 Yunyi Yang
Yunyi Yang Zhihong Wu1,2
Zhihong Wu1,2 Guangting Han
Guangting Han Haoxi Ben
Haoxi Ben- 1School of Energy and Environment, Southeast University, Nanjing, China
- 2Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, Nanjing, China
- 3School of Textile and Clothing, Qingdao University, Qingdao, China
- 4Laboratory of New Fiber Materials and Modern Textile, Qingdao, China
A key challenge in studying the upgrading process for the thermochemical conversion of biomass, such as lignin, is to understand the underlying mechanisms of catalytic conversion at the atomic scale. In this study, a method combined with in situ 2H NMR and GC-MS was proposed for investigating the conversion of phenethyl phenyl ether (PPE) in the hydrotreating process, as catalyzed by Pd, Ru, and Pt loaded onto C or γ-Al2O3. The results indicated that Pd/γ-Al2O3 prefers to produce more ether bond-cleaved products, while Pt prefers to produce more hydrogenation products from PPE. Furthermore, based on this new strategy, a possible reaction mechanism of PPE with Pd/γ-Al2O3 was presented from the atomic point of view, showing the potential of this in situ detection method for reaction mechanism studies. Besides, mechanistic investigations by GC-MS were accomplished for the hydrothermal treatment of PPE for comparison with the new method. The results showed that the in situ 2H NMR combined with GC-MS provided a deeper understanding of the catalytic mechanism compared to GC-MS alone.
Introduction
Lignin is a waste material from the paper industry and a by-product from second-generation bio-ethanol production processes. Lignin is also abundant in waste biomass from agriculture, as well as yard and forestry wastes (Barton et al., 2018). Furthermore, lignin is the only renewable aromatic resources in nature (Konnerth et al., 2015). However, as low-heat fuels or additives of concrete, the commercial utilization rate of lignin is still <10%, which is far lower than that of cellulose and hemicellulose (Li et al., 2015; Wang et al., 2019). Under this circumstance, by converting lignin into fuel or valuable chemicals through appropriate methods, reducing use of fossil fuel and lower emissions of carbon dioxide could be achieved (David and Ragauskas, 2010). Thus, how to make more reasonable use of lignin has attracted the attention of researchers around the world.
At present, common conversion methods of lignin include pyrolysis, acid or alkali catalysis, hydrocracking, gasification, and reductive transformation (Katahira et al., 2016; Shuai et al., 2016; Fan et al., 2018; Santana et al., 2018; Sirousrezaei et al., 2018). Among these, the premise of the efficient utilization of lignin is the depolymerization of lignin (Chio et al., 2019; Zhang and Wang, 2020). In terms of structure, lignin is a three-dimensional polymer which is mainly composed of basic phenylpropane units, such as p-hydroxyphenyl, guaiacyl and syringyl (Zakzeski et al., 2010). These basic units are connected by C-C or C-O bonds. From Table 1, the reported abundance of C-O bonds is more than that for C-C bonds in softwood and hardwood (Chakar and Ragauskas, 2004; Zakzeski et al., 2010). Besides, the theoretical calculation of a C-O bond's dissociation energy is usually smaller than that of a C-C bond in lignin model compounds, which indicates that the dissociation of lignin usually starts with a C-O bond. Therefore, the cleavage of C-O bonds is of great significance for the depolymerization of lignin (Elder and Beste, 2014).
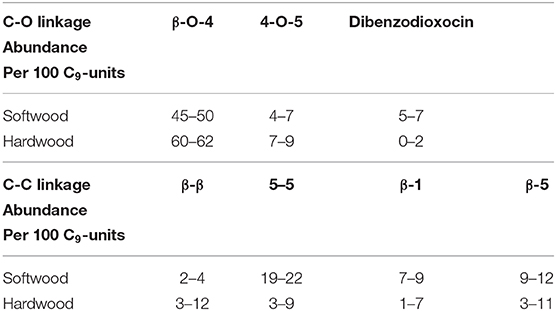
Table 1. Reported abundance of major linkages in softwood and hardwood lignin (Chakar and Ragauskas, 2004; Zakzeski et al., 2010).
Hydrogenolysis, where H2 is used at high temperature and high pressure with metal catalysts, is regarded as an effective way to cleave the bond of C-O for the purpose of converting lignin into high-value liquid precursors (Yan and Dyson, 2013). Thus, it is crucial to find a suitable catalyst for breaking C-O bonds effectively. However, lignin has a relatively complex structure albeit without a defined one. Thus, model compounds have been employed to represent some typical structures of lignin. Phenethyl phenyl ether (PPE) has been commonly used to represent the β-O-4 linkage, which is one of the major linkages in the lignin structure. Recently, Mauriello et al. (2018) applied the co-precipitation technique to synthesize Pd/Ni to catalyze the conversion of PPE. In 2013, Song et al. (2013) employed Pd/C, Ru/C, and Ni/C for hydrogenation and cracking reactions of PPE. Most of the research focuses on the catalytic effect of different catalysts on PPE but less on the catalytic mechanism. However, the study of catalytic mechanism is also of considerable importance in the synthesis of catalysts and the design of production processes (Shuang et al., 2019).
To study inner mechanisms, choosing appropriate techniques to monitor the reaction is important. GC-MS has been most widely used in various chemical reactions in order to speculate the reaction process. However, some specific reaction process and intermediate products cannot be effectively recorded and studied with GC-MS. Recently, in situ monitoring using NMR has attractive attention. Richter et al. (2019) used in situ 13C NMR spectra to identify final products, intermediates, and by-products during the whole reaction, which in turn helped to reveal the inner mechanism of electrocatalytic oxidation of alcohol. An in situ 1H NMR technique was also employed by Wang et al. in order to follow the reaction of inulin biomass (Wang et al., 2015). The above-mentioned studies showed that in situ detection is an effective way to understand the reaction mechanism. Isotope labeling combined with 2H NMR is another effective means for investigating the reaction mechanism. Wang et al. (2018) employed CD4 to trace the hydrogen atoms of methane in the co-pyrolysis of methane and cellulose, providing evidence of methane incorporation into aromatic compounds by 2H NMR characterization. In our previous research, the ring-opening mechanism of lignin model compounds was investigated by 2H NMR as well (Ben et al., 2016).
In this study, we proposed a strategy of isotopic (deuterium) labeling combined with in situ 2H NMR monitoring and GC-MS to detect and analyze the catalytic upgrading process of PPE. Pt, Pd, and Ru on two commonly used supporting materials (carbon and Al2O3) served as catalysts. For comparison, our strategy and GC-MS alone were separately applied in the hydrotreating process and hydrothermal treatment of PPE. Furthermore, based on the data from the whole reaction, a possible transformation mechanism at the atomic scale was proposed and discussed. This research showed the potential of a high-pressure in situ 2H NMR monitoring method using isotopic labeling for studying the upgrading process of biomass model compounds.
Materials and Methods
Materials
PPE, tetramethylsilane-d12(TMS-d12), chloroform, phenol, ethyl benzene, phenylethyl alcohol, cyclohexanone, cyclohexanol and cyclohexane ethanol were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Pt/Al2O3(5%), Pt/C(5%), Pd/Al2O3(5%), Pd/C(5%), Ru/Al2O3(5%), and Ru/C(5%) were purchased from Sigma-Aldrich and dried to remove moisture before use. Deuterium gas was purchased from Airgas.
Experiment and Experimental Procedure
Hydrotreating Process
A reaction mixture was prepared in a 5 mm NMR tube (Wilmad), with 100 μL of PPE, 0.5 μL of TMS-d12 and 2 mg of catalyst. The NMR tube was purged five times with D2 to remove the air. The initial pressure in the NMR tube was set to 700 kPa, after which it was inserted into the preheated NMR spectrometer where the temperature was 100°C. The spectra were recorded every hour for 16 h. Finally, the samples which reacted in the NMR tube were dissolved in chloroform for subsequent GC-MS analysis.
Hydrothermal Treatment
5 mg of PPE, 0.3 mg of catalyst and 5 ml of water were added to the reaction tube. The reactor was purged with H2 three times to exclude air and then pressurized to 700 kPa with H2. The reactor was kept at 100°C for 12 h. The products were filtered and extracted with 1 mL of chloroform for GC-MS analysis. The external label method was used for the quantitative analysis of products.
Characterization of Products
The products from hydrotreating process were analyzed by 2H NMR and GC-MS, while the products from hydrothermal treatment were analyzed by GC-MS.
All 2H NMR data were recorded with a Bruker Avance/DMX 600 MHz NMR spectrometer. 2H NMR spectra were detected with 1.5 s of acquisition time, 2 s of relaxation delay and 1,024 scans by Bruker's pulse program “zg2h” with the lock channel in a Bruker's BBO probe. Then, data were processed by MestReNova 11.0 with automatic phase correction and six-order Bernstein polynomial. The chemical shift for 2H NMR was referenced to 0.00 ppm of TMS-d12.
The analysis of GC-MS was accomplished by an Agilent G1530A gas chromatograph (GC) interfaced with a HP 5973 mass spectrometer. The GC injector was operated at 280°C. 61.3 mL/min of constant He flow was applied to the capillary column (Agilent 190915-433). The GC oven was programmed as follows: hold at 50°C for 1 min, heated up to 280°C with a ramp of 10.0°C/min and hold at 280°C for 1 min.
Theoretical Chemical Calculation Method
All theoretical chemical calculations were conducted by Gaussian09. For the calculation of chemical shifts, model compounds were optimized with B3LYP/6-31g+(d, p) while frequency calculations were performed on the optimized model to ensure that there was no virtual frequency. Gauge-independent atomic orbital (GIAO) methodology combined with B3LYP/6-311g+(2d, p) was used to acquire the NMR isotropic shielding tensors. In order to reduce the error, linear regression was employed to convert the calculation results to chemical shifts. The relevant formula is as follows where the intercept is 31.8884 and the slope is −1.0481 (Jain et al., 2009; Pierens, 2014).
The M06-2X method and the 6-31G+(d, p) basis set were employed to calculate the C-H bond dissociation energy (BDE) of PPE at 100°C and 700 kPa (Wang and Liu, 2016).
Results and Discussion
The hydrotreating process and hydrothermal treatment were conducted as the two common approaches to the lignin upgrading process. For insights into the catalytic mechanism of PEE, a milder condition (100°C and 700 kPa) was used in this study, which allowed for the preservation of potential intermediates.
Hydrotreating Process
Figures 1, 2 and Figures S1–S4 show the results of the deuterium-traced reaction of PPE as catalyzed by Pd, Ru, and Pt which were loaded onto C or γ-Al2O3.
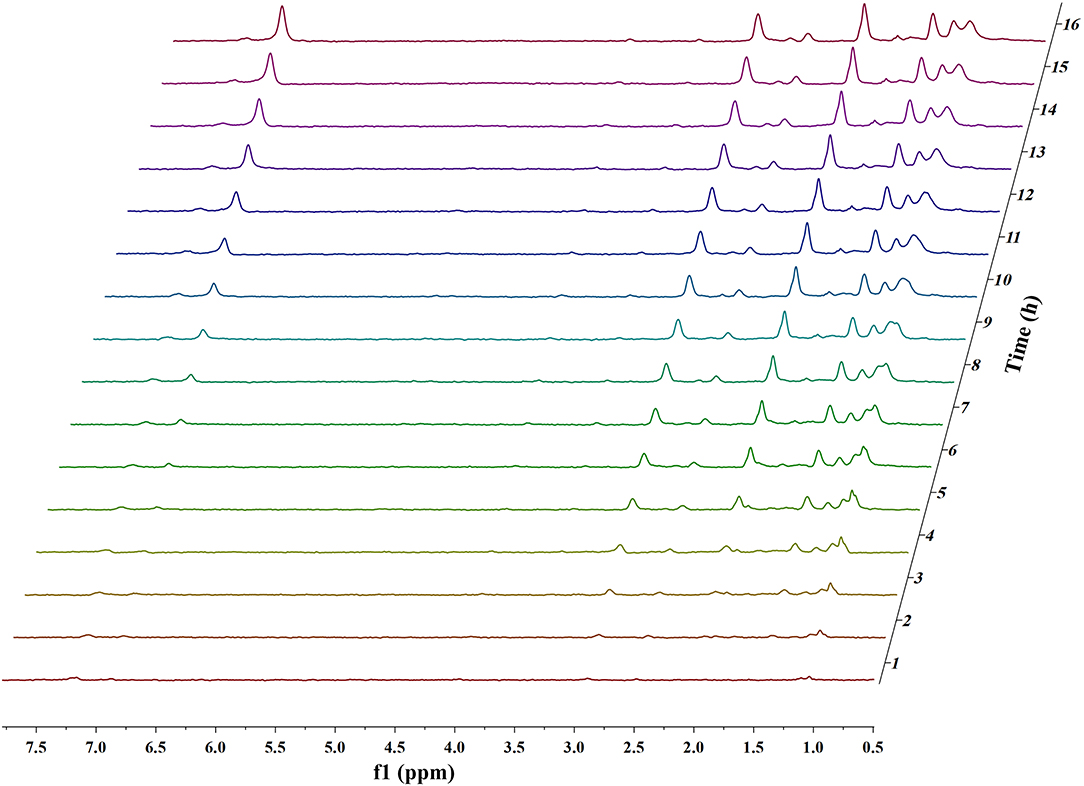
Figure 1. In situ 2H NMR monitoring of the deuterium traced upgrading reaction of PPE on Pd/γ-Al2O3 (5 wt%).
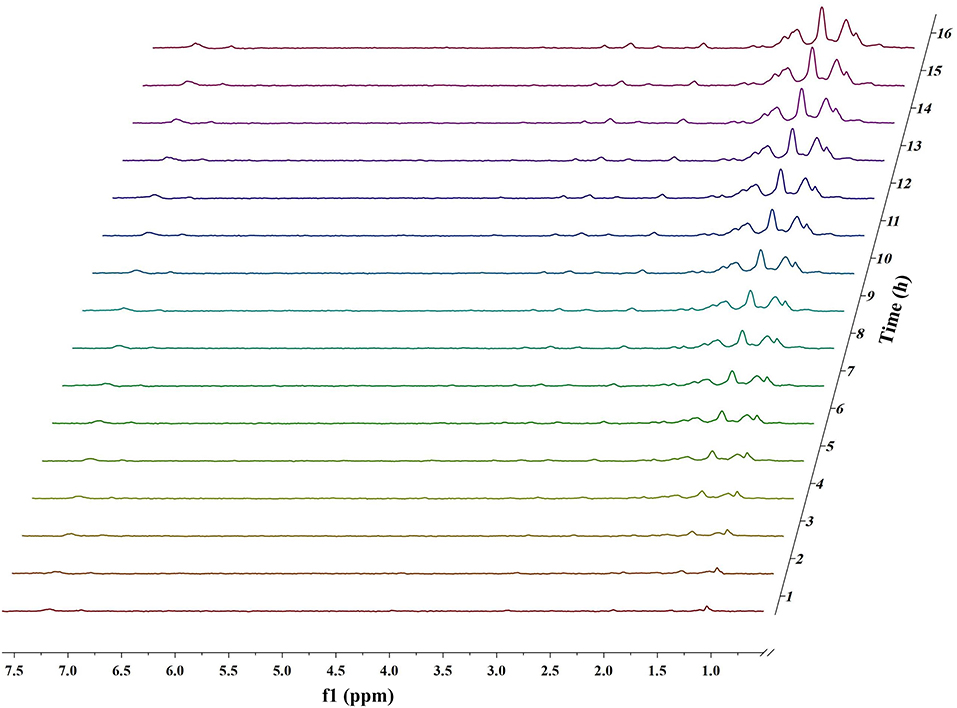
Figure 2. In situ 2H NMR monitoring of the deuterium traced upgrading reaction of PPE on Pt/γ-Al2O3 (5 wt%).
The Analysis of the Chemical Shift of Products
In 2H NMR analysis, the chemical shifts in the functional groups refer to Table S1 (Mullen et al., 2009; Naik et al., 2010; Bordoloi et al., 2015; Ardiyanti et al., 2016; Tessarolo et al., 2016). Accordingly, the peaks from 0.4 to 1.8 ppm belong to aliphatic deuterium. Besides, the possible reaction pathway of PPE in the hydrotreating process is shown in Figure S5, according to the products of GC-MS. Therefore, aliphatic deuterium should derive from deuterium labeled ethylbenzene, cyclohexanol, ethyl cyclohexane, and the hydrogenation products of PPE. Based on the GC-MS data (Figures S6–S8), deuterium labeled ethyl cyclohexane cannot be detected. Furthermore, the content of deuterium labeled cyclohexanol was limited. Thus, the detected aliphatic deuterium mainly belonged to deuterium labeled ethylbenzene and hydrogenation products of PPE. The existence of deuterium labeled ethylbenzene represents a break in the β-O-4 bond while the other products containing aliphatic deuterium point to the hydrogenation of PPE (Figure S5). By comparing the products catalyzed by Pd/γ-Al2O3 and Pt/γ-Al2O3 (Figures S6–S8), we see that Pd/γ-Al2O3 can promote the cleavage of ether bond better, while more hydrogenation products could be produced under the catalysis of Pt/γ-Al2O3.
The difference in the conversion products of PPE under different catalysts was also reflected in 2H NMR. The areas of the two peaks around 1.43 ppm and 2.01 ppm in Figure 1 (Pd/γ-Al2O3) were much larger than in the case of Figure 2 (Pt/γ-Al2O3). Besides, these two peaks had similar areas and appeared simultaneously 3 h after the start of reaction (Figure 1). From Table S1, the peak at 2.01 ppm did not belong to deuterium labeled ethylbenzene nor to the hydrogenation products of PPE. The peak at 1.43 ppm belonged to the range of β-CH3 attached to the aromatic ring, which may have been produced by deuterium labeled ethylbenzene. Meanwhile, according to the data from the Gaussian simulation (Table S2), the terminal aliphatic deuterium of ethylbenzene was assigned to the peak around 1.33 ppm, which is close to the chemical shift of 1.43 ppm. Combined with GC-MS data, the peak at 1.43 ppm should belong to deuterium labeled ethylbenzene and the peak at 2.01 ppm should belong to deuterium labeled phenol. Thus, the remaining peaks in the range from 0.4 ppm to 1.8 ppm were mainly assigned to the products from the hydrogenation of PPE. From Figure S9, the relative aliphatic deuterium content under Pt/γ-Al2O3 was greater than that under Pd/γ-Al2O3. This means that the Pt/γ-Al2O3 can better promote the hydrogenation of PPE. The result was also consistent with the GC-MS data (Figure S8).
Pd
The data above have shown that Pd/γ-Al2O3 can effectively catalyze the cleavage of the β-O-4 bond. Further study of the data reveals the mechanism. In Figure 1, there are two special peaks around 6.89 and 2.89 ppm which do not belong to the conversion products of PPE. First, the peak at 6.89 ppm belongs to aromatic deuterium (Table S1), which should come from the exchange of H/D. Consistently, the products from the H/D exchange of PPE molecules were detected in GC-MS as well (Figure S10). It can be inferred from Figure S10 that the hydrogen which was exchanged with deuterium was the hydrogen 13(14) (the number of atoms can be found in to the Figure 3) and the hydrogen of benzene.
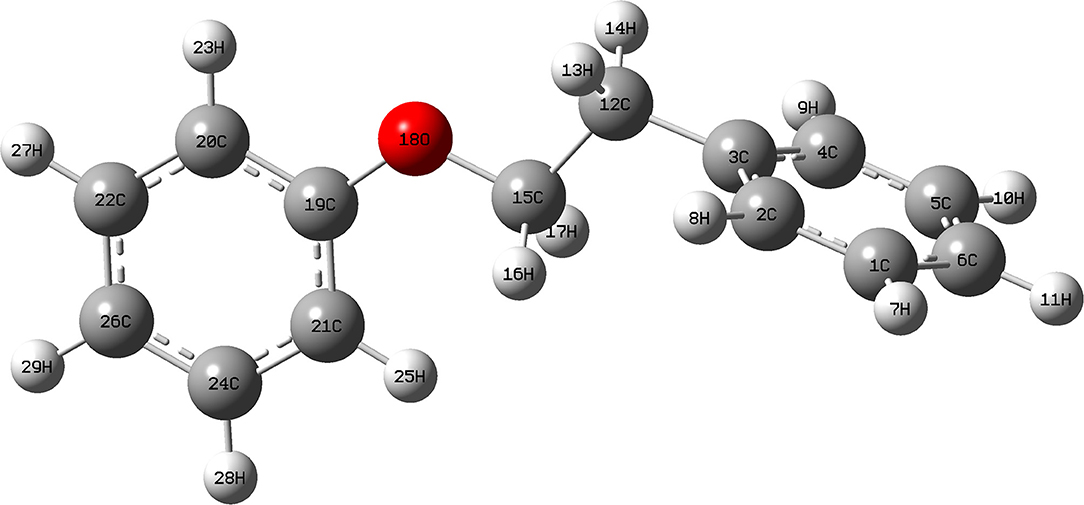
Figure 3. Model structure of PPE.
To further determine the location of H/D exchanges in PPE, Gaussian simulations of chemical shifts and BDE calculations were introduced to this study. The predicted chemical shift in hydrogen 13(14) was about 3.10 ppm, while that for hydrogen 16(17) was around 3.79 ppm (Table S3). Besides, Barton's research showed that the chemical shift in hydrogen 13(14) was 3.00 ppm, while that for hydrogen 16(17) was 4.05 ppm in 1H NMR (Barton et al., 1987). This means that hydrogen 13(14) might be exchanged with the deuterium. In addition, as the BDE of C-H13 is far lower than that of C-H16 according to Gaussian simulation, the bond of C-H13 can more easily perform an H/D exchange (Table S4). Therefore, the chemical shift of 2.89 ppm should be assigned to the deuterium exchanged with H13(14). Furthermore, based on the predicted value of the chemical shift (Table S3) and Barton's research, the peak at 6.89 ppm should belong to the deuterium exchanged with the hydrogen atom on the benzene ring attached to the oxygen atom (Barton et al., 1987). However, the exact hydrogen atom has not thus far been determined. The result of Gaussian optimization illustrated that the PPE molecules were usually configured as shown in Figure 4. Considering H13(14) exchanged with deuterium at the surface of Pd/γ-Al2O3, it can be inferred that the Pd/γ-Al2O3 could hold PPE molecules in this special position (Figure 4) to perform H/D exchanges. Hence, H23 was more likely exchanged with deuterium at the surface of Pd/γ-Al2O3 than the other hydrogen atom on the benzene ring. Besides, through semi-quantitative analysis, it was found that using Pd/γ-Al2O3 led to more H/D exchanges compared with other catalysts (Figure S11).
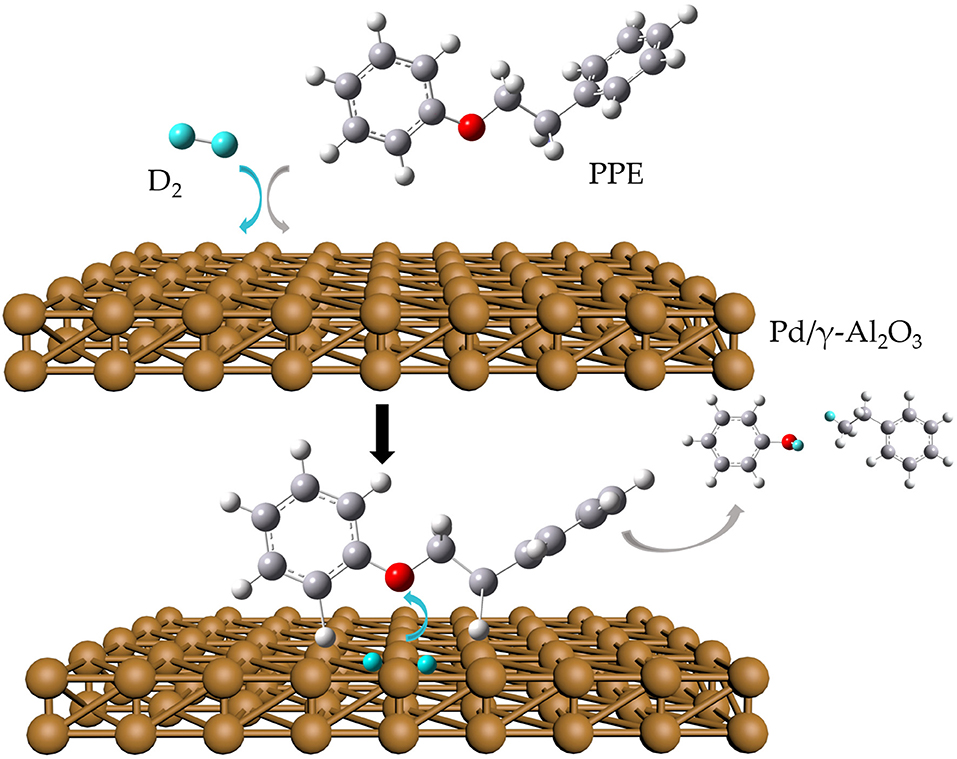
Figure 4. Possible cleavage mechanism of PPE Pd/γ-Al2O3.
Additionally, it is noteworthy that this special position in Figure 4 allows the β-O-4 bond to be fully exposed to the surface of Pd/γ-Al2O3, facilitating the cleavage of the ether bond. It is the possible mechanism that Pd/γ-Al2O3 can more effectively catalyze the cracking of the ether bond (Figure 4). This help us understand the catalytic mechanism from the aspect of atomic scale.
The in situ 2H NMR results from the deuterium traced upgrading reaction of PPE on Pd/C showed that the peaks were concentrated on the products from the hydrogenation of PPE (Figure S1). Compared with Pd/γ-Al2O3, which can catalyze the cleavage of ether bonds, Pd/C tended to exhibit catalytic hydrogenation. It is reported that the noble metals mainly show the function of hydrogenation while the acidic support could exhibit the function of cracking the C-O bond (Robinson et al., 2016; Funkenbusch et al., 2019; Kim et al., 2019). Therefore, Pd/γ-Al2O3 which has a noble metal on an acidic support should exhibit better cleavage function than Pd/C which has a noble metal on an inactive support. This may be the reason why Pd/γ-Al2O3 can catalyze the cleavage of ether bonds more effectively than Pd/C. Besides, the results show that the substrate of Pd had a certain influence on the upgrading process in this condition.
Ru
From Figures S3, S4, the chemical shift in the products catalyzed by Ru was mainly around 1.0 ppm, thus belonging to the hydrogenation products of PPE. By comparing the relative aliphatic deuterium content, the efficiency of Ru supported by carbon and γ-Al2O3 was both lower than for the two other noble metals in the upgrading of PPE (Figure S9). Meanwhile, the products from the H/D exchange by Ru were also limited (Figure S11). Thus, the catalytic activity of Ru was not satisfied under this reaction conditions.
Pt
For Pt/γ-Al2O3 and Pt/C, the detected results indicated that Pt can promote the hydrogenation of the benzene ring in PPE. Due to overlaps in the chemical shift between the hydrogenation products in 2H NMR, further detailed characterizations for the hydrogenation products were accomplished by GC-MS. The GC-MS results (Figure S8) indicated that, compared with Pd/γ-Al2O3, Pt/γ-Al2O3 performed a better hydrogenation process in realizing more partial and full hydrogenation products, while the conversion ratio of PPE was similar under Pd/γ-Al2O3 and Pt/γ-Al2O3 (Figure S12). Another interesting phenomenon was that Pd/γ-Al2O3 preferred to promote hydrogenation on the aliphatic attached aromatic ring (the left benzene ring in the PPE structure in Figure 5), while Pt/γ-Al2O3 preferred to promote hydrogenation on the oxygen-attached aromatic ring (the right benzene ring in the PPE structure in Figure 5) or on both rings.
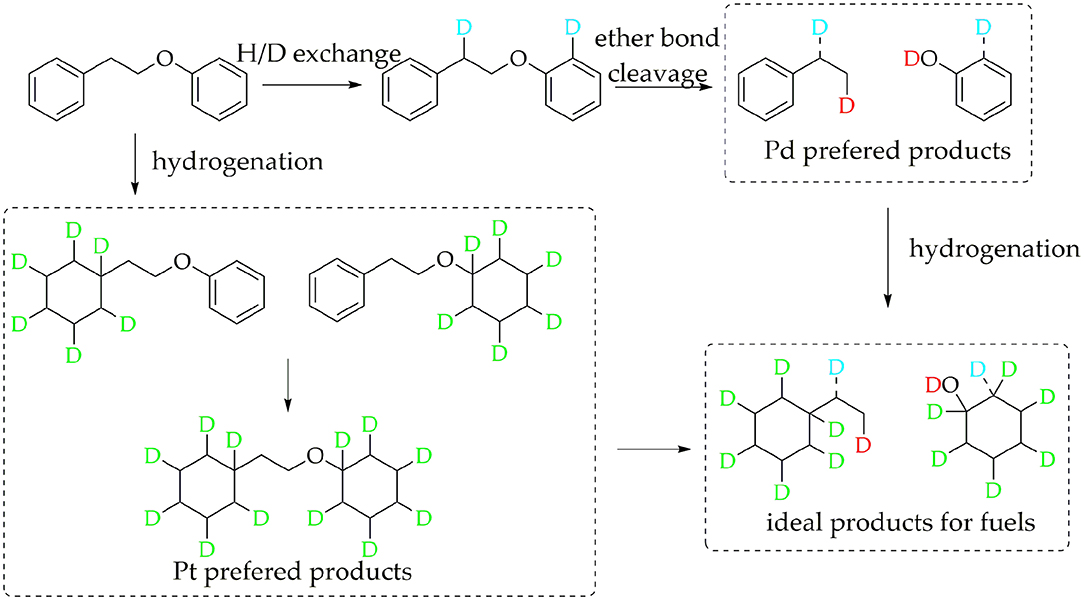
Figure 5. Tentative reaction pathways for in situ 2H NMR monitoring of deuterium traced upgrading reaction of PPE on Pd or Pt/γ-Al2O3.
Obviously, the products of Pd/γ-Al2O3 and Pd/C were different based on the data. However, the products upgraded by Pt/γ-Al2O3 (Figure 2) and Pt/C (Figure S2) did share similar peaks. Analogously, the products upgraded by Ru/γ-Al2O3 (Figure S3) and Ru/C (Figure S4) were nearly the same. This means that the supporting materials affect the reaction pathways differently for diverse catalysts. Besides, the carbon supported catalysts appeared to have lower activity to upgrade PPE in this condition compared with γ-Al2O3 supported catalysts (Figure S9).
Based on the discussion above, the tentative reaction pathways were proposed as Figure 5. In general, Pd/γ-Al2O3 promoted the cleavage of the ether bond, while Pt/γ-Al2O3 was beneficial for hydrogenation. Our detection strategy captured valuable information in the upgrading reaction of PPE, which is meaningful for understanding the catalytic mechanism and the design of new catalyst. For example, in order to achieve better hydrogenolysis of the ether bond, Pd/γ-Al2O3 can be used as a catalyst. Further, a bimetallic catalyst of Pd/γ-Al2O3 combined Pt/γ-Al2O3 may be able to realize an ideal upgrading for PPE or even real lignin, in turn promoting the production of cycloparaffin.
Hydrothermal Treatment
Although the effects of catalysts were different, the composition of the products was similar in the hydrothermal treatment of PPE catalyzed on Pd, Ru and Pt loaded onto C or γ-Al2O3 (Table S5). It was reported that the dissociation energy of C19-O was higher than that of C15-O (101.1 vs. 69.9 kJ/mol), which indicated that C15-O was more susceptible to be cleaved, producing phenol and ethylbenzene by hydrogenolysis (Gomez-Monedero et al., 2017). But, in this study, the yield of phenol was far more than for ethylbenzene. Meanwhile, phenethyl alcohol was detected. The research conducted by Gomez-Monedero et al. (2017) showed that the C19-O bond of PPE can be cleaved in methanol solution by hydrogenolysis generating benzene and phenethyl alcohol. However, benzene was not found in this reaction system, indicating that the hydrogenolysis of C19-O did not occur. Thus, phenylethyl alcohol in the product component was primarily derived from the hydrolysis of the ether bond. Therefore, both hydrogenolysis and hydrolysis reactions were involved in the hydrothermal process.
Among reaction products, there were also some products from hydrogenation. The hydrogenation ratio under hydrothermal conditions (Figure S13) shows the order of hydrogenation ability: Pd/C>Pt/γ-Al2O3>Pd/γ-Al2O3>Pt/C>Ru/C>Ru/γ-Al2O3. Although Pd/C had the highest conversion ratio for PPE, the hydrogenation ratio was more than 50%, which suggests that the reaction process was not selective. For Ru/γ-Al2O3, the hydrogenation ratio for PPE was the lowest and the cleavage ratio of ether bond was around 70%. This indicated that Ru/γ-Al2O3 had the best capacity for the cleavage of the ether bond under this condition. Besides, Pt/γ-Al2O3, Pd/γ-Al2O3, Ru/C and Pt/C had similar catalytic effects. For these catalysts, the ability to catalyze the cleavage of C-O bonds was better than the ability to promote hydrogenation. 4-(2-(cyclohexyloxy)ethyl)cyclohex-1-ene and 5-(2-(cyclohex-2-en-1-1-yl)ethoxy)cyclohexa-1,3-diene were detected in the reaction system, which indicated that the hydrogenation of the reactants was not sufficient. The yields of cyclohexanol, ethyl cyclohexane and 2-cyclohexylethanol were also limited. These may be explained by the low pressure of H2.
The possible reaction paths for PPE in the hydrothermal treatment upgrading reaction are shown in Figure 6. However, if merely based on the results of GC-MS, the initial reaction cannot be identified, neither can the microscopic mechanism. Compared with using GC-MS alone, in situ 2H NMR monitoring combined with GC-MS acquired much more information. For the hydrotreating process, we first proposed the mechanism for the catalytic cracking of PPE by Pd/γ-Al2O3 at the atomic scale, which underlines the superiority of this method in micro-mechanism analysis. Compared with the use of GC-MS alone, in situ 2H NMR monitoring combined with isotope labeling and GC-MS can predict the reaction process and the mechanism for the catalyst more effectively.
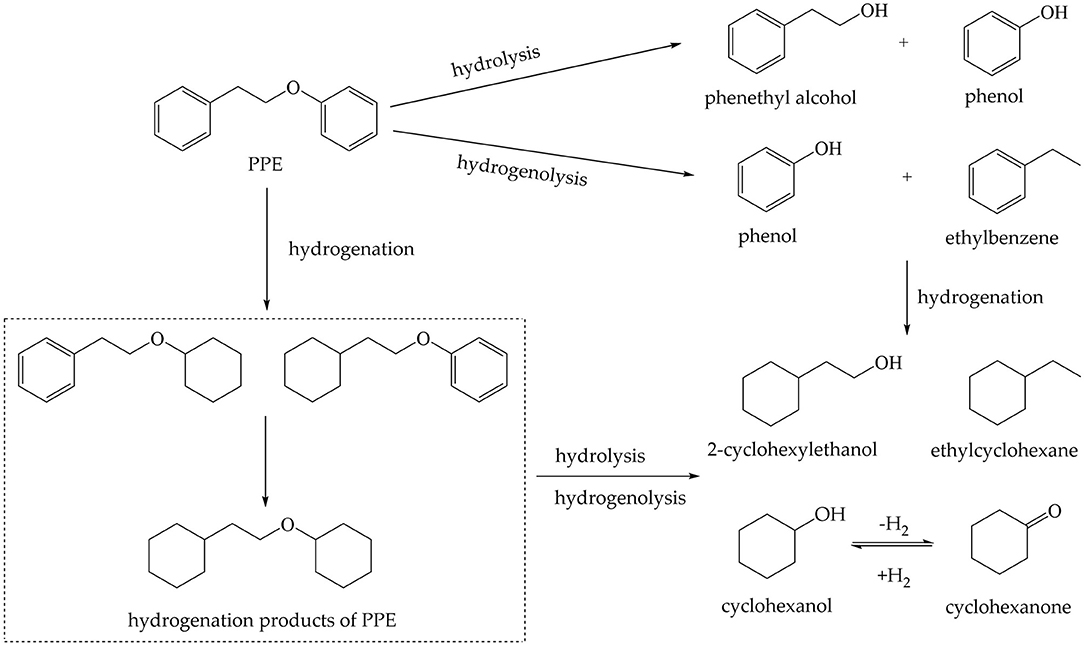
Figure 6. Tentative reaction pathways for the hydrothermal treatment upgrading reaction of PPE.
Conclusions
This study proposed a novel reaction monitoring strategy which is high-pressure in situ 2H NMR and GC-MS for the upgrading process. This strategy exhibited powerful capabilities for identifying reaction products and provided deeper insights into the mechanisms. By employing this method, investigations into catalytic mechanisms for the hydrotreating process of PPE using Pd, Ru and Pt loaded onto C or γ-Al2O3 were accomplished. The results indicated that Pd/γ-Al2O3 was found to have better capacity for catalyzing the break in ether bonds, while Pt tented to produce more hydrogenation products. In addition, two special positions in PPE have been found to have a significantly higher H/D exchange ratio when using Pd/γ-Al2O3, which indicated that Pd/γ-Al2O3 may be able to hold PPE molecules in this special conformation in order to better perform ether bond cleavage. The possible catalytic mechanism of PPE by Pd/γ-Al2O3 was given from an atomic point of view. By comparing with GC-MS used alone, the superiority of this method in predicting the reaction process and catalyst mechanism was further shown. We believe that this method could open up a new direction in exploring the mechanism behind many kinds of reactions.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
HB proposed the conception of the study and directed the project. HB and YY conducted the experiment. ZW and YL performed the statistical analysis. YY and HB wrote the original draft. GH, WJ, and MW reviewed the manuscript.
Funding
This research was funded by from the National Natural Science Foundation of China under Grand No. 51706044, the Natural Science Foundation of the Jiangsu of China under Grand No. BK20170666, and the Recruitment Program for Young Professionals in China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2020.00114/full#supplementary-material
References
Ardiyanti, A. R., Bykova, M. V., Khromova, S. A., Yin, W., Venderbosch, R. H., Yakovlev, V. A., et al. (2016). Ni-based catalysts for the hydrotreatment of fast pyrolysis oil. Energy Fuels 30, 1544–1554. doi: 10.1021/acs.energyfuels.5b02223
Barton, D. H. R., Finet, J. P., Motherwell, W. B., and Pichon, C. (1987). The chemistry of pentavalent organobismuth reagents. part 8. phenylation and oxidation of alcohols by tetraphenylbismuth esters. J. Chem. Soc. Perkin Trans. 1, 251–259. doi: 10.1039/P19870000251
Barton, R. R., Carrier, M., Segura, C., Fierro, J. L. G., Park, S., Lamb, H. H., et al. (2018). Ni/HZSM-5 catalyst preparation by deposition-precipitation. part 2. catalytic hydrodeoxygenation reactions of lignin model compounds in organic and aqueous systems. Appl. Cataly. A General 562, 294–309. doi: 10.1016/j.apcata.2018.06.012
Ben, H., Jarvis, M. W., Nimlos, M. R., Gjersing, E. L., Sturgeon, M. R., Foust, T. D., et al. (2016). Application of a pyroprobe–deuterium NMR system: deuterium tracing and mechanistic study of upgrading process for lignin model compounds. Energy Fuels 30, 2968–2974. doi: 10.1021/acs.energyfuels.5b02729
Bordoloi, N., Narzari, R., Chutia, R. S., Bhaskar, T., and Kataki, R. (2015). Pyrolysis of mesua ferrea and pongamia glabra seed cover: characterization of bio-oil and its sub-fractions. Bioresour. Technol. 178, 83–89. doi: 10.1016/j.biortech.2014.10.079
Chakar, F. S., and Ragauskas, A. J. (2004). Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 20, 131–141. doi: 10.1016/j.indcrop.2004.04.016
Chio, C., Sain, M., and Qin, W. (2019). Lignin utilization: a review of lignin depolymerization from various aspects. Renew. Sust. Energ. Rev. 107, 232–249. doi: 10.1016/j.rser.2019.03.008
David, K., and Ragauskas, A. J. (2010). Switchgrass as an energy crop for biofuel production: a review of its ligno-cellulosic chemical properties. Energy Environ. Sci. 3, 1182–1190. doi: 10.1039/B926617H
Elder, T., and Beste, A. (2014). Density functional theory study of the concerted pyrolysis mechanism for lignin models. Energy Fuels 28, 5229–5235. doi: 10.1021/ef5013648
Fan, L., Chen, P., Zhou, N., Liu, S., Zhang, Y., Liu, Y., et al. (2018). In-situ and ex-situ catalytic upgrading of vapors from microwave-assisted pyrolysis of lignin. Bioresour. Technol. 247, 851–858. doi: 10.1016/j.biortech.2017.09.200
Funkenbusch, L. T., Mullins, M. E., Salam, M. A., Creaser, D., and Olsson, L. (2019). Catalytic hydrotreatment of pyrolysis oil phenolic compounds over Pt/Al2O3 and Pd/C. Fuel 243, 441–448. doi: 10.1016/j.fuel.2019.01.139
Gomez-Monedero, B., Faria, J., Bimbela, F., and Ruiz, M. P. (2017). Catalytic hydroprocessing of lignin β-O-4 ether bond model compound phenethyl phenyl ether over ruthenium catalysts. Biomass Convers. Biorefin. 7, 385–398. doi: 10.1007/s13399-017-0275-5
Jain, R., Bally, T., and Rablen, P. R. (2009). Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem. 74, 4017–4023. doi: 10.1021/jo900482q
Katahira, R., Mittal, A., Mckinney, K., Chen, X., Tucker, M. P., Johnson, D. K., et al. (2016). Base-catalyzed depolymerization of biorefinery lignins. ACS Sustain. Chem. Eng. 4, 1474–1486. doi: 10.1021/acssuschemeng.5b01451
Kim, S., Kwon, E. E., Kim, Y. T., Jung, S., and Lee, J. (2019). Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem. 21, 3715–3743. doi: 10.1039/C9GC01210A
Konnerth, H., Zhang, J., Ma, D., Prechtl, M. H. G., and Yan, N. (2015). Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water. Chem. Eng. Sci. 123, 155–163. doi: 10.1016/j.ces.2014.10.045
Li, C., Zhao, X., Wang, A., Huber, G. W., and Zhang, T. (2015). Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 115, 11559–11624. doi: 10.1021/acs.chemrev.5b00155
Mauriello, F., Paone, E., Pietropaolo, R., Balu, A. M., and Luque, R. (2018). Catalytic transfer hydrogenolysis of lignin-derived aromatic ethers promoted by bimetallic Pd/Ni systems. ACS Sust. Chem. Eng. 6, 9269–9276. doi: 10.1021/acssuschemeng.8b01593
Mullen, C. A., Strahan, G. D., and Boateng, A. A. (2009). Characterization of various fast-pyrolysis bio-oils by NMR spectroscopy. Energy Fuels 23, 2707–2718. doi: 10.1021/ef801048b
Naik, S., Goud, V. V., Rout, P. K., and Dalai, A. K. (2010). Supercritical CO2 fractionation of bio-oil produced from wheat–hemlock biomass. Bioresour. Technol. 101, 7605–7613. doi: 10.1016/j.biortech.2010.04.024
Pierens, G. K. (2014). 1H and 13C NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J. Comput. Chem. 35, 1388–1394. doi: 10.1002/jcc.23638
Richter, J. B., Eßbach, C., Senkovska, I., Kaskel, S., and Brunner, E. (2019). Quantitative in situ13C NMR studies of the electro-catalytic oxidation of ethanol. Chem. Commun. 55, 6042–6045. doi: 10.1039/C9CC02660F
Robinson, A. M., Hensley, J. E., and Medlin, J. W. (2016). Bifunctional catalysts for upgrading of biomass-derived oxygenates: a review. Acs Catal. 8, 5026–5043. doi: 10.1021/acscatal.6b00923
Santana, J. A., Carvalho, W. S., and Ataide, C. H. (2018). Catalytic effect of ZSM-5 zeolite and HY-340 niobic acid on the pyrolysis of industrial kraft lignins. Ind. Crops Prod. 111, 126–132. doi: 10.1016/j.indcrop.2017.10.023
Shuai, L., Amiri, M. T., Questellsantiago, Y. M., Heroguel, F., Li, Y., Kim, H., et al. (2016). Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 354, 329–333. doi: 10.1126/science.aaf7810
Shuang, C., Guilin, Z., and Caixia, M. (2019). Green and renewable bio-diesel produce from oil hydrodeoxygenation: strategies for catalyst development and mechanism. Renew. Sust. Energy Rev. 101, 568–589. doi: 10.1016/j.rser.2018.11.027
Sirousrezaei, P., Jae, J., Ha, J., Ko, C. H., Kim, J. M., Jeon, J., et al. (2018). Mild hydrodeoxygenation of phenolic lignin model compounds over a FeReOx/ZrO2 catalyst: zirconia and rhenium oxide as efficient dehydration promoters. Green Chem. 20, 1472–1483. doi: 10.1039/C7GC03823B
Song, Q., Cai, J., Zhang, J., Weiqiang, Y. U., Wang, F., and Jie, X. (2013). Hydrogenation and cleavage of the C-O bonds in the lignin model compound phenethyl phenyl ether over a nickel-based catalyst. Chinese J. Catal. 34, 651–658. doi: 10.1016/S1872-2067(12)60535-X
Tessarolo, N. S., Silva, R. V. S., Vanini, G., Casilli, A., Ximenes, V. L., Mendes, F. L., et al. (2016). Characterization of thermal and catalytic pyrolysis bio-oils by high-resolution techniques: 1H NMR, GC × GC-TOFMS and FT-ICR MS. J. Anal. Appl. Pyrolysis 117, 257–267. doi: 10.1016/j.jaap.2015.11.007
Wang, A., Austin, D., He, P., Mao, X., Zeng, H., and Song, H. (2018). Direct catalytic co-conversion of cellulose and methane to renewable petrochemicals. Catal. Sci. Technol. 8, 5632–5645. doi: 10.1039/C8CY01749B
Wang, H., Pu, Y., Ragauskas, A., and Yang, B. (2019). From lignin to valuable products–strategies, challenges, and prospects. Bioresour. Technol. 271, 449–461. doi: 10.1016/j.biortech.2018.09.072
Wang, M., and Liu, C. (2016). Theoretic studies on decomposition mechanism of o -methoxy phenethyl phenyl ether: primary and secondary reactions. J. Anal. Appl. Pyrolysis 117, 325–333. doi: 10.1016/j.jaap.2015.10.016
Wang, Y., Pedersen, C. M., Qiao, Y., Deng, T., Shi, J., and Hou, X. (2015). In situ NMR spectroscopy: Inulin biomass conversion in ZnCl2 molten salt hydrate medium—SnCl4 addition controls product distribution. Carbohydr. Polym. 115, 439–443. doi: 10.1016/j.carbpol.2014.09.011
Yan, N., and Dyson, P. J. (2013). Transformation of biomass via the selective hydrogenolysis of CO bonds by nanoscale metal catalysts. Curr. Opin. Chem. Eng. 2, 178–183. doi: 10.1016/j.coche.2012.12.004
Zakzeski, J., Bruijnincx, P. C. A., Jongerius, A. L., and Weckhuysen, B. M. (2010). The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 110, 3552–3599. doi: 10.1021/cr900354u
Keywords: lignin, in situ 2H NMR, GC-MS, mechanism, upgrading process
Citation: Yang Y, Wu Z, Luo Y, Han G, Jiang W, Wang M and Ben H (2020) An Investigation Into the Upgrading Process of Lignin Model Dimer—Phenethyl Phenyl Ether by in situ 2H NMR and GC-MS. Front. Energy Res. 8:114. doi: 10.3389/fenrg.2020.00114
Received: 17 March 2020; Accepted: 13 May 2020;
Published: 11 June 2020.
Edited by:
Fengxia Yue, South China University of Technology, ChinaReviewed by:
Selhan Karagoz, Karabük University, TurkeyHalil Durak, Yüzüncü Yil University, Turkey
Yanding Li, Massachusetts Institute of Technology, United States
Copyright © 2020 Yang, Wu, Luo, Han, Jiang, Wang and Ben. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haoxi Ben, YmVuaGFveGlAZ21haWwuY29t