
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 14 February 2019
Sec. Advanced Clean Fuel Technologies
Volume 7 - 2019 | https://doi.org/10.3389/fenrg.2019.00008
This article is part of the Research Topic Natural Gas and Unconventional Methane-Rich Sources: Upgrading, Conversion and Valorization View all 4 articles
 Javier Lasobras1Jaime Soler1
Javier Lasobras1Jaime Soler1 Javier Herguido1
Javier Herguido1 Miguel Menéndez1*Alonso Jimenez2
Miguel Menéndez1*Alonso Jimenez2 Mariana da Silva2María J. Franco2Izaskun Barrio2Jesús Làzaro2
Mariana da Silva2María J. Franco2Izaskun Barrio2Jesús Làzaro2Methane aromatization is a promising technology for the transformation of natural gas into liquid products, but suffers from the problem of catalyst deactivation by coke. A two-zone fluidized bed reactor has been proposed as a tool to counteract the catalyst deactivation, by providing continuous catalyst regeneration in the same vessel where the main reaction is carried out. This work shows the effect of the main operating conditions (carburization temperature, reaction temperature, carburization time, nature of regenerating agent and feed flow and height of the hydrocarbon entry point). Optimal reduction time and temperature were 1 h and 350°C. Best conversion and selectivity were achieved at 700°C without catalyst deactivation in the TZFBR.
A problem for the use of methane from remote sources is the transportation from the reservoir, because gas pipelines are only functional for relatively short distances. Since many deposits of natural gas (NG) are in remote off-shore locations, in the mid-1990s investors started projects that envisioned the building of floating liquid natural gas (LNG) facilities (Pospíšila et al., 2019). British Petroleum (BP) forecasts that LNG will substitute pipeline gas as the most frequent form of inter-regionally traded natural gas in the early 2020s (Schach and Madlener, 2018).
However, liquefaction is an energy-demanding process. When considering all steps (production liquefaction, transport and regasification) the cost of liquefying and regasification represents about 40% of the process. To avoid these costs, gas transport in the form liquid substances at room temperature has been proposed. These liquids are obtained with processes known as “gas to liquid” (GTL), which transform methane, through chemical reactions, into a liquid fuel. One of the most promising GTL processes is based on the aromatization of methane. From the first study of Wang (Wang et al., 1993) research has been extensively carried out on this subject. However, achieving a technologically viable process involves improvements in the catalyst, increases in conversions and yields and stability of the process. Most of these investigations are collected in three reviews, those of Ma et al. (2013), Mamonov et al. (2013), and (Majhi et al., 2013). They show the large amount of research, and therefore interest, in the methane aromatization reaction.
It is commonly accepted that Mo/ZSM-5 catalyst is the most efficient for this process (Borry et al., 1999; Xu and Lin, 1999; Liu et al., 2006). The reaction takes place in two periods: an induction period (activation of the catalyst) and another in which the reaction of aromatization takes place. During the first period, a molybdenum carbide is formed. Despite this not being the main reaction, it is very important since it activates the catalyst to subsequently carry out the aromatization reaction. This period of induction occurs during the first minutes of the reaction and is the time when the active phase of the catalyst is formed (molybdenum carbide). In the second period we can distinguish the stages of methane activation, dimerization, oligomerization, and cyclization. In the methane activation, the first phenomenon that happens is the chemisorption of methane on the metal, as indicated by Xu et al. (2003), although there are authors who believe that methane adsorption is carried out through interaction of this one with the OH groups of the zeolite (Chen et al., 1996).
Once methane has been activated, the reaction to the other products is immediate (Kim et al., 2000). The next step of the reaction is the formation of the C2s (dimerization) which, as well as the activation of the methane, takes place in the metal part of the catalyst. The mechanism of this step is still under discussion. Some authors propose as being responsible for dimerization, either as , or CH3· (Chen et al., 1995; Wang et al., 1995; Xie et al., 1995; Solymosi et al., 1997; Jiang et al., 1999). In contrast, another study suggests that it is (Wang et al., 1995) and others associate it with carbide or indicate it as (Liu et al., 1999; Meriaudeau et al., 1999; Zhou et al., 2000; Ha et al., 2002; Xu et al., 2003; Wong et al., 2012).
After C2s formation, the next step is the formation of the aromatic compounds. The rapid disappearance of C2s eliminates the kinetic and thermodynamic limitations (Kim et al., 2000), which favor the reaction going forward. This last stage no longer occurs in molybdenum, but in acid sites of the zeolite. So, the compounds formed on the metallic sites migrate toward the acid centers of the zeolite (Liu et al., 1999, 2001). In this stage all the authors agree that the acidity of the zeolite allows for an increase in the chain length (oligomerization) and its subsequent cyclization. The mechanisms of reaction in this stage are by carbocations and by hydrogen transfer.
The formed C2s migrate toward the Brønsted acid center of the zeolite, where they interact with the protons, so that each proton passes from the acid center to one of the carbons forming the carbocation (or carbenium ion) C2. The formation of the carbocation is by physical adsorption of the hydrocarbon by van der Waals interactions with the structure of the zeolite. The alkenes increase the length of their chain within the zeolite channels. Carbocations are joined in different reactions until reaching C6H12, at which point there are two possible outcomes: that it reacts with C2, formed in previous steps, so that after its union it breaks, being responsible for C6 and ethane; or by protonation of C6H12 give C6 and H2. In either case C6 is obtained, which cyclizes instantaneously and, after successive transfers of hydrogen and protolysis, forms benzene.
In addition to benzene, which is a majority product because its geometry and size coincide with that of the zeolitic pore, toluene and naphthalene are also observed. Toluene can be formed from the methyl ion, formed in the initial steps, by reacting with benzene. Naphtalene can be formed from the reaction of benzene with the carbocation C4.
An important drawback to overcome in this process is the formation of coke which deactivates the catalyst (at 700°C the selectivity to coke is close to 30%). The first type of carbonaceous deposits comes from the activation of the catalyst, that is to say, they are produced when the molybdenum is carburized. In turn, during the carburization of molybdenum, two types of carbonaceous deposits can be distinguished: the carbides themselves and coke. Coke, unlike carbide, does not participate in the reaction, on the contrary, it is carbon that is placed on the catalyst preventing the reaction from being carried out. This type of coke stays in the metal, blocking the first step of the reaction.
The second type of deposits occurs in oligomerization processes and cyclization, while the hydrogen transfer stages and protolysis take place. This coke also prevents the reaction, but in the second step of it, and may be responsible for sealing the pores of the zeolite. However, a small amount of this coke allows a decrease in the presence of extremely acid centers that are capable of decomposing hydrocarbons (Wang et al., 1997; Liu et al., 1999).
Although deposits occur at two moments of the reaction and in two different locations, there are three different types of coke: a graphitic one located in the channels of the zeolite, a second graphitic coke, located in the carbide, and finally one coke poor in hydrogen (or pre-graphitic) located on the outer surface of the catalyst particles. The hydrogen-poor coke (CHx) is formed both on the metal carbides and on the zeolite, being the main responsible for the catalyst deactivation (Xu et al., 2003).
The Two Zone Fluidized Bed Reactor (TZFBR) is an effective way to counteract the effects of catalyst deactivation, as several previous studies have proved in a variety of reactions (Herguido et al., 2005; Herguido and Menéndez, 2017). In this type of reactor, two zones are created in a fluidized bed by the simple procedure of feeding into the hydrocarbon stream at an intermediate point of the bed and placing an oxidizing stream at the bottom of the reactor. The desired reaction and the coke formation occur in the upper zone and the catalyst regeneration in the lower zone. This reactor would be an advantageous alternative to the operation in packed bed reactors, which would need cyclic operations, with several reactors in parallel, some of them being employed for the reaction while others are being regenerated. If packed bed reactors were used, the operation would be unsteady, which makes the downstream processing more difficult. Another alternative is to use a circulating fluidized bed, where the catalyst is regenerated in a fluidized bed and the reaction is produced in another fluidized bed. This is a well-known system, widely employed in FCC, but keeping the catalyst circulation while avoiding the mixing of hydrocarbons and air is a complex task. The comparison of a conventional fluidized bed and the TZFBR for methane aromatization was made in a previous study (Gimeno et al., 2010), where we found that a stable operation was achieved. However, a study on the effect of operating conditions was not presented. In the present work, the TZFBR is proposed to carry out methane aromatization and the optimal work conditions will be studied by modifying several operation parameters.
The catalyst employed in this work was ZSM-5/bentonite/Mo with a mass ratio 70.7/23.6/5.67. The specific surface calculated by BET method was 222 m2/g and the particle size between 160 and 320 micrometers. More details about the preparation method were presented in a previous paper (Lasobras et al., 2017). The need to incorporate a binder material such as bentonite is a consequence of the small particle size of zeolite and the low hardness that would cause attrition under fluidization conditions. However, the zeolite-binder interaction can affect the structural and textural properties of the zeolite and modify the performance (Michels et al., 2014). So, an optimization of this catalyst was proposed with emphasis on the effects of thermal treatments during the main stages of the preparation procedure (Lasobras et al., 2017).
The fluidized bed reactor was a 27 mm ID, 300 mm long quartz cylindrical tube equipped with a porous quartz distributor plate located inside an electrical furnace. Reactants were fed using mass flow controllers (Alicat Scientific). The catalyst was reduced in situ under a hydrogen flow at 350°C or 450°C for 6 h before reaction. The operation ranges were as follows: temperature = 675–725°C, catalyst weight = 12 g and relative gas velocity for the fluidized bed reactor (ur = u0/umf) = 2–4. The exit gases of the reactor were maintained at 200°C in order to avoid condensation of products and were analyzed with an online gas chromatograph (Varian, CP 3,800). Benzene, toluene, xylene and naphthalene, were separated using a VF-624 ms capillary column (0.32 mm × 30 m) and determined by a flame ionization detector (FID). Unreacted methane, hydrogen, ethane, ethylene, acetylene, and nitrogen were separated by a molecular sieve and analyzed by a thermal conductivity detector (TCD). The total gas flow rate at the reactor exit was measured with a bubble flowmeter.
From the experimental data on total flow and composition of the products, the conversion and selectivity to each product were calculated with the following equations.
• Methane conversion
Where FCH4, i and FCH4,o are the molar flow of methane in the input and output streams, respectively.
• Selectivity to product i
Where Fi is the molar flow of product i in the exit stream and nC is the number of carbon atoms in this product.
In this article we will discuss mainly the selectivity to benzene, which is the main aromatic product. The selectivity to other products (xylene, naphthalene, ethane, ethane, and acetylene) is given in Supplementary Information.
In the case of TZFBR with CO2 as regenerating gas, the selectivity to CO is divided by two, in order to account for the fact that two molecules of CO are formed when CO2 gasifies coke.
The selectivity to coke was calculated by difference in the carbon balance. This value was checked by measuring the amount of coke formed in each experiment, which was calculated from the amount of CO and CO2 formed during the catalyst regeneration. We found that the difference between the amount of coke calculated by carbon balance and by measuring the gases formed during regeneration was small (typically <10%). This result confirms the feasibility of using the carbon balance to estimate the selectivity to coke.
Reproducibility was checked by repeated experiments under the same operating conditions and the difference between the mean values of benzene yield was smaller than 0.05%.
The effect of the reduction temperature was tested for two temperatures: 350 and 450°C (Figure 1).
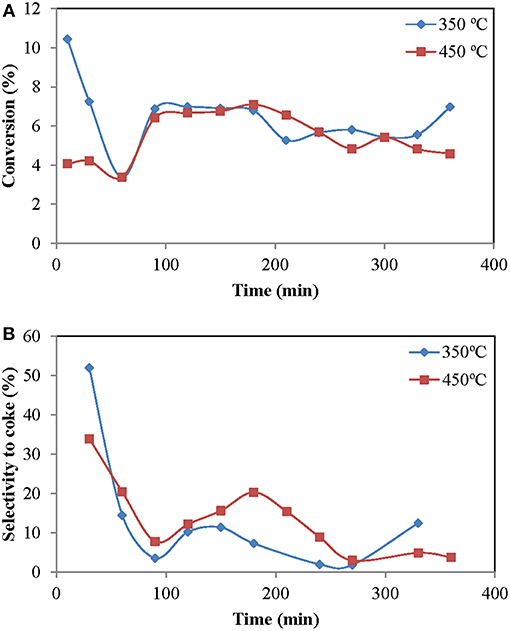
Figure 1. Influence of the reduction temperature in methane aromatization reaction (time on stream): (A) Methane conversion, (B) Selectivity to coke. Operating conditions: Wcat = 12 g, CH4:O2:N2 = 70:1.5: 28.5%; urlowerzone = 2.5 and T = 700°C.
Both conversion and selectivity to benzene were similar at both temperatures. However, the selectivity to coke was slightly higher at 450°C. This can be due to an excessive reduction of molybdenum oxide at 450°C, which causes a greater tendency to the formation of coke. If the reduction temperature was excessive and Mo° was obtained, it would explain this behavior, since metallic molybdenum is actively leading to conversion of methane, but into coke and hydrogen (Solymosi et al., 1997).
The reaction temperature is a very important parameter since it affects both the thermodynamics and the kinetics of the reaction and, therefore, the conversion and selectivity to desired (benzene) and undesired (coke) products. The reached conversion was very similar for the two higher temperatures, but this performance decreased at 650°C (Figure 2). Taking into account the values of the equilibrium, the value at 725°C should be higher than at 700°C and this higher than at 675°C. The fact that there was almost no difference between the first two can be due to the catalyst deactivation. The catalyst deactivation is due to the coke deposition, a reaction that is favored by increasing the temperature.
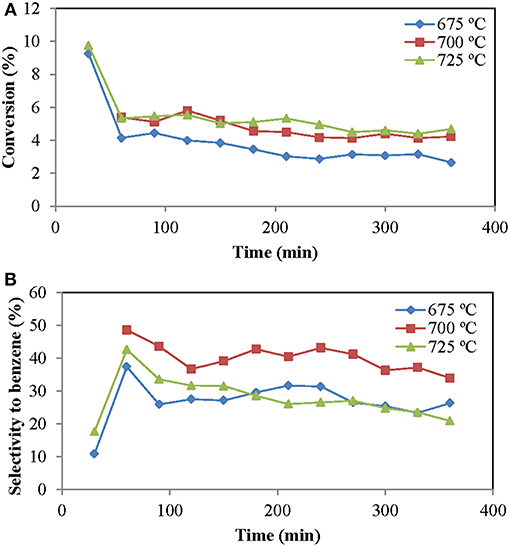
Figure 2. Influence of temperature on methane aromatization reaction (time on stream): (A) Methane conversion, (B) Selectivity to benzene. Operating conditions: Wcat = 12 g, CH4:O2:N2 = 70: 0.5:29.5; urlowerzone = 2. Equilibrium conversion, considering only the methane to benzene reaction, is c.a. 9.8% at 675°C, 12% at 700°C and 14% at 725°C.
Carburization is a phenomenon that always occurs in methane aromatization, being its starting point, since it gives rise to active species responsible for the reaction. In the case of the two-zone reactor it is necessary to provide a period of time for the formation of carbides. This period of carburization is the time in which the reaction is carried out without introducing oxidant, so that methane can react forming carbides, without removing them by the oxidizing agent. Only carbon oxides were formed in experiments where we had not any carburization time before starting to feed oxygen to the TZFBR. It is foreseeable that there is an optimal time prior to the incorporation of the oxidant into the reaction since with very short periods of time would not allow the formation of carbides, but long times would cause the catalyst to be excessively deactivated by coke formation. Figure 3 shows the results obtained with different carburization times. In our results, the best performance was achieved with the largest carburization time (60 min). In addition, we analyzed the formation of CO during carburization, which is due to the reduction of Mo oxides. We found that the CO formation rate was very low (<10% of the initial value) after 1 h, which suggests that carburization was almost complete after this time.
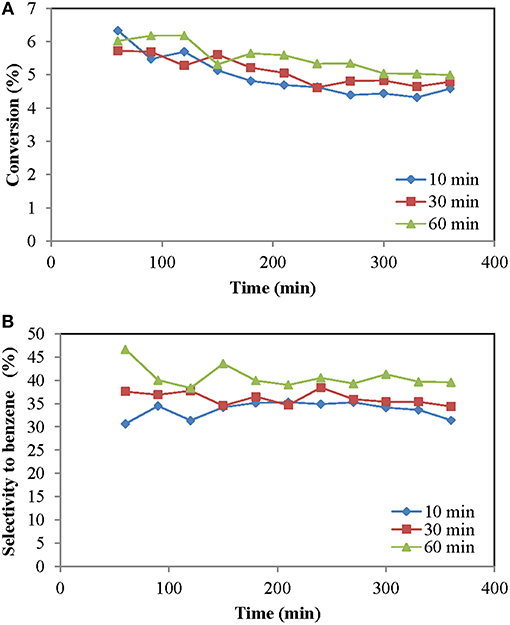
Figure 3. Influence of the carburization period (time on stream): (A) Methane conversion, (B) Selectivity to benzene. Operating conditions: Wcat = 18 g, CH4:O2:N2 = 70:29.25:0.75, urlowerzone = 2, urupperzone = 6.7 and T = 700°C.
The type of regenerating agent being fed is one of the most important parameters in TZFBR, since its proper functioning will depend on its capacity to regenerate the catalyst. Oxygen and carbon dioxide have been studied as oxidizing agents. O2 reacts faster with coke than CO2, which is good for the elimination of the coke deposited on the catalyst. But if this oxidation is excessive the metallic carbides formed in the first stage would be eliminated, so that one of the active phases of the catalyst would be lost.
The comparison is made under the same conditions, but with different percentage of oxidant. This is because, according to the stoichiometry, in order to eliminate the same amount of coke with O2 as with CO2, we need to double the amount of the second.
Oxygen is more reactive than CO2, so it is completely consumed when it comes into contact with the formed coke giving rise to CO. The CO formed in either of the two processes, despite being an inert species, seems to have a catalyst stabilization function (Ohnishi et al., 1999). Figure 4 shows that, although both have the same regenerative capacity according to stoichiometry, oxygen was a better regenerating gas than CO2. By regenerating the catalyst better, the conversion is greater, since the catalyst is more active. As for the selectivity, it was also higher for the case of O2.
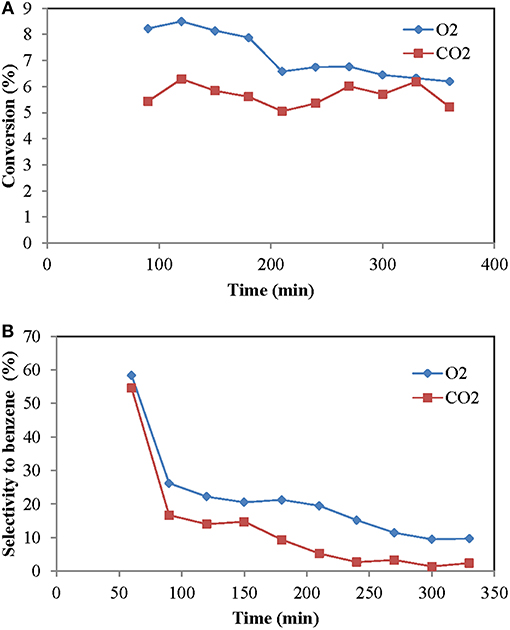
Figure 4. Influence of the oxidant type (time on stream): (A) Methane conversion, (B) selectivity to benzene. Operating conditions: Wcat = 12 g, CH4:O2:N2 = 70:2:28 and CH4:CO2:N2 = 70:4:26, urlowerzone = 2.5, urupperzone = 10.8 and T = 700°C.
There are discrepancies in the literature on the effects of CO2. Some authors find that benzene selectivity is favored by the addition of CO2 (Ohnishi et al., 1999), but others (Liu et al., 2002) observe that the selectivity decreases. In the case of TZFBR, the worsening could be due to a low conversion of CO2 in the lower zone, reaching the top of the two-zone reactor, where it would react with CH4 producing CO and H2. A similar behavior was described by Tan et al. (2002) in the case of a fixed bed, a fact that would explain why the reactor maintains a good conversion and low selectivity to benzene. Cook et al. (2009) observed that in a fluidized bed reactor there was no improvement with the CO2 co-fed or even a detriment of performance, which is in agreement with our results. Whether CO comes from the coke, or from the aforementioned reaction is something that, a priori, cannot be distinguished. In order to know the CO origin, CO2 (5%) was fed through a bed of coked catalyst with an ur = 3.4 at reaction temperature (700°C). The result was that part of the CO2 did not react, which is indicative of the fact that the parallel reactions of coke gasification and methane reforming with CO2 (dry reforming) can occur in the TZFBR. Although molybdenum carbide is a catalyst for dry reforming of methane (Brungs et al., 2000), we found that the selectivity to CO was similar when CO2 was used as regenerating agent (68%) and when O2 was used (62%). This results also indicate that the contribution of dry reforming to methane conversion in TZFBR was small.
Since aromatics are secondary products, a decrease in the space velocity favors the reaction and the selectivity to aromatics. By lowering the flow we get lower space velocities. The decrease in flow in a fixed-bed reactor is easy, however, in a fluidized reactor, and more if it is a TZFBR, this variation has direct influence on the reduced velocity (ur). If the reduced velocity is <1.5, it could happen that, although the solid remains in a fluidized state, there is not enough exchange of solids between the reducing and oxidizing zones of the reactor. Taking into account this limitation, several tests were made at different ur, but always keeping the ur in the lower zone above 2. The results are shown in Figure 5. The increase in the space velocity decreased the conversion, as it is expected in any reactor.
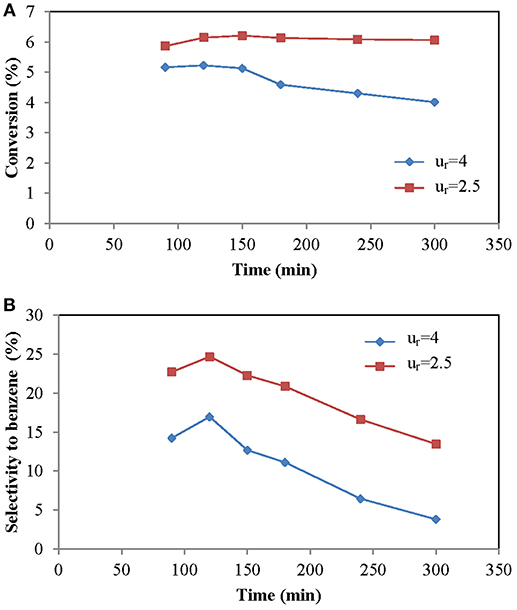
Figure 5. Influence of the variation of the feed flow (time on stream time). (A) Methane conversion, (B) Selectivity to benzene. Operating conditions: Wcat = 12 g, CH4:O2:N2 = 70:1.5:28.5 and T = 700°C.
Taking into account that increasing the flow rate fed to the reactor or the percentage of CH4 in the feed have similar effects, the molar flow rate of methane was increased. In addition, by increasing the flow, the formation of larger bubbles was observed (Julián et al., 2012, 2014) and, therefore, the transfer of bubble-emulsion matter will be slower, resulting in lower conversions. It was observed that the selectivity to benzene decreased with a high feed flow. An explanation for this decrease could be that the separation between the oxidizing zone and the reducing zone was lost. If the speed was too high, O2 could reach the upper zone, leading to the combustion of methane, which implies an increase in CO selectivity and a worse regeneration of the catalyst with the consequent decrease in conversion and also, since the yield to CO remains practically constant, a lower selectivity to benzene.
The height of the rod (i.e., the point where methane is fed in the TZFBR) is the distance between the porous plate and the exit point of the methane through the rod. It directly influences the amount of catalyst remaining in the regeneration zone (between the plate and the end of the rod) and, by difference, defines the amount of catalyst remaining in the reaction zone (from the outlet of the rod to the top of the catalyst bed). A small regeneration zone favors a greater reaction zone, and therefore, a longer residence time in this last zone. On the other hand, a small regeneration zone may be insufficient to properly regenerate the catalyst, or the oxidant may reach the reaction zone with all the drawbacks that this entails (higher selectivity to CO by direct combustion of the reagent, low selectivity to benzene and greater deactivation). If, on the contrary, the rod is too high, the amount of catalyst in the reaction zone is little, which decreases the conversion and selectivity to aromatics. In addition, in that situation there is an excess of catalyst in the regeneration zone, which does not actively participate in the reaction. Figure 6 shows the evolution of the conversion and selectivity over time for heights of the rod 2.6 and 5.5 cm.
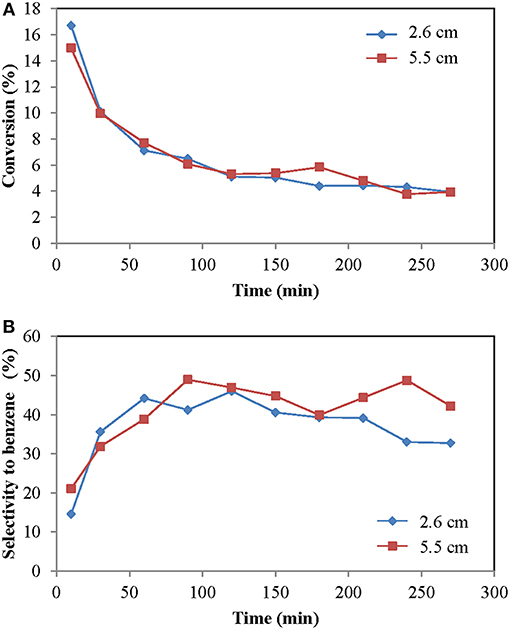
Figure 6. Influence of the variation of the height of the rod in the TZFBR (time on stream): (A) Methane conversion, (B) Selectivity to benzene. Operational conditions: Wcat = 14.5 g, CH4:CO2:N2 = 70:1:29, urlowerzone = 2.9, urupperzone = 5.6, and T = 700°C.
In both cases, the conversion was practically the same. In contrast, the selectivity showed different behaviors. In the case where the rod was at 2.6 cm there is a greater reaction zone. This fact does not entail a greater conversion, as would be expected. For the case of the rod at 5.5 cm, it is possible that the size of the regeneration zone allows for the recovery of the catalyst without any regenerating agent reaching the reaction zone, which allows all the oxidant to be used in the catalyst regeneration and not in the reaction with the hydrocarbons, thus improving the regeneration process. The existence of a reduced velocity at the entrance of the rod decreased the amount of C2s formed due to the longer residence time, which allows its conversion to benzene.
The main operational parameters for the aromatization reaction with Mo/HZSM-5/bentonite catalyst in a TZFBR have been studied. So, the optimal parameters for the catalyst reduction with H2 before methane aromatization were: reduction at 350°C and a period of carburization of 60 min. These conditions for the reduction and carburization steps allow for good conversions and selectivity, without a great deactivation by formation of coke in the reactor. On the other hand, the best conversion and selectivity to aromatic products without deactivation of the catalyst were achieved at 700°C.
The effect of the oxidizing agent varied depending on the species employed. O2 reacted quickly with the catalyst coke producing CO. If O2 is in excess, it forms CO2 or can remove the carbides. This reactivity prevents it from reaching the reaction zone, which makes it a good regenerating gas. CO2 reaches the reaction zone, something that was checked by passing CO2 through a coked bed and checking its existence in the exhaust gases. If the CO2 in excess reaches the reaction zone, it can inhibit the reaction. However, CO2 is not able to eliminate carbides, so it might be of interest if the amount of coke to be eliminated in the regeneration zone was small. The effect of changing the space velocity has also been studied. Increasing the flow rate leads to lower conversions and unstable behavior. It has also been observed that very high velocities can result in the oxidant reaching the reaction zone. The height of the rod mainly influenced the methane conversion since the amounts of catalyst in the reaction and regeneration zones were altered. In summary, the most influential factor when carrying out the reaction was the space velocity (or the residence time), since it changes the conversion and the product distribution.
JaL performed the experiments. JS, JH, MM, AJ, MdS, MF, IB, and JeL contributed to the planning, the interpretation of results and the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2019.00008/full#supplementary-material
Borry, R., Kim, Y., Huffsmith, A., Reimer, J., and Iglesia, E. (1999). Structure and density of Mo and acid sites in Mo-exchanged H-ZSM5 catalysts for nonoxidative methane conversion. J. Phys. Chem. B 103, 5787–5796. doi: 10.1021/jp990866v
Brungs, A. J., York, A. P. E., Claridge, J. B., Márquez-Alvarez, C., and Green, M. L. H. (2000). Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts. Catal. Lett. 70, 117–122. doi: 10.1023/A:1018829116093
Chen, L., Lin, L., Xu, Z., Zhang, T., Xin, Q., Ying, P., et al. (1996). Fourier transform-infrared investigation of adsorption of methane and carbon monoxide on HZSM-5 and Mo/HZSM-5 zeolites at low temperature. J. Catal. 161, 107–114. doi: 10.1006/jcat.1996.0167
Chen, L. Y., Lin, L. W., Xu, Z. S., Li, X. S., and Zhang, T. (1995). Dehydro-Oligomerization of Methane to Ethylene and Aromatics over Molybdenum/HZSM-5 Catalyst. J. Catal. 157, 190–200. doi: 10.1006/jcat.1995.1279
Cook, B., Mousko, D., Hoelderich, W., and Zennaro, R. (2009). Conversion of methane to aromatics over Mo2C/ZSM-5 catalyst in different reactor types. Appl. Catal. A Gen. 365, 34–41. doi: 10.1016/j.apcata.2009.05.037
Gimeno, M. P., Soler, J., Herguido, J., and Menéndez, M. (2010). Counteracting catalyst deactivation in methane aromatization with a two zone fluidized bed reactor. Ind. Eng. Chem. Res. 49, 996–1000. doi: 10.1021/ie900682y
Ha, V. T. T., Tiep, L. V., Meriaudeau, P., and Naccache, C. (2002). Aromatization of methane over zeolite supported molybdenum: active sites and reaction mechanism. J. Mol. Catal. A Chem. 181. 283–290. doi: 10.1016/S1381-1169(01)00373-9
Herguido, J., and Menéndez, M. (2017). Advances and trends in two-zone fluidized-bed reactors. Curr. Opin. Chem. Eng. 17,15–21. doi: 10.1016/j.coche.2017.05.002
Herguido, J., Menéndez, M., and Santamaría, J. (2005). On the use of fluidized bed catalytic reactors where reduction and oxidation zones are present simultaneously. Catal. Today 100. 181–189. doi: 10.1016/j.cattod.2004.11.004
Jiang, H., Wang, L., Cui, W., and Xu, Y. (1999). Study on the induction period of methane aromatization over Mo/HZSM-5: partial reduction of Mo species and formation of carbonaceous deposit. Catal. Lett. 57, 95–102. doi: 10.1023/A:1019087313679
Julián, I., Gallucci, F., Annaland, M. S., Herguido, J., and Menéndez, M. (2012). Coupled PIV/DIA for fluid dynamics studies on a Two-Section Two-Zone Fluidized Bed Reactor. Chem. Eng. J. 207, 122–132. doi: 10.1016/j.cej.2012.06.015
Julián, I., Herguido, J., and Menéndez, M. (2014). A non-parametric bubble size correlation for a Two-Section Two-Zone Fluidized Bed Reactor (TS-TZFBR). Powder Technol. 256, 146–157. doi: 10.1016/j.powtec.2014.02.004
Kim, Y., Borry, R. W. III., and Iglesia, E. (2000). Genesis of methane activation sites in Mo-exchanged H–ZSM-5 catalysts. Micropor. Mesopor. Mat. 35–36, 495–509. doi: 10.1016/S1387-1811(99)00245-0
Lasobras, J., Medrano, J. A., Soler, J., Herguido, J., Menéndez, M., Jimenez, A., et al. (2017). Preparation of Mo/HZSM-5/bentonite catalyst for methane aromatization in a fluidized bed reactor. Int. J. Chem. Reactor Eng. 0081, 1–9. doi: 10.1515/ijcre-2017-0081
Liu, H., Bao, X., and Xu, Y. (2006). Methane dehydroaromatization under nonoxidative conditions over Mo/HZSM-5 catalysts: identification and preparation of the Mo active species. J. Catal. 239, 441–450. doi: 10.1016/j.jcat.2006.02.018
Liu, H., Li, T., Tian, B., and Xu, Y. (2001). Study of the carbonaceous deposits formed on a Mo/HZSM-5 catalyst in methane dehydro-aromatization by using TG and temperature-programmed techniques. Appl. Catal. A Gen. 213, 103–112. doi: 10.1016/S0926-860X(00)00883-8
Liu, S., Wang, L., Ohnishi, R., and Ichikawa, M. (1999). Bifunctional Catalysis of Mo/HZSM-5 in the dehydroaromatization of methane to benzene and naphthalene XAFS/TG/DTA/MASS/FTIR characterization and supporting effects. J. Catal. 181, 175–188. doi: 10.1006/jcat.1998.2310
Liu, Z., Nutt, M., and Iglesia, E. (2002). The effects of CO2, CO and H2 co-reactants on methane reactions catalyzed by Mo/H-ZSM-5. Catal. Lett. 81, 271–279. doi: 10.1023/A:1016553828814
Ma, S., Guo, X., Zhao, L., Scott, S., and Bao, X. (2013). Recent progress in methane dehydroaromatization: from laboratory curiosities to promising technology. J. Energy Chem. 22, 1–20. doi: 10.1016/S2095-4956(13)60001-7
Majhi, S., Mohanty, P., Wang, H., and Pant, K. K. (2013). Direct conversion of natural gas to higher hydrocarbons: a review. J. Energy Chem. 22, 543–554. doi: 10.1016/S2095-4956(13)60071-6
Mamonov, N. A., Fadeeva, E. V., Grigoriev, D. A., Mikhailov, M. N., Kustov, L. M., and Alkhimov, S. A. (2013). Metal/zeolite catalysts of methane dehydroaromatization. Russ. Chem. Rev. 82, 567–585. doi: 10.1070/RC2013v082n06ABEH004346
Meriaudeau, P., Tiep, L., Ha, V., Naccache, C., and Szabo, G. (1999). Aromatization of methane over Mo/H-ZSM-5 catalyst: on the possible reaction intermediates. J. Mol. Catal. A Chem. 144, 469–471. doi: 10.1016/S1381-1169(99)00050-3
Michels, N.-L., Mitchell, S., and Pérez-Ramírez, J. (2014). Effects of binders on the performance of shaped hierarchical MFI zeolites in methanol-to-hydrocarbons. ACS Catal. 4, 2409–2417. doi: 10.1021/cs500353b
Ohnishi, R., Liu, S., Dong, Q., Wang, L., and Ichikawa, M. (1999). Catalytic dehydrocondensation of methane with CO and CO2 toward benzene and naphthalene on Mo/HZSM-5 and Fe/Co-modified Mo/HZSM-5. J. Catal. 182, 92–103. doi: 10.1006/jcat.1998.2319
Pospíšila, J., Charvátb, P., Arsenyevac, O., Klimeša, L., Špiláčeka, M., and Klemeš, J.J. (2019). Energy demand of liquefaction and regasification of natural gas and the potential of LNG for operative thermal energy storage. Renew. Sustain. Energy Rev. 99, 1–15. doi: 10.1016/j.rser.2018.09.027
Schach, M., and Madlener, R. (2018). Impacts of an ice-free Northeast Passage on LNG markets and geopolitics. Energy Policy 122, 238–448. doi: 10.1016/j.enpol.2018.07.009
Solymosi, F., Cserényi, J., Szöke, A., Bánsági, T., and Oszk,ó, A. (1997). Aromatization of methane over supported and unsupported Mo-based catalysts. J. Catal. 165, 150–161. doi: 10.1006/jcat.1997.1478
Tan, P., Leung, Y., Lai, S., and Au, C. (2002). Methane aromatization over 2 wt% Mo/HZSM-5 in the presence of O2 and NO. Catal. Lett. 78, 251–258. doi: 10.1023/A:1014956501472
Wang, D., Lunsford, J. H., and Rosynek, M. P. (1997). Characterization of a Mo/ZSM-5 catalyst for the conversion of methane to benzene. J. Catal. 169, 347–358. doi: 10.1006/jcat.1997.1712
Wang, L. S., Tao, L. X., Xie, M. S., Xu, G. F., Huang, J. S., and Xu, Y. D. (1993). Dehydrogenation and Aromatization of Methane under Nonoxidizing Conditions. Catal. Lett. 21, 35–41. doi: 10.1007/BF00767368
Wang, L. S., Xu, Y. D., Xie, M. S., Liu, S. T., Tao, L. X., and Xu, G. F. (1995). “Activation and aromatization of methane and ethane over Mo(VI)/HZSM-5 and W(VI)/HZSM-5 zeolites catalysts,” in: Catalysis by Microporous Materials, eds H. K. Beyer, H. G. Karge, I. Kiricsi, and J. B. Nagy (Amsterdam: Elsevier Science Publ.), 495. doi: 10.1016/S0167-2991(06)81260-7
Wong, K. S., Thybaut, J. W., Tangstad, E., Stöcker, M. W., and Marin, G. B. (2012). Methane aromatisation based upon elementary steps: kinetic and catalyst descriptors. Microporous Mesoporous Mater. 164, 302–312. doi: 10.1016/j.micromeso.2012.07.002
Xie, M. S., Liu, S. T., Wang, L. S., Xie, M. S., and Guo, X. X. (1995). Methane Activation without using Oxidants over Mo/HZSM-5 Zeolite catalysts. Catal. Lett. 30, 135–149. doi: 10.1007/BF00813680
Xu, Y. D., Bao, X. H., and Lin, L. W. (2003). Direct conversion of methane under nonoxidative conditions. J. Catal. 216, 386–395. doi: 10.1016/S0021-9517(02)00124-0
Xu, Y. D., and Lin, L. W. (1999). Recent advances in methane dehydro-aromatization over transition metal ion-modified zeolite catalysts under non-oxidative conditions. Appl. Catal. A Gen. 188, 53–67. doi: 10.1016/S0926-860X(99)00210-0
Keywords: methane aromatization, BTX, fluidized bed reactor, TZFBR, MoHZSM-5
Citation: Lasobras J, Soler J, Herguido J, Menéndez M, Jimenez A, da Silva M, Franco MJ, Barrio I and Làzaro J (2019) Methane Aromatization in a Fluidized Bed Reactor: Parametric Study. Front. Energy Res. 7:8. doi: 10.3389/fenrg.2019.00008
Received: 10 December 2018; Accepted: 24 January 2019;
Published: 14 February 2019.
Edited by:
Fernando Bimbela, Universidad Pública de Navarra, SpainReviewed by:
Jordi Llorca, Universitat Politecnica de Catalunya, SpainCopyright © 2019 Lasobras, Soler, Herguido, Menéndez, Jimenez, da Silva, Franco, Barrio and Làzaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Menéndez, TWlndWVsLm1lbmVuZGV6QHVuaXphci5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.