 Yi-Ting Lu
Yi-Ting Lu Alex R. Neale
Alex R. Neale Chi-Chang Hu2
Chi-Chang Hu2 Laurence J. Hardwick
Laurence J. Hardwick
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Energy Res. , 12 February 2021
Sec. Electrochemical Energy Storage
Volume 8 - 2020 | https://doi.org/10.3389/fenrg.2020.602918
This article is part of the Research Topic Energy Storage Systems Beyond Li-Ion Intercalation Chemistry View all 11 articles
In the field of secondary batteries, the growing diversity of possible applications for energy storage has led to the investigation of numerous alternative systems to the state-of-the-art lithium-ion battery. Metal-air batteries are one such technology, due to promising specific energies that could reach beyond the theoretical maximum of lithium-ion. Much focus over the past decade has been on lithium and sodium-air, and, only in recent years, efforts have been stepped up in the study of divalent metal-air batteries. Within this article, the opportunities, progress, and challenges in nonaqueous rechargeable magnesium and calcium-air batteries will be examined and critically reviewed. In particular, attention will be focused on the electrolyte development for reversible metal deposition and the positive electrode chemistries (frequently referred to as the “air cathode”). Synergies between two cell chemistries will be described, along with the present impediments required to be overcome. Scientific advances in understanding fundamental cell (electro)chemistry and electrolyte development are crucial to surmount these barriers in order to edge these technologies toward practical application.
Energy storage technologies are under extensive investigation because they could contribute towards resolving a major challenge encountered by modern society, that is, the increasing demand for energy. Batteries can be used to achieve this goal by storing energy from intermittent renewable energy sources (such as solar and wind) and releasing the energy at the point of use when required. Lithium-ion batteries are the most advanced technology, which can store and deliver energy through thousands of cycles or even more (Li et al., 2020b; Ng et al., 2020). However, though enormous progress has been made improving specific energy (Wh kg−1) and reducing costs of Li-ion over the past decade, the specific energy of this cell chemistry is nearing its theoretical limit. Therefore, alternative battery chemistries such as metal-air batteries are becoming attractive for their much higher theoretical specific energies than those of Li-ion, as shown in Table 1. Much attention has been focused on the Li-air cell since Abraham et al. reintroduced the concept (Abraham and Jiang, 1996), and Bruce’s group demonstrated that lithium peroxide (Li2O2) could be oxidized from the cathode (Ogasawara et al., 2006). Since then, Li and its analogs such as Na and K have been studied to make metal-air batteries (Hartmann et al., 2013; Ren and Wu, 2013; Luo et al., 2019b; Gilmore and Sundaresan, 2019; Li et al., 2020a; Ha et al., 2020; Han et al., 2020; Hu et al., 2020). Until now, many issues remain unsolved for these metal-air batteries. For example, Li, Na, and K anodes are plagued with poor reversibility because of their high reactivity with the electrolyte and the severe dendrite formation, which may trigger short circuits within the cell and even lead to serious fires or explosions (Yu et al., 2017; Hardwick and De León, 2018; Xiao et al., 2018). Some strategies may improve the cyclability of these alkali metals; for instance, Liu and coworkers performed a cell-level analysis to provide suggestions that integrate the best material properties with optimal cell design parameters. Following these design tips, a lithium-metal cell having specific energy beyond 500 Wh kg−1 can be achieved with good cycling (Liu et al., 2019). Jiao’s group demonstrated stable Na cycling with an average Coulombic efficiency of 97% over 400 cycles by adding sodium hexafluoroarsenate (Na[AsF6]) as an additive in organic carbonate-based electrolytes (Wang et al., 2019b). Xu’s group fabricated a SnO2-coated porous carbon nanofiber as the host for K metal anodes, wherein SnO2 facilitates uniform K nucleation and deposition to suppress the dendritic growth of K (Zhao et al., 2020). Such design of a K host enabled dendrite-free K deposition/stripping with ultralong cycling for over 1700 h.

TABLE 1. Theoretical specific energies and cell voltages for various nonaqueous metal-air batteries and conventional Li-ion batteries.
More recently, divalent metal anodes (Mg, Ca) have received increased attention due to their natural abundance, low cost, high theoretical capacity, low redox potential (see Table 2), and anticipated improved safety. Note that literature about the electrochemistry of Be, Sr, and Ba is rarely seen (Leisegang et al., 2019); to the best of our knowledge, demonstration of metal-air cells using these metals has not so far been reported. Beyond the Group 2 elements, Cu has a comparably high reduction potential (Cu2+/Cu, 0.34 vs. SHE), and the resultant cell voltage is not comparable to those common anodes Li, Na, and K and therefore would be an impractical battery chemistry. Zn is mainly applied in aqueous rechargeable Zn-air battery chemistries because of its high theoretical specific energy and safety (Lu et al., 2017; Fu et al., 2019). Conversely, research into the nonaqueous Zn-air battery is still quite limited (Gelman et al., 2019; Chen et al., 2020). Some works have studied the Zn deposition in nonaqueous electrolytes (Liu et al., 2016); however, nonaqueous Zn-ion batteries, rather than nonaqueous Zn-air, are the primary subjects of these recent investigations (Han et al., 2016; Zhang et al., 2019). Therefore, this work focuses primarily on the electrochemistry of metallic Mg and Ca and their use in metal-air batteries. Furthermore, both Mg and Ca are thought to have lower propensity toward dendrite formation during battery cycling, possibly resulting from their lower self-diffusion barrier for adatoms during the plating process (Jäckle et al., 2018; Ponrouch et al., 2019; Biria et al., 2020). In addition, divalent metal-air batteries have superior theoretical specific energies compared to Na-air and K-air and can be comparable to Li-air, as summarized in Table 1. Additionally, the natural abundance of Mg and Ca greatly outweighs that of Li (Table 2), indicating that lower costs per unit energy stored could be more readily achieved for Mg and Ca (for assumed similarly performing practical systems). Despite these advantages, realizing divalent metal-air batteries using earth-abundant anodes still remains rather challenging with respect to all aspects of the cell configuration, the anode, cathode, and electrolyte, as summarized in Figure 1. By comparison to monovalent Li+/Na+-based electrolytes, the higher charge density of divalent Mg2+/Ca2+ cations yields increased interaction strength with counter-anion and surrounding environment and, thus, can inhibit solubilities, ionic transport/conductivity, and electrochemical rate capability in conventional solvent media. In terms of the anode, the solid electrolyte interphase (SEI) formed spontaneously on Mg and Ca metals is mostly impermeable to the cation in many conventional electrolyte materials, unlike the SEI on Li, Na, and K metal anodes, hampering the deposition/dissolution processes (Muldoon et al., 2014; Yao et al., 2019). Regarding the air cathode, the discharge products (MO and/or MO2, where M = Mg, Ca) generated in the oxygen reduction reaction (ORR) are usually electrochemically very stable and electrically insulating and therefore cannot be decomposed without significant applied overpotentials via the oxygen evolution reaction (OER) upon charging (Shiga et al., 2013; Reinsberg et al., 2016b; Smith et al., 2016; Li et al., 2017; Shiga et al., 2017). For the electrolyte, the most important issue is its mutual compatibility with both electrode interfaces (Hardwick and De León, 2018). Therefore, an electrolyte with a wide electrochemical window (i.e., good stability against electrochemical oxidation and reduction) is highly desirable. However, at present, an electrolyte that allows high-efficiency electrochemical reactions on both the anode (negative electrode) and cathode (positive electrode) is yet to be developed. One strategy would be using Mg2+/Ca2+ permeable membranes to separate the anolyte and catholyte compartments, as can be seen in other metal-air systems (Leng et al., 2015; Liang and Hayashi, 2015; Hwang et al., 2016), although extra cost, technical challenges, and rate capability of the cell need to be considered. On the other hand, issues concerning CO2/moisture crossover will require consideration when practical cells are fabricated.
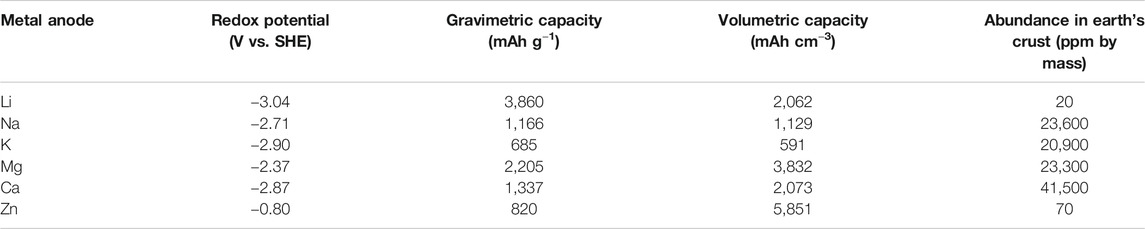
TABLE 2. Theoretical capacities (gravimetric and volumetric), redox potentials under standard aqueous conditions, and abundance of various metal anodes. Abundance values are taken from reference (Haynes, 2014).
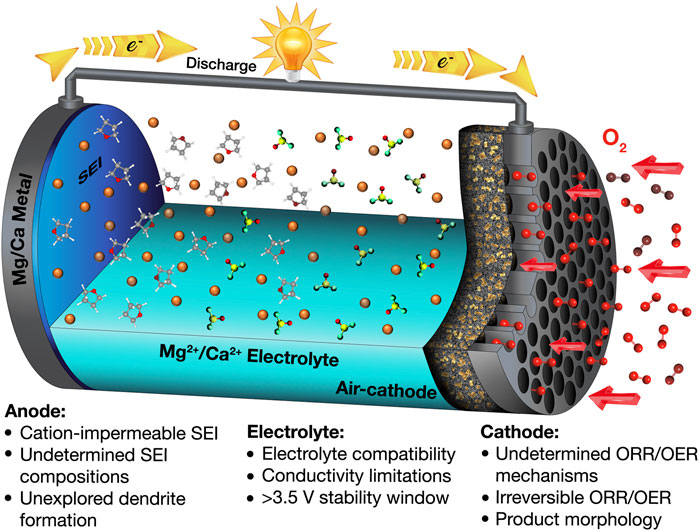
FIGURE 1. Schematic of a discharging Mg/Ca-O2 cell and the challenges for nonaqueous alkaline-earth metal-air batteries.
This review will critically discuss advances in the electrolyte development for the electrochemistry at both the anode and cathode for Mg/Ca-O2 batteries. Starting from the development of electrolytes for reversible divalent metal deposition, various established solution systems are reviewed. Although most of these electrolyte formulations are originally designed for reversible Mg/Ca deposition or rechargeable Mg/Ca-ion batteries, some can be possible candidates to accommodate suitable oxygen electrochemistry on the cathode after modification. On the other hand, the investigations concerning the cathode electrochemistry of divalent cations that have been reported in various electrolytes are also explored. Insights into the potential ORR/OER mechanisms can be gleaned by applying various electrochemical/material characterizations. A further aspect of divalent metal-air batteries that may become critical, similar to challenges within Li-air cells, is the use of redox mediators that may address the challenge of irreversible ORR/OER. Hence, by employing an efficient redox mediator that provides an alternate lower energy pathway toward oxidation of discharge products, a rechargeable metal-air battery could be achieved with high round-trip efficiency and avoid likely decomposition reactions commonly observed at large overpotentials. Divalent metal-air battery research is still in its infancy, and most of the recent works only target the processes at one electrode interface. Therefore, this review will discuss what has been reported previously for the anode and cathode, respectively, and aims to inspire possible future research directions for the full divalent metal-air cell (and/or battery).
Electrolyte development for divalent cation-based metal-air batteries creates different challenges from the analogous monovalent alkali metals, primarily attributed to the nature of divalent cations. Owing to their high charge density (i.e., chemical hardness), divalent cations suffer from significantly stronger cation-anion (ion pairing) and cation-solvent interactions, resulting in lower cation mobility and higher energy penalty for desolvation during the process of electroplating (Tchitchekova et al., 2017). Much of the electrolyte development for Mg- and Ca-based electrolytes has concerned the stable plating and stripping processes at the metal anodes. This work and the current state of the art are explored and discussed in the following sections. Furthermore, the realization of an electrolyte formulation with suitable properties for good performance at the anode, as well as at the air cathode, is likely to prove a serious challenge. Consideration is given to the applicability of these systems for use at the air cathodes, and recommendations made for systems deserve further investigations or considerable modifications before use.
Early research on Mg2+-containing electrolytes was predominantly focused on the electrodeposition of Mg metal. The combination of simple Mg salts (magnesium bromide, ethoxide, methoxide, perchlorate, and thiocyanate) and commonly used solvents (acetonitrile, aniline, benzonitrile, bromoethane, dimethylaniline, ether, formamide, o-toluidine, and pyridine) did not result in any Mg electrodeposition. By contrast, the use of organomagnesium halides (so-called Grignard reagent, RMgX, R = alkyl or aryl group and X = halide like Cl or Br) in ethereal solutions enabled the electroplating of Mg (Overcash and Mathers, 1933). Connor and coworkers investigated the Mg electrodeposition in Grignard reagents, wherein the deposit obtained from Grignard reagent ethylmagnesium bromide (EtMgBr in diethyl ether, 2.5 M) contained 71% Mg (Connor et al., 1957). Although these deposits were white and metallic, they were not pure and very brittle and the reversibility of the Mg plating process was not explored in this article. On the other hand, it could be concluded that this Grignard reagent as a deposition bath is unsatisfactory because of the irreversible deposition, byproducts, and the short life of the Grignard solution. More recently, Liebenow reported the reversible deposition of Mg on Ag and Au substrates using EtMgBr in tetrahydrofuran (THF) (Liebenow, 1997). Conversely, at Ni and Cu substrates, great losses of electrochemically active Mg were observed, indicating the substrate-dependent reversibility of Mg plating in this class of electrolyte. Even though the reversible Mg plating was observed, the EtMgBr/THF electrolyte had low ionic conductivity and poor anodic stability, rendering it unsuitable in full-cell applications. Some studies have studied the direct use of Grignard reagents for Mg deposition (Haas and Gedanken, 2008; Guo et al., 2010; Zhao et al., 2011; Cheng et al., 2013; Chang et al., 2015). However, their limited anodic stability remains a major issue, which precludes practical applications.
Apart from pure Grignard-type electrolytes (RMgX in ethereal solution), other organomagnesium-based systems have also been extensively studied, and, for instance, the combination of organomagnesium (MgR2 or RMgX) as Lewis bases with aluminum- or boron-based Lewis acids (Liebenow et al., 2000; Muldoon et al., 2012; Carter et al., 2014; Zhu et al., 2014; Dongmo et al., 2020). Brenner found that the addition of boron-containing compounds (boranes, alkylboranes, and boron halides) to Grignard reagents improves the Mg deposition, although a small amount of B could be codeposited with Mg (Brenner, 1971; Brenner and Sligh, 1971). Later, Gregory and coworkers made a breakthrough and showed that the addition of aluminum halides, which are less expensive and less toxic than boron-type compounds, to the Grignard solutions resulted in high current efficiencies of deposition and gave good Mg deposits, with the purity higher than 99.9% (Gregory et al., 1990). In this work, the RMgCl-AlCl3/THF (R = methyl, ethyl, and butyl) electrolytes were prepared at various ratios of RMgCl:AlCl3, and all gave high Coulombic efficiencies (over 90%) and bright, crystalline deposits. This work also demonstrated a simple synthesis of magnesium organoborates for the first time. Based on these exciting improvements as a result of combining the Lewis acid and Lewis base, many efforts have been made to design new aluminum- and boron-based Mg electrolytes and a variety of well-performing systems have been invented. For example, Aurbach’s group optimized the PhMgCl-AlCl3/THF (Ph = phenyl group) electrolyte, which exhibited 100% reversibility combined with oxidative stability of approximately 3 V vs. Mg metal for the anodic scan (Mizrahi et al., 2008). Guo and coworkers reported the Mes3B-(PhMgCl)2/THF electrolyte (Mes = 3,5-dimethylphenyl), which showed greater anodic stability up to 3.5 V vs. Mg metal (Figure 2A-i), leading to almost 100% Coulombic efficiency for Mg deposition (Guo et al., 2012). Also, cyclic voltammograms (CVs) of Mes3B-(PhMgCl)2/THF electrolyte on various electrodes revealed that the anodic stability could reach up to 3 V (vs. Mg) at Pt, Ni, and Al electrodes, enabling a large operation window with such current collector materials (Figure 2A-ii).
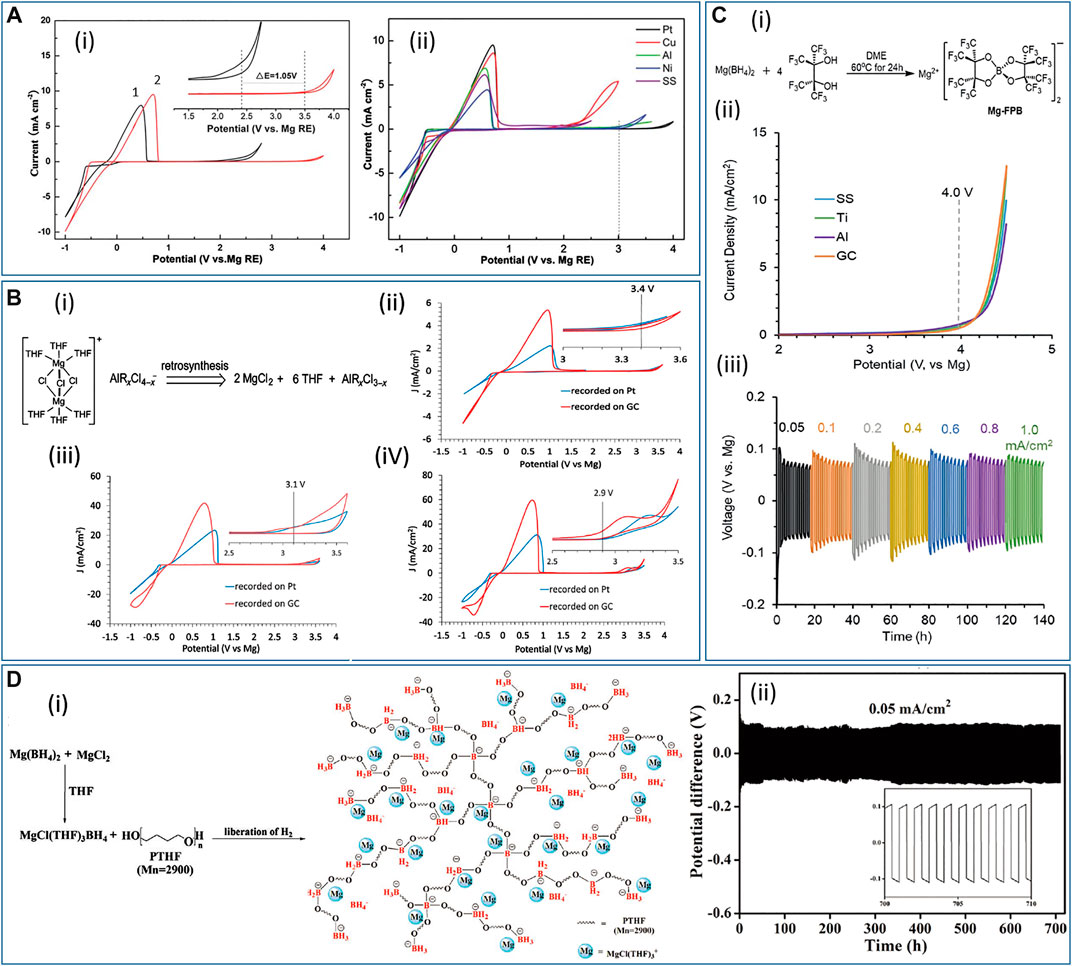
FIGURE 2. (A‐i) Cyclic voltammograms of Pt electrodes at 50 mV s−1 in (1) 0.4 M Mes3B-PhMgCl and 2) 0.5 M Mes3B-(PhMgCl)2. Inset is an enlargement of the 1.5–4 V region. (A‐ii) Cyclic voltammograms of Mes3B-(PhMgCl)2/THF on several working electrodes. Reproduced with permission from (Guo et al., 2012). (B‐i) Retrosynthesis of electroactive [(μ-Cl)3Mg2(THF)6]+ species. Cyclic voltammograms of (B‐ii) MgCl2-AlCl3/THF, (B-iii) MgCl2-AlPh3/THF, and (B-iV) MgCl2-AlEtCl2/THF at 50 mV s−1 on GC and Pt. All insets show the enlargement of the oxidation region. Reproduced with permission from (Liu et al., 2014). (C‐i) Synthesis of Mg-FPB. (C‐ii) Linear sweep voltammograms of Mg-FPB at 50 mV s−1 on different working electrodes. (C‐iii) Mg plating/stripping at current densities from 0.05 to 1 mA cm−2 in Mg/0.5 M Mg-FPB (in diglyme)/Mg symmetric cell. Reproduced with permission from (Luo et al., 2019a). (D‐i) Synthetic route of PTB-GPE based on the reaction of Mg[BH4]2 with MgCl2 and PTHF. (D‐ii) Mg plating/stripping at 0.05 mA cm−2 over 700 h in Mg/PTB@GF-GPE/Mg symmetric cell. Reproduced with permission from (Du et al., 2019).
Another emerging class of Mg electrolytes is the magnesium halide-based electrolyte (Doe et al., 2014; Canepa et al., 2015; Ha et al., 2016; See et al., 2017; Hegemann et al., 2019). Replacing the organomagnesium, organoborate- and organoaluminate-type electrolytes with inorganic magnesium halide can be more favorable for practical Mg rechargeable batteries since the nucleophilic organic ligands in the former materials can be too reactive and unstable. The use of magnesium halide can be dated back to 1957. Conner and coworkers found that 2.5 M MgBr2/diethyl ether as the electrolyte yielded dark and brittle Mg deposits, with the purity being 60–70%, whereas a metallic Mg deposit with a purity of 90% was observed when a small amount of Li[BH4] was added to the MgBr2/ether electrolyte (Connor et al., 1957). Another Mg electrolyte of MgBr2/ether with the addition of Li[AlH4] also showed enhanced Mg deposition. In this solution, the Mg deposit coexisted with Al as an alloy, with Al making up 1–10% of the deposited layer. On the other hand, early attempts to apply MgCl2 as the source of Mg were studied by Aurbach’s group and the first use of MgCl2-AlCl3/THF electrolyte was proposed (Viestfrid et al., 2005). In this study, the resulting CV exhibited a Coulombic efficiency of 34% and a large cycling overpotential over 1 V. The same group claimed in a later work that enhancing the electrochemical performance of Mg deposition and dissolution could be achieved by an electrochemical conditioning process consisting of a repeated cyclic voltammetry treatment (Shterenberg et al., 2014). However, such a conditioning process is not reported in many other publications. The explanation is probably related to contaminants (e.g., water, oxygen, and reactive organic molecules) that affect the electrochemistry, giving rise to poor Mg deposition behaviors (He et al., 2017). On the other hand, a dimer species [(μ-Cl)3Mg2(THF)6]+ has been identified as the active species in electrochemically active electrolytes derived from organomagnesium precursors with Lewis acids in THF (Pour et al., 2011; Guo et al., 2012). In 2014, based on a retrosynthesis rationale (Figure 2B-i), Liu’s group designed a simple synthesis that produces the electrochemically active dimer [(μ-Cl)3Mg2(THF)6]+ in THF as electrolytes (Liu et al., 2014). In this work, MgCl2 was used as the Mg source in combination with various aluminum-based Lewis acids (AlCl3, AlPh3, AlEtCl2), and these three electrolytes revealed Coulombic efficiencies above 90%. The MgCl2-AlCl3 electrolyte showed improved oxidation stability (3.4 V vs. Mg, Figure 2B-ii) compared to MgCl2-AlPh3 (3.1 V, Figure 2B-iii) and MgCl2-AlEtCl2 (2.9 V, Figure 2B-iV). Nevertheless, MgCl2-AlCl3 had a low conductivity (0.26 mS cm−1) and poor solubility (0.04 M), whereas MgCl2-AlPh3 and MgCl2-AlEtCl2 displayed better conductivities (2.96 and 6.99 mS cm−1, respectively) and solubilities (0.43 and 0.67 M, respectively). Although each of the electrolyte formulations had some disadvantages, it is suggested that properties of the MgCl2-AlRxCl3-x electrolytes, including anodic stability, conductivity, and solubility, can be modulated by the Lewis acid components.
Mg deposition in ionic liquids (ILs) has also been under intense investigation due to their negligible vapor pressure, nonflammability, moderate ionic conductivity, good thermal stability, and electrochemical stability (Abdallah et al., 2012). For example, Morita’s group reported an optimized cation structure of imidazolium-based ILs as “ionic solvents” to accommodate the Grignard reagent MeMgBr/THF complex (Kakibe et al., 2010). Introducing a methyl substituent group at the 2-position of the imidazolium ring cation can suppress the reaction of imidazolium cation with MeMgBr, stabilizing the electrolyte formulation. The authors demonstrated that utilizing an asymmetric structure of imidazolium cation, thereby lowering viscosity and increasing ionic conductivity, leads to the improved reversibility of Mg deposition. The optimal Coulombic efficiency in this work was 71%, and the electrochemical window had a range between −1 and 3.7 V (vs. Mg). Later, the same group studied binary IL electrolytes [DEME][TFSI]n[FSI]1-n (DEME = N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium, TFSI = bis{(trifluoromethyl)sulfonyl}imide, FSI = bis(fluorosulfonyl)imide) containing MeMgBr/THF (Kakibe et al., 2012). The authors described the use of binary IL with mixed anions inspired by the good rechargeability and electrochemical properties demonstrated in lithium-ion battery electrolytes. CVs of Mg electrodeposition exhibited that [DEME][TFSI]0.5[FSI]0.5 has the highest deposition charge and lowest overpotentials of plating/stripping compared to other binary IL formulations, which could be the preferable ionic structure of this IL composition for Mg deposition to proceed. A Coulombic efficiency above 90% could be achieved over 100 cycles in the galvanostatic Mg deposition/dissolution test. These works revealed that ILs have primarily been used to dissolve Mg salts as solvents, suggesting that ILs can potentially be applied to various electrolyte systems aside from Grignard reagents; for example, IL-based electrolytes containing Mg[BH4]2, Mg[TFSI]2, or Mg[ClO4]2 salts can be seen in the literature (Narayanan et al., 2009; Watkins et al., 2016; Lee et al., 2018; Gao et al., 2019; Ma et al., 2019). Since the characteristic properties of ILs can be modified by controlling the ion structures, ILs with improved (electro)chemical properties could potentially be designed to facilitate high-performance Mg deposition/dissolution.
Besides liquid electrolytes, the use of solid-state electrolytes for Mg deposition has also been explored (Kumar and Munichandraiah, 1999; Pandey and Hashmi, 2009; Sarangika et al., 2017; Deivanayagam et al., 2019; Fan et al., 2020). Despite their low ionic conductivities, solid-state electrolytes are in general safer because they are leak-proof and nonflammable (Jaschin et al., 2020). For example, Higashi and coworkers invented an inorganic solid-state electrolyte of Mg[BH4][NH2] (Higashi et al., 2014). Mg[BH4][NH2] was derived from Mg[BH4]2 and possessed much higher ionic conductivity (10–3 and 10–6 mS cm−1 for Mg[BH4][NH2] and Mg[BH4]2, respectively, at 150 °C). Although the Coulombic efficiency is not high (ca. 50%), negligible onset potential difference between plating and stripping processes indicates the reversibility could be acceptable at 150 °C, and the anodic stability is shown to be ca. 3 V (vs. Mg). In addition to acting as the electrolyte, the authors commented that such a thin solid electrolyte can be utilized as a coating layer for Mg metal, which separates the Mg metal from the conventional liquid electrolytes, therefore allowing for versatile applications. Polymer electrolytes, which may combine the high conductivity of liquid electrolytes and the above-mentioned advantages of solid electrolytes, can also be utilized as solid-state electrolytes for Mg plating (Kim et al., 2013). In 2003, Aurbach’s group first reported a polymer electrolyte Mg[AlCl2EtBu]2/tetraglyme/PVdF tested in a rechargeable Mg-ion cell that had good cyclability in a wide temperature range between 0 and 80 °C (Chusid et al., 2003). This polymer electrolyte had good anodic stability up to 2.5 V (vs. Mg) and ionic conductivity of 3.7 mS cm−1 at 25 °C. In 2015, Shao and coworkers fabricated a nanocomposite electrolyte based on polyethylene oxide (PEO), MgO, and Mg[BH4]2 with a Coulombic efficiency of 98% at 100 °C, and the voltage gap between the onset potentials for stripping and plating was only 0.2 V (Shao et al., 2015). The PEO has electron lone pairs on its ether-type oxygen, which exhibits strong coordinating capability. Therefore, even though complete dissociation is proven unlikely, the dissociation of Mg[BH4]2 in PEO could be enhanced, leading to better deposition performance.
As the development of electrolytes proceeds, some state-of-the-art electrolytes have been reported with new design concepts. In 2019, Liu’s group developed a chloride-free, electrochemically active, and anodically stable magnesium fluorinated pinacolatoborate electrolyte, Mg[B(O2C2(CF3)4)2]2 (abbreviated as Mg-FPB, Figure 2C-i) in diglyme, in order to replace chloride-based electrolytes that are possibly corrosive at positive potentials (Luo et al., 2019a). The strong chelating effect of bidentate alkyloxide ligands can stabilize the B center and, therefore, the resultant anion can withstand larger positive polarization. The Mg-FPB/diglyme electrolyte delivered a Coulombic efficiency of 95% for the Mg plating/stripping processes, and the anodic stability was up to 4 V (vs. Mg) on stainless steel, Ti, Al, and glassy carbon (GC) electrodes (Figure 2C-ii). The weakly coordinating B[O2C2(CF3)4]2- anion plays a pivotal role in providing a wide potential window, which is potentially suitable to high-voltage cathode materials. In addition, the weak cation-anion interaction gives higher solubility and good ionic conductivity in the electrolyte (0.5 M and 3.95 mS cm−1). A symmetric Mg/0.5 M Mg-FPB (in diglyme)/Mg cell was assembled to evaluate the long-term electrochemical performance at various current densities (Figure 2C-iii), and the polarization under galvanostatic control was below 100 mV for all current densities measured. More surprisingly, nuclear magnetic resonance (NMR) results suggested negligible hydrolysis of the electrolyte two days after the addition of water (10,000 ppm). This observation implies that Mg-FPB/diglyme is a promising choice for practical metal-air batteries where moisture impurities from the external gas supply are possible and likely.
Another work presented by Wang and coworkers demonstrated reversible Mg deposition in the dual-salt electrolyte 0.4 M Mg[TFSI]2/0.1 M Mg[BH4]2/diglyme (Wang et al., 2019a). The addition of a relatively small amount of Mg[BH4]2 prevents the decomposition of [TFSI]-, rendering the electrodeposition reversible. This article proposed that although Mg can be deposited from the conventional electrolyte without Mg[BH4]2, the deposited Mg reacts rapidly with the [TFSI]- anion in the [Mg[TFSI](diglyme)2]+ cluster present in the bulk electrolyte. In contrast, introducing Mg[BH4]2 neutralizes the solvation shell of the [Mg[TFSI](diglyme)2]+ cluster, turning it into [Mg[BH4][TFSI](diglyme)], since [BH4]- is a rather strong coordinating ligand. This neutralized cation cluster somehow inhibits the decomposition of [TFSI]- in the solvation shell. With this advantageous dual-salt formulation, a Coulombic efficiency of 99% was achieved, and X-ray photoelectron spectroscopy (XPS) results displayed that most of the Mg deposits were Mg metal instead of Mg2+. Besides, no B, N, F, or S signals were found on the deposited Mg. As for solid-state electrolytes, Du and coworkers developed a crosslinked polytetrahydrofuran-borate-based gel polymer electrolyte (Figure 2D-i) with the addition of MgCl2 inside a glass fiber membrane (PTB@GF-GPE), which allowed not only reversible Mg plating/stripping for 700 h at 0.05 mA cm−2 (Figure 2D-ii), but also a wide operation temperature range between −20 and 60 °C (Du et al., 2019). Differing from other works using Mg[BH4]2/THF liquid electrolyte, this work attempted to design a crosslinked polymer matrix, which enables facile Mg2+ transfer, to act as a gel polymer electrolyte with improved mechanical strength and thermal stability. To achieve this goal, polytetrahydrofuran (PTHF, average molecular weight = 2,900) and Mg[BH4]2 were dissolved in THF, and a reaction between [BH4]- and the hydroxyl functional groups occurred in situ on a glass fiber membrane. After the crosslinking, the anion [BH4]− reacted with the PTHF polymer, generating a PTHF-borate-based gel polymer electrolyte, PTB@GF-GPE. In such structure, the anion can be incorporated into the large, immobilized polymer matrix, resulting in a high transference number of 0.73 for Mg2+ cations at the room temperature.
For rechargeable Mg-air batteries, irreversible Mg plating/stripping should be overcome first. Also, the oxidative stability of electrolytes should be high enough to be compatible with the electrochemistry of oxygen and reaction intermediates generated at the positive electrode interface. To be more specific, the electrolyte must, at least, keep stable at ca. 3 V (vs. Mg), considering the theoretical cell voltage of Mg-O2 cell (refer to Table 1). In practicality, significant overpotentials for the recharging (oxidative decomposition) of any MgO/MgO2 discharge products are likely. Consequently, anodic stability limits exceeding ca. 3.5 V would likely be required. A variety of electrolyte systems have been investigated to achieve these goals, and progress has been summarized in Figure 3, along with comparative information for Ca-based systems discussed in the next subsection. Many sorts of Grignard reagents are sufficiently electroactive; nevertheless, their instability caused by the nucleophilic nature restricts their practical application. Lewis acids as additives could be an effective solution to the instability of Grignard reagents, as demonstrated by the aluminum- or boron-based Lewis acids. Meanwhile, a retrosynthesis strategy suggests that it is possible to use Mg halides as Mg sources for the Mg electrodeposition with high reversibility. Substituting Mg halides for any possible organic species as the Mg source can be used to enhance the electrochemical stability of electrolyte. However, the relatively low conductivity and salt solubility of such systems still need to be resolved. Another concern is that the electrochemical oxidation stability of electrolytes containing chloride substantially depends on the current collector used (Liu et al., 2014). The incompatibility of chloride-containing electrolytes with certain current collectors (e.g., Al or stainless steel) could easily cause electrolyte degradation during cycling.
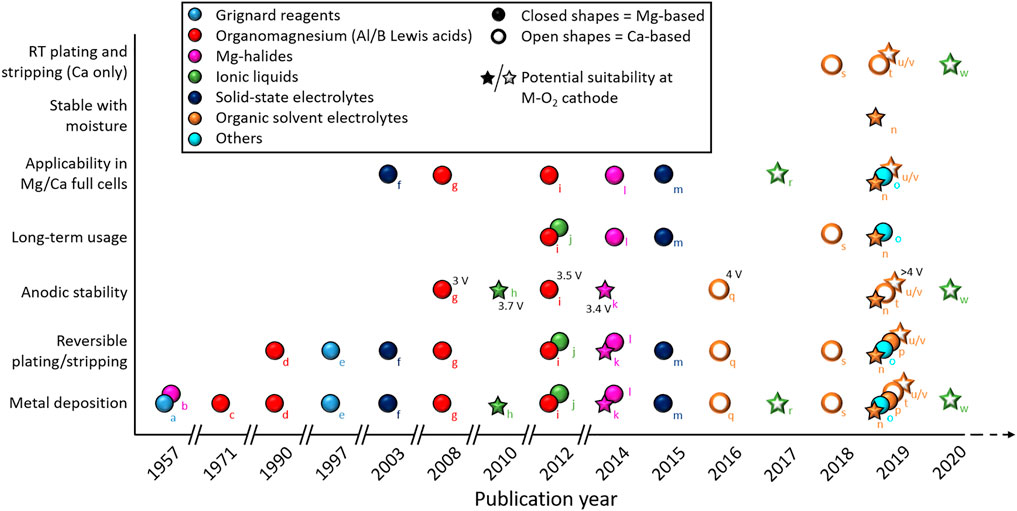
FIGURE 3. Summary figure of the progress of electrolyte development, highlighting various technical achievements, for Mg (closed shapes) and Ca (open shapes) plating/stripping cycling. Electrolytes are grouped by color to show the type of materials used and the star symbols highlight particular formulations that possess properties making them promising candidates for future investigations in Mg/Ca-O2 batteries. Labels a-w refer to literature cited in this section and are summarized in the supporting information (RT = room temperature).
The appeal of ILs as candidate electrolyte solvents is related to the excellent safety, negligible volatility, and good ionic conductivity of many different cation and anion combinations. As the cation and anion structures both contribute to the properties of ILs, modification of ILs for better properties tailored toward electrochemical deposition can be achieved through structure optimization. Given that metal-air batteries require an electrolyte compatible with the cathode and the ORR intermediates and products (i.e., superoxide and peroxide), ILs could be a potential candidate owing to their typically good (electro)chemical stability. More investigations on IL blends (with another ionic liquid or another organic solvent) are also recommended, considering many synergistic effects on electrochemistry in such blends have been reported in the literature (Lair et al., 2010; Giridhar et al., 2012; Grande et al., 2015; Neale et al., 2017). Solid-state electrolytes are also promising electrolyte systems for metal-air batteries, taking into account their mechanical strength, leak-free feature, and nonflammability. Gel polymer electrolytes are, in particular, a famous subject to be applied in Li-air cells (Yi et al., 2015). In recent decades, an emerging class of materials called organic ionic plastic crystals (OIPCs) have been studied as solid-state electrolytes. Such plastic crystals have a definite three-dimensional lattice, but some fraction can be allowed to flow under stress due to the structural disorder stemming from rotational or reorientational motions of cations and/or anions (Pringle et al., 2010; Goossens et al., 2019). Some research about OIPCs applied in Li-battery applications can be found in the literature (MacFarlane et al., 1999; Zhou and Matsumoto, 2007; Jin et al., 2014). Nonetheless, the direct use of OIPCs for efficient Mg stripping/plating is yet to be reported to the best of our knowledge. On the other hand, it will also be valuable to continue working on electrolytes with innovative design concepts. As discussed, Mg-FPB/diglyme shows good anodic stability and good reversibility, while the addition of Mg[BH4]2 can allow reversible Mg deposition in conventional electrolyte Mg[TFSI]2/diglyme that is unstable with Mg metal. Also, glyme-based electrolytes have been used widely in metal-air battery applications (Aldous and Hardwick, 2016; Yu et al., 2017; Carbone et al., 2018). Considering new approaches that enable the utilization of conventional electrolytes in Mg deposition, some traditional electrolytes could be worthy of a reevaluation, suggesting another possible research direction.
Staniewicz reported the earliest work on Ca-metal based cells, based on a thionyl-chloride (SOCl2) system, utilizing a tetrachloroaluminate calcium salt (Ca[AlCl4]2) in SOCl2 (Staniewicz, 1980). The Ca-metal surface suffered serious corrosion under discharge and storage, generating CaCl2 as a primary product and electroplating of Ca (onto a Ni substrate) could not be observed in this electrolyte system. Later, the important work of Aurbach et al. demonstrated the practical impossibility of achieving Ca electroplating in a series of more conventional nonaqueous electrolyte systems (Aurbach et al., 1991). Therein, reducing currents were ascribed to the reductive decomposition of the electrolyte solvent or salt and no evidence for electroplated Ca was observed. The authors attributed this to surface passivation and the lack of Ca2+ ion mobility in the surface films formed.
Ponrouch and coworkers demonstrated the first evidence for the electroplating of Ca metal four years ago (Ponrouch et al., 2016). By employing raised temperatures (75–100 °C), plating/stripping redox was achieved on stainless steel electrodes using a Ca[BF4]2 salt in a 1:1 mixture of propylene carbonate (PC) and ethylene carbonate (EC, see Figure 4A). The authors demonstrated some evidence that Ca[ClO4]2 salts also support plating and stripping but with poorer reversibility, whereas Ca[TFSI]2 electrolytes showed no evidence of stripping electrochemistry. Deposited layers were shown to contain primarily phases of Ca and CaF2, where the fraction of the latter decreased on continued cycling, attributed to initial SEI formation (of CaF2 rich phases) in early stages of plating. Through the optimization of Ca[BF4]2 electrolyte conductivity, the authors were even able to demonstrate some stable electroplating/stripping cycling within symmetrical Ca/Ca cells at 100 °C. More recently, Biria and coworkers reported on the room temperature Ca plating/stripping in Ca[BF4]2 in PC/EC electrolytes by utilizing Cu as an inert substrate (Biria et al., 2019). Therein, the process appears to be of good Coulombic efficiency (>96%) but requires large overpotentials to support plating and stripping (>1 V vs. Ca2+/Ca at 0.55 mA cm−2). The resulting surface films contained no observable CaF2 phase (by X-ray diffraction), which was attributed to reduced electrolyte reactivity at lower temperatures relative to the earlier work. In the absence of additional crystalline phases, components of the carbonate solvents and entrapped [BF4]−, observed by FTIR spectroscopy, composed the Ca2+ conducting layer.
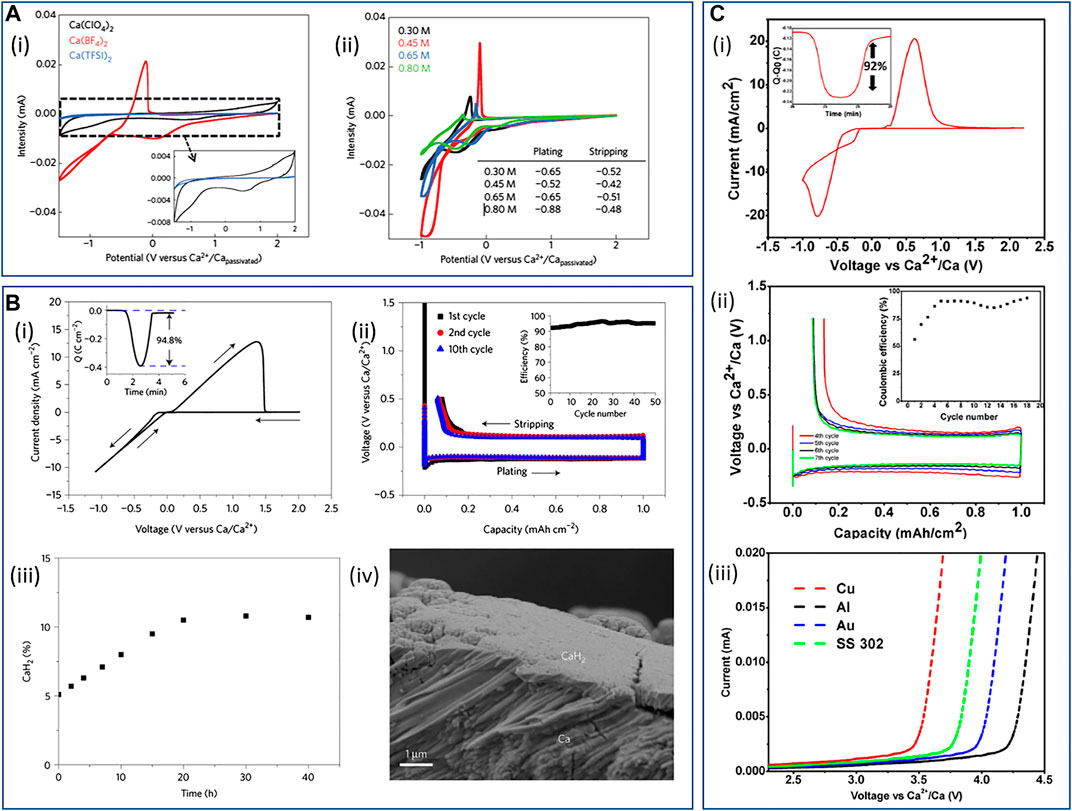
FIGURE 4. (A) Cyclic voltammograms of EC:PC-based electrolytes (0.5 mV s−1 scan rate) at 100 °C with (i) a 0.3 M concentration of different Ca2+ salts and (ii) a range of Ca[BF4]2 concentrations from 0.3 to 0.8 M. Reproduced with permission from (Ponrouch et al., 2016). (B) Calcium plating/stripping in 1.5 M Ca[BH4]2/THF; (i) Cyclic voltammogram of calcium plating/stripping. The working, reference, and counter electrode are Au, Ca, and Pt, respectively. Scan rate 25 mV s−1. Inset shows charge passed on plating/stripping from the CV. (ii) Galvanostatic calcium plating/stripping at a rate of 1 mA cm−2 (iR-corrected). The working, reference, and counter electrode are Au, Ca, and Ca, respectively. (iii) Accumulation of CaH2 at open circuit after depositing 1 mAh cm−2 of calcium on the electrode, expressed as a mole percentage of the charge passed and (iv) the cross-sectional image of the electrode after 20 h rest at open circuit. Reproduced with permission from (Wang et al., 2018a). (C) Electrochemical measurements in Ca[B(Ohfip)4]2/DME electrolyte: (i) fourth CV cycle on an Au substrate with calcium as the reference and counter electrode (scan rate of 25 mV s−1); (ii) galvanostatic plating/stripping of calcium on an Au electrode with Ca counter- and reference electrodes at 0.2 mA cm−2 to a capacity of 1 mAh cm−2; (iii) linear sweep voltammograms recorded on various substrates at a scan rate of 0.05 mV s−1 with Ca as both the reference and counterelectrodes. Reproduced with permission from (Shyamsunder et al., 2019). Copyright 2019 American Chemical Society.
The first demonstration of reversible plating and stripping of Ca at room temperature utilized the formulation of a 1.5 M Ca[BH4]2/THF electrolyte (Wang et al., 2018a). Following an electrochemical reducing cleaning step, the borohydride electrolyte facilitated minimal overpotentials (0.1 V at 1 mA cm−2) and resulted in Coulombic efficiencies of 93–95% of Ca plating/stripping at a gold substrate (Figures 4B-i,ii). CaH2 films form spontaneously from contact between Ca metal and the Ca[BH4]2/THF electrolyte and throughout deposited films during plating, contributing in part to the suppression of continued electrolyte decomposition. The accumulation of CaH2 was demonstrated to saturate at the surface at open-circuit potential (OCP) (Figures 4B-iii,iv), but the reasonably thick films also contributed to increased overpotentials of stripping and losses in Coulombic efficiency. Ta and coworkers demonstrated that the deposition of Ca in Ca[BH4]2/THF electrolytes at inert Au and Pt substrates likely involves an initial slow chemical step in a chemical-electrochemical deposition mechanism (Ta et al., 2019). Unlike the above work of Wang et al., wherein spontaneous reaction between deposited Ca metal and THF in the electrolyte led to CaH2 formation, the preceding chemical step at inert Au/Pt surfaces was ascribed to a hydride abstraction from the [BH4]− anions (not THF) and was found to be substrate-dependent. By simulation of voltammetry at Au and Pt microelectrodes, the rate constant of the chemical step at Au was estimated as ca. 10 times smaller than that for Pt, contributing to smooth/uniform or island-like deposits at Au and Pt, respectively. This is attributed to the slower rate of the chemical step allowing time for lateral diffusion and well-distributed hydride on the Au surface, whereas less lateral diffusion can occur during the more rapid accumulation of the adsorbed hydride on Pt.
While favorable Ca plating/stripping is achieved on the Au surface, demonstrating the requirement for a stable SEI-type phase that supports Ca2+ mobility, the anodic stability of a Ca[BH4]2/THF electrolyte is only ca. 3 V vs. Ca2+/Ca (Wang et al., 2018a). As such, due to the reducing nature of [BH4]− anions, such a formulation is likely to be incompatible with a Ca-O2 electrochemical system. In this context, Li et al. and Shyamsunder et al. both demonstrated good room temperature Ca plating/stripping using a Ca[B(Ohfip)4]2 (Ohfip = hexafluroisopropoxide) salt in dimethoxyethane (DME) (Li et al., 2019; Shyamsunder et al., 2019). Shyamsunder et al. demonstrated the electrolyte can facilitate a Coulombic efficiency of Ca plating/stripping up to 92% at Au (Figure 4C-i) but suffered from moderately higher overpotentials (0.3 V at 0.2 mA cm−2, Figure 4C-ii) and short circuit failure owing to dendritic growth of Ca. Critically, both research groups reported good anodic stabilities for these electrolytes: 4.2 and 4.8 V vs. Ca2+/Ca for stainless steel and Al, respectively, at 0.25 M (Li et al.); 3.8 and 4.1 V vs. Ca2+/Ca for Au and Al, respectively, at 0.5 M (Shyamsunder et al., Figure 4C-iii). Both groups reported significant CaF2 phases and, importantly, no evidence of CaH2 in the SEI films on Ca deposits. However, upon prolonged cycling (Shyamsunder et al.), persistent growth of CaF2 resulted in the surface passivation of the substrate electrodes. Furthermore, Shyamsunder et al. reported on the improved plating/stripping performance by doping the electrolyte with 0.1 M of tetrabutylammonium chloride ([Bu4N]Cl), facilitating a conductivity enhancement that supported enhanced deposition and stripping rate capabilities, improving Coulombic efficiencies and improving the cycle lifetime of the cells. This effect was demonstrated by Ta et al. in their mechanistic study of Ca plating/stripping in Ca[BH4]2/THF electrolytes (Ta et al., 2019). In addition to improving electrolyte conductivity, the authors highlighted similar plating/stripping enhancements by chloride additive that have been seen in Mg systems; however, the underlying explanation for improved plating/stripping and cell performances is yet to be fully explored. While this electrolyte formulation is limited with respect to growing surface passivation and solvent volatility, the application of a novel salt toward supporting Ca deposition/stripping with good oxidative stability represents an important step for the use of Ca metal anodes in high-voltage systems. The reduction in susceptibility of electrolyte components to continued reductive decomposition and the introduction of additives to understand and control SEI formation should be key strategies toward developing Ca anode viability.
The utilization of ILs as alternative nonaqueous electrolyte solvents to support Ca plating/stripping has also been explored in recent years. Shiga et al. explored the fabrication of a full Ca-O2 cell with an ether functionalized quaternary ammonium-based IL, [DEME][TFSI] with 0.1 M Ca[TFSI]2 (Shiga et al., 2017). Therein, while the full cell showed some evidence of reversibility with the use of a 2,2,6,6-tetramethylpiperidine-1-oxyl- (TEMPO) functionalized cathode and combined with parasitic decomposition reactions on charging (discussed in the following sections), the IL-based electrolyte did present evidence of Ca plating/stripping at a Pt substrate at 60 °C with faster scan rates. Under slower scan rates, where freshly deposited Ca can react with the electrolyte to a greater extent, stripping was reduced even further or not observed at all. However, even at higher scan rates (100–200 mV s−1), the Coulombic efficiency of the plating/stripping process was less than 10% and required very high overpotentials (ca. 2 V vs. Ca2+/Ca).
Subsequently, Biria et al. reported the room temperature plating/stripping of Ca in an ionic liquid electrolyte using 1-ethyl-3-methylimidazolium trifluoromethanesulfonate with 1 M Ca[BF4]2 salt (Biria et al., 2020). Plating and stripping overpotentials of the Ca at a Cu substrate under galvanostatic control were initially substantial (ca. ±4 V vs. Ca2+/Ca) but stabilized with successive cycling to 1 V vs. Ca2+/Ca, attributed to SEI formation by the authors. Therein, persistent quantities of CaF2 and CaS are identified before and after stripping steps. Additionally, Coulombic efficiencies of Ca plating/stripping in this formulation stabilized to approximately 56% after the initial three stabilization cycles. Owing to the attractive properties presented by the potential use of ILs as electrolyte solvents in metal-air batteries, these works represent noteworthy preliminary work in this area. Further development of IL-based electrolytes, in the context of Ca-O2 cells, should concern the realization of low overpotential plating and stripping with considerable improvements to Coulombic efficiencies.
Within the general context of electrolyte development for electrochemical cells based on the use of Ca-metal anodes, significant developments have been achieved in only the last 4–5 years. While previously considered as somewhat a practical impossibility, these recent developments revealed the feasibility of electrodeposition and dissolution with moderate overpotentials and good efficiencies. The timeline for various scientific achievements for Ca (and Mg) plating/stripping is summarized in Figure 3. Electrolytes based on borate-based anions have been demonstrated with the most success at calcium anodes, supporting the formation of Ca2+ conductive surface films on the metal. While high temperatures were critical in the initial demonstration of more conventional [BF4]--based salts, achieving reversible Ca plating/stripping at room temperature was an important step in understanding the viability of Ca cells. Depending on the salt/solvent combinations selected, CaF2 and CaH2 have been found to be important SEI components arising from electrolyte reducing reactions at Ca anodes. However, continuous accumulation of CaF2 appears to eventually build passivation of the interface; thus controlling the first stages of SEI formation with, for example, the introduction of reactive additives or pretreatment steps should be considered. Further understanding of critical SEI components in different electrolytes and their effects on deposition/dissolution efficiency, stability, and morphology will be an important next step to aid in the design of new strategies for improving the reliability of the Ca/electrolyte interface. Additionally, methods for the promotion of salt solubility and electrolytic conductivity are vital for future material development.
However, applying the context of Ca-O2 battery chemistry (and the wider general context of utilizing high-voltage cathodes), limitations in oxidative stability of the electrolyte materials are important to consider. Due to the challenges in recharging various M-oxide deposits, cell voltages in excess of 3.5 V are expected (even with potential redox mediators). In addition, stability toward reactive intermediates and products (not currently fully understood for Ca) is essential for any candidate electrolyte. Consequently, electrolytes based on the [BH4]- anion are highly unlikely to be suitable for use in Ca-O2 cells as oxidative decomposition of the THF/Ca[BH4]2 occurs just below 3 V vs. Ca2+/Ca. Likewise, the understood reactivity of organic carbonates toward superoxide radical intermediates negates any formulations based on these solvents for further application in Ca-O2.
Of the materials studied and discussed for Ca plating/stripping in this work, the fluorinated alkyl borate salt Ca[B(Ohfip)4]2 and its use in glyme solvents could be an interesting candidate material for initial studies of the ORR/OER. A reasonably high oxidative stability (>3.5 V depending on the electrode material) combined with no directly obvious protons susceptible to attack by superoxide radical intermediates is encouraging. Furthermore, owing to the good anodic stability (4 V vs. Mg2+/Mg) of the Mg-FPB/diglyme electrolyte, discussed in a previous section (Luo et al., 2019a), as well as the improved plating/stripping efficiency and reductive stability at Mg metal compared to the Mg[B(Ohfip)4]2 salt, the related Ca analog (i.e., Ca-FPB) should be considered for future investigations of both Ca plating/stripping and Ca-O2 electrochemistry. However, for extended practical full-cell use, electrolyte volatility must be a considered factor. In this regard, low chain length glymes (like dimethoxyethane) are unlikely to be practically suitable. Conversely, while much effort is required to improve the reversibility of plating/stripping in the IL-based electrolytes discussed here, the nonvolatility of ILs addresses the critical aspect of electrolyte evaporation under conventional cell operation.
Understanding the fundamental electrochemistry and ORR/OER mechanisms is essential for the development of nonaqueous metal-air batteries (Johnson et al., 2014). The ORR in many nonaqueous electrolytes, in the absence of Mn+ cations, can be quite reversible (or quasi reversible) via a one-electron transfer process (Sawyer et al., 1983; Aldous and Hardwick, 2014). When the alkali metal cation, including lithium, sodium, or potassium, is present, the oxygen reduction and evolution behaviors change significantly and the reversibility can decrease. This change can be ascribed to the surface passivation by the insoluble metal oxygen species formed by various reactions between the metal cation and superoxide radical anions (and various intermediary species) (De Giorgio et al., 2011; Khan and Zhao, 2014; Wang et al., 2018b; Sheng et al., 2018). However, the utilization of these monovalent alkali metal cations in rechargeable metal-air batteries is still promising with the assistance of appropriate electrolytes (Johnson et al., 2014), electrocatalysts (Yang et al., 2013), or additives like redox mediators (Kundu et al., 2015), which lead to improved ORR and OER performance. However, for divalent alkaline-earth cations, the reversibility of the ORR and OER can be significantly different and research is relatively limited to date. The ORR in Mg2+/Ca2+-containing electrolytes is scarcely reversible, as described in the introduction.
Investigations of the ORR and OER in ILs have been carried out in the past decade. AlNashef et al. first demonstrated that the superoxide ion can be stable in ILs without the presence of impurities (AlNashef et al., 2001). Among various ILs, [Pyrr14][TFSI] (Pyrr14 = 1-butyl-1-methylpyrrolidinium) receives much attention as it exhibits relatively good stability with respect to the superoxide ion (enabling good reversibility of the ORR on the timescale of conventional cyclic voltammetry experiments) and has been studied in Li-O2 applications (Katayama et al., 2005; Monaco et al., 2013; Das et al., 2015; Neale et al., 2016). Behm’s group probed the oxygen reduction electrochemistry on Au and GC electrodes in 0.1 M Mg[TFSI]2 in [Pyrr14][TFSI], employing a three-electrode cell with forced electrolyte convection (Law et al., 2016). They reported that the addition of Mg salt renders the ORR irreversible, with the ORR current decaying quickly in the first few cycles. However, the appearance of a small anodic peak was observed after some CV cycles. When CVs were recorded in an expanded potential window (where the negative potential limit was extended), a more pronounced anodic peak appeared already in the first cycle. When combined with a series of CV experiments in varied potential ranges, the anodic peak, which is thought of as the OER, is related to the reduction process in a more negative potential region. Therein, the ORR is coupled with the formation of a surface film of MgF2 and other [TFSI]- decomposition products that were found to build passivation at the electrodes interface and impede reaction kinetics. In this work, the authors do not observe any MgO or MgO2 ORR products by the postmortem XPS and they attributed this to a more favorable formation of MgF2 generated from reaction with the electrolyte.
Behm’s group later published another work about 0.1 M Mg[TFSI]2 in [Pyrr14][TFSI] IL, where they studied the ORR and OER on a range of electrode materials, namely, Pt, Au, GC, Mn2O3, and MnO2, in a rotating ring-disc electrode (RRDE) setup to gain more general insights (Bozorgchenani et al., 2018). However, in the Mg2+-containing IL, none of these electrode materials exhibited any OER feature and the ring electrode (held at 1 V vs. Mg/MgO) did not capture any intermediates either. During the cycling, the cathodic current dropped quickly as a result of the passivation layer. On the other hand, it is noticeable that the electrode surface was not totally passivated after some cycles, indicated by the emergence of a small OER peak related to superoxide reoxidation. The XPS results revealed that MgO2 composed a great portion of the passivation layer, which seems contradictory to the previous work where MgF2 was the main component of the passivation layer (Law et al., 2016). However, although a number of additional tests were conducted, the authors were still not able to find parameters/conditions that resulted in the discrepancy between these two works.
In 2019, Behm’s group reported a more comprehensive research work on the same IL, [Pyrr14][TFSI], where differential electrochemical mass spectrometry (DEMS) and in situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) measurements were simultaneously carried out in a thin-layer flow cell (Jusys et al., 2019). In the O2-saturated [Pyrr14][TFSI], two well-defined mass transport-controlled reduction current plateaus can be observed during the ORR, and the DEMS result suggests superoxide and peroxide species are the products. By contrast, only a tiny OER peak can be seen, which is attributed to the removal of ORR products by the flowing electrolyte. On the other hand, comparing the ATR-FTIR results of N2- and O2-saturated [Pyrr14][TFSI], the –OH-stretching mode (from water impurities) is more intense in the O2-enriched [Pyrr14][TFSI] on the negative scan. The authors suggest that it may result from the adsorbed (super)oxide anions formed during the ORR since water tends to adsorb preferably with adsorbed anions. Furthermore, two potential-dependent IR absorption bands, with one located between 1,000 and 1,100 cm−1 and the other at ca. 1,200 cm−1, are both related to superoxide and/or peroxide. After carefully studying Mg2+-free [Pyrr14][TFSI] with saturated N2/O2, the O2-saturated [Pyrr14][TFSI] containing 0.1 M Mg[TFSI]2 was examined under the same experimental conditions. The CV results are in agreement with previous results (see discussion above), where a serious passivation layer on the electrode is suggested. Again, DEMS results support that the main ORR product is MgO2, consistent with the previous work using ex situ XPS (Bozorgchenani et al., 2018). In addition, in situ IR reveals an interaction of water with MgOx or MgF2 formation (the latter observed in reference (Law et al., 2016)). This article, in many aspects, is in agreement with the previous two articles about the ORR in Mg2+-containing [Pyrr14][TFSI]. Besides, utilization of in situ IR spectroscopy is demonstrated to be beneficial to study the interplay between IL ions and the ORR intermediates.
Dimethyl sulfoxide (DMSO) is commonly selected as a suitable solvent for O2 electrochemistry due to its appreciable ability to stabilize reactive intermediates of ORR (Trahan et al., 2012), but it should be noted that the solvent, as well as the anions and electrode substrates, may be expected to influence the ORR/OER electrochemistry, including the onset potential, reaction mechanism, and product distribution. Baltruschat’s group carried out a detailed investigation in order to elucidate the underlying mechanism of ORR in 0.4 M Mg[ClO4]2/DMSO (Reinsberg et al., 2016a). CVs and corresponding mass spectrometric CV (MSCV) were recorded with the DEMS technique for a few cycles without obvious deactivation of the electrode. However, no oxygen evolution is visible during the positive sweep, indicating the irreversibility of ORR (Figure 5A-i). The authors inferred this phenomenon could be explained by the decomposition of ORR products or intermediates by unknown chemical reactions. It is worth noticing that the ORR onset potential is close to the O2− formation rather than MgO2 formation (see Figure 5A-i). In this work, however, MgO2 or other peroxide species (instead of MgO) was suggested to be the main ORR product during the cathodic polarization of CV on GC, Au, and Pt, whereas the partial formation of MgO was found on Ru and Rh, according to the DEMS results (Figure 5A-ii). Combining the DEMS results and some kinetics investigation, the authors proposed an ORR mechanism (Figure 5A-iii) where O2− forms at the initial step, followed by the hypothesized MgO2+ formation. Then, MgO2 forms via the second reduction, in competition with other chemical reactions. Herein, the authors suggested that it is improbable for the cations to have a strong impact on the noncharged O2 molecule as the onset of the ORR appears to be unaffected. In contrast, the cation does have a strong impact on the second reduction step, considering the ORR product distribution in electrolytes containing [Bu4N]+ or Li+, wherein peroxide cannot form in the former case but can be found in the latter. Therefore, the existence of a hypothetical superoxide-cation complex (MgO2+) intermediate can rationalize why the type of cation has an impact on the reduction potential of the second electron transfer (corresponding to peroxide formation) instead of the ORR onset potential (corresponding to superoxide formation). Later, Baltruschat’s group published another work concerning the influence of divalent cations on the ORR (Reinsberg et al., 2018). The ORR and OER in 0.4 M M[ClO4]2 (M = Mg, Ca, Sr, Ba) electrolytes were investigated on Au, Pt, and GC electrodes. Different from its analogs (Ca2+, Sr2+, Ba2+), peroxide forms in Mg2+-containing DMSO on three different electrode materials according to the DEMS results. Conversely, the other divalent cations show some superoxide formation to various degrees on Pt and GC. Further elucidation of the difference in product distribution will be delivered in the section on Ca-O2 electrochemistry. These two articles from the same group reported that the ORR/OER is scarcely reversible in the Mg[ClO4]2/DMSO electrolyte.
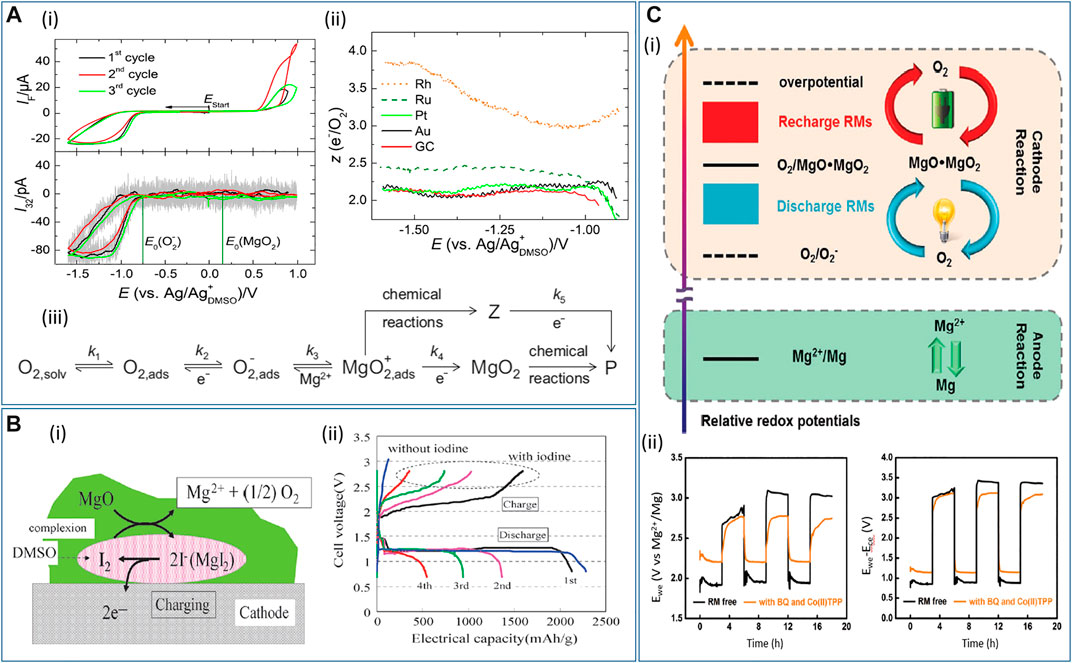
FIGURE 5. (A‐i) Subsequent CVs at 10 mV s−1 and corresponding mass spectrometric CVs for O2 in 0.4 M Mg[ClO4]2/DMSO saturated with a mixture of Ar and O2 (80 : 20) on Pt. (A‐ii) Number of electrons z transferred per oxygen molecule on different electrode materials in 0.4 M Mg[ClO4]2/DMSO with 20% O2 saturation. (A‐iii) Proposed ORR mechanism on Pt in Mg2+-containing DMSO electrolyte with ki being the rate constant of step i, P being the unknown final products, and Z being an intermediate. Reproduced with permission from (Reinsberg et al., 2016a). (B‐i) Proposed reaction mechanism of the charging process for Mg-O2 cell using I2 as the redox mediator. (B‐ii) Discharge-charge curves of Mg-O2 cell with I2 at 60 °C. Reproduced with permission from (Shiga et al., 2013). (C‐i) Operation principles of systems with dual redox mediators. (C‐ii) Galvanostatic cycling of Mg-O2 cell using 1,4-benzoquinone and 5,10,15,20-tetraphenyl-21H,23H-porphine cobalt(II) as dual redox mediators. The left panel shows the voltage profile measured against the Mg reference electrode in a three-electrode configuration, whereas the right panel shows the voltage profile against the counter electrode in a two-electrode configuration. Reproduced with permission from (Dong et al., 2016).
In 2015, Vardar et al. reported their study on the ORR discharge products in Mg-O2 cell using PhMgCl-Al[OPh]3/THF electrolyte (Vardar et al., 2015), since this electrolyte not only allows reversible plating/stripping at room temperature but has high oxidation stability above 4 V vs. Mg2+/Mg. The measured OCP (ca. 2 V) and operating cell voltage (ca. 1.5 V) suggest free superoxide (O2−) forms in the first electrochemical step, followed by a chemical step where MgO2 forms and an O2 molecule is liberated. Then, MgO2 undergoes disproportionation into MgO. The whole process is described in the following equations:
Thus, an ECC (electrochemical-chemical-chemical) mechanism is hypothesized. If MgO or MgO2 is generated directly via electrochemical reactions (i.e., four-/two-electron transfer processes), the recorded OCP and operating cell potential must be larger and thus closer to the theoretical voltages of MgOx (2.91 V for MgO2 and 2.95 V for MgO, see Table 1). The postmortem characterization of discharged air electrodes by Auger electron spectroscopy (AES) showed that the discharge product was a mixture of MgO and MgO2, with the volume percentage being 70% and 30%, respectively. After the air electrode was recharged, Raman and AES both revealed the preferential decomposition for MgO2 but rather limited decomposition for MgO. All these works suggest that MgO2/MgO are not directly generated via electrochemical reactions but via multistep processes involving initial O2− formation.
In contrast to the ongoing research of the fundamental ORR/OER electrochemistry, redox mediators (RMs) can be incorporated into the electrolytes to construct practical rechargeable Mg-O2 batteries because of the resulting stable and efficient battery cycling. The redox mediator is a molecule dissolved in a solution that is oxidized into RM+ (or other oxidized forms) upon recharging, which in turn oxidizes the ORR products MxOy, with itself being reduced back to RM. RMs have been widely demonstrated in metal-air battery research at the Li-O2 and Na-O2 cathode (Chen et al., 2013; Yin et al., 2015). Shiga and coworkers first demonstrated a rechargeable Mg-O2 cell at an elevated temperature (60 °C), using iodide (I−) as the RM (Shiga et al., 2013). In this work, the proposed catalysis mechanism suggests that I− is oxidized to I3− upon charge and then I3− decomposes the discharge product MgO (Figure 5B-i). Although obvious capacity fading was manifested over merely four cycles (Figure 5B-ii), the rechargeability of the Mg-O2 cell proved to be possible. Herein, it is worth noting that the authors only investigated the decomposition of MgO rather than other possible reaction intermediates (superoxide and peroxide species). Although MgO is the final discharge product and is the most thermodynamically favorable compound, many recent studies also suggested (su)peroxide species as possible products during the ORR, as discussed above. Therefore, the interaction between Mg(O2)2 or MgO2 and RMs and the resultant electrochemical response is also of great importance.
Another research article explored the simultaneous use of two RMs in a Mg-O2 cell (working principles shown in Figure 5C-i), one for discharge and the other for recharge (Dong et al., 2016). More specifically, the discharge redox mediator (1,4-benzoquinone, BQ) promotes the formation of MgO2 or MgO and limits the superoxide formation, which thereby alleviates the ORR overpotentials. Additionally, the presence of BQ as a discharge RM can produce small-sized, uniform MgO2/MgO particles on the cathode, which could be able to increase the available capacity of Mg-O2 cells. On the other hand, the recharge RM (5,10,15,20-tetraphenyl-21H,23H-porphine cobalt(II), Co(II)TPP) facilitates the decomposition of discharge products, confirmed by SEM and XPS. Overall, the charge-discharge gap is decreased by 0.6 V (0.3 V for each process, see the left panel in Figure 5C-ii), saving much energy wasted for sluggish kinetics of both oxygen electrochemistries. Note that, in a two-electrode configuration (the right panel in Figure 5C-ii), some overpotentials can be caused by the passivation of Mg counter electrode, which results in an underestimated round-trip efficiency. Furthermore, the long-term stability of any RMs at the anode interface requires significant consideration when investigating their application. However, this research gives the proof-of-concept demonstration that two different RMs can be applied at once to enhance the full-cell performance.
The earliest report of a system based on electrochemical reactions of Ca and O2 detailed the use of a molten salt electrolyte (29 mol% CaO, 71 mol% CaCl2 with ZrO) at 850 °C, a CaSi alloy anode, and a perovskite positive electrode (Pujare, 1988). Fabricated cells were cycled at 850–900 °C presenting cell voltages in the range of ca. 1–2 V; while no characterization of products is reported, the authors describe the full-cell electrochemistry as in the following equation:
More recently, while remaining relatively unexplored compared to analogous Mg-O2 systems, several works focusing on the effect of Ca2+ on the ORR/OER and the investigation into nonaqueous Ca-O2 full cells have been reported. Baltruschat’s group conducted the first detailed investigations of the nature of electrochemical oxygen reduction within liquid nonaqueous Ca2+-containing electrolytes, namely, Ca[ClO4]2 salts in DMSO (Reinsberg et al., 2016b). In this work, a combined series of CV, DEMS, and RRDE experiments are described at different electrode substrates to assess the ORR/OER. While the formation of CaO2 (calcium peroxide) is favored at Au, with no evidence of any reversibility, Ca(O2)2 (calcium superoxide) is the dominant product of ORR at GC and Pt (as well as Rh and Ru). Interestingly, the two-electron (per mole O2) reduction at Au, attributed to peroxide formation, is not coupled with passivation and deactivation of the surface (i.e., no large hysteresis after the potential sweep is reversed), unlike Li+/DMSO systems. However, the reasons for the preferential formation of CaO2 at Au is yet to be explored. For the Pt and GC electrodes, DEMS measurements combined with a secondary detection electrode held at oxidative potentials, and the analogous RRDE configuration experiments, confirmed the reoxidation of dissolved superoxide species to be approaching 90% of O2 species reduced during the ORR at the reducing Pt/GC electrodes (Figure 6A). The secondary working electrode, an Au-sputtered Teflon membrane held close to the mass-spectrometer, reduced the total quantity of O2 consumed during cyclic voltammetry (Figure 6A-iii), indicating significant regeneration of molecular O2 from the oxidation of dissolved species. In combination with a degree of reoxidation of precipitated species at high voltages, these results indicate high Coulombic efficiency and good chemical reversibility of the ORR at Pt/GC. However, the significant overpotentials required for oxidation of the dissolved superoxide indicates that this is not free O2− (or loosely interacting) in solution, but rather the authors describe likely formation of contact ion pairs in solution with the dissolved Ca2+.
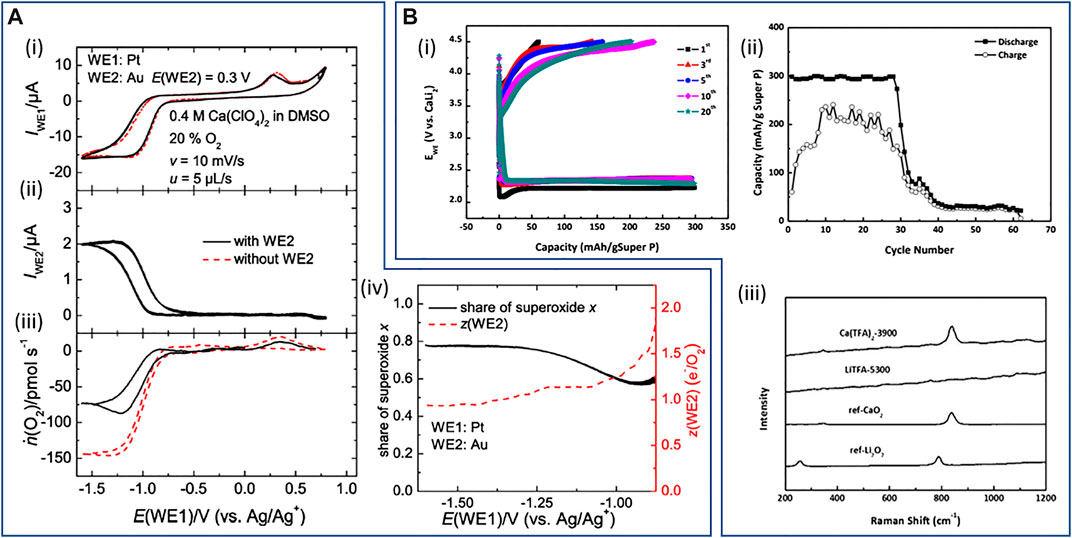
FIGURE 6. (A) Identification of the soluble products via DEMS using 0.4 M Ca[ClO4]2/DMSO saturated with a mixture of 80% Ar and 20% O: (i) cyclic voltammograms at the Pt-working electrode (WE) with (black) and without (red) the use of an Au-sputtered Teflon membrane (E(WE2) = 0.3 V) in the detection compartment. (ii) Faradaic currents at the sputtered Teflon membrane. (iii) Corresponding flow of oxygen ṅ(O2). (iv) Resulting electron numbers at the second working electrode (black) and share of superoxide x (red) calculated. (A) Reproduced with permission from (Reinsberg et al., 2016b). Copyright 2016 American Chemical Society. (B) Intermetallic CaLi2-O2 batteries: (i) voltage profile of galvanostatic cycling and (ii) capacity retention of CaLi2-O2 cell using 0.1 M Ca[OTf]2/TEGDME at a current density of 50 mA g−1SuperP and a capacity of 300 mAh g−1SuperP. (iii) Raman spectra of the fully discharged cathodes without PVDF of CaLi2-O2 cells where label the salt used and the number indicate the discharge capacity (mAh g−1SuperP). Discharged Ca[OTf]2 (or Ca[TFA]2) cell shows a clear band at 840 cm−1 in agreement with the reference spectra of CaO2. (B) Reproduced with permission from (Kim et al., 2020).
In a subsequent study by Baltruschat’s research group, the effect of different alkaline-earth cations on the ORR/OER in DMSO-type electrolytes was demonstrated, as discussed initially in the context of Mg2+ electrolytes in the previous section (Reinsberg et al., 2018). As per the previous investigation, peroxide remains the dominant product at Au electrodes in DMSO electrolytes containing Ca2+ as well as Mg2+, Sr2+, and Ba2+ (where [ClO4]- was used as the salt counter-anion in all electrochemical measurements). However, while the onset of O2 reduction at Pt and GC remained consistent, the products of ORR were found to be highly dependent on the divalent cation. Therein, the tendency for peroxide formation over superoxide (as per the number of electrons per mole of O2 consumed) increases from Ca2+<Sr2+<Ba2+≈Mg2+, such that superoxide dominates in Ca2+-containing systems and exclusively peroxide forms in the presence of Mg2+. The prevalence of superoxide or peroxide formation in these DMSO-based electrolytes is attributed, in part, to the type of interactions between superoxide generated in the first electron transfer and the cation at the electrode interface. The authors suggest that strongly polarizing cations with a large acceptor number (AN) may, by the withdrawal of electron density, polarize the superoxide radical anion and facilitate the second electron transfer. However, since there are no AN data available for Ca2+ (or Sr2+), these trends were only compared for Mg2+/Ba2+ with Li+ and Na+ cations and it remains to be seen if this parameter would also describe the very different behavior of the Ca2+ electrolyte.
The construction of Ca-O2 full cells relies on the ability to stabilize and control both challenging interfaces for Ca-metal plating/stripping and the ORR/OER. Formulation of electrolytes suitable for both environments remains a challenge for all of the metal-O2 battery chemistries; however, given the previous discussions of recent developments in these fields, such examples pertaining to Ca are still limited. The first such example made use of an IL-based electrolyte coupled with a RM-functionalized cathode framework and a Ca-metal chip anode, all held at 60 °C (Shiga et al., 2017). The electrolyte was a [DEME][TFSI] IL with 0.1 M Ca[TFSI]2 and the redox mediator was TEMPO grafted to a polymer backbone. Herein, the reaction on discharge under an O2 atmosphere was attributed to the formation of CaO at the cathode with greater than 1,500 mAh g−1 capacity at ca. 1.8 V on the first discharge. Upon charging, gas evolution analysis (by gas chromatography–mass spectrometry, GC-MS) demonstrated that oxidation of discharge products formed under O2 was possible in the presence of the TEMPO-group RM. However, the charge/discharge efficiency fell in the range of 60–80%, and the observed significant capacity fade was attributed primarily to anode stability/reversibility issues. While the IL-based electrolyte somewhat facilitates plating/stripping, as discussed in the earlier section, large overpotentials (ca. 2 V) and poor efficiencies (<10%) at the Ca-metal interface reduced cell voltages and led to rapid cell failure. Significant volumes of H2 gas were also evolved during charging of the cell, likely originating from important routes to the decomposition of the electrolyte that would impact cell operation.
In a recent attempt to alleviate issues pertaining to the reactivity and conductivity of Ca interfaces, while enabling the use of more conventional electrolyte systems for the benefit of cathode processes, the use of an intermetallic CaLi2 alloy electrode has been employed in full Ca/Li-O2 cells (Kim et al., 2020). In conjunction with a simple carbon black (Super P)-based cathode, the CaLi2 alloy anode supports full-cell cycling under O2 with a tetraethylene glycol dimethyl ether (TEGDME)/trifluoromethanesulfone ([OTf]−) electrolyte with 1 M Li[OTf] or 0.1 M Ca[OTf]2. While the majority of testing relates to the use of the Li-salt electrolytes, wherein the CaLi2 alloy anode supports cycling performances comparable to Li-metal, CaLi2-O2 cells employing Ca-electrolytes were able to cycle for more than 20 discharge/charge cycles (Figure 6B-i,ii). Reversible cycling was, therein, attributed to the formation of CaO2 at the cathode confirmed by Raman spectroscopy (Figure 6B-iii) and XRD and XPS analysis. Notably, cycling was reported under comparatively low current density and capacity regimes due to the poor conductivity of this Ca-based electrolyte formulation. The full CaLi2-O2 cell employing Ca[OTf]2 did, however, support a large single discharge capacity up to 5,300 mAh g−1 (based on mass of carbon cathode with a 2 V (vs. CaLi2) cut-off limit). For both single salt electrolyte systems, the authors present evidence of the other cation at the cathode after cycling in various reaction products, demonstrating dissolution of both Ca and Li from the alloy anode occurs. While this factor may complicate prolonged cycling of full cells, the application of dual electrolytes containing Ca2+ and Li+ warrants further investigations. Critically, this work highlights how the CaLi2 alloy anode enables the possibility of employing commercially available electrolyte components to facilitate both the Ca plating/stripping steps and the ORR/OER in the same cell. Dedicated optimization of the electrolyte components to increase Ca2+ solubility (beyond 0.1 M) and ionic conductivity could further expand the performance and cyclability of such CaLi2-O2 systems.
The electrochemistry of the ORR and OER in aprotic electrolytes containing Mg2+and Ca2+ divalent metal cations remains in the early stages of investigation. So far, Baltruschat’s group has conducted a series of important fundamental investigations on the effects of divalent metal ions (mainly Mg/Ca and one including Sr/Ba) on the O2 electrochemistry in DMSO. Therein, reaction mechanisms and product formation were shown to be dependent on substrate and cation species within the DMSO. Additionally, analogous to Li- and Na-based M-O2 chemistry, the chosen solvent is also expected to affect these processes via solvation effects on the metal cations and the reaction intermediates at the electrode interface. Such mechanistic investigations still need to be expanded in more electrolyte systems to explore any general applicability and, subsequently, to inform the design of new materials/substrates toward minimizing passivation and promoting reversibility of the reactions. Furthermore, ILs have been demonstrated as promising candidates to accommodate the oxygen electrochemistry for Mg/Ca; however, in addition to solubility and transport limitations, such investigations suggest that the incorporation of RMs or effective electrocatalysts into the air electrode would be required to overcome passivation of the interface.
The nature of the discharge process and the final product at the cathode interface is critical for determining cell capacity and rechargeability in M-O2 cells. For Li/Na/K-O2 chemistries, the monovalent cation superoxides (MO2, M = Li+, Na+, K+) play a pivotal role in determining, to different degrees, the discharge performance (ORR). For example, the solvation of LiO2 reaction intermediate has been found to affect how the final product Li2O2 can precipitate on the air cathode (Johnson et al., 2014); NaO2 can make up a significant part of the discharge products (Ortiz-Vitoriano et al., 2015); KO2 is the sole discharge product (Yu et al., 2017). In contrast, the dependence of the ORR on the superoxide (as product or reaction intermediate) has not been demonstrated in the comparative divalent Mg/Ca systems, and the formation of the peroxide and oxide species are primary products. The only exception is Ca(O2)2, which has been reported to form as the primary product on various working electrodes (aside from Au). However, this is only observed in the DMSO-based electrolyte, and the explanation is not yet fully understood. In order to obtain more information about the effect of electrolytes on the ORR/OER, the desired product distribution, and to ascertain critical stability information, operando spectroscopic investigations of the air-cathode interface in more electrolyte systems (DMSO, acetonitrile, ether, glyme, etc.) should be the focus of future research. Nevertheless, progress is highly dependent on the development of suitable Mg and Ca salts that have an appreciable solubility in these solvents to provide sufficient ionic conductivity.
On the other hand, the oxygen reduction is still rather irreversible in Mg/Ca systems compared with the Li/Na/K analogs, probably due to both the thermodynamically less favorable decomposition of ORR products and the more sluggish OER kinetics. To be more specific, MgO2/MgO and CaO2/CaO, main discharge products in Mg/Ca systems, are thermodynamically much more stable than LiO2/Li2O2, NaO2/Na2O2, and KO2 (∆G° = −230/−570.8 kJ mol−1 for LiO2/Li2O2, ∆G° = −218.8/−449.7 kJ mol−1 for NaO2/Na2O2, ∆G° = −239.4 kJ mol−1 for KO2, ∆G° = −567.8/−568.9 kJ mol−1 for MgO2/MgO, and ∆H° = −652* kJ mol−1 for CaO2 and ∆G° = 603.5* kJ mol−1 for CaO; asterisk labeled value is for formation enthalpy since no ∆G° is available). Also, multiple-electron transfer (at least two electrons) during the OER can result in sluggish kinetics. Although rechargeable metal-air batteries in these systems can be realized using RMs, understanding the underlying mechanisms and how they are mediated by solvents, substrates, overpotentials, and discharge/charge rates is still essential to develop practically viable batteries. For instance, some studies suggested that the ORR in Mg2+-containing electrolytes proceeds via an inner-sphere route (Reinsberg et al., 2016a), while other articles observed an opposite behavior similar to the outer-sphere mechanism (Law et al., 2016). This difference in the observed ORR/OER mechanisms can cause confusion whether or not the electrocatalyst is needed, therefore impeding the design of proper air electrodes. On the other hand, having metal oxide as the final product in Mg-air or Ca-air batteries may not be ideal, although it delivers the largest discharge capacity and energy density. Considering the reversibility of the discharge product, the metal peroxide could be more desirable for rechargeable metal-air batteries (Smith et al., 2016). It has been well accepted that the properties of discharge products (composition, morphology, etc.), as well as their interaction with the electrolyte, can strongly influence the cell capacity and cycle life (Vardar et al., 2015). Therefore, designing the air cathode and electrolyte, which allows the targeted formation of desired ORR products, will be a future direction for investigations.
Plating and stripping at Mg anodes have been studied for many years, with much progress being made within the past 2 decades. However, many reported electrolyte classes are expected to be incompatible in M-O2 systems and for alternative high-voltage chemistries. Conversely, much of the evidence of successful Ca plating/stripping has arrived only in the previous 5 years and has been realized without comparative organometallic- or halide-based chemistries that may be expected to yield stability and corrosion issues. In light of the discussed developments, suggestions and recommendations have been made to inspire possible applications of some potentially suitable electrolytes in M-O2 full cells, wherein pairing good oxidative stabilities and conductivities with low overpotential and efficient plating/stripping is a critical target for candidate electrolytes. Further understanding of the components that make for a good SEI on Mg/Ca anodes should be prioritized to support design of improved materials. Recent works on the cathode (electro)chemistries have then been discussed to glean information from these mechanistic studies. Such fundamental investigations, in the very early stages compared to the ongoing discoveries in comparative Li/Na-O2 chemistries, are important especially for the continued optimization and control of the discharge and charge processes and, subsequently, designing suitable air cathodes therein. However, while degrees of reversibility have been observed in both Mg and Ca systems, only redox mediators show the potential ability to facilitate efficient ORR/OER during battery cycling. Consequently, the parallel development of electroactive redox mediator additives may be vital to support rechargeability of the ORR/OER in a meaningful way and their effects on the anode processes should be considered. To date, research on Mg/Ca has been still quite limited in comparison with Li/Na/K, particularly for the air cathode. More electrolyte systems containing various salts/solvents are required to be probed, in order to obtain more generalized understandings of important processes in divalent metal-air batteries and deeper insights into their underlying chemical nature.
Y-TL: writing original draft and review, editing, and visualization. AN: writing original draft and review, editing, and visualization. C-CH: supervision, writing review, and editing. LH: funding acquisition, project administration, supervision, writing review, and editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the funding from EPSRC (EP/R020744/1 and EP/R000441/1). Additionally, the authors would like to acknowledge Callum Shields for the artistic contribution to create Figure 1.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2020.602918/full#supplementary-material.
Abdallah, T., Lemordant, D., and Claude-Montigny, B. (2012). Are room temperature ionic liquids able to improve the safety of supercapacitors organic electrolytes without degrading the performances?. J. Power Sources 201, 353–359. doi:10.1016/j.jpowsour.2011.10.115
Abraham, K., and Jiang, Z. (1996). A polymer electrolyte based rechargeable lithium/oxygen battery. J. Electrochem. Soc. 143, 1–5. doi:10.1149/1.1836378
Aldous, I. M., and Hardwick, L. J. (2014). Influence of tetraalkylammonium cation chain length on gold and glassy carbon electrode interfaces for alkali metal-oxygen batteries. J. Phys. Chem. Lett. 5, 3924–3930. doi:10.1021/jz501850u
Aldous, I. M., and Hardwick, L. J. (2016). Solvent-mediated control of the electrochemical discharge products of non-aqueous sodium-oxygen electrochemistry. Angew Chem. Int. Ed. Engl. 55, 8254–8257. doi:10.1002/anie.201601615
Alnashef, I. M., Leonard, M. L., Kittle, M. C., Matthews, M. A., and Weidner, J. W. (2001). Electrochemical generation of superoxide in room-temperature ionic liquids. ECS Solid State Lett. 4, D16–D18. doi:10.1149/1.1406997
Aurbach, D., Skaletsky, R., and Gofer, Y. (1991). The electrochemical behavior of calcium electrodes in a few organic electrolytes. J. Electrochem. Soc. 138, 3536. doi:10.1149/1.2085455
Biria, S., Pathreeker, S., Li, H., and Hosein, I. D. (2019). Plating and stripping of calcium in an alkyl carbonate electrolyte at room temperature. ACS Appl. Energy Mater. 2, 7738–7743. doi:10.1021/acsaem.9b01670
Biria, S., Pathreeker, S., Genier, F. S., Li, H., and Hosein, I. D. (2020). Plating and stripping calcium at room temperature in an ionic-liquid electrolyte. ACS Appl. Energy Mater. 3, 2310–2314. doi:10.1021/acsaem.9b02529
Bozorgchenani, M., Fischer, P., Schnaidt, J., Diemant, T., Schwarz, R. M., Marinaro, M., et al. (2018). Electrocatalytic oxygen reduction and oxygen evolution in Mg free and Mg–containing ionic liquid 1 butyl 1 methylpyrrolidinium bis (trifluoromethanesulfonyl) imide. ChemElectroChem. 5, 2600–2611. doi:10.1002/celc.201800508
Brenner, A., and Sligh, J. (1971). Electrodeposition of magnesium and beryllium from organic baths. Trans. Inst. Met. Finish. 49, 71–78. doi:10.1080/00202967.1971.11870170
Brenner, A. (1971). Note on the electrodeposition of magnesium from an organic solution of a magnesium boron complex. J. Electrochem. Soc. 118, 99. doi:10.1149/1.2407964
Canepa, P., Jayaraman, S., Cheng, L., Rajput, N. N., Richards, W. D., Gautam, G. S., et al. (2015). Elucidating the structure of the magnesium aluminum chloride complex electrolyte for magnesium-ion batteries. Energy Environ. Sci. 8, 3718–3730. doi:10.1039/c5ee02340h
Carbone, L., Moro, P. T., Gobet, M., Munoz, S., Devany, M., Greenbaum, S. G., et al. (2018). Enhanced lithium oxygen battery using a glyme electrolyte and carbon nanotubes. ACS Appl. Mater. Interfaces. 10, 16367–16375. doi:10.1021/acsami.7b19544
Carter, T. J., Mohtadi, R., Arthur, T. S., Mizuno, F., Zhang, R., Shirai, S., et al. (2014). Boron clusters as highly stable magnesium-battery electrolytes. Angew Chem. Int. Ed. Engl. 53, 3173–3177. doi:10.1002/anie.201310317
Chang, Z., Yang, Y., Wang, X., Li, M., Fu, Z., Wu, Y., et al. (2015). Hybrid system for rechargeable magnesium battery with high energy density. Sci. Rep. 5, 11931–11938. doi:10.1038/srep11931
Chen, Y., Freunberger, S. A., Peng, Z., Fontaine, O., and Bruce, P. G. (2013). Charging a Li-O₂ battery using a redox mediator. Nat. Chem. 5, 489. doi:10.1038/nchem.1646
Chen, P., Zhang, K., Tang, D., Liu, W., Meng, F., Huang, Q., et al. (2020). Recent progress in electrolytes for Zn–air batteries. Front. Chem. 8. doi:10.3389/fchem.2020.00372
Cheng, G., Xu, Q., Zhang, M., Ding, F., Liu, X., and Jiao, L. (2013). Electrochemical reversibility of magnesium deposition-dissolution on aluminum substrates in Grignard reagent/THF solutions. Chin. Sci. Bull. 58, 3385–3389. doi:10.1007/s11434-013-6008-7
Chusid, O., Gofer, Y., Gizbar, H., Vestfrid, Y., Levi, E., Aurbach, D., et al. (2003). Solid state rechargeable magnesium batteries. Adv. Mater. 15, 627–630. doi:10.1002/adma.200304415
Connor, J. H., Reid, W. E., and Wood, G. B. (1957). Electrodeposition of metals from organic solutions: V. Electrodeposition of magnesium and magnesium alloys. J. Electrochem. Soc. 104, 38. doi:10.1149/1.2428492
Das, S., Højberg, J., Knudsen, K. B., Younesi, R., Johansson, P., Norby, P., et al. (2015). Instability of ionic liquid-based electrolytes in Li–O2 batteries. J. Phys. Chem. C. 119, 18084–18090. doi:10.1021/acs.jpcc.5b04950
De Giorgio, F., Soavi, F., and Mastragostino, M. (2011). Effect of lithium ions on oxygen reduction in ionic liquid-based electrolytes. Electrochem. Commun. 13, 1090–1093. doi:10.1016/j.elecom.2011.07.004
Deivanayagam, R., Cheng, M., Wang, M., Vasudevan, V., Foroozan, T., Medhekar, N. V., et al. (2019). Composite polymer electrolyte for highly cyclable room-temperature solid-state magnesium batteries. ACS Appl. Energy Mater. 2, 7980–7990. doi:10.1021/acsaem.9b01455
Doe, R. E., Han, R., Hwang, J., Gmitter, A. J., Shterenberg, I., Yoo, H. D., et al. (2014). Novel, electrolyte solutions comprising fully inorganic salts with high anodic stability for rechargeable magnesium batteries. Chem. Commun. 50, 243–245. doi:10.1039/C3CC47896C
Dong, Q., Yao, X., Luo, J., Zhang, X., Hwang, H., and Wang, D. (2016). Enabling rechargeable non-aqueous Mg-O. Chem. Commun. 52, 13753–13756. doi:10.1039/C6CC07818D
Dongmo, S., Zaubitzer, S., Schüler, P., Krieck, S., Jörissen, L., Wohlfahrt‐Mehrens, M., et al. (2020). Stripping and plating a magnesium metal anode in bromide‐based non‐nucleophilic electrolytes. ChemSusChem. 13, 3530–3538. doi:10.1002/cssc.202000249
Du, A., Zhang, H., Zhang, Z., Zhao, J., Cui, Z., Zhao, Y., et al. (2019). A crosslinked polytetrahydrofuran‐borate‐based polymer electrolyte enabling wide‐working‐temperature‐range rechargeable magnesium batteries. Adv. Mater. 31, 1805930. doi:10.1002/adma.201805930
Fan, H., Zhao, Y., Xiao, J., Zhang, J., Wang, M., and Zhang, Y. (2020). A non-nucleophilic gel polymer magnesium electrolyte compatible with sulfur cathode. Nano Res., 1–6. doi:10.1007/s12274-020-2923-5
Fu, J., Liang, R., Liu, G., Yu, A., Bai, Z., Yang, L., et al. (2019). Recent progress in electrically rechargeable zinc–air batteries. Adv. Mater. 31, 1805230. doi:10.1002/adma.201805230
Gao, X., Mariani, A., Jeong, S., Liu, X., Dou, X., Ding, M., et al. (2019). Prototype rechargeable magnesium batteries using ionic liquid electrolytes. J. Power Sources. 423, 52–59. doi:10.1016/j.jpowsour.2019.03.049
Gelman, D., Shvartsev, B., and Ein-Eli, Y. (2019). “Challenges and prospect of non–aqueous non-alkali (NANA) metal–air batteries,” in Electrochemical energy storage: next generation battery concepts. Editor R.-A. Eichel (Cham, Switzerland: Springer International Publishing), 127–168.
Gilmore, P., and Sundaresan, V. B. (2019). A functionally graded cathode architecture for extending the cycle‐life of potassium‐oxygen batteries. Batteries Supercaps. 2, 678–687. doi:10.1002/batt.201900025
Giridhar, P., El Abedin, S. Z., and Endres, F. (2012). Electrodeposition of aluminium from 1-butyl-1-methylpyrrolidinium chloride/AlCl3 and mixtures with 1-ethyl-3-methylimidazolium chloride/AlCl3. Electrochim. Acta. 70, 210–214. doi:10.1016/j.electacta.2012.03.056
Goossens, K., Rakers, L., Heinrich, B., Ahumada, G., Ichikawa, T., Donnio, B., et al. (2019). Anisotropic, organic ionic plastic crystal mesophases from persubstituted imidazolium pentacyanocyclopentadienide salts. Chem. Mater. 31, 9593–9603. doi:10.1021/acs.chemmater.9b02338
Grande, L., Von Zamory, J., Koch, S. L., Kalhoff, J., Paillard, E., and Passerini, S. (2015). Homogeneous lithium electrodeposition with pyrrolidinium-based ionic liquid electrolytes. ACS Appl. Mater. Interfaces. 7, 5950–5958. doi:10.1021/acsami.5b00209
Gregory, T. D., Hoffman, R. J., and Winterton, R. C. (1990). Nonaqueous electrochemistry of magnesium: applications to energy storage. J. Electrochem. Soc. 137, 775. doi:10.1149/1.2086553
Guo, Y., Yang, J., Nuli, Y., and Wang, J. (2010). Study of electronic effect of Grignard reagents on their electrochemical behavior. Electrochem. Commun. 12, 1671–1673. doi:10.1016/j.elecom.2010.08.015
Guo, Y.-S., Zhang, F., Yang, J., Wang, F.-F., Nuli, Y., and Hirano, S.-I. (2012). Boron-based electrolyte solutions with wide electrochemical windows for rechargeable magnesium batteries. Energy Environ. Sci. 5, 9100–9106. doi:10.1039/C2EE22509C
Ha, J. H., Adams, B., Cho, J.-H., Duffort, V., Kim, J. H., Chung, K. Y., et al. (2016). A conditioning-free magnesium chloride complex electrolyte for rechargeable magnesium batteries. J. Mater. Chem. 4, 7160–7164. doi:10.1039/C6TA01684G
Ha, T. A., Pozo-Gonzalo, C., Nairn, K., Macfarlane, D. R., Forsyth, M., and Howlett, P. C. (2020). An investigation of commercial carbon air cathode structure in ionic liquid based sodium oxygen batteries. Sci. Rep. 10, 1–10. doi:10.1038/s41598-020-63473-y
Haas, I., and Gedanken, A. (2008). Synthesis of metallic magnesium nanoparticles by sonoelectrochemistry. Chem. Commun., 1795–1797. doi:10.1039/B717670H
Han, S.-D., Rajput, N. N., Qu, X., Pan, B., He, M., Ferrandon, M. S., et al. (2016). Origin of electrochemical, structural, and transport properties in nonaqueous zinc electrolytes. ACS Appl. Mater. Interfaces. 8, 3021–3031. doi:10.1021/acsami.5b10024
Han, S., Cai, C., Yang, F., Zhu, Y., Sun, Q., Zhu, Y. G., et al. (2020). Interrogation of the reaction mechanism in a Na–O2 battery using in situ transmission electron microscopy. ACS Nano. 14, 3669–3677. doi:10.1021/acsnano.0c00283
Hardwick, L. J., and De León, C. P. (2018). Rechargeable multi-valent metal-air batteries. Johnson Matthey Technol. Rev. 62, 134–149. doi:10.1595/205651318X696729
Hartmann, P., Bender, C. L., Vračar, M., Dürr, A. K., Garsuch, A., Janek, J., et al. (2013). A rechargeable room-temperature sodium superoxide (NaO2) battery. Nat. Mater. 12, 228–232. doi:10.1038/NMAT3486
He, S., Nielson, K. V., Luo, J., and Liu, T. L. (2017). Recent advances on MgCl2 based electrolytes for rechargeable Mg batteries. Energy Stor. Mater. 8, 184–188. doi:10.1016/j.ensm.2016.12.001
Hegemann, P., Hegemann, M., Zan, L., and Baltruschat, H. (2019). Stability of tetraglyme for reversible magnesium deposition from a magnesium aluminum chloride complex. J. Electrochem. Soc. 166, A245. doi:10.1149/2.0871902jes
Higashi, S., Miwa, K., Aoki, M., and Takechi, K. (2014). A novel inorganic solid state ion conductor for rechargeable Mg batteries. Chem. Commun. 50, 1320–1322. doi:10.1039/C3CC47097K
Hu, K., Qin, L., Zhang, S., Zheng, J., Sun, J., Ito, Y., et al. (2020). Building a reactive armor using S-doped graphene for protecting potassium metal anodes from oxygen crossover in K−O2 batteries. ACS Energy Lett. doi:10.1021/acsenergylett.0c00715
Hwang, H. J., Chi, W. S., Kwon, O., Lee, J. G., Kim, J. H., and Shul, Y.-G. (2016). Selective ion transporting polymerized ionic liquid membrane separator for enhancing cycle stability and durability in secondary zinc–air battery systems. ACS Appl. Mater. Interfaces. 8, 26298–26308. doi:10.1021/acsami.6b07841
Jäckle, M., Helmbrecht, K., Smits, M., Stottmeister, D., and Groβ, A. (2018). Self-diffusion barriers: possible descriptors for dendrite growth in batteries? Energy Environ. Sci. 11, 3400–3407. doi:10.1039/C8EE01448E
Jaschin, P. W., Gao, Y., Li, Y., and Bo, S.-H. (2020). A materials perspective on magnesium-ion-based solid-state electrolytes. J. Mater. Chem. 8, 2875–2897. doi:10.1039/C9TA11729F
Jin, L., Howlett, P. C., Pringle, J. M., Janikowski, J., Armand, M., Macfarlane, D. R., et al. (2014). An organic ionic plastic crystal electrolyte for rate capability and stability of ambient temperature lithium batteries. Energy Environ. Sci. 7, 3352–3361. doi:10.1039/C4EE01085J
Johnson, L., Li, C., Liu, Z., Chen, Y., Freunberger, S. A., Ashok, P. C., et al. (2014). The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li–O2 batteries. Nat. Chem. 6, 1091. doi:10.1038/nchem.2101
Jusys, Z., Schnaidt, J., and Behm, R. J. (2019). O2 reduction on a Au film electrode in an ionic liquid in the absence and presence of Mg2+ ions: product formation and adlayer dynamics. J. Chem. Phys. 150, 041724. doi:10.1063/1.5051982
Kakibe, T., Yoshimoto, N., Egashira, M., and Morita, M. (2010). Optimization of cation structure of imidazolium-based ionic liquids as ionic solvents for rechargeable magnesium batteries. Electrochem. Commun. 12, 1630–1633. doi:10.1016/j.elecom.2010.09.012
Kakibe, T., Hishii, J.-Y., Yoshimoto, N., Egashira, M., and Morita, M. (2012). Binary ionic liquid electrolytes containing organo-magnesium complex for rechargeable magnesium batteries. J. Power Sources. 203, 195–200. doi:10.1016/j.jpowsour.2011.10.127
Katayama, Y., Sekiguchi, K., Yamagata, M., and Miura, T. (2005). Electrochemical behavior of oxygen/superoxide ion couple in 1-butyl-1-methylpyrrolidinium bis (trifluoromethylsulfonyl) imide room-temperature molten salt. J. Electrochem. Soc. 152, E247–E250. doi:10.1149/1.1946530
Khan, A., and Zhao, C. (2014). Enhanced performance in mixture DMSO/ionic liquid electrolytes: toward rechargeable Li–O2 batteries. Electrochem. Commun. 49, 1–4. doi:10.1016/j.elecom.2014.09.014
Kim, Y.-S., Cho, Y.-G., Odkhuu, D., Park, N., and Song, H.-K. (2013). A physical organogel electrolyte: characterized by in situ thermo-irreversible gelation and single-ion-predominent conduction. Sci. Rep. 3, 1–6. doi:10.1038/srep01917
Kim, M.-J., Kang, H.-J., Im, W. B., and Jun, Y.-S. (2020). Rechargeable intermetallic calcium–lithium–O2 batteries. ChemSusChem. 13, 574–581. doi:10.1002/cssc.201902925
Kumar, G. G., and Munichandraiah, N. (1999). Reversibility of Mg/Mg2+ couple in a gel polymer electrolyte. Electrochim. Acta. 44, 2663–2666. doi:10.1016/S0013-4686(98)00388-0
Kundu, D., Black, R., Adams, B., and Nazar, L. F. (2015). A highly active low voltage redox mediator for enhanced rechargeability of lithium–oxygen batteries. ACS Cent. Sci. 1, 510–515. doi:10.1021/acscentsci.5b00267
Lair, V., Sirieix-Plenet, J., Gaillon, L., Rizzi, C., and Ringuedé, A. (2010). Mixtures of room temperature ionic liquid/ethanol solutions as electrolytic media for cerium oxide thin layer electrodeposition. Electrochim. Acta. 56, 784–789. doi:10.1016/j.electacta.2010.09.102
Law, Y. T., Schnaidt, J., Brimaud, S., and Behm, R. J. (2016). Oxygen reduction and evolution in an ionic liquid ([BMP][TFSA]) based electrolyte: a model study of the cathode reactions in Mg-air batteries. J. Power Sources. 333, 173–183. doi:10.1016/j.jpowsour.2016.09.025
Lee, B., Cho, J.-H., Seo, H. R., Na, S. B., Kim, J. H., Cho, B. W., et al. (2018). Strategic combination of Grignard reagents and allyl-functionalized ionic liquids as an advanced electrolyte for rechargeable magnesium batteries. J. Mater. Chem. 6, 3126–3133. doi:10.1039/C7TA09330F
Leisegang, T., Meutzner, F., Zschornak, M., Münchgesang, W., Schmid, R., Nestler, T., et al. (2019). The aluminum-ion battery: a sustainable and seminal concept?. Front. Chem. 7, 268. doi:10.3389/fchem.2019.00268
Leng, L., Zeng, X., Chen, P., Shu, T., Song, H., Fu, Z., et al. (2015). A novel stability-enhanced lithium-oxygen battery with cellulose-based composite polymer gel as the electrolyte. Electrochim. Acta. 176, 1108–1115. doi:10.1016/j.electacta.2015.07.111
Li, C. S., Sun, Y., Gebert, F., and Chou, S. L. (2017). Current progress on rechargeable magnesium–air battery. Adv. Energy Mater. 7, 1700869. doi:10.1002/aenm.201700869
Li, Z., Fuhr, O., Fichtner, M., and Zhao-Karger, Z. (2019). Towards stable and efficient electrolytes for room-temperature rechargeable calcium batteries. Energy Environ. Sci. 12, 3496–3501. doi:10.1039/C9EE01699F
Li, C., Wei, J.-S., Qiu, K., and Wang, Y.-G. (2020a). Li−air battery with a super-hydrophobic Li-protective layer. ACS Appl. Mater. Interfaces. doi:10.1021/acsami.0c05494
Li, X., Xing, Y., Xu, J., Deng, Q., and Shao, L.-H. (2020b). Uniform yolk–shell structured Si–C nanoparticles as a high performance anode material for the Li-ion battery. Chem. Commun. doi:10.1039/C9CC07997A
Liang, F., and Hayashi, K. (2015). A high-energy-density mixed-aprotic-aqueous sodium-air cell with a ceramic separator and a porous carbon electrode. J. Electrochem. Soc. 162, A1215. doi:10.1149/2.0421507jes
Liebenow, C., Yang, Z., and Lobitz, P. (2000). The electrodeposition of magnesium using solutions of organomagnesium halides, amidomagnesium halides and magnesium organoborates. Electrochem. Commun. 2, 641–645. doi:10.1016/S1388-2481(00)00094-1
Liebenow, C. (1997). Reversibility of electrochemical magnesium deposition from Grignard solutions. J. Appl. Electrochem. 27, 221–225. doi:10.1023/A:1018464210084
Liu, T., Shao, Y., Li, G., Gu, M., Hu, J., Xu, S., et al. (2014). A facile approach using MgCl2 to formulate high performance Mg2+ electrolytes for rechargeable Mg batteries. J. Mater. Chem. 2, 3430–3438. doi:10.1039/C3TA14825D
Liu, Z., Cui, T., Pulletikurthi, G., Lahiri, A., Carstens, T., Olschewski, M., et al. (2016). Dendrite‐free nanocrystalline zinc electrodeposition from an ionic liquid containing nickel triflate for rechargeable Zn‐based batteries. Angew. Chem. Int. Ed. 55, 2889–2893. doi:10.1002/anie.201509364
Liu, J., Bao, Z., Cui, Y., Dufek, E. J., Goodenough, J. B., Khalifah, P., et al. (2019). Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy. 4, 180–186. doi:10.1038/s41560-019-0338-x
Lu, Y.-T., Chien, Y.-J., Liu, C.-F., You, T.-H., and Hu, C.-C. (2017). Active site-engineered bifunctional electrocatalysts of ternary spinel oxides, M0.1Ni0.9Co2O4 (M: Mn, Fe, Cu, Zn) for the air electrode of rechargeable zinc–air batteries. J. Mater. Chem. 5, 21016–21026. doi:10.1039/C7TA06302D
Luo, J., Bi, Y., Zhang, L., Zhang, X., and Liu, T. L. (2019a). A stable, non‐corrosive perfluorinated pinacolatoborate Mg electrolyte for rechargeable Mg batteries. Angew. Chem. Int. Ed. 58, 6967–6971. doi:10.1002/anie.201902009
Luo, Z., Li, Y., Liu, Z., Pan, L., Guan, W., Liu, P., et al. (2019b). Prolonging the cycle life of a lithium–air battery by alleviating electrolyte degradation with a ceramic–carbon composite cathode. ChemSusChem. 12, 4962–4967. doi:10.1002/cssc.201901629
Ma, Z., Forsyth, M., Macfarlane, D. R., and Kar, M. (2019). Ionic liquid/tetraglyme hybrid Mg[TFSI]2 electrolytes for rechargeable Mg batteries. Green Energy Environ. 4, 146–153. doi:10.1016/j.gee.2018.10.003
Macfarlane, D. R., Huang, J., and Forsyth, M. (1999). Lithium-doped plastic crystal electrolytes exhibiting fast ion conduction for secondary batteries. Nature. 402, 792–794. doi:10.1038/45514
Mizrahi, O., Amir, N., Pollak, E., Chusid, O., Marks, V., Gottlieb, H., et al. (2008). Electrolyte solutions with a wide electrochemical window for rechargeable magnesium batteries. J. Electrochem. Soc. 155, A103–A109. doi:10.1149/1.2806175
Monaco, S., Soavi, F., and Mastragostino, M. (2013). Role of oxygen mass transport in rechargeable Li/O2 batteries operating with ionic liquids. J. Phys. Chem. Lett. 4, 1379–1382. doi:10.1021/jz4006256
Muldoon, J., Bucur, C. B., Oliver, A. G., Sugimoto, T., Matsui, M., Kim, H. S., et al. (2012). Electrolyte roadblocks to a magnesium rechargeable battery. Energy Environ. Sci. 5, 5941–5950. doi:10.1039/C2EE03029B
Muldoon, J., Bucur, C. B., and Gregory, T. (2014). Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem. Rev. 114, 11683–11720. doi:10.1021/cr500049y
Narayanan, N. V., Raj, B. A., and Sampath, S. (2009). Magnesium ion conducting, room temperature molten electrolytes. Electrochem. Commun. 11, 2027–2031. doi:10.1016/j.elecom.2009.08.045
Neale, A. R., Li, P., Jacquemin, J., Goodrich, P., Ball, S. C., Compton, R. G., et al. (2016). Effect of cation structure on the oxygen solubility and diffusivity in a range of bis{(trifluoromethyl)sulfonyl}imide anion based ionic liquids for lithium–air battery electrolytes. Phys. Chem. Chem. Phys. 18, 11251–11262. doi:10.1039/C5CP07160G
Neale, A. R., Goodrich, P., Hughes, T.-L., Hardacre, C., Ball, S. C., and Jacquemin, J. (2017). Physical and electrochemical investigations into blended electrolytes containing a glyme solvent and two bis {(trifluoromethyl) sulfonyl} imide-based ionic liquids. J. Electrochem. Soc. 164, H5124. doi:10.1149/2.0141708jes
Ng, B., Peng, X., Faegh, E., and Mustain, W. E. (2020). Using nanoconfinement to inhibit the degradation pathways of conversion-metal oxide anodes for highly stable fast-charging Li-ion batteries. J. Mater. Chem. 8, 2712–2727. doi:10.1039/C9TA11708C
Ogasawara, T., Débart, A., Holzapfel, M., Novák, P., and Bruce, P. G. (2006). Rechargeable Li2O2 electrode for lithium batteries. J. Am. Chem. Soc. 128, 1390–1393. doi:10.1021/ja056811q
Ortiz-Vitoriano, N., Batcho, T. P., Kwabi, D. G., Han, B., Pour, N., Yao, K. P. C., et al. (2015). Rate-dependent nucleation and growth of NaO2 in Na–O2 batteries. J. Phys. Chem. Lett. 6, 2636–2643. doi:10.1021/acs.jpclett.5b00919
Overcash, D. M., and Mathers, F. (1933). The electrodeposition of magnesium. Trans. Electrochem. Soc. 64, 305. doi:10.1149/1.3504531
Pandey, G., and Hashmi, S. (2009). Experimental investigations of an ionic-liquid-based, magnesium ion conducting, polymer gel electrolyte. J. Power Sources. 187, 627–634. doi:10.1016/j.jpowsour.2008.10.112
Ponrouch, A., Frontera, C., Bardé, F., and Palacín, M. R. (2016). Towards a calcium-based rechargeable battery. Nat. Mater. 15, 169–172. doi:10.1038/nmat4462
Ponrouch, A., Bitenc, J., Dominko, R., Lindahl, N., Johansson, P., and Palacin, M. R. (2019). Multivalent rechargeable batteries. Energy Stor. Mater. doi:10.1016/j.ensm.2019.04.012
Pour, N., Gofer, Y., Major, D. T., and Aurbach, D. (2011). Structural analysis of electrolyte solutions for rechargeable Mg batteries by stereoscopic means and DFT calculations. J. Am. Chem. Soc. 133, 6270–6278. doi:10.1021/ja1098512
Pringle, J. M., Howlett, P. C., Macfarlane, D. R., and Forsyth, M. (2010). Organic ionic plastic crystals: recent advances. J. Mater. Chem. 20, 2056–2062. doi:10.1039/B920406G
Pujare, N. U. (1988). A calcium oxygen secondary battery. J. Electrochem. Soc. 135, 260. doi:10.1149/1.2095574
Reinsberg, P., Bondue, C., and Baltruschat, H. (2016a). Mechanistic investigation of the oxygen reduction in magnesium ion-containing dimethyl sulfoxide. Electrochim. Acta. 200, 214–221. doi:10.1016/j.electacta.2016.03.157
Reinsberg, P., Bondue, C. J., and Baltruschat, H. (2016b). Calcium–oxygen batteries as a promising alternative to sodium–oxygen batteries. J. Phys. Chem. C. 120, 22179–22185. doi:10.1021/acs.jpcc.6b06674
Reinsberg, P., Abd-El-Latif, A.-E.-a. A., and Baltruschat, H. (2018). Investigation of the complex influence of divalent cations on the oxygen reduction reaction in aprotic solvents. Electrochim. Acta. 273, 424–431. doi:10.1016/j.electacta.2018.03.123
Ren, X., and Wu, Y. (2013). A low-overpotential potassium–oxygen battery based on potassium superoxide. J. Am. Chem. Soc. 135, 2923–2926. doi:10.1021/ja312059q
Sarangika, H., Dissanayake, M., Senadeera, G., Rathnayake, R., and Pitawala, H. (2017). Polyethylene oxide and ionic liquid-based solid polymer electrolyte for rechargeable magnesium batteries. Ionics. 23, 2829–2835. doi:10.1007/s11581-016-1870-3
Sawyer, D. T., Calderwood, T. S., Yamaguchi, K., and Angelis, C. T. (1983). Synthesis and characterization of tetramethylammonium superoxide. Inorg. Chem. 22, 2577–2583. doi:10.1021/ic00160a022
See, K. A., Liu, Y.-M., Ha, Y., Barile, C. J., and Gewirth, A. A. (2017). Effect of concentration on the electrochemistry and speciation of the magnesium aluminum chloride complex electrolyte solution. ACS Appl. Mater. Interfaces. 9, 35729–35739. doi:10.1021/acsami.7b08088
Shao, Y., Rajput, N. N., Hu, J., Hu, M., Liu, T., Wei, Z., et al. (2015). Nanocomposite polymer electrolyte for rechargeable magnesium batteries. Nanomater. Energy. 12, 750–759. doi:10.1016/j.nanoen.2014.12.028
Sheng, C., Yu, F., Wu, Y., Peng, Z., and Chen, Y. (2018). Disproportionation of sodium superoxide in metal–air batteries. Angew. Chem. Int. Ed. 57, 9906–9910. doi:10.1002/anie.201804726
Shiga, T., Hase, Y., Kato, Y., Inoue, M., and Takechi, K. (2013). A rechargeable non-aqueous Mg–O2 battery. Chem. Commun. 49, 9152–9154. doi:10.1039/C3CC43477J
Shiga, T., Kato, Y., and Hase, Y. (2017). Coupling of nitroxyl radical as an electrochemical charging catalyst and ionic liquid for calcium plating/stripping toward a rechargeable calcium–oxygen battery. J. Mater. Chem. 5, 13212–13219. doi:10.1039/C7TA03422A
Shterenberg, I., Salama, M., Gofer, Y., Levi, E., and Aurbach, D. (2014). The challenge of developing rechargeable magnesium batteries. MRS Bull. 39, 453–460. doi:10.1557/mrs.2014.61
Shyamsunder, A., Blanc, L. E., Assoud, A., and Nazar, L. F. (2019). Reversible calcium plating and stripping at room temperature using a borate salt. ACS Energy Lett. 4, 2271–2276. doi:10.1021/acsenergylett.9b01550
Smith, J. G., Naruse, J., Hiramatsu, H., and Siegel, D. J. (2016). Theoretical limiting potentials in Mg/O2 batteries. Chem. Mater. 28, 1390–1401. doi:10.1021/acs.chemmater.5b04501
Staniewicz, R. J. (1980). A study of the calcium-thionyl chloride electrochemical system. J. Electrochem. Soc. 127, 782. doi:10.1149/1.2129758
Ta, K., Zhang, R., Shin, M., Rooney, R. T., Neumann, E. K., and Gewirth, A. A. (2019). Understanding Ca electrodeposition and speciation processes in nonaqueous electrolytes for next-generation Ca-ion batteries. ACS Appl. Mater. Interfaces. 11, 21536–21542. doi:10.1021/acsami.9b04926
Tchitchekova, D., Monti, D., Johansson, P., Bardé, F., Randon-Vitanova, A., Palacín, M., et al. (2017). On the reliability of half-cell tests for monovalent (Li+, Na+) and divalent (Mg2+, Ca2+) cation based batteries. J. Electrochem. Soc. 164, A1384–A1392. doi:10.1149/2.0411707jes
Trahan, M. J., Mukerjee, S., Plichta, E. J., Hendrickson, M. A., and Abraham, K. (2012). Studies of Li-air cells utilizing dimethyl sulfoxide-based electrolyte. J. Electrochem. Soc. 160, A259. doi:10.1149/2.048302jes
Vardar, G., Nelson, E. G., Smith, J. G., Naruse, J., Hiramatsu, H., Bartlett, B. M., et al. (2015). Identifying the discharge product and reaction pathway for a secondary Mg/O2 battery. Chem. Mater. 27, 7564–7568. doi:10.1021/acs.chemmater.5b03608
Viestfrid, Y., Levi, M., Gofer, Y., and Aurbach, D. (2005). Microelectrode studies of reversible Mg deposition in THF solutions containing complexes of alkylaluminum chlorides and dialkylmagnesium. J. Electroanal. Chem. 576, 183–195. doi:10.1016/j.jelechem.2004.09.034
Wang, D., Gao, X., Chen, Y., Jin, L., Kuss, C., and Bruce, P. G. (2018a). Plating and stripping calcium in an organic electrolyte. Nat. Mater. 17, 16–20. doi:10.1038/nmat5036
Wang, W., Lai, N. C., Liang, Z., Wang, Y., and Lu, Y. C. (2018b). Superoxide stabilization and a universal KO2 growth mechanism in potassium–oxygen batteries. Angew. Chem. Int. Ed. 57, 5042–5046. doi:10.1002/anie.201801344
Wang, H., Feng, X., Chen, Y., Liu, Y.-S., Han, K. S., Zhou, M., et al. (2019a). Reversible electrochemical interface of Mg metal and conventional electrolyte enabled by intermediate adsorption. ACS Energy Lett. 5, 200–206. doi:10.1021/acsenergylett.9b02211
Wang, S., Cai, W., Sun, Z., Huang, F., Jie, Y., Liu, Y., et al. (2019b). Stable cycling of Na metal anodes in a carbonate electrolyte. Chem. Commun. 55, 14375–14378. doi:10.1039/C9CC07419H
Watkins, T., Kumar, A., and Buttry, D. A. (2016). Designer ionic liquids for reversible electrochemical deposition/dissolution of magnesium. J. Am. Chem. Soc. 138, 641–650. doi:10.1021/jacs.5b11031
Xiao, N., Gourdin, G., and Wu, Y. (2018). Simultaneous stabilization of potassium metal and superoxide in K–O2 batteries on the basis of electrolyte reactivity. Angew. Chem. Int. Ed. 57, 10864–10867. doi:10.1002/anie.201804115
Yang, W., Salim, J., Ma, C., Ma, Z., Sun, C., Li, J., et al. (2013). Flowerlike Co3O4 microspheres loaded with copper nanoparticle as an efficient bifunctional catalyst for lithium–air batteries. Electrochem. Commun. 28, 13–16. doi:10.1016/j.elecom.2012.12.007
Yao, Z., Hegde, V. I., Aspuru‐Guzik, A., and Wolverton, C. (2019). Discovery of calcium‐metal alloy anodes for reversible Ca‐ion batteries. Adv. Energy Mater. 9, 1802994. doi:10.1002/aenm.201802994
Yi, J., Liu, X., Guo, S., Zhu, K., Xue, H., and Zhou, H. (2015). Novel stable gel polymer electrolyte: toward a high safety and long life Li–air battery. ACS Appl. Mater. Interfaces. 7, 23798–23804. doi:10.1021/acsami.5b08462
Yin, W.-W., Yue, J.-L., Cao, M.-H., Liu, W., Ding, J.-J., Ding, F., et al. (2015). Dual catalytic behavior of a soluble ferrocene as an electrocatalyst and in the electrochemistry for Na–air batteries. J. Mater. Chem. 3, 19027–19032. doi:10.1039/C5TA04647E
Yu, W., Lau, K. C., Lei, Y., Liu, R., Qin, L., Yang, W., et al. (2017). Dendrite-free potassium–oxygen battery based on a liquid alloy anode. ACS Appl. Mater. Interfaces. 9, 31871–31878. doi:10.1021/acsami.7b08962
Zhang, J., Zhou, Q., Tang, Y., Zhang, L., and Li, Y. (2019). Zinc–air batteries: are they ready for prime time?. Chem. Sci. 10, 8924–8929. doi:10.1039/c9sc04221k
Zhao, Q. S., Nuli, Y. N., Guo, Y. S., Yang, J., and Wang, J. L. (2011). Reversibility of electrochemical magnesium deposition from tetrahydrofuran solutions containing pyrrolidinyl magnesium halide. Electrochim. Acta. 56, 6530–6535. doi:10.1016/j.electacta.2011.04.114
Zhao, X., Chen, F., Liu, J., Cheng, M., Su, H., Liu, J., et al. (2020). Enhanced surface binding energy regulates uniform potassium deposition for stable potassium metal anodes. J. Mater. Chem. 8, 5671–5678. doi:10.1039/C9TA14226F
Zhou, Z.-B., and Matsumoto, H. (2007). Lithium-doped, organic ionic plastic crystal electrolytes exhibiting high ambient-temperature conductivities. Electrochem. Commun. 9, 1017–1022. doi:10.1016/j.elecom.2006.12.012
Keywords: metal-air batteries, divalent cations, magnesium batteries, calcium batteries, metal electroplating, oxygen electrochemistry
Citation: Lu Y-T, Neale AR, Hu C-C and Hardwick LJ (2021) Divalent Nonaqueous Metal-Air Batteries. Front. Energy Res. 8:602918. doi: 10.3389/fenrg.2020.602918
Received: 04 September 2020; Accepted: 20 November 2020;
Published: 12 February 2021.
Edited by:
Jian Liu, University of British Columbia Okanagan, CanadaReviewed by:
Liqiang Mai, Wuhan University of Technology, ChinaCopyright © 2021 Lu, Neale, Hu and Hardwick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurence J. Hardwick, aGFyZHdpY2tAbGl2ZXJwb29sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.