 Hongcan Liu
Hongcan Liu Shun Ma
Shun Ma Junjun Wu
Junjun Wu Yingkai Wang
Yingkai Wang Xinghui Wang
Xinghui Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Energy Res., 10 February 2021
Sec. Electrochemical Energy Storage
Volume 9 - 2021 | https://doi.org/10.3389/fenrg.2021.639741
This article is part of the Research TopicEmerging Technologies for Safe Rechargeable Energy StorageView all 4 articles
Compared to liquid electrolytes, lithium solid-state electrolytes have received increased attention in the field of all-solid-state lithium ion batteries due to safety requirements and higher energy density. However, solid-state electrolytes face many challenges, including lower ionic conductivity, complex interfaces, and unstable physical or electrochemical properties. One of the most effective strategies is to find a new type of lithium solid-state electrolyte with improved properties. Traditional trial and error methods require resources and time to verify the new solid-state electrolytes. Recently, new lithium solid-state electrolytes were predicted through machine learning (ML), which has proved to be an efficient and reliable method for screening new functional materials. This paper reviews the lithium solid-state electrolytes that have been discovered based on ML algorithms. The selection and preprocessing of datasets in ML technology are initially discussed before describing the latest developments in screening lithium solid-state electrolytes through different ML algorithms in detail. Lastly, the stability of candidate solid-state electrolytes and the challenges of discovering new lithium solid-state electrolytes through ML are highlighted.
Air pollution control has become an increasingly severe problem in recent years. Some countries aim to ban new gasoline and diesel-powered vehicles by 2030, and governments have implemented policies that develop renewable energy. However, most renewable energy sources, such as wind and solar power, are intermittent energy sources that use rechargeable batteries for storage to enable daily usage. Rechargeable batteries, especially lithium ion batteries (LIBs) with an organic liquid electrolyte, are used in portable consumer electronics and electric vehicles owing to their high energy density and long life span. Even though the energy density of the LIBs has improved tremendously over recent decades, it still needs to be further enhanced to meet the high requirements of the consumers. Moreover, safety is another vital issue that has impeded the application of these types of batteries after LIB safety accidents caused by organic liquid electrolytes (Qu et al., 2015). An efficient solution to this problem is to replace flammable organic electrolytes with solid-state electrolytes, which could significantly reduce the risk of leaks, evaporation, and decomposition and ensure higher safety. Solid-state lithium ion batteries (ASSLIB) are being explored in next-generation LIBs. This research faces the challenge of creating stable solid-state electrolytes with high ionic conductivity (Borodin et al., 2015).
Ideal solid-state electrolytes should ideally have the following merits: high ionic conductivity, a low migration energy barrier, a wide electrochemical window, strong electrochemical stability, and high mechanical rigidity to suppress dendrite growth on the anode. In the past few years, Li10GeP2S12 (Kamaya et al., 2011), Li7P3S11 (Yamane et al., 2007), and some solid polymer electrolytes were widely investigated. However, the lithium solid-state electrolytes reported to-date do not meet these commercial requirements and properties (Do et al., 2013; Sun et al., 2014; Zhang et al., 2014; Chai et al., 2017; Yang et al., 2017) and new solid-state electrolytes with better performance are required. Traditional searching tactics involve trial-and-error methods that require a lot of time to fabricate and verify the new solid-state electrolytes, with slow progress and low efficiency.
The Materials Genome Initiative (founded in 2011) has successfully used a machine learning (ML) algorithm to screen new solid-state electrolytes (Holdren, 2011; Sendek et al., 2019; Hatakeyama-Sato et al., 2018; Xie and Grossman, 2018; Cubuk et al., 2019; Zhang et al., 2019). ML is a statistical method involving different algorithm structures that can learn the key information from available data and extract useful characteristics. The computation speed of ML algorithms is much faster than density functional theory (DFT) (Li et al., 2017; Sendek et al., 2018). Conventional methods (e.g., the first principle-based DFT) may take over four weeks to predict, while ML only takes 1 s (Saal et al., 2013). ML technology is usually applied in forecasting attributes, aiming to reflect molecular materials and targeted attributes. Figure 1A shows the typical workflow used in screening lithium solid-state electrolytes through ML algorithms. The whole screening process begins by selecting suitable descriptors (datasets) divided into three parts: training data, testing data, and validating data. They then use different ML algorithms, including supervised, semi-supervised, and unsupervised learning, to calculate and predict potential candidates. The ionic conductivity of candidates is further verified by the DFT or Vienna Ab-initio Simulation Package (VASP) simulation.

FIGURE 1. (A) Workflow of screening solid-state electrolytes through the ML algorithm. (B) The distribution of Li-contained compounds from the Material Project database. Inset: histogram of materials conductivity over standard value. (C) Computationally observed ionic conductivity in candidate materials and extrapolation to room temperature. Reprinted (adapted) with permission from (Sendek., et al 2018). Copyright (2019) American Chemical Society. (D) Predicted ionic conductivities for the system Li8-cAaBbO4. (E) Relationship between experimental and predicted ionic conductivity.
Different ML algorithms with advantages could lead to varying results in screening lithium solid-state electrolytes. Generally, supervised learning (e.g., support vector machine, neural network, decision tree, regression method) needs huge data with an effective label to support the calculation and more accurate data. Unsupervised learning (e.g., clustering) could calculate the results by itself without labeling. Semi-supervised learning inherits the previous frame and is trained in many datasets. There is little cost involved in training new data, resulting in increasing calculation speed. This review offers a systematic comparison and discussion of the different algorithms used to boost discovery and further advance this new technology in screening the solid-state electrolytes.
The review first illustrates how others address and prepare with datasets, then introduces and discusses the latest developments in screening lithium solid-state electrolytes through different ML algorithms including Neural Network (NN), Support Vector Machine (SVM), Regression, and Clustering in detail. The review finally verifies the stability of some ML predicted materials and highlights the limitations and current challenges faced by ML-based screening for solid-state materials.
ML technology prioritizes choosing an appropriate dataset and then transferring it to a shape that an algorithm can handle. The Material Project is a database of materials and core program of the Materials Genome Initiative (Jain et al., 2013). It contains information about compounds from the Inorganic Crystal Structure Database (ICSD). Figure 1B gives the lithium ionic conductivity distribution of all Li-contained compounds by pymatgen (Ong et al., 2013). The data can be computed and analyzed by the Materials Application Programming Interface. The database includes information on the lattice structure, band structure, density of state, space group, energy, and phase diagram, etc. Different ML algorithms may use diverse descriptors and various representations as an input (Curtarolo et al., 2012; Jain et al., 2015; Kiran and Joseph, 2017). The more suitable the representation of the input data, the more accurately an algorithm can map it to the output data. For this reason, suitable input data should be easier to acquire than the output attributes that will be predicted, and it is better when the dimensionality is lower (Ghiringhelli et al., 2015).
To solve simulation errors, data should be preprocessed deliberatively. The preprocessing stage extracts raw data features and gives the subset unique characteristics, consisting of a continuous curve or discrete data. Feature extraction involves the transformation of the original data to essential characteristics. Its transforms, maps, or changes the dimensions of the original data into new data that reflects the input information more clearly. A large number of descriptors encode structures and properties, including: Coulomb Matrix (Rupp et al., 2012); Finger Prints; Graph Theory (Bonchev and Rouvray, 1991); 3D Geometry, Voronoi Tessellations (Ward et al., 2017); Simplified Molecular Input Line Entry Systems (SMILES) based on radial distribution functions (Schütt et al., 2014); and property-labeled material fragments (Isayev et al., 2017).
Neural networks, like the human brain, have many layers and units. Weights store knowledge and then form a completed net. It can process the input data layer by layer, thus converting the initial input representation into a more closely related representation to the output target. Learning is the process of adjusting the weights so that the training data is more accurate.
Sendek et al. reported a neural network for predicting the fast Li-ion conduction at room temperature (RT). They found that Li5B7S13, Li3InCl6, and Li2B2S5 had high ionic conductivity (Sendek et al., 2018). As shown in Figure 1C, 11 crystalline compounds were identified from 317 candidate materials by the ML-guide method. The ionic conductivity of Li5B7S13 was predicted to be 0.074 S/cm, six times higher than the best-known material (Ding et al., 2009; Zhao and Daemen, 2012; Chou and Hwang, 2014; Cubuk and Kaxiras, 2014; Xiao et al., 2015). This study built a convolution neural network to train the atomistic structure, and five descriptors were used as input data. The ML-based simulation revealed a 44-times improvement in the log-average of conductivity compared to random guesswork and manual computation. However, the input data contained less information about properties, the number of materials screened was small, and the training precision was low.
Zeeshan et al. predicted that LiAuI4 and Ba38Na58Li26N were superionic conductors with ionic conductivity of 9.4 × 10–4 and 10–3 S/cm respectively through crystal graph convolutional neural network (CGCNN) (Xie and Grossman, 2018; Zeeshan et al., 2018). The 100 networks model was used to predict shear and bulk moduli as well as some mechanical properties. The accuracy of the screening results was further confirmed by measuring electronic conductivity and thermodynamic stability. Compared to the usual models, the CGCNN method was more general but required more data to train. This method significantly decreased the costs involved with first-principles calculations.
A support vector machine classifies various samples by different labels to find a partition hyperplane. However, there have many partition hyperplanes that can separate the training samples. The boundary should classify the input samples robustly and have the strongest generalization ability to the unseen examples. The Fujimura team employed the support vector regression (SVR) method to predict ionic conductivity (Fujimura et al., 2013). Figure 1D shows the predicted 72 compositions. This work indicated that Li4GeO4 had the highest ionic conductivity. They used the SVM method with a Gaussian kernel to predict the low-temperature conductivities of the compounds. The phase transition temperature (Tc), diffusivity at 1600 K (D1600), the average volume of the disordered structures (Vdis), and experimental temperature T were regarded as independent variables, while the logarithm of ionic conductivity as the dependent variable. To confirm which descriptor combinations can show the best performance, they undertook many experiments and included different numbers of variables to estimate the prediction error. The bootstrapping error was lowest when it chose a combination of these three: D1600, T.
Cubuk et al. discovered that LiN5P3O, Li3Na4O3, LiPO3, LiMg3K2O4, LiNaMg3O5, Li2K3GaO4, Li5Na2O3, Li4NaGaO4, Li2MgO2, Li5K2O3, and Li5Na2NO2 satisfied all of the screening criteria, including stability, high Li conductivity, low cost and weight, and a large window of electrochemical stability (Cubuk et al., 2019). They used 30 elemental descriptors from 40 materials to train a linear SVM through the leave-one-out cross-validation (Sendek et al., 2016). The prediction accuracy is low because the selected 40 materials are out of Material Project or ICSD. In order to solve the trade-off between ML model accuracy and previously uncharacterized materials, they took the outputs of the structure model in Sendek’s research (Sendek et al., 2018) as the labels to train the generic descriptors model, named transfer learning (Pan and Yang, 2010). Through this transfer learning, a new generic model was trained using three datasets, including 40 data, Material Project data, and 21 of the 12,716 lithium-containing materials, with an accuracy of 87.5%, 92.0%, and 85.7%, respectively.
Because of these complex chemical systems, it is still difficult to completely calculate total conductivity by existing computing capacity. Most conductors have added polymers, such as plasticizers, which makes the calculations more complicated. To solve this complex interaction computing, Kan Hatakeyama-Sato’s team used a polymer database to train a gradient boosting model, which shows a 90% accuracy for training data and 81% accuracy for the testing data (Hatakeyama-Sato et al., 2018). As shown in Figure 1E, experiment data were similar to the predicted ionic conductivity. The analysis indicated that electronegativity and polarity of the monomer units were dominant variables in determining ionic conductivity.
The regression model studies the relationship between dependent variables (targets) and independent variables (predictors), used in predictive analysis, time series models, and finding causal relationships between variables. The kernel ridge regression and gradient boosting regression algorithm model were used to find the potential solid-state electrolytes LiOH, LiAuI4, LiBH4, Li2WS4, and Ba38Na58Li26N, etc. (Zeeshan et al., 2018), through training the elastic tensor from the materials project database. An elastic tensor can assess the interface stability between Li anode and solid-state electrolytes. Sendek’s team related the crystallographic directions with the elastic tensor. They took DFT-computed information on 482 electrolyte materials as input data and set up an ‘rbf’ kernel ridge model with α and γ equal to 0.01, and two gradient boosting models with different max depth, minimum samples per leaf, and minimum samples split. This study predicted the elastic tensor of 548 materials with a cubic crystal structure. Although the training data was small, the regression model was useful. They found a relationship between material stiffness and other characteristics like mass density, the ratio of bond ionicity, volume per atom, and sub lattice electronegativity.
Another study employed logistic regression (LR) to identify superionic (σ ≥ 10–4 S/cm) and non-superionic (σ < 10–4 S/cm) structures (Sendek et al., 2016). Austin’s team found that these compounds (BaLiBS3, Li5B7S13, Sr2LiCBr3N2, SrLi(BS2)3, LiErSe2, Li2B2S5, Li3ErCl6, Li2HIO, Li2GePbS4, LiSO3F, LiCl, LiSmS2, Li3InCl6, CsLi2BS3, LiMgB3(H9N)2, RbLiS, LiHoS2, LiErS2, LiHo3Ge2(O4F)2, KLiS) had superionic probability ≥50%, band gap ≥1 eV, upper bound oxidation potential ≥4 V, no transition metals, energy above the convex hull = 0 eV per atom. The dataset consisted of 40 materials with 20 diverse feature characteristics shown in Table 1. It is worth noting that the Li–B–S system was also selected using NN methods (Sendek et al., 2018). The regression illustrated how a multi-descriptor could build a model with better predictive power than a single feature alone. They used LR to build all possible models and then calculated the training misclassification rate (TMR) and the cross-validated misclassification rate (CVMR). As shown in Figure 2A, the 5-feature model had minimal CVMR, proving that the model had statistical significance. The X-randomization performance metric parameter was above the conventional threshold of 0.5 for the optimal LR model. Figure 2B shows the classification performance. The y-axis presents the conductivity higher than 10–4 S/cm, and four materials were misclassified.
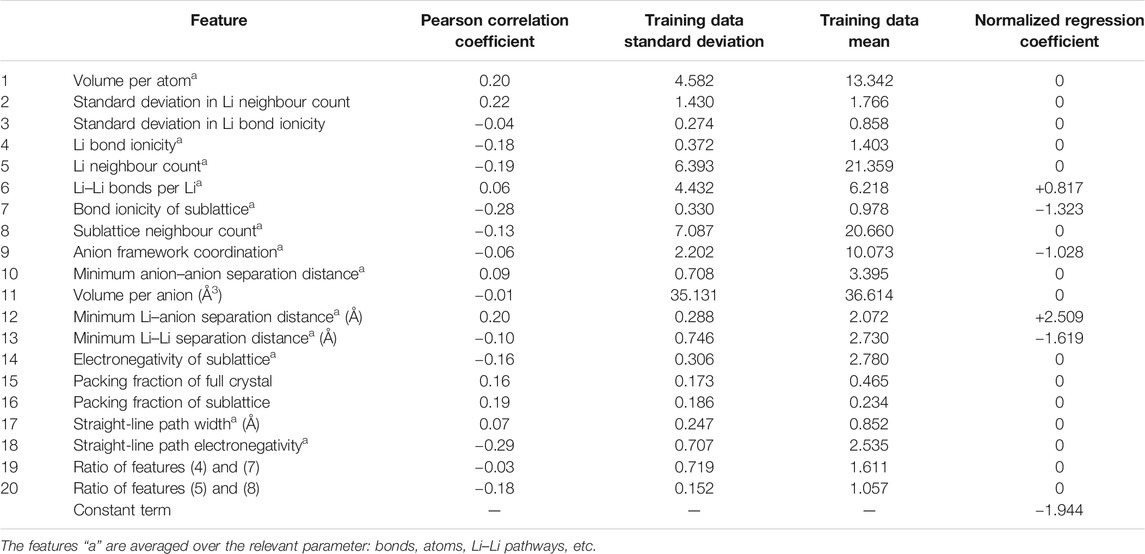
TABLE 1. Pearson correlation coefficient, training data standard deviation, training data mean, and normalized regression coefficient of 20 features.

FIGURE 2. (A) The cross-validation misclassification rate (CVMR) curve and the training misclassification rate (TMR) curve. (B) Classification performance of the training data under leave-one-out cross-validation using logistic regression. (C) Unsupervised clustering of all Li-containing compounds, seven groups divided by algorithm. (D) The results of research through the ML method, displaying compound formula and ionic conductivity, respectively. (E) The parameters of energy formation/Atom (eV) and the energy above the hull/Atom (eV). The red line indicates the formation of energy, and the blue line indicates the energy above the hull.
It is difficult and time-consuming to calculate the migration barriers and ion diffusion of the solid-state electrolytes, especially considering the huge material space. The unsupervised learning algorithm can discover the relationship between properties and X-ray diffraction (XRD) intensities have emerged in screening lithium solid-state electrolytes. Zhang et al. found that Li8N2Se, Li6KBiO6, and Li5P2N5 (i.e., three new materials systems) had an ionic conductivity higher than 10–2 S/cm (Zhang et al., 2019). Figure 1C shows the tree dendrogram generated using the agglomerative hierarchical clustering method. The highest result appears in groups Ⅴ and Ⅵ. These new materials comprise new structures, chemistries, and compositions that are significantly different from existing chemistries. Using clustering methods to classify modified X-ray diffraction (mXRD) can define every anion lattice and fully capture the anionic crystal structure information. The data contained 528 compounds, and three different models were generated to classify the results. The latter two models were set to evaluate the robustness of the clustering algorithm.
The unsupervised learning rising algorithm discovered a quantitative correlation between group and conductivity. The clustering model captured the physical dependence of fast solid-state Li-ion diffusion on anion lattice and gathered excellent materials together. The unsupervised method solves the problem created by scarce datasets, unlike supervised learning models, which need label training data. In unsupervised learning, the label of training data is unknown. It relies on learning non-labeled data to reveal intrinsic properties and laws of data, which form the basis of data analysis. The variances and errors of model parameters are rarely affected by the experimental method.
This study used different ML algorithms to screen and discover typical solid-state electrolytes with high ionic conductivity. As shown in Figure 2D, most materials were concentrated in the range of 10–4 S/cm. In particular, Li5B7S13, Li8SeN2, KLi6BiO6, and Li5P2N5 exhibit a high ionic conductivity near 10–2 S/cm. As a screening method for solid-state electrolytes, the NN structure had the most robust ability in precision or recall. The neural units can learn about the strange input layer by layer, resulting in a high learning rate, meaning that classification tasks often use the NN model. As for the clustering method, it can discover the complex unseen patterns hiding behind multi-dimensional data. If the clustering is sufficiently subdivided, the conductivity of the same species will be very similar. SVR has higher prediction accuracy and stability when there is less label data. Compared to the SVR, the LR algorithm has advantages in prediction efficiency, but the prediction accuracy fluctuates greatly. Gradient Boosting, Kernel Ridge Regression, and Crystal Graph Convolution Neural Network were distributed at lower ionic conductivity. Thus, clustering and the NN method provide robust prediction and a highly efficient screening method for solid-state electrolytes. Our recent work illustrates ML methods in applications, advantages, challenges, and typical references, as summarized in Table 2.

TABLE 2. Comparison of different computational methods, the table shows the mentioned methods in advantages, challenges, applications, and recent typical references.
It is important to verify the stability of the predicted materials, which could be determined by their formation energy and energy above hull: Ehull (Ong et al., 2008; Jain et al., 2011). Theoretically, the formation of energy should be negative and Ehull = 0 eV for thermodynamically stable materials. First principles were used to calculate the formation of energy and Ehull of the screening results from previous research via VASP. As shown in Figure 2E, the typical predicted materials have negative formation energy, and most of the samples exhibit a low Ehull, almost equal to zero, confirming a stable thermodynamically. It is worth to mention that Li6Ho(BO3)3, with ionic conductivity of up to 5.1 × 10−3 S/cm in (Sendek et al., 2018) shows both low formation energy and zero energy above the hull, which could be a promising material. In addition to the ionic conductivity, the electrochemical stable window (Wang et al., 2015) is another crucial feature in screening excellent solid-state electrolytes. Yizhou Zhu’s team (Zhu et al., 2015) used first principles to analyze thermodynamics and found that solid-state electrolyte materials have a limited electrochemical window. Thus, when considering candidate solid-state electrolytes, formation energy, energy above the hull, and the electrochemical window should also be included.
There are still many factors limiting the process of screening new solid-state electrolytes. 1) The training data is still not large enough to satisfy the big structure model, which affects the accuracy of the screening results. 2) The constructed model is still too complicated and cannot adapt to the scarcity of material data, and the model should be as simple as possible. 3) Suitable descriptors have not yet been precisely selected, and there is still no quantitative connection between performance and the parameters of the material. To tackle these challenges, we need greater effects both from the material and algorithmic scientists. On the one hand, material scientists should fabricate and verify the lithium solid-state electrolytes found through ML, which will increase the dataset and provide better guidance for the further optimization of the algorithm. On the other hand, more advanced or updated methodologies could be applied to screen lithium solid-state electrolytes according to data availability. For example, neural networks (e.g., Auto Encoders, Generative Adversarial Networks, etc.) were recently used to predict crystal and material properties (Ryan et al., 2018; Zheng et al., 2018), even in a scenario with little training data. Active learning (Gao et al., 2020) combines data labeling and model training to minimize labeling costs by prioritizing high-value data and classifying the materials based on small material data. These new algorithms have potential applications in screening for lithium solid-state electrolytes.
This review discusses the latest developments in the use of ML algorithms in screening for solid-state electrolyte materials. Some potential lithium solid-state electrolytes were predicted with high ionic conductivity and stability. We focused on demonstrating various ML algorithms, including clustering, support vector machine, neural networks, and regression, used in screening for solid-state electrolytes. In general, neural networks showed a significant advantage in screening. The challenge of using ML in screening for solid-state electrolytes include a lack of data and complicated algorithms. These could be resolved by experimentally verifying the predicted candidates and developing new ML algorithms. As a powerful simulation method, ML have accelerated the discovery of lithium solid-state electrolytes and could be developed to screen for other functional materials.
HL, SM, JW, YW - College of Physics and Information Engineering, Institute of Micro-Nano Devices and Solar Cells, Fuzhou University, Fuzhou, Fujian 350108, People's Republic of China; XW ‐ College of Physics and Information Engineering, Institute of Micro-Nano Devices and Solar Cells, Fuzhou University, Fuzhou, Fujian 350108, People's Republic of China; Jiangsu Collaborative Innovation Center of Photovolatic Science and Engineering, Changzhou, Jiangsu 213164, People's Republic of China.
This work was supported by the National Natural Science Foundation of China (Grant 11704071), the Excellent Youth Foundation of the Fujian Scientific Committee (Grant 2019J06008), and the Award Program, Fujian Minjiang Scholar Professorship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bonchev, D., and Rouvray, D. H. (1991). Chemical graph theory: introduction and fundamentals. New York: Abacus Press.
Borodin, O., Olguin, M., Spear, C. E., Leiter, K. W., and Knap, J. (2015). Towards high throughput screening of electrochemical stability of battery electrolytes. Nanotechnology 26 (35), 354. doi:10.1088/0957-4484/26/35/354003
Chai, J., Liu, Z., Ma, J., Liu, C., Miao, J., Liu, D., et al. (2017). In Situ generation of poly (Vinylene carbonate) based solid electrolyte with interfacial stability for LiCoO2 lithium batteries. Adv. Sci. 4 (2), 377. doi:10.1002/advs.201600377
Chou, C.-Y., and Hwang, G. S. (2014). On the origin of the significant difference in lithiation behavior between silicon and germanium. J. Power Sources 263, 252–258. doi:10.1016/j.jpowsour.2014.04.011
Cubuk, E. D., and Kaxiras, E. (2014). Theory of structural transformation in lithiated amorphous silicon. Nano Lett. 14 (7), 4065–4070. doi:10.1021/nl5015525
Cubuk, E. D., Sendek, A. D., and Reed, E. J. (2019). Screening billions of candidates for solid lithium-ion conductors: a transfer learning approach for small data. J. Chem. Phys. 150, 214701. doi:10.1063/1.5093220
Curtarolo, S., Setyawan, W., Wang, S., Xue, J., Yang, K., Taylor, R. H., et al. (2012). AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235. doi:10.1016/j.commatsci.2012.02.002
Ding, N., Xu, J., Yao, Y. X., Wegner, G., Fang, X., Chen, C. H., et al. (2009). Determination of the diffusion coefficient of lithium ions in nano-Si. Solid State Ionics 180 (2–3), 222–225. doi:10.1016/j.ssi.2008.12.015
Do, C., Lunkenheimer, P., Diddens, D., Götz, M., Weiss, M., Loidl, A., et al. (2013). Li+ transport in poly(ethylene oxide) based electrolytes: neutron scattering, dielectric spectroscopy, and molecular dynamics simulations. Phys. Rev. Lett. 111 (1), 018301. doi:10.1103/PhysRevLett.111.018301
Fujimura, K., Seko, A., Koyama, Y., Kuwabara, A., Kishida, I., Shitara, K., et al. (2013). Accelerated materials design of lithium superionic conductors based on first-principles calculations and machine learning algorithms. Adv. Energy Mater. 3, 980–985. doi:10.1002/aenm.201300060
Gao, M., Zhang, Z., Guo, Y., Arık, S. Ö., Davis, L. S., and Pfister, T. (2020). “Consistency-based semi-supervised active learning: towards minimizing labeling cost,” in Computer Vision – ECCV 2020. Eccv 2020. Lecture Notes in Computer Science. Editors A. Vedaldi, H. Bischof, T. Brox, and JM. Frahm (Cham: Springer).
Ghiringhelli, L. M., Vybiral, J., Levchenko, S. V., Draxl, C., and Scheffler, M. (2015). Big data of materials science: critical role of the descriptor. Phys. Rev. Lett. 114 (10), 105503. doi:10.1103/PhysRevLett.114.105503
Hatakeyama-Sato, K., Tezuka, T., Nishikitani, Y., Nishide, H., and Oyaizu, K. (2019). Synthesis of lithium-ion conducting polymers designed by machine learning-based prediction and screening. Chem. Lett. 48, 130. doi:10.1246/cl.180847
Holdren, J. P. (2011). Materials genome initiative for global competitiveness, Report. United States: White House Office of Science and Technology Policy.
Isayev, O., Oses, C., Toher, C., Gossett, E., Curtarolo, S., and Tropsha, A. (2017). Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 8, 15679. doi:10.1038/ncomms15679
Jain, A., Hautier, G., Ong, S. P., Moore, C. J., Fischer, C. C., Persson, K. A., et al. (2011). Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B Condens. Matter 84 (4), 045115. doi:10.1103/physrevb.84.045115
Jain, A., Ong, S. P., Hautier, G., Chen, W., Richards, W. D., Dacek, S., et al. (2013). Commentary: the materials project: a materials genome approach to accelerating materials innovation. Appl. Mater. 1 (1), 011002. doi:10.1063/1.4812323
Jain, A., Ong, S. P., Chen, W., Medasani, B., Qu, X., Kocher, M., et al. (2015). FireWorks: a dynamic workflow system designed for high-throughput applications. Concurr. Comput. Pract. Ex. 27 (17), 5037–5059. doi:10.1002/cpe.3505
Kamaya, N., Homma, K., Yamakawa, Y., Hirayama, M., Kanno, R., Yonemura, M., et al. (2011). A lithium superionic conductor. Nat. Mater. 10, 682–686. doi:10.1038/nmat3066
Kiran, M., and Joseph, S. (2017). Atomate: a high-level interface to generate, execute, and analyze computational materials science workflows. Comput. Mater. Sci. 10, 64.
Li, W., Ando, Y., Minamitani, E., and Watanabe, S. (2017). Study of Li atom diffusion in amorphous Li3PO4 with neural network potential. J. Chem. Phys. 147 (21), 214106. doi:10.1063/1.4997242
Ong, S. P., Richards, W. D., Jain, A., Hautier, G., Kocher, M., Cholia, S., et al. (2013). Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319. doi:10.1016/j.commatsci.2012.10.028
Ong, S. P., Wang, L., Kang, B., and Ceder, G. (2008). Li–Fe–P–O2 phase diagram from first principles calculations. Chem. Mater. 20 (5), 1798–1807. doi:10.1021/cm702327g
Pan, S. J., and Yang, Q. (2010). A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 22, 1345–1359. doi:10.1109/TKDE.2009.191
Qu, A., Jain, X., and Rajput, N. N. (2015). The Electrolyte Genome project: a big data approach in battery materials discovery. Comput. Mater. Sci. 103, 56–67.
Rupp, M., Tkatchenko, A., Müller, K. R., and von Lilienfeld, O. A. (2012). Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301. doi:10.1103/PhysRevLett.108.058301
Ryan, K., Lengyel, J., and Shatruk, M. (2018). Crystal structure prediction via deep learning. J. Am. Chem. Soc. 140, 10158–10168. doi:10.1021/jacs.8b03913
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B., and Wolverton, C. (2013). Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65 (11), 1501–1509. doi:10.1007/s11837-013-0755-4
Schütt, K. T., Glawe, H., Brockherde, F., Sanna, A., Müller, K. R., Gross, E., et al. (2014). How to represent crystal structures for machine learning: towards fast prediction of electronic properties. Phys. Rev. B 89, 205118. doi:10.1103/physrevb.89.205118
Sendek, A. D., Cubuk, E. D., Antoniuk, E. R., et al. (2018). Machine learning-assisted discovery of solid Li-ion conducting materials. Chem. Mater. 31 (2), 3272. doi:10.1021/acs.chemmater.8b03272
Sendek, A. D., Yang, Q., Cubuk, E. D., et al. (2016). Holistic computational structure screening of more than 12,000 candidates for solid lithium-ion conductor materials. Energy Environ. Sci. 10, 1039. doi:10.1021/acs.chemmater.8b03272
Sendek, A. D., Cubuk Ekin, D., van Antoniuk, R., Cheon, G., Cui, Y., and Reed, E. J. (2019). Machine learning-assisted discovery of many new solid Li-ion conducting materials. Mater. Sci. 8, 750. doi:10.1021/acs.chemmater.8b03272
Sun, B., Mindemark, J., Edström, K., and Brandell, D. (2014). Polycarbonate-based solid polymer electrolytes for Li-ion batteries. Solid State Ionics 262, 738–742. doi:10.1016/j.ssi.2013.08.014
Wang, Y., Richards, W. D., Ong, S. P., Miara, L. J., Kim, J. C., Mo, Y., et al. (2015). Design principles for solid-state lithium superionic conductors. Nat. Mater. 14, 1026–1031. doi:10.1038/nmat4369
Ward, L., Liu, B., Krishna, A., Hegde, V. I., Agrawal, A., Choudhary, A., Wolverton, C., et al. (2017). Including crystal structure attributes in machine learning models of formation energies via Voronoi tessellations. Phys. Rev. B 96, 024104. doi:10.1103/physrevb.96.024104
Xiao, R., Li, H., and Chen, L. (2015). Candidate structures for inorganic lithium solid-state electrolytes identified by high-throughput bond-valence calculations. J. Materiom. 1 (4), 235. doi:10.1016/j.jmat.2015.08.001
Xie, T., and Grossman, J. C. (2018). Crystal graph convolutional neural networks for an accurate, and interpretable prediction of material properties. Phys. Rev. Lett. 120 (14), 145301. doi:10.1103/physrevlett.120.145301
Yamane, H., Shibata, M., Shimane, Y., Junke, T., Seino, Y., Adams, S., et al. (2007). Crystal structure of a superionic conductor, Li7P3S11, Solid State Ionics. 178, 1163–1167. doi:10.1016/j.ssi.2007.05.020
Yang, J., Pistorino, J. C., Pratt, R. C., and Eitouni, H. B. (2017). Copolymers of peo and fluorinated polymers as electrolyte for lithium batteries. US.
Zeeshan, A., Tian, X., and Chinmay, M. (2018). Machine learning enabled computational screening of inorganic solid electrolytes for suppression of dendrite formation in lithium metal anodes. ACS Cent. Sci. Acscentsci. 8, 996–1006. doi:10.1021/acscentsci.8b00229
Zhang, J., Yue, L., Hu, P., Liu, Z., Qin, B., Zhang, B., et al. (2014). Taichi-inspired rigid-flexible coupling cellulose-supported solid polymer electrolyte for high-performance lithium batteries. Sci. Rep. 4 (1), 6272. doi:10.1038/srep06272
Zhang, Y., He, X., and Chen, Z. (2019). Unsupervised discovery of solid-state lithium ion conductors. Nat. Commun. 10, 5260. doi:10.1038/s41467-019-13214-1
Zhao, Y., and Daemen, L. L. (2012). Superionic conductivity in lithium-rich anti-perovskites. J. Am. Chem. Soc. 134 (36), 15042–15047. doi:10.1021/ja305709z
Zheng, X., Zheng, P., and Zhang, R. Z. (2018). Machine learning material properties from the periodic table using convolutional neural networks. Chem. Sci. 9, 8426. doi:10.1039/c8sc02648c
Keywords: lithium ion battery, solid-state electrolyte, machine learning, simulating calculation, material
Citation: Liu H, Ma S, Wu J, Wang Y and Wang X (2021) Recent Advances in Screening Lithium Solid-State Electrolytes Through Machine Learning. Front. Energy Res. 9:639741. doi: 10.3389/fenrg.2021.639741
Received: 09 December 2020; Accepted: 07 January 2021;
Published: 10 February 2021.
Edited by:
Jiajun Wang, Harbin Institute of Technology, ChinaCopyright © 2021 Liu, Ma, Wu, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinghui Wang, c2VhcGh5MjNAZnp1LmVkdS5jbg==, †ORCID: Xinghui Wang, b3JjaWQub3JnLzAwMDAtMDAwMi01Mzg0LTA2NjY=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.