy state for pyrochlore lanthanide-zirconates with lanthanide cations after Eu is responsible for the maximum ionic conductivity of Eu2Zr2O7 pyrochlore (Yamamura, 2003).
3.4. Solid Solutions and Solubility
Formation and thermodynamic stability of solid solutions is a topic of studies in various research fields, including battery research (Meethong et al., 2007; Li et al., 2018; Phan et al., 2019). In the past we published a series of studies on lanthanide phosphate ceramic-type solid solutions in the context of geothermometry and nuclear waste management (Li et al., 2014; Kowalski and Li, 2016; Hirsch et al., 2017; Kowalski et al., 2017b; Neumeier et al., 2017b; Ji et al., 2019a). This is because the information gained allows for the assessment of long term stability of ceramic nuclear waste forms against phase separation. It was shown experimentally (Popa et al., 2007; Li et al., 2018; Phan et al., 2019) and by ab initio simulations (Li et al., 2014; Kowalski and Li, 2016) that single phase orthophosphate-based solid solutions are highly regular. The excess enthalpy of mixing, HE, of a A1−xBxPO4 mixed cation compound could be described by a simple equation (Popa et al., 2007):
where W is a Margules interaction parameters (Prieto, 2009). A solid solution is stable against formation of a miscibility gap if W < 2RT, where R is the gas constant. It has been demonstrated experimentally (Yamada et al., 2006; Li et al., 2018; Phan et al., 2019) that LixFePO4 solid solution has a wide miscibility gap, for x between 0.05 and 0.89 at room temperature. However, there is a problem with derivation of consistent model for thermodynamic parameters of mixing for this system, including mixing enthalpy or Margules interaction parameters (Phan et al., 2019). It is thus of great interest to characterize W parameter for LixFePO4 system using the computational methods that were proven by us for lanthanide phosphates (Kowalski and Li, 2016; Ji et al., 2019a).
The first systematic ab initio calculations of W parameters for monazite-type, orthophosphate solid solutions were performed by Li et al. (2014). In follow-up studies (Kowalski and Li, 2016), we explained the quadratic dependence of W parameter on the difference in molar volumes of endmembers (ΔV) by a strain energy-based model, in which
where E is the Young's modulus and V is the volume. These studies show that ΔV is an important parameter that determines the value of W parameter. Considering the elastic properties of both endmember phases, this equation can be also written as (Kowalski and Li, 2016):
where GA is the shear modulus of the doped phase and BB is the bulk modulus of the dopant phase. The Young's, shear and bulk moduli of FePO4 and LiFePO4 phases as computed by Maxisch and Ceder (2006) are given in Table 5. The values of Margules interaction parameter computed with the Equations (4) and (5) are also reported. These values are too low and would result in thermodynamically stable solid solution at all Li content (such forms at room temperature for W < 5 kJ/mol, Li et al., 2014), which is inconsistent with experimental data of Yamada et al. (2006).
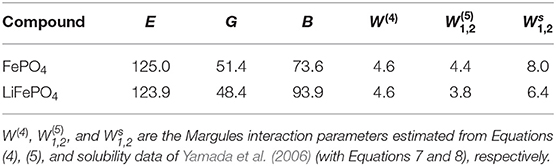
Table 5. The computed by Maxisch and Ceder (2006) with the DFT+U method Young's (E), shear (G) and bulk moduli (B) for FePO4 and LiFePO4 compounds. The values are given in GPa.
For lanthanide orthophosphates, Neumeier et al. (2017b) compared the derived ab initio W parameters with the calorimetric measurements of La1−xLnxPO4 (Ln = Eu, Gd) solid solutions. The measured values are smaller than the computed ones. The reason for this discrepancy is the difference in the value of measured and computed ΔV. When Neumeier et al. (2017b) used the measured ΔV values and rescaled the ab initio computed values according to Equation (4) as:
they obtained a good match to the measured values. However, in our estimate we used the experimental volumes (Table 2) and any such a correction would require significant error in one of the reported volumes. In order to obtain the Margules interaction parameters that are consistent with the measured solubilities (Yamada et al., 2006, see below), instead of the reported ΔV = 20 Å3 between the two endmember phases it should be ΔV = 26 Å3. Our computed volumes, however, give even smaller difference of ΔV = 14 Å3 (Table 2).
The experimental maximum solubilities of Yamada et al. (2006) show slightly asymmetric solid solution with maximum solubility of Li in FePO4 of x1 = 0.05 and content/depletion of Li in LiFePO4 of x2 = 0.89. Such an offset between both solubilities is consistent with the prediction of Equation (5). With these measured solubilities, the Margules interaction parameters can be derived. The maximum solubilities x1 and x2 at the two ends of slightly asymmetric solid solution or the relevant Margules interaction parameters W1 and W2 can be derived by solving self-consistently two equations (Mogilevsky, 2007; Ji et al., 2019a):
These equations realize chemical equilibrium between Li (equilibrium concentration of x1 in FePO4) and Li-vacancy (equilibrium concentration of x2 in LiFePO4) in FePO4 (first equation) and LixFePO4 (second equation) phases, respectively, and are derived by equality of the respected chemical potentials (Mogilevsky, 2007). The self-consistent solution of Equations (7) and (8) can be easily done in a numerical way. The resulted Margules interaction parameters for LixFePO4 solid solution obtained taking the measured solubilities as an input (Yamada et al., 2006) and assuming room temperature are given in Table 5. The derived W1 = 8.0 kJ/mol and W2 = 6.4 kJ/mol would result in an excess mixing enthalpy, HE of 1.7 kJ/mol at x = 0.6 [computed as , Li et al., 2014], which is in perfect agreement with the experimental measurements (~2.5±1.0 kJ/mol, Stevens et al., 2006; Phan et al., 2019). Interestingly, the same values can be derived from Equation (5) and elastic moduli reported in Table 5, assuming ΔV = 26 Å3. We note that such an offset is plausible because we consider a solid solution between Li cation and a vacant site, while the considered models were designed strictly for mixing of two cation species. We also attempted direct computation of Margules interaction parameter by ab initio methods. We obtained values between 13 and 28 kJ/mol, depending on the computational approach. However, we notice that such a small value of energy (enthalpy) is very sensitive to the computational setup, and can not be derived here precisely because of the inability of computing forces with the Wannier projectors scheme (see section 3.2). Other studies also result in large spread of predicted values (Figure 2 in Phan et al., 2019) and indicate sensitivity of this parameter to other effects, including the electronic entropy of Fe in different redox state in the LixFePO4 solid solution (Zhou et al., 2006).
In Figure 3, we plot the free energy of mixing at room temperature, using the Margules interaction parameters extracted form the maximum solubilities measurements of Yamada et al. (2006). With the horizontal line we indicate the widths of wide miscibility and spinodal gaps. This information is important for understanding of charging relationships in the LixFePO4 system (Phan et al., 2019).
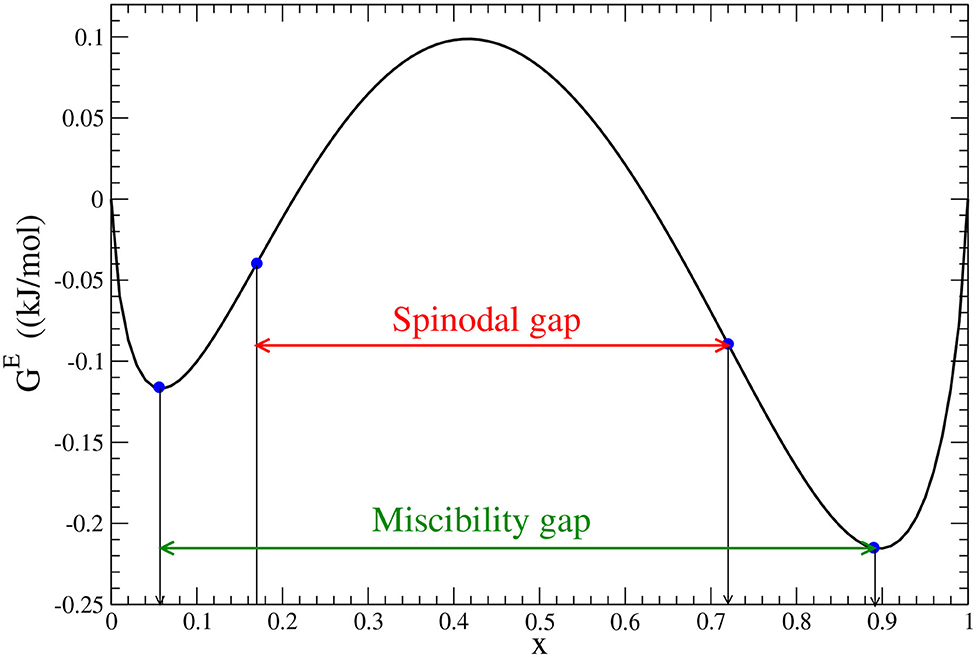
Figure 3. The mixing free energy for LixFePO4 solid solution computed at room temperature, T, as (Li et al., 2014), assuming configurational entropy of ideal mixing and neglecting other entropy contributions. R is the gas constant. W1 and W2 Margules interaction parameters are these from last column of Table 5. The miscibility and spinodal gaps are indicated by minima and inflection points marked with blue circles.
3.5. Ionic Conductivity
Because of high ionic conductivity, yttria-stabilized zirconia (see Figure 4 for the structure) is commonly used as solid state electrolyte. Its ionic conduction properties have been investigated in many studies, including experimental (Strickler and Carlson, 1964, 1965; Ioffe et al., 1978; Li et al., 1994; Lee et al., 2001, 2003; Kilo et al., 2003; Zhang et al., 2007) as well as theoretical and atomistic modeling approaches (Kilo et al., 2003; Krishnamurthy et al., 2005b; Sizov et al., 2014; Bukaemskiy et al., 2021). The conductivity is usually described using Arrhenius-type equation (Ioffe et al., 1978; Ahamer et al., 2017) and the ionic conductivity can be written in the following simple form:
where σ0 is the pre-exponential factor and Ea is the activation energy. In Table 6, we report the available experimental and computed data on Ea together with the results of our simulations. We note that molecular dynamics simulations could be also used for simulation of ionic diffusion, but for systems with activation barriers close to ~1 eV such a method requires long simulation times (e.g., 10 ns as applied by Sizov et al., 2014). Such simulation times are beyond the capability of ab initio molecular dynamics methods (capable of simulations at ps time scales) and could be performed only with less accurate description of interatomic interactions by simple interatomic potentials, like in studies of Sizov et al. (2014).
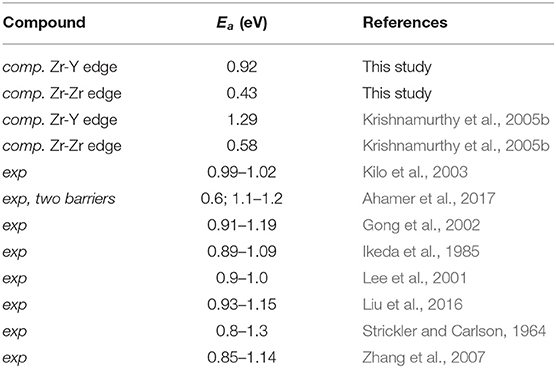
Table 6. The computed here and previously published data on Ea for ionic conductivity in Y-stabilized zirconia.
The computed activation energy of 0.92 eV is well-consistent with the measured values as well as with some previous theoretical predictions. Here, following the studies of Ahamer et al. (2017) and Guan et al. (2020) we assume diffusion along Y-Zr edge as a diffusion rate determining step. Interestingly, the activation barrier for transition along Zr-Zr edge is twice smaller as the one for transition along Y-Zr edge, which is in line with previous findings (Krishnamurthy et al., 2005b). The diffusion paths along these two edges are depicted in Figure 4. In the performed simulations with Eu as dopant, the activation barrier is comparable (0.97eV). This is consistent with previous studies showing similar effects of other tri-valent cations on stabilization of cubic zirconia and its ionic conductivity (Krishnamurthy et al., 2005a). With the derived activation barrier we also computed the attempt frequency, ν0, using the transition state theory (Equation 2). We obtained value of 2.37 · 1012s−1. With the computed activation energy and the attempt frequency, using Equation (1) we derived temperature and Y-content dependent ionic conductivity. The results are plotted in Figure 5 against the experimental data. Our model matches the measured values reasonably well, including temperature and Y content dependence. This is in great part due to inclusion of vacancy migration probability as estimated by Bukaemskiy et al. (2021) (see Equation 1). This shows that atomistic modeling and appropriate theoretical consideration can deliver accurate prediction for ionic conductivity in solid state electrolyte candidate materials. Nevertheless, as discussed by Ahamer et al. (2017) and Guan et al. (2020), the oxygen diffusion in yttria-stabilized zirconia is a complex process, which details, however, could be revealed by combination of computed and measured data.
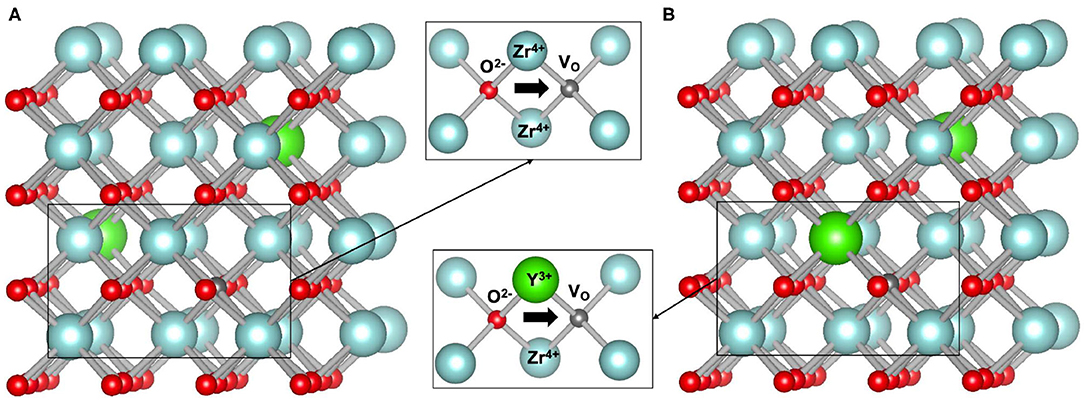
Figure 4. The structure of yttria-stabilized zirconia (YO1.5 − ZrO2 system) with indication of the oxygen diffusion mechanism along possible Zr-Zr (A) and Y-Zr (B) edges.
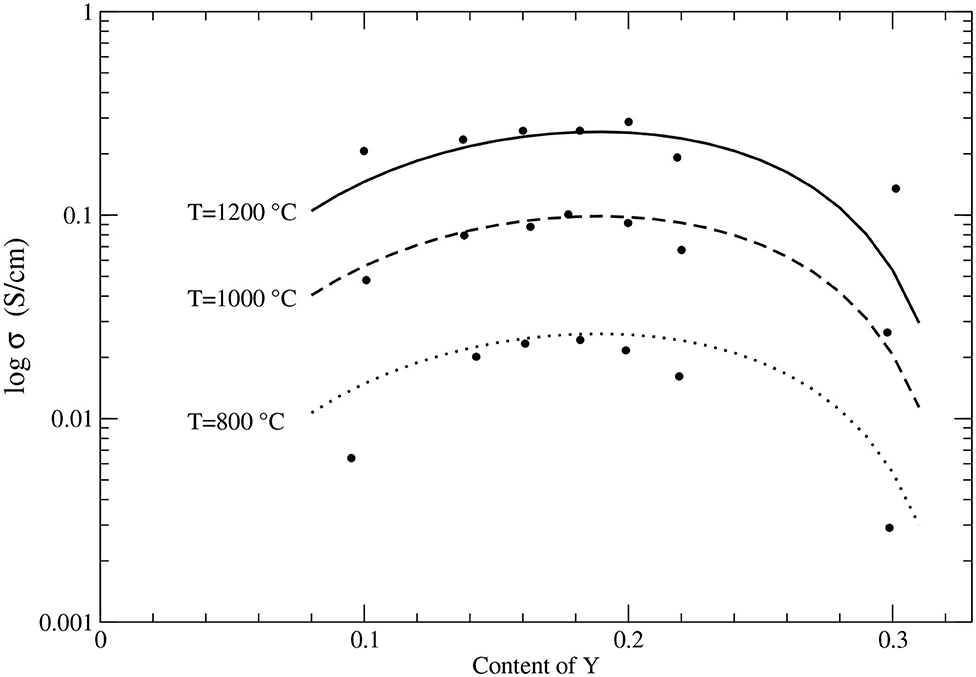
Figure 5. The computed (lines) ionic conductivity in YO1.5 − ZrO2 system. The data (black filled circles) come from Strickler and Carlson (1964) and Liu et al. (2016).
4. Conclusions
In this contribution we presented an overview of our decade-long atomistic modeling contribution to the research on orthophosphates and zirconates type ceramics. We discussed the atomistic modeling derivation of structural, thermodynamic, and diffusion properties that are of importance for application of these materials as compounds in energy storage devices. In particular, we discussed the importance of the application of parameter free DFT+U approach and selection of realistic projector functions for counting the occupations of strongly correlated d and f orbitals for the Hubbard model-based DFT+U scheme. Only with this approach we were able to correctly reproduce the measured formation enthalpies of FePO4 and LiFePO4 phases. The consideration of the thermodynamic properties of LixFePO4 solid solutions indicates a system with a wide miscibility gap, which values and slight asymmetry are qualitatively consistent with the existing experimental data. Based on the measured solubility data we derived set of Margules interaction parameters that describe this solid solution. The resulting excess free energy of mixing shows wide miscibility and spinodal gaps at room temperature. We also discussed our studies of zirconium-based ceramics. In particular, we derived the stability diagram of yttrium-doped zirconia, showing that it stabilizes in cubic phase at Y content of ~8%, well in line with the experimental measurements. The computed formation enthalpies along YO1.5 − ZrO2 solid solution are also well consistent with the measured data. With the computation of transition state and application of vacancy distribution model we were able to derive activation energies and temperature-dependent ionic conductivities for oxygen diffusion in this material, that are well-consistent with the measured data. This shows the power of carefully set up atomistic modeling for computation of various properties of ceramic materials as compounds for energy storage devices.
We discussed various successfully studies of application of atomistic modeling to prediction of a set of physical and chemical properties of orthophosphates and zirconates. In most cases, the best results have been obtained by a joint computational and experimental approach, or at least by extensive testing and comparison to the available experimental data. Application of a reliable, state-of-the-art ab initio approach is also a crucial factor contributing to this success. With the steady increase in the availability of computational power we expect that atomistic modeling research will be applied to tackle even more complex problems, such as kinetically driven dissolution or corrosion processes, and for effective screening of materials for energy applications.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
PK, ZH, and OC contributed equally to computing the data. PK performed collective analysis of the data and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the computing time granted by the JARA Vergabegremium and provided on the JARA Partition part of the supercomputers JURECA at Forschungszentrum Jülich and CLAIX at RWTH Aachen University.
Footnotes
1. ^In this contribution we call DFT methods an ab initio approach as the exchange-correlation functionals utilized in our studies were designed based on pure-theoretical considerations.
References
Ahamer, C., Opitz, A. K., Rupp, G. M., and Fleig, J. (2017). Revisiting the temperature dependent ionic conductivity of yttria stabilized zirconia (YSZ). J. Electrochem. Soc. 164, F790–F803. doi: 10.1149/2.0641707jes
Ali, K., Arya, A., Ghosh, P. S., and Dey, G. K. (2016). A first principle study of the pressure dependent elastic properties of monazite LaPO4. AIP Conf. Proc. 1728:020090. doi: 10.1063/1.4946141
Andrehs, G., and Heinrich, W. (1998). Experimental determination of REE distributions between monazite and xenotime: potential for temperature-calibrated geochronology. Chem. Geol. 149, 83–96. doi: 10.1016/S0009-2541(98)00039-4
Anithakumari, P., Grover, V., Nandi, C., Bhattacharyya, K., and Tyagi, A. K. (2016). Utilizing non-stoichiometry in Nd2Zr2O7 pyrochlore: exploring superior ionic conductors. RSC Adv. 6, 97566–97579. doi: 10.1039/C6RA08722A
Beridze, G., Birnie, A., Koniski, S., Ji, Y., and Kowalski, P. M. (2016). DFT+U as a reliable method for efficient ab initio calculations of nuclear materials. Prog. Nucl. Energ. 92, 142–146. doi: 10.1016/j.pnucene.2016.07.012
Beridze, G., and Kowalski, P. M. (2014). Benchmarking the DFT+U method for thermochemical calculations of uranium molecular compounds and solids. J. Phys. Chem. A 118, 11797–11810. doi: 10.1021/jp5101126
Blanca-Romero, A., Kowalski, P. M., Beridze, G., Schlenz, H., and Bosbach, D. (2014). Performance of DFT+U method for prediction of structural and thermodynamic parameters of monazite-type ceramics. J. Comput. Chem. 35, 1339–1346. doi: 10.1002/jcc.23618
Bukaemskiy, A. A., Vinograd, V. L., and Kowalski, P. M. (2021). Ion distribution models for defect fluorite ZrO2-AO1.5 (A=Ln, Y) solid solutions: I. Relationship between lattice parameter and composition. Acta Mater. 202, 99–111. doi: 10.1016/j.actamat.2020.10.045
Cerdas, F., Titscher, P., Bognar, N., Schmuch, R., Winter, M., Kwade, A., et al. (2018). Exploring the effect of increased energy density on the environmental impacts of traction aatteries: a comparison of energy optimized lithium-ion and lithium-sulfur batteries for mobility applications. Energies 11:150. doi: 10.3390/en11010150
Chroneos, A., Rushton, M., Jiang, C., and Tsoukalas, L. (2013). Nuclear wasteform materials: atomistic simulation case studies. J. Nucl. Mater. 441, 29–39. doi: 10.1016/j.jnucmat.2013.05.012
Cococcioni, M., and de Gironcoli, S. (2005). Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71:035105. doi: 10.1103/PhysRevB.71.035105
Diazguillen, J., Diazguillen, M., Padmasree, K., Fuentes, A., Santamaria, J., and Leon, C. (2008). High ionic conductivity in the pyrochlore-type Gd2 − yLayZr2O7 solid solution (0 ≤ y ≤ 1). Solid State Ion. 179, 2160–2164. doi: 10.1016/j.ssi.2008.07.015
Dong, H., Guo, H., He, Y., Gao, J., Han, W., Lu, X., et al. (2017). Structural stability and Li-ion transport property of LiFePO4 under high-pressure. Solid State Ion. 301, 133–137. doi: 10.1016/j.ssi.2017.01.026
Drey, D. L., O'Quinn, E. C., Subramani, T., Lilova, K., Baldinozzi, G., Gussev, I. M., et al. (2020). Disorder in Ho2Ti2-xZrxO7: pyrochlore to defect fluorite solid solution series. RSC Adv. 10, 34632–34650. doi: 10.1039/D0RA07118H
Dunn, J. B., Gaines, L., Kelly, J. C., James, C., and Gallagher, K. G. (2015). The significance of li-ion batteries in electric vehicle life-cycle energy and emissions and recycling's role in its reduction. Energy Environ. Sci. 8, 158–168. doi: 10.1039/C4EE03029J
Eremin, N. N., Marchenko, E. I., Petrov, V. G., Mitrofanov, A. A., and Ulanova, A. S. (2019). Solid solutions of monazites and xenotimes of lanthanides and plutonium: atomistic model of crystal structures, point defects and mixing properties. Comput. Mater. Sci. 157, 43–50. doi: 10.1016/j.commatsci.2018.10.025
Ewing, R., and Wang, L. (2002). Phosphates as nuclear waste forms. Rev. Mineral. Geochem. 48, 673–699. doi: 10.2138/rmg.2002.48.18
Feng, J., Xiao, B., Zhou, R., and Pan, W. (2013). Anisotropy in elasticity and thermal conductivity of monazite-type REPO4 (RE=La, Ce, Nd, Sm, Eu and Gd) from first-principles calculations. Acta Mater. 61, 7364–7383. doi: 10.1016/j.actamat.2013.08.043
Finger, L. W., and Hazen, R. M. (1980). Crystal structure and isothermal compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 51, 5362–5367. doi: 10.1063/1.327451
Finkeldei, S., Kegler, P., Kowalski, P., Schreinemachers, C., Brandt, F., Bukaemskiy, A., et al. (2017). Composition dependent order-disorder transition in NdxZr1 − xO2 − 0.5x pyrochlores: a combined structural, calorimetric and ab initio modeling study. Acta Mater. 125, 166–176. doi: 10.1016/j.actamat.2016.11.059
Finkeldei, S., Stennett, M. C., Kowalski, P. M., Ji, Y., de Visser-Týnová, E., Hyatt, N. C., et al. (2020). Insights into the fabrication and structure of plutonium pyrochlores. J. Mater. Chem. A 8, 2387–2403. doi: 10.1039/C9TA05795A
García-Moreno, O., Alvarez-Vega, M., García-Jaca, J., Gallardo-Amores, J. M., Sanjuán, M. L., and Amador, U. Influence of the structure on the electrochemical performance of lithium transition metal phosphates as cathodic materials in rechargeable lithium batteries: A new high-pressure form of LiMPO4 (M = Fe Ni). Chem. Mater. (2001) 13, 1570–1576. doi: 10.1021/cm000596p
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., et al. (2009). Quantum espresso: a modular and open-source software project for quantum simulations of materials. J. Phys. 21:395502. doi: 10.1088/0953-8984/21/39/395502
Gomis, O., Lavina, B., Rodríguez-Hernández, P., Muñoz, A., Errandonea, R., Errandonea, D., et al. (2017). High-pressure structural, elastic, and thermodynamic properties of zircon-type HoPO4 and TmPO4. J. Phys. 29:095401. doi: 10.1088/1361-648X/aa516a
Gong, J., Li, Y., Tang, Z., Xie, Y., and Zhang, Z. (2002). Temperature-dependence of the lattice conductivity of mixed calcia/Yttria-stabilized zirconia. Mater. Chem. Phys. 76, 212–216. doi: 10.1016/S0254-0584(01)00522-3
Götsch, T., Wallisch, W., Stöger-Pollach, M., Klötzer, B., and Penner, S. (2016). From zirconia to Yttria: sampling the YSZ phase diagram using sputter-deposited thin films. AIP Adv. 6:025119. doi: 10.1063/1.4942818
Guan, S.-H., Zhang, K.-X., Shang, C., and Liu, Z.-P. (2020). Stability and anion diffusion kinetics of Yttria-stabilized zirconia resolved from machine learning global potential energy surface exploration. J. Chem. Phys. 152:094703. doi: 10.1063/1.5142591
Hirsch, A., Kegler, P., Alencar, I., Ruiz-Fuertes, J., Shelyug, A., Peters, L., et al. (2017). Structural, vibrational, and thermochemical properties of the monazite-type solid solution La1 − xPrxO4. J. Solid State Chem. 245, 82–88. doi: 10.1016/j.jssc.2016.09.032
Huittinen, N., Arinicheva, Y., Kowalski, P., Vinograd, V., Neumeier, S., and Bosbach, D. (2017). Probing structural homogeneity of La1 − xGdxPO4 monazite-type solid solutions by combined spectroscopic and computational studies. J. Nucl. Mater. 486, 148–157. doi: 10.1016/j.jnucmat.2017.01.024
Huittinen, N., Scheinost, A. C., Ji, Y., Kowalski, P. M., Arinicheva, Y., Wilden, A., et al. (2018). A spectroscopic and computational study of Cm3+ Incorporation in lanthanide phosphate rhabdophane (LnPO4 · 0.67H2O) and monazite (LnPO4). Inorg. Chem. 57, 6252–6265. doi: 10.1021/acs.inorgchem.8b00095
Ikeda, S., Sakurai, O., Uematsu, K., Mizutani, N., and Kato, M. (1985). Electrical-conductivity of yttria-stabilized zirconia single-crystals. J. Mater. Sci. 20, 4593–4600. doi: 10.1007/BF00559349
Ioffe, A., Rutman, D., and Karpachov, S. (1978). On the nature of the conductivity maximum in zirconia-based solid electrolytes. Electrochim. Acta 23, 141–142. doi: 10.1016/0013-4686(78)80110-8
Iyer, R. G., Delacourt, C., Masquelier, C., Tarascon, J.-M., and Navrotsky, A. (2006). Energetics of LiFePO4 and polymorphs of its delithiated form, FePO4. Electrochem. Solid-State Lett. 9, A46–A48. doi: 10.1149/1.2140496
Jahn, S., and Kowalski, P. M. (2014). Theoretical approaches to structure and spectroscopy of earth materials. Rev. Mineral. Geochem. 78:691. doi: 10.2138/rmg.2014.78.17
Ji, Y., Beridze, G., Bosbach, D., and Kowalski, P. M. (2017a). Heat capacities of xenotime-type ceramics: an accurate ab initio prediction. J. Nucl. Mater. 494, 172–181. doi: 10.1016/j.jnucmat.2017.07.026
Ji, Y., Beridze, G., Li, Y., and Kowalski, P. M. (2017b). Large scale simulation of nuclear waste materials. Energy Proc. 127, 416–424. doi: 10.1016/j.egypro.2017.08.108
Ji, Y., Kowalski, P. M., Kegler, P., Huittinen, N., Marks, N. A., Vinograd, V. L., et al. (2019a). Rare-earth orthophosphates from atomistic simulations. Front. Chem. 7:197. doi: 10.3389/fchem.2019.00197
Ji, Y., Kowalski, P. M., Neumeier, S., Deissmann, G., Kulriya, P. K., and Gale, J. D. (2017c). Atomistic modeling and experimental studies of radiation damage in monazite-type LaPO4 ceramics. Nucl. Instrum. Methods Phys. Res. Sect. B 393, 54–58. doi: 10.1016/j.nimb.2016.09.031
Ji, Y., Marks, N. A., Bosbach, D., and Kowalski, P. M. (2019b). Elastic and thermal parameters of lanthanide-orthophosphate (LnPO4) ceramics from atomistic simulations. J. Eur. Ceram. 39, 4264–4274. doi: 10.1016/j.jeurceramsoc.2019.05.038
Jin, B., Qin Zhang, R., and Jiang, Q. (2013). Electronic and atomic structures of LiMPO4 (M= Fe, Fe1/4Mn1/4Co1/4Ni1/4): a DFT study. Adv. Electrochem. 1, 22–26. doi: 10.1166/adel.2013.1005
Jolley, K., Asuvathraman, R., and Smith, R. (2017). Inter-atomic potentials for radiation damage studies in CePO4 monazite. Nucl. Instrum. Methods Phys. Res. B 393, 93–96. doi: 10.1016/j.nimb.2016.10.016
Kick, M., Reuter, K., and Oberhofer, H. (2019). Intricacies of DFT+U, not only in a numeric atom centered orbital framework. J. Chem. Theory Comput. 15, 1705–1718. doi: 10.1021/acs.jctc.8b01211
Kilo, M., Argirusis, C., Borchardt, G., and Jackson, R. A. (2003). Oxygen diffusion in yttria stabilised zirconia-experimental results and molecular dynamics calculations. Phys. Chem. Chem. Phys. 5, 2219–2224. doi: 10.1039/B300151M
Kowalski, P. M. (2020). Formation enthalpy of Ln2B2O7-type (B=Ti,Sn,Hf,Zr) compounds. Scr. Mater. 189, 7–10. doi: 10.1016/j.scriptamat.2020.07.048
Kowalski, P. M., Beridze, G., Ji, Y., and Li, Y. (2017a). Towards reliable modeling of challenging f electrons bearing materials: experience from modeling of nuclear materials. MRS Adv. 2, 491–497. doi: 10.1557/adv.2017.46
Kowalski, P. M., Beridze, G., Li, Y., Ji, Y., Friedrich, C., Sasioglu, E., et al. (2016). Feasible and reliable ab initio approach to computation of materials relevant for nuclear waste management. Ceram. Trans. 258, 205–217. doi: 10.1002/9781119236016.ch21
Kowalski, P. M., Beridze, G., Vinograd, V. L., and Bosbach, D. (2015). Heat capacities of lanthanide and actinide monazite-type ceramics. J. Nucl. Mater. 464, 147–154. doi: 10.1016/j.jnucmat.2015.04.032
Kowalski, P. M., Ji, Y., Li, Y., Arinicheva, Y., Beridze, G., Neumeier, S., et al. (2017b). Simulation of ceramic materials relevant for nuclear waste management: case of La1 − xEuxPO4 solid solution. Nucl. Instrum. Methods Phys. Res. Sect. B 393, 68–72. doi: 10.1016/j.nimb.2016.09.029
Kowalski, P. M., and Li, Y. (2016). Relationship between the thermodynamic excess properties of mixing and the elastic moduli in the monazite-type ceramics. J. Eur. Ceram. Soc. 36, 2093–2096. doi: 10.1016/j.jeurceramsoc.2016.01.051
Krishnamurthy, R., Srolovitz, D. J., Kudin, K. N., and Car, R. (2005a). Effects of lanthanide dopants on oxygen diffusion in Yttria-stabilized zirconia. J. Am. Ceram. 88, 2143–2151. doi: 10.1111/j.1551-2916.2005.00353.x
Krishnamurthy, R., Yoon, Y.-G., Srolovitz, D. J., and Car, R. (2005b). Oxygen diffusion in yttria-stabilized zirconia: a new simulation model. J. Am. Ceram. 87, 1821–1830. doi: 10.1111/j.1151-2916.2004.tb06325.x
Krogstad, J. A., Lepple, M., Gao, Y., Lipkin, D. M., and Levi, C. G. (2011). Effect of yttria content on the zirconia unit cell parameters. J. Am. Ceram. 94, 4548–4555. doi: 10.1111/j.1551-2916.2011.04862.x
Kvashnina, K. O., Kowalski, P. M., Butorin, S. M., Leinders, G., Pakarinen, J., Bès, R., et al. (2018). Trends in the valence band electronic structures of mixed uranium oxides. Chem. Commun. 54, 9757–9760. doi: 10.1039/C8CC05464A
Lee, J.-H., Yoon, S., Kim, B.-K., Kim, J., Lee, H.-W., and Song, H.-S. (2001). Electrical conductivity and defect structure of yttria-doped ceria-stabilized zirconia. Solid State Ion. 144, 175–184. doi: 10.1016/S0167-2738(01)00903-1
Lee, K., Youn, Y., and Han, S. (2017). Identification of ground-state spin ordering in antiferromagnetic transition metal oxides using the Ising model and a genetic algorithm. Sci. Technol. Adv. Mater 18, 246–252. doi: 10.1080/14686996.2017.1300046
Lee, T. A., Navrotsky, A., and Molodetsky, I. (2003). Enthalpy of formation of cubic yttria-stabilized zirconia. J. Mater. Res. 18, 908–918. doi: 10.1557/JMR.2003.0125
Li, P., Chen, I.-W., and Penner-Hahn, J. E. (1994). Effect of dopants on zirconia stabilization - an x-ray absorption study: I, trivalent dopants. J. Am. Ceram. 77, 118–128. doi: 10.1111/j.1151-2916.1994.tb06964.x
Li, Y., and Kowalski, P. M. (2018). Energetics of defects formation and oxygen migration in pyrochlore compounds from first principles calculations. J. Nucl. Mater. 505, 255–261. doi: 10.1016/j.jnucmat.2017.11.005
Li, Y., Kowalski, P. M., Beridze, G., Birnie, A. R., Finkeldei, S., and Bosbach, D. (2015). Defect formation energies in A2B2O7 pyrochlores. Scr. Mater. 107, 18–21. doi: 10.1016/j.scriptamat.2015.05.010
Li, Y., Kowalski, P. M., Beridze, G., Blanca-Romero, A., Ji, Y., Vinograd, V. L., et al. (2016). Atomistic simulations of ceramic materials relevant for nuclear waste management: cases of monazite and pyrochlore. Ceram. Trans. 255:165. doi: 10.1002/9781119234531.ch15
Li, Y., Kowalski, P. M., Blanca-Romero, A., Vinograd, V., and Bosbach, D. (2014). Ab initio calculation of excess properties of solid solutions. J. Solid State Chem. 220, 137–141. doi: 10.1016/j.jssc.2014.08.005
Li, Z., Yang, J., Li, C., Wang, S., Zhang, L., Zhu, K., et al. (2018). Orientation-dependent lithium miscibility gap in LiFePO4. Chem. Mater. 30, 874–878. doi: 10.1021/acs.chemmater.7b04463
Liu, T., Zhang, X., Wang, X., Yu, J., and Li, L. (2016). A review of zirconia-based solid electrolytes. Ionics 22, 2249–2262. doi: 10.1007/s11581-016-1880-1
López-Solano, J., Rodríguez-Hernández, P., Munoz, A., Gomis, O., Santamaría-Perez, D., Errandonea, D., et al. (2010). Theoretical and experimental study of the structural stability of TbPO4 at high pressures. Phys. Rev. B 81:144126. doi: 10.1103/PhysRevB.81.144126
Mandal, B., Deshpande, S., and Tyagi, A. (2008). Ionic conductivity enhancement in Gd2Zr2O7 pyrochlore by Nd doping. J. Mater. Res. 23, 911–916. doi: 10.1557/jmr.2008.0112
Maxisch, T., and Ceder, G. (2006). Elastic properties of olivine LixFePO4 from first principles. Phys. Rev. B 73:174112. doi: 10.1103/PhysRevB.73.174112
Meethong, N., Huang, H.-Y. S., Carter, W. C., and Chiang, Y.-M. (2007). Size-dependent lithium miscibility gap in nanoscale Li1 − xFePO4. Electrochem. Solid-State Lett. 10:A134. doi: 10.1149/1.2710960
Mogilevsky, P. (2007). On the miscibility gap in monazite-xenotime systems. Phys. Chem. Miner. 34, 201–214. doi: 10.1007/s00269-006-0139-1
Moynihan, C. T., Gavin, D. L., and Syed, R. (1982). Pre-exponential term in the arrhenius equation for electrical conductivity of glass. J. Phys. Colloq. 43, C9-395–C9-398. doi: 10.1051/jphyscol:1982975
Murphy, G. L., Zhang, Z., Tesch, R., Kowalski, P. M., Avdeev, M., Kuo, E. Y., et al. (2021). Tilting and distortion in rutile-related mixed metal ternary uranium oxides: a structural, spectroscopic, and theoretical investigation. Inorgan. Chem. 60, 2246–2260. doi: 10.1021/acs.inorgchem.0c03077
Neumeier, S., Arinicheva, Y., Ji, Y., Heuser, J. M., Kowalski, P. M., Kegler, P., et al. (2017a). New insights into phosphate based materials for the immobilisation of actinides. Radiochim. Acta 105, 961–984. doi: 10.1515/ract-2017-2819
Neumeier, S., Kegler, P., Arinicheva, Y., Shelyug, A., Kowalski, P. M., Schreinemachers, C., et al. (2017b). Thermochemistry of La1 − xLnxPO4 monazites (Ln=Gd, Eu). J. Chem. Thermodyn. 105, 396–403. doi: 10.1016/j.jct.2016.11.003
Padhi, A. K., Nanjundaswamy, K. S., and Goodenough, J. B. (1997). Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144:1188. Available online at: https://iopscience.iop.org/article/10.1149/1.1837571
Perdew, J. P., Ruzsinszky, A., Csonka, G. I., Vydrov, O. A., Scuseria, G. E., Constantin, L. A., et al. (2008). Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100:136406. doi: 10.1103/PhysRevLett.100.136406
Phan, A. T., Gheribi, A. E., and Chartrand, P. (2019). Modelling of phase equilibria of LiFePO4−FePO4 olivine join for cathode material. Can. J. Chem. 97, 2224–2233. doi: 10.1002/cjce.23416
Ploc, R. A. (1981). The lattice parameter of cubic ZrO2 formed on zirconium. J. Nucl. Mater. 99, 124–128. doi: 10.1016/0022-3115(81)90146-X
Pomfret, M. B., Stoltz, C., Varughese, B., and Walker, R. A. (2005). Structural and compositional characterization of yttria-stabilized zirconia: evidence of surface-stabilized, low-valence metal species. Anal. Chem. 77, 1791–1795. doi: 10.1021/ac048600u
Popa, K., Konings, R. J. M., and Geisler, T. (2007). High-temperature calorimetry of (La1 − xLnx)PO4 solid solutions. J. Chem. Thermodyn. 39, 236–239. doi: 10.1016/j.jct.2006.07.010
Prieto, M. (2009). Thermodynamics of solid solution-aqueous solution systems. Rev. Mineral. Geochem. 70, 47–85. doi: 10.2138/rmg.2009.70.2
Prosini, P. P., Lisi, M., Zane, D., and Pasquali, M. (2002). Determination of the chemical diffusion coefficient of lithium in LiFePO4. Solid State Ion. 148, 45–51. doi: 10.1016/S0167-2738(02)00134-0
Rustad, J. R. (2012). Density functional calculations of the enthalpies of formation of rare-earth orthophosphates. Am. Mineral. 97:791. doi: 10.2138/am.2012.3948
Schlenz, H., Peters, L., Roth, G., Hirsch, A., and Neumeier, S. (2018). 9. Phosphates as Safe Containers for Radionuclides. Technical report, Nukleare Entsorgung und Reaktorsicherheit.
Shamblin, J., Feygenson, M., Neuefeind, J., Tracy, C., Zhang, F., Finkeldei, S., et al. (2016). Probing disorder in isometric pyrochlore and related complex oxides. Nat. Mater. 15, 507–511. doi: 10.1038/nmat4581
Shein, I. R., and Shalaeva, E. V. (2016). Pressure-induced zircon to monazite phase transition in Y1 − xLaxPO4: first-principles calculations. J. Struct. Chem. 57, 1513–1518. doi: 10.1134/S0022476616080047
Sizov, V. V., Lampinen, M. J., and Laaksonen, A. (2014). Molecular dynamics simulation of oxygen diffusion in cubic yttria-stabilized zirconia: effects of temperature and composition. Solid State Ionics 266, 29–35. doi: 10.1016/j.ssi.2014.08.003
Stavrou, E., Tatsi, A., Raptis, C., Efthimiopoulos, I., Syassen, K., Mu noz, A., et al. (2012). Effects of pressure on the structure and lattice dynamics of TmPO4: experiments and calculations. Phys. Rev. B 85:024117. doi: 10.1103/PhysRevB.85.024117
Stevens, R., Dodd, J. L., Kresch, M. G., Yazami, R., Fultz, B., Ellis, B., et al. (2006). Phonons and thermodynamics of unmixed and disordered Li0.6FePO4. J. Phys. Chem. B 110, 22732–22735. doi: 10.1021/jp063831l
Strickler, D. W., and Carlson, W. G. (1964). Ionic conductivity of cubic solid solutions in the system CaO|Y2O3|ZrO2. J. Am. Ceram. 47, 122–127. doi: 10.1111/j.1151-2916.1964.tb14368.x
Strickler, D. W., and Carlson, W. G. (1965). Electrical conductivity in the ZrO2-rich region of several M2O3 − ZrO2 Systems. J. Am. Ceram. 48, 286–289. doi: 10.1111/j.1151-2916.1965.tb14742.x
Sun, M., Stackhouse, J., and Kowalski, P. (2020). The +2 oxidation state of cr incorporated into the crystal lattice of UO2. Commun. Mater. 1:13. doi: 10.1038/s43246-020-0014-5
Tang, P., and Holzwarth, N. (2003). Electronic structure of FePO4, LiFePO4, and related materials. Phys. Rev. B 68:165107. doi: 10.1103/PhysRevB.68.165107
Ushakov, S., Helean, K., and Navrotsky, A. (2001). Thermochemistry of rare-earth orthophosphates. J. Mater. Res. 16:2623. doi: 10.1557/JMR.2001.0361
Vanderbilt, D. (1990). Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41:7892. doi: 10.1103/PhysRevB.41.7892
Wang, J., Zhou, Y., and Lin, Z. (2005). First-principles elastic stiffness of LaPO4 monazite. Appl. Phys. Lett. 87:051902. doi: 10.1063/1.2005392
Whittingham, M. S., Song, Y., Lutta, S., Zavalij, P. Y., and Chernova, N. A. (2005). Some transition metal (oxy) phosphates and vanadium oxides for lithium batteries. J. Mater. Chem. 15, 3362–3379. doi: 10.1039/b501961c
Williams, M. L., Jercinovic, M. J., and Hetherington, C. J. (2007). Microprobe monazite geochronology: understanding geologic processes by integrating composition and chronology. Annu. Rev. Earth Planet. Sci. 35, 137–175. doi: 10.1146/annurev.earth.35.031306.140228
Wu, X., Kang, F., Duan, W., and Li, J. (2019). Density functional theory calculations: a powerful tool to simulate and design high-performance energy storage and conversion materials. Proc. Nat. Sci. Mater. 29, 247–255. doi: 10.1016/j.pnsc.2019.04.003
Wuensch, B. (2000). Connection between oxygen-ion conductivity of pyrochlore fuel-cell materials and structural change with composition and temperature. Solid State Ion 129, 111–133. doi: 10.1016/S0167-2738(99)00320-3
Xia, X.-L., Ouyang, J.-H., and Liu, Z.-G. (2010). Electrical properties of gadolinium-europium zirconate eramics. J. Am. Ceram. 93, 1074–1080. doi: 10.1111/j.1551-2916.2009.03505.x
Yamada, A., Koizumi, H., Nishimura, S.-I., Sonoyama, N., Kanno, R., Yonemura, M., et al. (2006). Room-temperature miscibility gap in LixFePO4. Nat. Mater. 5, 357–360. doi: 10.1038/nmat1634
Yamamura, H. (2003). Electrical conductivity anomaly around fluorite-pyrochlore phase boundary. Solid State Ion. 158, 359–365. doi: 10.1016/S0167-2738(02)00874-3
Zakaria, Z., Abu Hassan, S. H., Shaari, N., Yahaya, A. Z., and Boon Kar, Y. (2020). A review on recent status and challenges of yttria stabilized zirconia modification to lowering the temperature of solid oxide fuel cells operation. Int. J. Energy Res. 44, 631–650. doi: 10.1002/er.4944
Zhang, C., Li, C.-J., Zhang, G., Ning, X.-J., Li, C.-X., Liao, H., et al. (2007). Ionic conductivity and its temperature dependence of atmospheric plasma-sprayed yttria stabilized zirconia electrolyte. Mater. Sci. Eng. B 137, 24–30. doi: 10.1016/j.mseb.2006.10.005
Zhao, X., and Vanderbilt, D. (2002). Phonons and lattice dielectric properties of zirconia. Phys. Rev. B 65:075105. doi: 10.1103/PhysRevB.65.075105
Zhou, F., Maxisch, T., and Ceder, G. (2006). Configurational electronic entropy and the phase diagram of mixed-valence oxides: the case of LixFePO4. Phys. Rev. Lett. 97:155704. doi: 10.1103/PhysRevLett.97.155704
Keywords: orthophosphates, atomistic simulations (ab-initio calculations), energy storage materials, ceramics, thermodynamics, solid solution, solid-state electrolyte
Citation: Kowalski PM, He Z and Cheong O (2021) Electrode and Electrolyte Materials From Atomistic Simulations: Properties of LiXFEPO4 Electrode and Zircon-Based Ionic Conductors. Front. Energy Res. 9:653542. doi: 10.3389/fenrg.2021.653542
Received: 14 January 2021; Accepted: 05 March 2021;
Published: 30 March 2021.
Edited by:
Jun Huang, Central South University, ChinaCopyright © 2021 Kowalski, He and Cheong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piotr M. Kowalski, cC5rb3dhbHNraUBmei1qdWVsaWNoLmRl