 Emily Sahadeo
Emily Sahadeo Gary Rubloff2,3
Gary Rubloff2,3 Chuan-Fu Lin
Chuan-Fu Lin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res., 25 February 2021
Sec. Electrochemical Energy Storage
Volume 9 - 2021 | https://doi.org/10.3389/fenrg.2021.618368
This article is part of the Research TopicThin Film Technology for Advanced Energy Storage SystemsView all 7 articles
Among the many emerging technologies under investigation as alternatives to the successful Lithium-ion battery, the magnesium battery is promising due to the wide availability of magnesium, its high volumetric capacity, and the possibility for safety improvements. One of the largest challenges facing rechargeable magnesium batteries is the formation of a passivation layer at the Mg metal anode interface when reactive species in the electrolyte are reduced at the electrode-electrolyte interface. To control the solid electrolyte interphase in Lithium batteries, protective layers called artificial solid electrolyte interphase (ASEI) layers have been successful in improving Li metal anode performance. The approach of protecting Mg metal anodes from electrolyte degradation has been demonstrated by fewer studies in the literature than Li systems. In this work, we discuss the properties of Al2O3 thin films deposited using atomic layer deposition as an artificial solid electrolyte interphase at the Mg anode. Our results demonstrate that Al2O3 does prevent electrolyte degradation due to the reductive nature of Mg. However, undesirable properties such as defects and layer breakdown lead to Mg growth that causes soft-shorting. The soft-shorting occurs with and without the protection layer, indicating the ALD layer does not prevent it and hinders Al2O3 from being an ideal candidate for a protection layer. Crucial effects of this layer on Mg electrochemistry at the interface were observed, including growth of Mg deposits leading to soft-shorting of the cell whose morphology showed a dependence on the Al2O3 layer. These results may provide guidelines for the future design and development of protective ASEI layers for Mg anodes.
In advancing energy storage technologies beyond the lithium-ion (Li-ion) battery, emerging systems have challenges and scientific questions which emphasize how new chemistries deviate from the more established understandings of the Li-ion battery. One of these promising alternatives to the Li-ion battery is the Magnesium (Mg) battery. Rechargeable Mg batteries (RMB), in which Mg metal is used as the anode, have sparked increased interest over the past 2 decades. Some advantages of Mg include its higher abundance than Li and Mg metal’s larger volumetric capacity (3,832 mAh/cm3) compared to both Li metal (2,061 mAh/cm3) and graphite (777 mAh/cm3), the latter of which is commercially used in Li-ion batteries. A critical component of battery electrodes, and anodes in particular, is the solid electrolyte interphase (SEI) layer that forms at the anode/electrolyte interface due to electrode reactions with electrolyte components (Aurbach et al., 2001). SEI layers have been extensively studied in Li-ion batteries and the understanding of the properties of these layers has been advanced significantly since the Li-ion battery’s creation (Peled and Menkin, 2017). In general, SEI layers do not completely hinder Li ion diffusion at the Li metal electrode or prevent the batteries from cycling, and in the case of graphite anodes SEI layers are critical to preventing solvent co-intercalation into the anode (Erickson et al., 2015). Conversely, the SEI layer that forms on Mg metal when using conventional carbonate electrolytes, whose components degrade and react at the electrode interface, is referred to as a passivation layer. This impermeable layer composed of products that have low Mg ion conductivity can be detrimental to Mg battery performance, impeding electrochemical reactions from occurring at the anode after it forms (Aurbach et al., 2003).
Due to this issue, significant research has focused on developing new Mg electrolytes to circumvent passivation layer formation on Mg metal anodes. This research has been successful in the development of electrolytes and complex Mg salts that can be paired with a Mg metal anode; however, their synthesis is often time-consuming and until recently many of these electrolytes lacked certain desirable properties such as a wide electrochemical stability window. Many reviews in the literature have summarized the great advances made in magnesium electrolytes in recent years that are beyond the scope of this work (Muldoon et al., 2014; Song et al., 2016; Attias et al., 2019). While the ultimate goal of Mg anode protection is to utilize more conventional carbonate-based electrolytes, some other simple salt-based electrolytes are common which are more compatible with Mg metal but also need improved understanding of their interfacial characteristics at the anode interface to help make progress toward Mg anode protection.
One electrolyte of interest utilizes magnesium bis(trifluoromethanesulfonylimide) (Mg (TFSI)2) salt in ether solvents. Mg (TFSI)2 in a mix of 1,2-dimethoxyethane (DME) and diglyme was one of the first compositions with this salt demonstrated as an electrolyte for RMB (Ha et al., 2014). Further studies of Mg (TFSI)2/DME electrolyte demonstrated that there was a high overpotential for both Mg deposition (0.6 V) and stripping (1.5 V) using this electrolyte (Shterenberg et al., 2015). The high overpotential in Mg (TFSI)2 electrolyte was attributed in some studies to the TFSI− anion’s instability at the Mg anode which causes it to degrade and form MgS as well as MgF2 (Yoo et al., 2017; Ding et al., 2018; Jay et al., 2019). Electrolytes containing the Mg (TFSI)2 salt also demonstrate sensitivity to water, which can aid solvent decomposition (Yu et al., 2017) and passivate the Mg anode interface (Bachhav et al., 2016). While the degradation layer is not completely passivating, it causes large voltage hysteresis issues in full-cell systems (Meng et al., 2019). However, more recent studies have suggested that this overpotential likely does not come from passivation, due to the observation of high overpotential for Mg plating after nucleation of the Mg deposits had already been observed (Eaves-Rathert et al., 2020). Some work has also demonstrated that cycling Mg metal in this electrolyte can cause dendrite growth (Ding et al., 2018), although the deposits are not long and branch-like and have other formation characteristics that are different from dendrites. However, it is now more well-known that spherical Mg deposits can grow through separators and create soft-shorts in many different Mg electrolytes (Yoo et al., 2017; Merrill and Schaefer, 2019; Eaves-Rathert et al., 2020; Song et al., 2020). Results in the current work support the observation that soft-shorting is the origin of the overpotential decrease in Mg-Mg symmetric cells.
Previous work by our group and others has demonstrated that adding water into organic electrolyte improves Mg2+ insertion/conversion in metal oxide cathode materials such as MnO2. However, these cathodes cannot be paired with a Mg metal anode while water is in the electrode or electrolyte and the electrolytes themselves, such as Mg(ClO4)2/PC, are also not compatible with the Mg metal anode. One solution to both compatibility issues is to create a protection layer on the Mg metal surface to prevent water, electrolyte components, and other potential contaminants from reacting with Mg metal and passivating the electrode. Another advantage of this method is that complex Mg electrolytes would not be needed. The application of protection layers has been extensively studied in Li-ion batteries (Wei et al., 2018; Wang et al., 2019) as well as other battery chemistries such as Na batteries (Luo et al., 2017). Protection layers are also called artificial SEI layers, as these interface layers generally serve to protect the anode interface from unwanted reactions by preemptively creating an engineered SEI layer with the desired properties needed to improve the anode’s compatibility with the electrolyte as well as its performance. The goal is to try to employ more simple, conventional electrolytes in Mg rechargeable batteries and achieve a high voltage system without complex electrolytes and salts.
Aluminum oxide (Al2O3) has a robust and well-known atomic layer deposition (ALD) chemistry (George, 2010). Al2O3 has been effective as a protection layer in both Li (Kozen et al., 2015) and Na (Luo et al., 2017) metal anode systems, protecting these interfaces from significant electrolyte degradation on the surface of metal anodes and helping prevent the formation of dendrites, which are an issue for both Li and Na. However, utilizing Al2O3 for Mg would serve different purposes. While the prevention of the electrolyte degradation is the major goal, understanding more about the Mg spherical growth and its effects on the interfacial chemistry is crucial. Further, Mg has poor diffusion kinetics in micron-scale natural SEIs, so using an ultrathin protection layer (artificial SEI) with good Mg mobility that is also electronically insulating is critical. ALD Al2O3 could potentially mitigate the poor Mg2+ transport by the creation of an extremely thin layer. While Mg2+ conductivity in Al2O3 may be low, the effective ionic conductance for Mg2+ to travel a few nanometers through Al2O3 may be sufficient despite low intrinsic Mg2+ ionic conductivity. Recent computational work has indicated that MgAl2O4 has an electrochemical stability window of 3.13 V, and suggests that although it is not stable at 0 V vs. Mg/Mg2+, it could exhibit some metastability that would enable it to function as a protective coating (Chen et al., 2019a). Further, another study demonstrated that MgAl2O4 has a low migration barrier for Mg2+, calculated to be 491 meV (Chen et al., 2019b). While ALD deposits non-magnesiated and amorphous Al2O3, if the layer can be magnesiated during the electrochemical process, the results from this computational work are encouraging. Research done using Al2O3 could also help inform experimental design for future studies interested in investigating MgAl2O4.
In this work, we investigate if an ultrathin, conformal protection layer can help mitigate the degradation of Mg (TFSI)2/DME electrolyte at the Mg interface. To study this electrolyte system, ALD Al2O3 was deposited on Mg foil and evaporated Mg metal substrates. The surface chemistry, overpotential evolution, impedance, and surface morphology were examined to investigate the effect of the Al2O3 layer on Mg deposition and stripping. The goal of this work is to demonstrate different methods and substrates which could be utilized to study the possibility for ALD materials to be used to protect Mg metal anodes, describe how Al2O3 affects the electrochemical properties and surface chemistry, and elucidate properties important for protection layers in rechargeable Mg batteries.
The magnesium metal foil used was >99.9% purity, 0.1 mm thick (MTI corporation). Prior to use, all foil was mechanically polished with silicon carbide sandpaper with three grits, going from lowest to highest (600, 1,200, and 2000) and then wiped with a Kim Wipe. For use in coin cells, foils were punched out with a hammer-driven punch with the desired diameters, either 3/8” or 1/2”. Thermal evaporation was also used to deposit Mg on stainless steel spacers (15 mm diameter, MTI) in a homemade high-vacuum evaporation chamber attached to a glovebox filled with inert Ar to minimize air and moisture exposure. The Mg source for evaporation was Mg ribbon (99.5%, Sigma Aldrich) scraped with a razor blade to remove the surface MgO layer. All Mg preparation, both foil and evaporation, was performed in either MBraun or LC technology gloveboxes with <0.5 ppm H2O and O2.
All ALD on Mg metal was performed in a Cambridge Ultratech Fiji reactor attached to an MBraun glovebox with inert Ar atmosphere, allowing for no exposure to the ambient atmosphere. The Al2O3 ALD process used trimethylaluminum (TMA, Sigma Aldrich) as the aluminum source and oxygen plasma as the oxidant. The ALD process consisted of a 0.06 s TMA pulse, Ar purge, 20 s oxygen plasma pulse, and an Ar purge at a reactor temperature of 150°C, giving a growth rate of ∼1 Å/cycle on Si wafer. Approximate thickness of the layers will be indicated in the text.
All electrochemical tests were performed in 2032 coin cells (MTI) in a symmetric configuration where both electrodes in the two-electrode cell were Mg metal (foil or evaporated). The separators used were glass fiber (Whatman GF-A) and were wet with 125 μL of electrolyte. The electrolyte was 0.25 M Mg (TFSI)2 in 1,2-dimethoxyethane (Mg (TFSI)2 salt from Solvionic was dried under vacuum at 200°C for 24 h, DME was used as-received). Galvanostatic cycling tests were run at fixed amounts of time (30 min) alternating positive and negative currents for oxidation and reduction, respectively, to monitor the overpotential. Electrochemical Impedance spectroscopy was performed an a Biologic VMP Potentiostat with 10 mV amplitude between frequencies of 200 kHz-10 mHz, either at the open circuit potential or at the potential value obtained during galvanostatic cycling. Details for alternate electrochemical impedance spectroscopy (EIS) voltages, frequency ranges, or voltage amplitudes will be indicated in the text.
Ex-situ XPS analysis was performed using a Kratos Ultra DLD XPS spectrometer using monochromatic aluminum x-rays. Cases where non-monochromatic magnesium x-rays needed to be used will be specified in the text. The spectra were calibrated to the Mg metal peak at 49.7 eV. The curves were fitted using CasaXPS software. Spectra were processed using a 70%–30% Gaussian-Lorentzian product function and a Shirley-type background (Shirley, 1972).
Before protecting the Mg metal, the surfaces of both Mg metal foil and evaporated Mg thin film were characterized. The surface of Mg foil needs to be mechanically polished due to the significant surface contamination that naturally forms on the surface during processing and shipping. From the SEM in Figure 1A, it is apparent that the surface is rough after polishing. This observation is one important difference from evaporated Mg in Figure 1C, but due to the high conformality of ALD and its ability to coat high aspect-ratio structures the roughness should not prevent coating by ALD. The evaporated Mg thin film surface in Figure 1C has a more uniform surface, where small Mg domains are visible, which is promising for a Mg substrate.
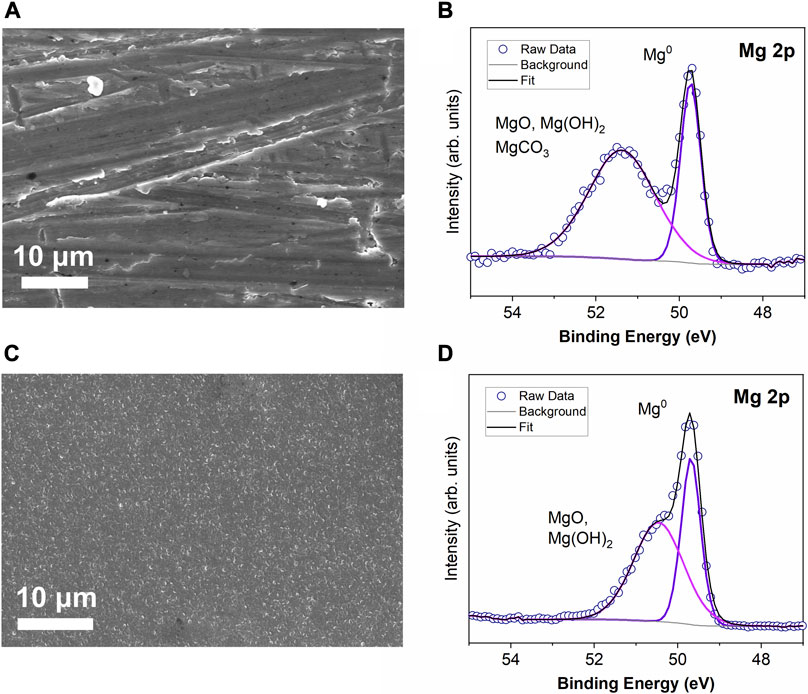
FIGURE 1. Comparison of SEM images and XPS high resolution spectra for (A) and (B) polished Mg foil substrates and (C) and (D) evaporated Mg metal on stainless steel.
The XPS high resolution spectra for the Mg 2p region Mg foil and evaporated Mg surface are depicted in Figures 1B,D. The only detectable elements after Mg evaporation are carbon, oxygen, and magnesium (Supplementary Figure 1). Although the surface contamination layers (typically MgO and MgCO3) were removed as much as possible from the Mg ribbon source used for the evaporation, there is still enough present to deposit with the Mg thin film. To further understand the chemical speciation at the Mg surface, the high resolution XPS spectra are shown for both types of Mg substrates in Figures 1B,D. In both cases, the Mg metal peaks are clearly apparent at 49.7 eV, but there are also significant amounts of MgO, Mg(OH)2, and MgCO3 species at the surface around 50–52 eV. The main difference between the Mg foil and evaporated Mg thin film surfaces is that the Mg foil retains a higher amount of carbon contamination, even after polishing, which is indicated by the broader Mg2+ peak at ∼51 eV due to the higher amount of MgCO3 on the surface of the Mg foil.
To get a baseline for the electrochemical performance, symmetric coin cells were made with 0.25 M Mg (TFSI)2/DME electrolyte and with bare Mg foil or evaporated Mg electrodes. The galvanostatic cycling and corresponding EIS of bare Mg foil is in Figures 2A,B. In the following data, 1 galvanostatic cycle consists of 30 min of negative current of −30 μA/cm2 then 30 min of positive current of 30 μA/cm2. In Figure 2A, the Mg foil cell exhibits a few cycles with a high overpotential around 2 V, but it stabilizes with an overpotential around 500 mV, which shows more stable and lower overpotential behavior than the case of the bare evaporated Mg in Figure 2C. The EIS in the inset of Figure 2B shows a large initial interfacial impedance measured at the open circuit voltage (OCV) in the inset, which supports that an SEI or passivation layer formed at the interface. As this Mg is not protected, the electrolyte is likely degraded at the interface, as demonstrated by the XPS results in Figures 3C,D, increasing the impedance. The remaining EIS data are different from the inset because they are performed at the overpotential value from the preceding galvanostatic cycle, so the AC voltage is centered at the voltage. The main semicircle, representing the interfacial resistance, evolves over the course of the galvanostatic cycling. Examining the EIS, the initial impedance at the OCV before cycling for Mg foil is less than the evaporated Mg, but it is still in the hundreds of kilo-ohms range, indicating the interfacial resistance is high. At the overpotential around 500 mV, after 10 cycles the impedance decreases significantly. Over successive cycles, the impedance begins to increase going from 30 to 40 galvanostatic cycles. This observation is likely due to the passivation of new Mg deposits which causes higher impedance at the interface.
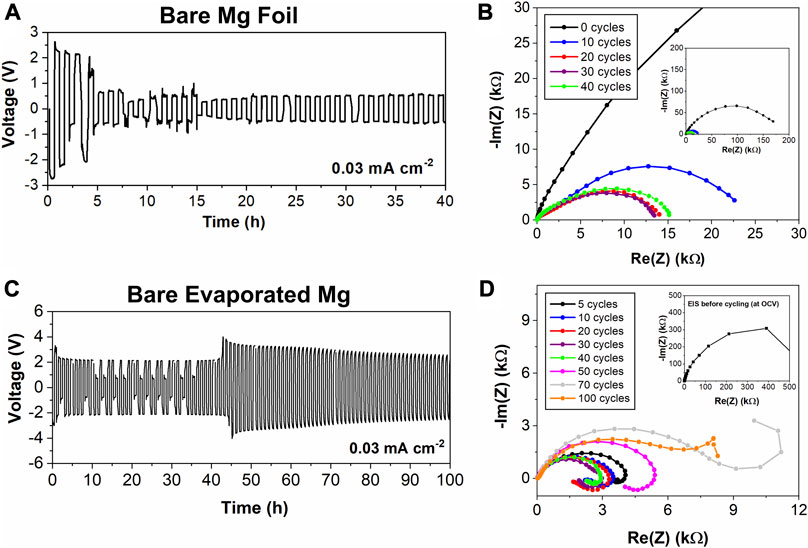
FIGURE 2. Electrochemical analysis of bare Mg anodes for Mg foil in (A) and (B), evaporated Mg in (C) and (D) (A) and (C) show galvanostatic cycling curves for symmetric Mg cells with 30 min constant current applied and (B) and (D) depict EIS analysis at the overpotential values reached in the galvanostatic cycling (EIS at the OCV in the inset).
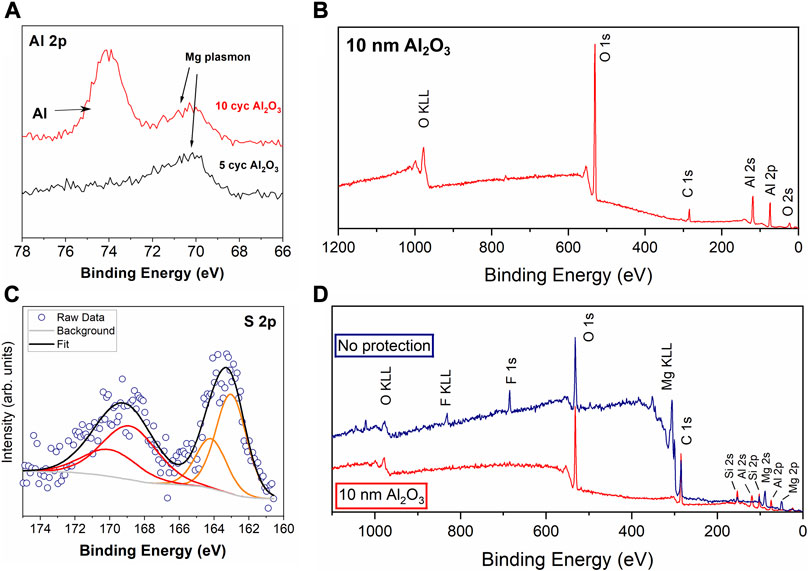
FIGURE 3. XPS of (A) evaporated Mg after 5 and 10 Al2O3 ALD cycles and (B) Mg metal foil after 100 cycles Al2O3 (∼10 nm) (C) high resolution S 2p spectrum from XPS of the soaked Mg foil sample in (D), where (D) shows XPS survey spectra of Mg metal electrodes soaked in 0.25 M Mg (TFSI)2 in DME electrolyte for 24 h.
Evaporated Mg was studied under the same conditions as Mg foil to determine if the differences in the electrochemistry could be attributed to the different methods of preparation and surface characteristics of the two types of Mg. The galvanostatic and EIS data for a bare evaporated Mg cell are depicted in Figures 2C,D. The overpotential starts high, around 3 V, then settles to 2 V. There are a few cycles where the overpotential drops below 1 V, indicating that the current at certain times could be maintained at a lower voltage. This high overpotential value is larger than some reported in the literature for Mg with this electrolyte (Tutusaus et al., 2017), and is consistent with the high overpotential region at the start of galvanostatic cycling of Mg foil. These high overpotentials are likely the true overpotential needed to deposit and strip Mg at the interface, while the small overpotential observed for Mg foil symmetric cells is due to soft-shorting of the cell recently observed by a few groups (Ding et al., 2018; Eaves-Rathert et al., 2020). Lower overpotentials were observed for a different trial with evaporated Mg thin films (Supplementary Figure S4), but in that case the overpotential does ultimately increase and reach the upper limit set for the measurement (4 V), the opposite of what would be expected for soft-shorting.
The impedance in Figure 2D decreases going from 5 to 30 cycles, then slowly increases up to 70 cycles, then decreases again at the 100th cycle, which showed agreement with the overpotentials observed in the cycling data in Figure 2C. The initial decrease in impedance could be due to fresh deposition and stripping sites being created during cycling. However, if those sites become passivated by reacting with the electrolyte the impedance may reach a point where it starts to increase again as the passivation layer increases in thickness. SEM imaging and XPS of the cycled evaporated Mg electrodes was difficult because the Mg deposits grew into the glass fiber separator, meaning they could not be separated from the Mg as shown in Supplementary Figure S5B. With the addition of an Al2O3 layer, we expect there to be changes in overpotential and impedance, likely increases for both. However, improvements in stability of the overpotential and impedance values are some outcomes that could indicate improvement of Mg deposition and stripping at the interface.
For ALD growth, the functional groups on the surface of the Mg need to be reactive with the Al precursor, trimethylaluminum (TMA). Since the XPS spectra in Figure 1demonstrated that the surface contains MgO and Mg(OH)2, it was determined that Al2O3 growth should not be an issue on Mg metal. Figure 3A shows the Al 2p high resolution spectrum of an evaporated Mg surface after 5 cycles and 10 cycles of Al2O3 ALD. This data illustrates that the Al2O3 coating does not grow enough to be detected on the surface until after 10 cycles, where the Al 2p peak appears at 74 eV. At 10 ALD cycles, the thickness of Al2O3 based on the growth rate on Si is about 1 nm. The Mg metal peak is still visible, which is apparent from the presence of the Mg plasmon peak around 70 eV. ALD deposition was also performed on Mg metal foil, and Figure 3B shows the XPS survey spectrum for Mg metal foil after 100 cycles of Al2O3 ALD. The thickness of the Al2O3 with this number of cycles is 9–10 nm, and at this thickness it completely covers the Mg metal—no signal in the XPS for Mg is apparent. We will refer to samples by their thickness of Al2O3 throughout this paper. Generally, 6–10 nm is the depth of the surface that XPS can examine, although this depends on the materials and the inelastic mean free path of the photoelectrons (Tilinin, 1996). This observation supports that the thickness of the Mg is likely consistent with the ALD growth rate on Si, with slightly thinner Al2O3 possible on the Mg based on the 10-cycle delay in detecting the Al signal.
Mg metal has a sufficiently low reduction potential that degradation can occur on the surface from electrolyte being reduced at the metal interface (Jay et al., 2019). To test the ability of Al2O3 to protect Mg metal from initial contact with the electrolyte, Mg metal foil electrodes, both bare and protected with 10 nm Al2O3, were soaked in 0.25 M Mg (TFSI)2/DME electrolyte for 24 h. The XPS survey spectra of these electrodes are shown in Figure 3D. The sample without protection has degradation products on the surface from the electrolyte, with both F and S appearing in the XPS spectra. The high resolution XPS spectrum in Figure 3C shows magnesium sulfide species at 162.5 eV and a sulfonyl/sulfoxide species around 168 eV. Both samples contain a small amount of Si contamination that likely comes from contact with residue from the latex gloves used inside of the glovebox. Most importantly, the sample with 10 nm of Al2O3 deposited does not show any degradation on the surface, as there are no F or S signals in the XPS, only Al, O, C, and the Si. These results confirm that the ALD layer can keep the Mg metal underneath from reducing the electrolyte components.
Mg foil was investigated to determine if the Mg foil substrate showed improved electrochemical properties or similar behavior to evaporated Mg samples. Evaporated Mg was tested with thin Al2O3 layers, 1 and 2 nm, and these results are included in Supplementary Figure 5. The data did not change significantly from Figures 2C,D, indicating the ALD layer did not have a strong influence on the electrochemistry with evaporated Mg. The electrochemical data for Mg foil protected with 2 nm Al2O3 is provided in Figure 4. While at the same current density of the bare sample from Figure 2A, the overpotential stays higher for many more cycles around 2 V, similar to the value for evaporated Mg. However, over time the overpotential value starts to decrease, and stabilizes around 325 mV, like the bare Mg foil sample in Figure 2A. The decreased overpotential again is likely due to a soft-short, thought it took longer to occur than the bare Mg foil cells. The EIS data follows the same overall trend previously exhibited by both the evaporated Mg samples and the control sample of the bare Mg foil. The initial value of the impedance at the OCV is high, but after cycling and probing the impedance at the overpotential for the reaction there is a decrease in interfacial impedance. The EIS measurements at the high overpotential of 2 V in Figure 4A showed unstable behavior at low frequencies, as demonstrated in the sample at 5 cycles in Figure 4B, so the EIS is not shown until cycle 70 when the overpotential decreased. Galvanostatic cycling and EIS data for Mg foil coated with 5 nm of Al2O3 showing similar behavior are in the supplementary information.
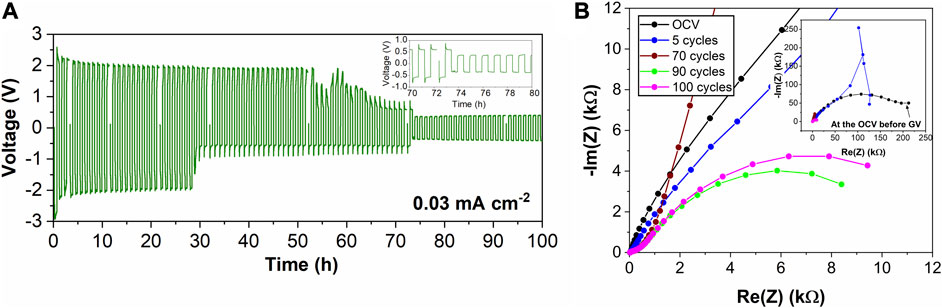
FIGURE 4. Mg foil with 2 nm Al2O3 in 0.25 M Mg (TFSI)2/DME electrolyte (A) galvanostatic cycling and (B) EIS at the overpotential values during cycling.
The electrochemistry of symmetric Mg metal electrodes gives some insight into the effect of Al2O3 on Mg redox reactions at the metal anode interface. Overall, all the samples, both bare and Al2O3 coated Mg, demonstrate similar trends in overpotential as well as impedance. The Mg foil samples differ from evaporated Mg in that they settle to a lower overpotential of a couple hundred mV, whereas the overpotential of evaporated Mg stays around 2 V regardless if it is protection with Al2O3. The low overpotential values for the Mg metal foil symmetric cells is likely due to soft-shorting of the cells. The next section will tie these observations into further SEM and XPS characterization to propose possible explanations for these behaviors and relate them to current findings in the literature.
It is challenging to definitively determine the origins of some of the electrochemical characteristics, but explanations are proposed in this section. In Figure 5A, galvanostatic curves for a bare sample of Mg metal foil and a sample with 10 nm Al2O3 are depicted. The higher current density (0.1 mA/cm2) was used for this comparison to speed up the soft-shorting effect. The lower current density of 0.03 mA/cm2 for a bare Mg foil electrode does show the soft-shorting as shown in Figure 2A and the supplementary information, but it can take a longer cycling time to achieve the effect. The corresponding post-cycling SEM images of the overall surface for these samples are shown in Figures 5B,C, respectively. Similar to examples in the last section, the bare Mg sample shown in black in Figure 5A initially has a high overpotential which gradually decreases. The cell was stopped immediately after the overpotential dropped from ∼1 V to <0.1 V, which is usually indicative of a soft-short (Ding et al., 2018). A soft-short is characterized by Mg deposits that grow through the separator and contact the opposite electrode, but due to the electronically insulating passivation layer on the deposits there is not a direct electron path, enabling capacitive contributions to be measured using EIS. This characteristic of the symmetric cells is being discussed in the literature—some researchers initially said that a sharp overpotential decrease may be due to stabilization of the interface, not necessarily shorting (Tutusaus et al., 2017). However, more recent studies have demonstrated Mg growth through separators indicating clearer soft-shorting phenomena (Yoo et al., 2017; Merrill and Schaefer, 2019; Eaves-Rathert et al., 2020; Song et al., 2020). For the samples here, we also believe that a soft-short may have occurred as pieces of the glass fiber separator can be seen attached to deposits in Figure 5B.
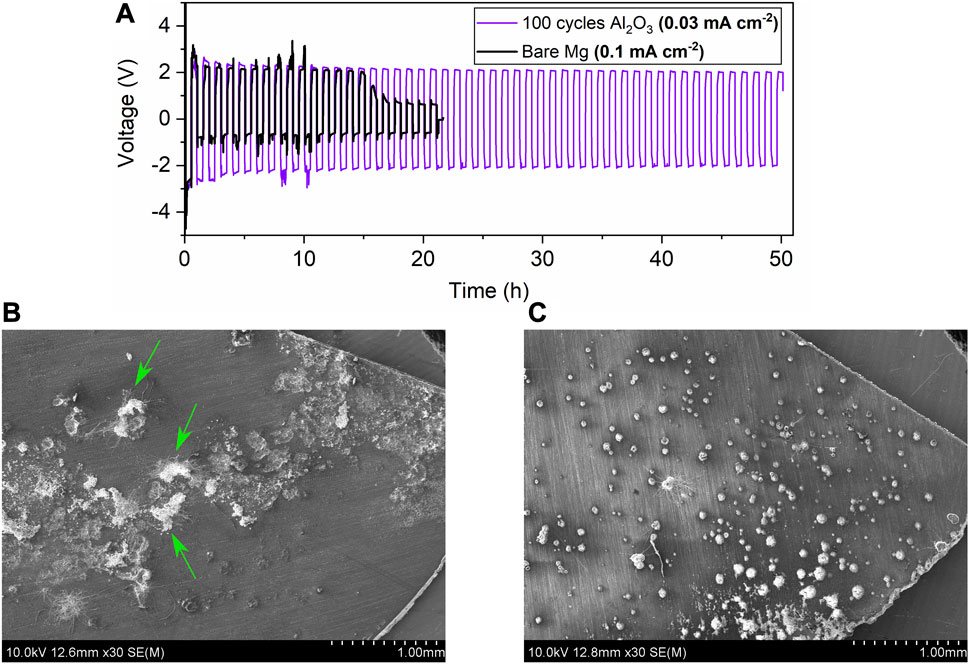
FIGURE 5. Galvanostatic and SEM data for both bare Mg and 10 nm Al2O3 on Mg (A) Galvanostatic cycling for both samples (B) SEM of bare Mg after 20 cycles with arrows pointing to pieces of the separator, and (C) SEM of 100 cycle covered sample after 50 cycles. The green arrows indicate pieces of glass fiber separator attached to the deposits on the Mg foil electrode.
On the other hand, the sample shown in purple in Figure 5A, Mg foil with 10 nm Al2O3, remains at a high stable overpotential right around 2 V, like the evaporated Mg samples. Unlike the bare sample which seems to have large, clustered areas of Mg deposits, the sample with Al2O3 seems to have a more regular distribution of sites where Mg is depositing and stripping, shown in Figure 5C. The stable overpotential and well-distributed deposits help support the idea that the low overpotential values are tied to soft-shorting. If the deposits are occurring more uniformly across the surface and not building up on top of one another (like 3D dendrites), they are less likely to go through the separator and make contact at the other electrode. These two different cases are illustrated in Figure 6. A soft-short may come from Mg deposits which grow from one electrode to the other, but because it is covered by a degradation layer that is slightly passivating, the passivation layer may act as an electrolyte and keep the cell from completely shorting. The more regular morphology in Figure 5C could also help explain the behavior for evaporated Mg electrodes because there was not significant evidence for soft-shorting in the evaporated electrodes.
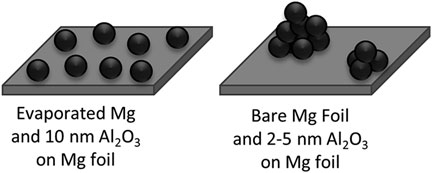
FIGURE 6. Schematic showing possible deposition morphologies based on Mg substrate and ALD film thickness.
To further understand the surface chemistry, different characterization methods were utilized to analyze how the electrolyte reacts on different surfaces of the cycled Mg electrodes. In Figure 7, SEM-EDS and XPS analysis is presented to demonstrate the surface chemical species on an electrode with 2 nm Al2O3 after it has been cycled for 250 h. The SEM images show both Mg deposits and regions where Mg has been stripped away on a previous cycle. The charging on the SEM image in Figure 7 is common for SEI layers on metals, as the degraded products are not electronically conductive. In the corresponding EDS map in Figure 7B, the Mg deposits as well as the stripped areas have significant amounts of O, F, S, and C species, while the pristine areas are mostly Mg and Al.
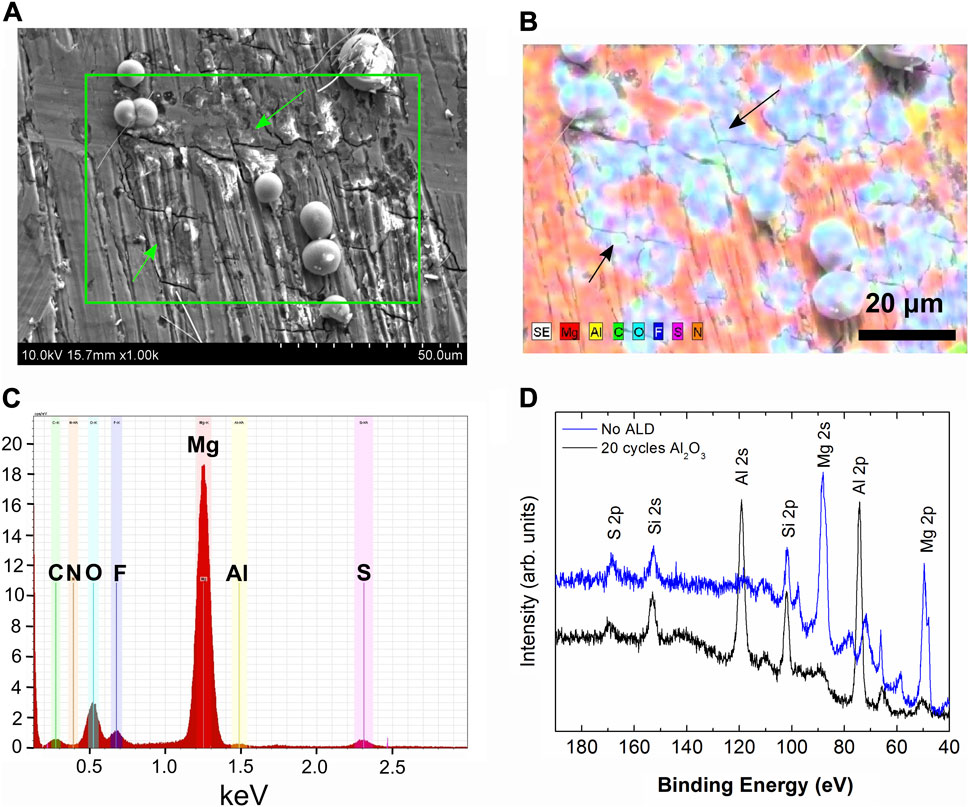
FIGURE 7. Analysis of a cycled Mg foil electrode with 2 nm Al2O3(A) SEM image showing Mg deposits and stripping regions, with stripped regions indicated by green arrows (B) EDS map of the region in the green box from (A) with stripped regions indicated with black arrows (C) EDS spectrum corresponding to the map in (B), and (D) XPS spectrum of the low binding energy region containing Mg and Al signals taken using a non-monochromatic Mg x-ray source.
Overall, the electrolyte degrades on newly formed Mg deposits that are not protected as S and F degradation products are apparent from Table 1. These spherical deposits are present on both bare and protected Mg foil surfaces upon cycling. The EDS mapping agrees well with the earlier results where Al2O3 protected electrodes were soaked in the electrolyte. The map shows that the degradation does not happen on the Mg regions where Al2O3 remains intact, but the electrolyte does degrade on the regions where Mg has been stripped from the surface, removing the Al2O3 layer and exposing fresh Mg, or on new fresh deposits above the Al2O3 layer. While EDS is not effective for detecting the Al2O3 due to the large amount of bulk Mg and very thin (∼2 nm) Al2O3 layer, apparent from the small Al signal in Figure 7C, the XPS in Figure 7D shows the Al signal is present on the surface, even after 250 h of cycling. From these results, it appears that Al2O3 can protect Mg from the electrolyte marginally, and the layer stays partially intact during cycling. However, the Mg deposits and strips above the layer likely due to defects in the Al2O3 or regions where the coating is very thin on a rough or sharp protrusion of Mg, therefore creating a pathway for electrons to the electrode/electrolyte. Over time, as Mg strips and deposits, the Al2O3 layer is degraded and no longer is able to completely prevent electrolyte from reacting with fresh Mg.
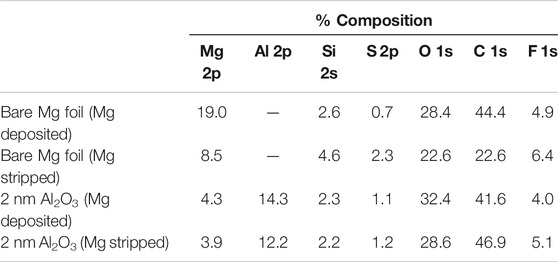
TABLE 1. Atomic composition from XPS analysis of cycled Mg foil electrodes.
Early studies on characterizing this electrolyte demonstrated similar spherical deposits to those observed in this study (Ha et al., 2014), which were identified as non-dendritic. Other works at the time demonstrated the high overpotential for deposition and stripping (Shterenberg et al., 2015) which was the inspiration for this work—we initially sought to determine if an artificial SEI could decrease this overpotential. Study of a modified version of the electrolyte, containing Mg (TFSI)2 and MgCl2 salts, demonstrated more reversible deposition and stripping as Cl− has been identified as a species that improves interfacial characteristics of Mg metal by blocking contaminants from reacting at the interface (Connell et al., 2016). However, even this Cl− containing electrolyte still demonstrated passivation and spherical Mg deposits, as well as pitting and roughened Mg surfaces upon cycling at high current densities (Yoo et al., 2017) and with sulfur containing electrolyte (Gao et al., 2018). The results in this work build upon these studies in the literature, while also helping to inform what properties are necessary for a protection layer to be effective.
From this work, Al2O3 protection layers deposited via ALD that are <10 nm in thickness are not effective at protecting the Mg anode interface from electrolyte degradation and do not help to decrease the overpotential for Mg deposition and stripping. Some other factors to consider are the roughness of the Mg anode surface, as there were differences in the performance of the evaporated Mg and Mg foil samples. However, these differences can not solely be attributed to the surface roughness as the evaporated Mg also had some MgO within the film.. Less rough surfaces, such as evaporated Mg and Mg foil with thicker ALD layers appeared to have more regularly dispersed deposits of Mg compared to Mg foils with thin or no Al2O3. This result may be related to surface roughness, as the mechanically cleaned surfaces are very rough and more electrochemical hot spots experiencing higher current density may be present. If the thicker Al2O3 layer decreases the effect of the surface roughness leading to certain spots having preferential deposition, that may lead to more regular distribution of the Mg deposits as shown in Figure 5C.
For future efforts in Mg anode protection, the results of this work give insight into important properties for the protection layer materials. The Al2O3 was unable to withstand the volume change induced by Mg deposition and stripping, likely leading to breakdown and cracking of the Al2O3, enabling easier electron conduction, and deposition of Mg on top of it. This observation means that more flexible layers such as polymers may be more feasible for Mg anodes, and this has been recently demonstrated using polyacrylonitrile (Son et al., 2018). Further, since it is not clear whether the Al2O3 was magnesiated during these tests, one issue with this protective layer could have been poor Mg diffusion, highlighting the importance of having an interphase layer with good Mg2+ ion conductivity. Additionally, if defects ultimately form in a protection layer, another method is to put additives in the electrolyte that react at the anode interface to form a Mg2+ conductive layer that prevents electrolyte degradation and passivation. This strategy has been demonstrated using iodine in a RMB system, and the additive can continually form the protection layer on new Mg deposits during cell operation (Li et al., 2018). While Al2O3 was not found to be effective in this study, further experiments could determine the exact cause of the failure, such as looking more closely at the surface roughness, the Mg2+ ion mobility, and formation of MgAl2O4. Other ALD chemistries for Mg materials should also be explored, as there are not many reported materials synthesized using ALD that are useful as Mg battery electrolytes or protection layers.
The effect of Al2O3 ALD layers on the electrochemical properties and surface chemistry of Mg metal anodes was investigated using a range of characterization techniques and Mg substrates. Two Mg metal substrates, one made via Mg evaporation and the other a polished Mg foil, were studied in symmetric coin cells in 0.25 M Mg (TFSI)2/DME electrolyte. Al2O3 was deposited on both substrates using ALD, and it was determined that the ALD layer prevented electrolyte decomposition when protected electrodes were soaked in the electrolyte. The study demonstrated that a thin layer (∼10 nm) of Al2O3 on Mg foil altered the growth behavior of Mg deposit during electrochemical cycling, which prevented a “3D growth” of Mg deposits that resulted in penetration into the separator and soft-short between electrodes for Mg electrodes without 10 nm ALD Al2O3. The further understanding of the Mg interface with an ALD layer at the surface and the alteration of the growth of Mg deposits provide constructive insights for developing an effective artificial SEI that may enable the rechargeable Mg-metal battery.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SL and ES conceived the project. ES, C-FL, and SL designed the experiments. ES performed the experiments, collected and analyzed the data, and wrote the manuscript. ES, GR, C-FL, and SL contributed to the discussion and analysis of the results and finalizing the manuscript.
This work was supported in all the experiments as part of the Nanostructures for Electrical Energy Storage (NEES), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001160, and continued in data analysis, manuscript writing and discussions supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021070.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank David Stewart for help running XPS. We also acknowledge the Maryland NanoCenter and the AIMLab for use of SEM.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2021.618368/full#supplementary-material.
Attias, R., Salama, M., Hirsch, B., Goffer, Y., and Aurbach, D. (2019). Anode-electrolyte interfaces in secondary magnesium batteries. Joule 3 (1), 27–52. doi:10.1016/j.joule.2018.10.028
Aurbach, D., Weissman, I., Gofer, Y., and Levi, E. (2003). Nonaqueous magnesium electrochemistry and its application in secondary batteries. Chem. Rec. 3 (1), 61–73. doi:10.1002/tcr.10051
Aurbach, D., Gofer, Y., Schechter, A., Chusid, O., Gizbar, H., Cohen, Y., et al. (2001). A comparison between the electrochemical behavior of reversible magnesium and lithium electrodes. J. Power Sourc. 97–98, 269–273. doi:10.1016/S0378-7753(01)00622-X
Bachhav, M. N., Hahn, N. T., Zavadil, K. R., Nelson, E. G., Crowe, A. J., Bartlett, B. M., et al. (2016). Microstructure and chemistry of electrodeposited Mg films. J. Electrochem. Soc. 163 (13), D645–D650. doi:10.1149/2.0181613jes
Chen, T., Ceder, G., Sai Gautam, G., and Canepa, P. (2019a). Evaluation of Mg compounds as coating materials in Mg batteries. Front. Chem. 7, 24. doi:10.3389/fchem.2019.00024
Chen, T., Sai Gautam, G., and Canepa, P. (2019b). Ionic transport in potential coating materials for Mg batteries. Chem. Mater. 31 (19), 8087–8099. doi:10.1021/acs.chemmater.9b02692
Connell, J. G., Genorio, B., Lopes, P. P., Strmcnik, D., Stamenkovic, V. R., and Markovic, N. M. (2016). Tuning the reversibility of Mg anodes via controlled surface passivation by H2O/Cl– in organic electrolytes. Chem. Mater. 28 (22), 8268–8277. doi:10.1021/acs.chemmater.6b03227
Ding, M. S., Diemant, T., Behm, R. J., Passerini, S., and Giffin, G. A. (2018). Dendrite growth in Mg metal cells containing Mg(TFSI)2/Glyme electrolytes. J. Electrochem. Soc. 165 (10), A1983–A1990. doi:10.1149/2.1471809jes
Eaves-Rathert, J., Moyer, K., Zohair, M., and Pint, C. L. (2020). Kinetic- versus diffusion-driven three-dimensional growth in magnesium metal battery anodes. Joule 4 (6), 1324–1336. doi:10.1016/j.joule.2020.05.007
Erickson, E. M., Markevich, E., Salitra, G., Sharon, D., Hirshberg, D., de la Llave, E., et al. (2015). Review—development of advanced rechargeable batteries: a continuous challenge in the choice of suitable electrolyte solutions. J. Electrochem. Soc. 162 (14), A2424–A2438. doi:10.1149/2.0051514jes
Gao, T., Hou, S., Huynh, K., Wang, F., Eidson, N., Fan, X., et al. (2018). Existence of solid electrolyte interphase in Mg batteries: Mg/S chemistry as an example. ACS Appl. Mater. Inter. doi:10.1021/acsami.8b02425
George, S. M. (2010). Atomic layer deposition: an overview. Chem. Rev. 110 (1), 111–131. doi:10.1021/cr900056b
Ha, S. Y., Lee, Y. W., Woo, S. W., Koo, B., Kim, J. S., Cho, J., et al. (2014). Magnesium(II) bis(trifluoromethane sulfonyl) imide-based electrolytes with wide electrochemical windows for rechargeable magnesium batteries. ACS Appl. Mater. Inter. 6 (6), 4063–4073. doi:10.1021/am405619v
Jay, R., Tomich, A. W., Zhang, J., Zhao, Y., De Gorostiza, A., Lavallo, V., et al. (2019). Comparative study of Mg(CB11H12)2 and Mg(TFSI)2 at the magnesium/electrolyte interface. ACS Appl. Mater. Inter. 11 (12), 11414–11420. doi:10.1021/acsami.9b00037
Kozen, A. C., Lin, C. F., Pearse, A. J., Schroeder, M. A., Han, X., Hu, L., et al. (2015). Next-generation lithium metal anode engineering via atomic layer deposition. ACS Nano. 9 (6), 5884–5892. doi:10.1021/acsnano.5b02166
Li, X., Gao, T., Han, F., Ma, Z., Fan, X., Hou, S., et al. (2018). Reducing Mg anode overpotential via ion conductive surface layer formation by iodine additive. Adv. Energ. Mater. 8 (7), 1701728. doi:10.1002/aenm.201701728
Luo, W., Lin, C.-F., Zhao, O., Noked, M., Zhang, Y., Rubloff, G. W., et al. (2017). Ultrathin surface coating enables the stable sodium metal anode. Adv. Energ. Mater. 7 (2), 1601526. doi:10.1002/aenm.201601526
Meng, Z., Foix, D., Brun, N., Dedryvère, R., Stievano, L., Morcrette, M., et al. (2019). Alloys to replace Mg anodes in efficient and practical Mg-Ion/Sulfur batteries. ACS Energ. Lett. 4 (9), 2040–2044. doi:10.1021/acsenergylett.9b01389
Merrill, L. C., and Schaefer, J. L. (2019). The influence of interfacial chemistry on magnesium electrodeposition in non-nucleophilic electrolytes using sulfone-ether mixtures. Front. Chem. 7, 194. doi:10.3389/fchem.2019.00194
Muldoon, J., Bucur, C. B., and Gregory, T. (2014). Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem. Rev. 114 (23), 11683–11720. doi:10.1021/cr500049y
Peled, E., and Menkin, S. (2017). Review—SEI: past, present and future. J. Electrochem. Soc. 164 (7), A1703–A1719. doi:10.1149/2.1441707jes
Shirley, D. A. (1972). High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 5 (12), 4709–4714.
Shterenberg, I., Salama, M., Yoo, H. D., Gofer, Y., Park, J.-B., Sun, Y.-K., et al. (2015). Evaluation of (CF3SO2)2N− (TFSI) based electrolyte solutions for Mg batteries. J. Electrochem. Soc. 162 (13), A7118–A7128. doi:10.1149/2.0161513jes
Son, S.-B., Gao, T., Harvey, S. P., Steirer, K. X., Stokes, A., Norman, A., et al. (2018). An artificial interphase enables reversible magnesium chemistry in carbonate electrolytes. Nat. Chem. 10, 532–539. doi:10.1038/s41557-018-0019-6
Song, J., Sahadeo, E., Noked, M., and Lee, S. B. (2016). Mapping the challenges of magnesium battery. J. Phys. Chem. Lett. 7 (9), 1736–1749. doi:10.1021/acs.jpclett.6b00384
Song, Z., Zhang, Z., Du, A., Dong, S., Li, G., and Cui, G. (2020). Insights into interfacial speciation and deposition morphology evolution at Mg-electrolyte interfaces under practical conditions. J. Energ. Chem. 48, 299–307. doi:10.1016/j.jechem.2020.02.019
Tilinin, I. S. (1996). Mean escape depth of signal photoelectrons ejected from solids by polarized x rays. Phys. Rev. B Condens Matter. 53 (2), 547–555. doi:10.1103/PhysRevB.53.547
Tutusaus, O., Mohtadi, R., Singh, N., Arthur, T. S., and Mizuno, F. (2017). Study of electrochemical phenomena observed at the Mg metal/electrolyte interface. ACS Energ. Lett. 2 (1), 224–229. doi:10.1021/acsenergylett.6b00549
Wang, Y., Sahadeo, E., Rubloff, G., Lin, C.-F., and Lee, S. B. (2019). High-capacity lithium sulfur battery and beyond: a review of metal anode protection layers and perspective of solid-state electrolytes. J. Mater. Sci. 54 (5), 3671–3693. doi:10.1007/s10853-018-3093-7
Wei, S., Choudhury, S., Tu, Z., Zhang, K., and Archer, L. A. (2018). Electrochemical interphases for high-energy storage using reactive metal anodes. Acc. Chem. Res. 51 (1), 80–88. doi:10.1021/acs.accounts.7b00484
Yoo, H. D., Han, S.-D., Bolotin, I. L., Nolis, G. M., Bayliss, R. D., Burrell, A. K., et al. (2017). Degradation mechanisms of magnesium metal anodes in electrolytes based on (CF3SO2)2N–at high current densities. Langmuir 33 (37), 9398–9406. doi:10.1021/acs.langmuir.7b01051
Keywords: magnesium battery, atomic layer deposition, solid electrolyte interphase, protection layer, thin film
Citation: Sahadeo E, Rubloff G, Lee SB and Lin C-F (2021) Al2O3 Thin Films on Magnesium: Assessing the Impact of an Artificial Solid Electrolyte Interphase. Front. Energy Res. 9:618368. doi: 10.3389/fenrg.2021.618368
Received: 16 October 2020; Accepted: 20 January 2021;
Published: 25 February 2021.
Edited by:
Jun Yan, Harbin Engineering University, ChinaReviewed by:
Chen Liao, Argonne National Laboratory (DOE), United StatesCopyright © 2021 Sahadeo, Rubloff, Lee and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily Sahadeo, ZXNhaGFkZW9AdW1kLmVkdQ==; Chuan-Fu Lin, bGluY0BjdWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.