 Daniel G. Reed
Daniel G. Reed George R. M. Dowson
George R. M. Dowson Peter Styring
Peter Styring
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 07 July 2017
Sec. Carbon Capture, Utilization and Storage
Volume 5 - 2017 | https://doi.org/10.3389/fenrg.2017.00013
This article is part of the Research Topic Carbon Dioxide Utilisation View all 10 articles
Reducing the cost of capturing CO2 from point source emitters is a major challenge facing carbon capture, utilization, and storage. While solid ionic liquids (SoILs) have been shown to allow selective and rapid CO2 capture by pressure swing separation of flue gases, expectations of their high cost hinders their potential application. Cellulose is found to be a reliable, cheap, and sustainable support for a range of SoILs, reducing the total sorbent cost by improving the efficiency of the ionic liquid (IL) through increased ionic surface area that results from coating. It was also found that cellulose support imparts surface characteristics, which increased total sorbent uptake. Combined, these effects allowed a fourfold to eightfold improvement in uptake per gram of IL for SoILs that have previously shown high uptake and a 9- to 39-fold improvement for those with previously poor uptake. This offers the potential to drastically reduce the amount of IL required to separate a given gas volume. Furthermore, the fast kinetics are retained, with adsorb–desorb cycles taking place over a matter of seconds. This means that rapid cycling can be achieved, which results in high cumulative separation capacity relative to a conventional temperature swing process. The supported materials show an optimum at 75% cellulose:25% IL as a result of even coating of the cellulose surface. The projected reduction in plant size and operational costs represents a potentially ground-breaking step forward in carbon dioxide capture technologies.
Carbon dioxide (CO2) has been shown to be the major contributing gas to global warming and detrimental climate change (IPCC, 2014). Carbon capture utilization and storage (CCUS) is one of the key frameworks being developed to counter this threat. Without further development and subsequent deployment of CCUS and associated technologies, models indicate that warming will not be limited to the previous 2°C target, let alone 1.5°C target contained in the COP21 Paris Agreement (IPCC, 2014; United Nations, 2015).
Within CCUS, once the CO2 is captured, it may be either sequestered into geological storage or utilized as a feedstock in other products through CO2 utilization. Both approaches require significant energy input. Either way, both methods first require a CO2 capture step, although in the utilization processes, this may be a reactive capture directly to a product. One of the key challenges facing CCUS remains the question of which approach to use in order to capture dilute CO2 from exhaust gases when over 90% of stationary emitters produce emissions concentrations of 15% by volume of CO2 or even less (Gale et al., 2005).
Removal and capture of CO2 from these point sources is an area of intense research. Even when only post-combustion removal of CO2 is considered, approaches range from adsorption and absorption, membrane and cryogenic separation, and microbial or algal biosystems (Figure 1) (Chou and Chen, 2004; Figueroa et al., 2008; Stolaroff et al., 2008; Brennecke and Gurkan, 2010; Karadas et al., 2010). Of the overarching technologies listed in Figure 1, adsorption and absorption systems are the most advanced and are themselves split into two main types; temperature swing absorption (TSA) and pressure swing adsorption (PSA). TSA processes typically rely on high temperatures in order to regenerate a solvent that readily captures CO2 liberating a high-concentration CO2 stream, whereas PSA processes rely on a drop in pressure, most usually from near-atmosphere to vacuum to release the more weakly bound CO2 (MacDowell et al., 2010; Samanta and Zhao, 2011).
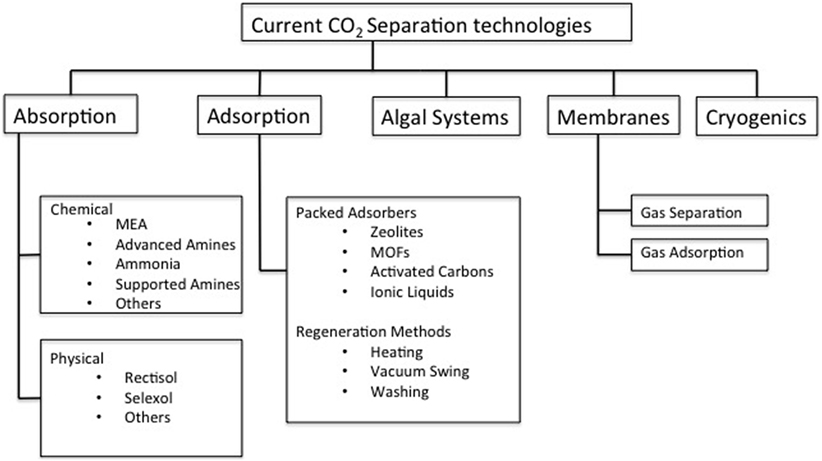
Figure 1. Current leading CO2 separation technologies.
The aforementioned choice between the capture approaches will likely be decided upon relative economic viability of the processes. This hinges on their energy requirements, prospective plant footprint area, and the balance of capital and operating costs. In turn, these economic elements will depend on both the nature of the process and the sorbent taken together. For example, a low-cost sorbent that has exorbitant energy requirements for CO2 capture and that will require a large plant footprint may struggle to compete with a far higher-cost sorbent that has lower energy requirements and smaller footprint. Location also needs to be considered. If there is surplus low-grade heat available, this can be used in the TSA process to afford desorption of the captured CO2. However, there are many potential emission sources where surplus heat is not available. In these cases, the PSA process using renewable energy becomes more economically efficient as the heating duty on solvent regeneration by fat outweighs compression duty.
Ionic liquids (ILs) and particularly room temperature ionic liquids (RTILs) are particularly expensive sorbents. Some of these show very promising uptake performance for CO2 capture such as high selectivity and inherent flexibility, allowing task-specific sorbents to be synthesized (Bates et al., 2002; Ramdin et al., 2012; Moen and Stene, 2014). These RTILs can also bridge both the physisorption and chemisorption classes of sorbent, potentially allowing both high uptake capacity and low regeneration costs in typically TSA processes.
One of the main drawbacks when using RTILs is the very slow rate of CO2 diffusion through the bulk liquid. Compared with benchmark amines, the diffusion rates of gas through the RTILs is up to 19 orders of magnitude slower (Jassim et al., 2007; Moya et al., 2014). This largely prevents rapid CO2 sorption and cycling and, therefore, increases the total IL inventory required to treat a given flow rate or volume of gas. Compounding this problem is the high cost of the RTILs. However, in some cases, this might be erroneous as only a few cases of cost modeling have been carried out for IL production. In addition, these are frequently estimates that are based on laboratory-scale syntheses or small-scale production, which can easily imply costs approaching £1 million per tonne (Baltus et al., 2005). A recent full techno-economic analysis of larger scale syntheses have given more likely IL prices ranging from £1,700 to £35,000 per tonne, based on manufacturers estimates but with a strong dependence on the IL structure (Klein-Marcuschamer, 2011). For comparison, the benchmark amine, MEA, has a market cost of £1,100 per tonne. Naturally, it should be noted that the wide range of results is due to scaling assumptions (Chen et al., 2014).
However, what is more certain is that even if the lower-cost figure for the production of ILs is the more accurate, the slow diffusion rate will require infeasibly large inventory supplies to be available at any point source emitter.
One potential solution to the twin problems of low-gas diffusion rates and high sorbent costs may be to use SoILs as sorbents. While these may appear to be a contradictory name, ILs are typically defined as salts with melting points below 120–140°C, allowing organic salts that have high melting points to be entirely solid at room temperature.
Previously published work (Styring et al., 2016) by this group has investigated SoILs and molecular organic salts as an alternative IL approach to CO2 capture, where PSA is used on finely ground ILs with melting points above room temperature. These solids are often structurally simpler and cheaper to synthesize than RTILs and naturally avoid gas diffusion issues by allowing the gas immediate access to a large IL surface area. They can also potentially avoid sorbent pumping issues as fine powders rather than typically viscous fluids and are significantly easier to dry (Styring et al., 2016).
Through this work, it was found that the high selectivity to CO2 over nitrogen, which is typical of RTILs, is maintained in the SoILs. However, in contrast with the RTIL capture methods that typically use TSA, PSA is required to counteract the fact that typically crystalline and impermeable SoILs will not allow full gas–solid contacting. As a result, since not all of the IL is available for gas–solid interaction, uptake capacity is limited compared to RTILs. By applying a high pressure swing process, where adsorption and separation occurs at high pressures (5–30 bar) and desorption occurs at atmospheric pressure, uptake capacities can be boosted. Once again, this method reduces the inventory of IL required to treat a given gas stream. An additional benefit of using a high pressure swing process is a potentially reduced capture plant footprint made up of less complex unit operations than those found in a TSA process, and operation at ambient temperature. Overall, there is a small temperature change of less than 1°C over the compression–decompression cycles. Furthermore, it should be noted that if the captured CO2 is to be transported by pipeline, large-scale compression and pumping equipment will already be present (Pershad et al., 2010).
These positive effects come at the price of the energy costs of pressurization. Costs have been estimated for a model pressure swing capture system using a range of operating pressures and separation performances based on experimental data. Costs estimated by this model compare well with literature examples of high pressure swing systems for CO2 separation. Table 1 compares model results with the most prominent separation technologies shown Figure 1. This table shows that when considering total energy costs, high pressure swing processes have a decided advantage over other separation technologies.

Table 1. Comparison of average reported energy costs for several post-combustion CO2 capture processes including maximum and minimum reported values (Desideri and Paolucci, 1999; Wong and Bioletti, 2002; Romeo et al., 2008; Zhang et al., 2008; Harkin et al., 2009; Rameshni, 2010; Yang and Zhai, 2010; Belaissaoui et al., 2012, 2013; Krishnamurthy et al., 2014; Kundu et al., 2014; Liu et al., 2014; Xie et al., 2014).
A frequent comment on this observation is that these overall energy comparisons can be misleading when different energy sources are used for each of the various capture methods (Wilson et al., 2016). For example, in a power plant context, temperature swing processes (MEA and amines primarily) could in principle be powered by low grade and low value heat. This would render the overall energy cost less relevant, especially if assuming this energy might otherwise be wasted. In comparison, the costs of gas compression or vacuum generation, while representing far less total energy, could have a larger associated energy penalty to the power plant if the assumption is made that higher-value electricity is required. This position naturally ignores the possibility that the same low-grade heat that putatively powers MEA capture could be used to power pressurization equipment, which fundamentally requires only mechanical motion and does not necessarily require electricity. Nor does it allow for situations where such waste low-grade heat is unavailable or insufficient for MEA capture to proceed without large economic costs. In such situations, overall energy costs of each approach, as shown in Table 1, is a fair basis of comparison. If renewable energy is used for the pressure swing then the situation is further improved.
The action of supporting ILs has previously been used in applications such as supported IL catalysis, a concept that combines the advantages of ILs with those of heterogeneous support materials; and gas separation, where ILs are supported on membranes to improve permeability (Mehnert, 2004; Ilconich et al., 2007). Supporting SoILs to specifically aid CO2 capture is a relatively unexplored area of research and may allow significantly improved CO2 uptake per unit mass of IL in the total sorbent. This will further reduce the quantity of IL needed to treat a given gas stream. This would primarily be achieved by coating a thin layer of the SoIL on a support particle with which it has good adhesion and full surface coverage. Assuming the support material is low cost, this in turn may substantially reduce sorbent inventory costs. Further benefits may include unique combinations of properties from both the IL and the support itself.
Supports such as activated carbon, alumina, and cellulose among others, could be considered as viable options. Of these, there is a history of using ILs with cellulose, typically in efforts to separate cellulose from biomaterials as cellulose is known to dissolve well in RTILs (Swatloski et al., 2002). It would, therefore, seem reasonable to assume that SoILs could have a good surface interaction and adhesion to the cellulose particles, allowing the desired thin layer to be deposited, giving total coverage of the support particle. The insolubility of cellulose in other solvents allows easy recovery of any supported SoIL. Furthermore, cellulose is also very cheap, is an abundant bio-renewable material and has only weak interaction with CO2. The tough fibrous structure of the cellulose support also leads to high mechanical strength, preventing the sorbent particles from breaking up under pressure swing conditions.
As with previous work (Styring et al., 2016), the cations and anions selected for the SoILs focused on low complexity cations with abundant precursor materials. Those chosen for cellulose support were tetraalkylammonium, pyridinium, and imidazolium cations. For the anions, acetate and bromide were chosen. RTILs using acetate have a track-record of high uptake capacity. Bromides, which typically have very low uptake capacities, have a facile synthetic pathway and were chosen to see if supporting them on cellulose would help improve sorption capacity (Shiflett and Yokozeki, 2008). The use of a cellulose support lowers the cost of the materials relative to a pure SoIL while also reducing the inventory of the SoIL. This also improves uptake capacity so has a double benefit. The materials are more expensive than activated carbons but comparable or cheaper than advanced solids such as molecular sieves/zeolites. We have chosen low-cost SoIL materials with market values close to pure MEA for example. These are not highly complex ILs needing exotic anions and cations.
All reagents were purchased at highest available purity from Sigma-Aldrich and used without further purification, other than drying. Amine quaternization reactions were carried out using Schlenk-line techniques under an inert (N2) atmosphere. Reactions using methyl iodide were carried out using aluminum foil protection on the exterior of the reaction vessel and low lighting. All solvents were HPLC grade. CO2 and N2 and were supplied by BOC-Linde. 1-Butyl-4-methylimidazolium bromide ([Bmim][Br]) and tetra-octylammonium bromide ([N8888][Br]) were directly purchased from Sigma-Aldrich and thoroughly dried under a flow of 20 mL/min nitrogen at 60°C for 24 h before uptake tests were carried out.
High pressure adsorption experiments were carried out using a bespoke packed-bed adsorption column constructed from Swagelok™ piping and fittings (Figures 2 and 3) using a Jasco PU-1580-CO2 supercritical CO2 pump, a Jasco BP-1580-81 back pressure regulator, an Omega PX409USB High Accuracy Pressure Transducer, a 42AAV48 Midwest Pressure Systems Gas Pressure Booster, and an AND GF-1000 High Capacity 3 decimal place balance. The reactor was isolated from the system using valves and the assembly weighed on the balance. Desorption was measured by slowly opening the valves while still on the balance. Supported sorbent packed densities were measured using a Micromeritics AccuPyc 1340 Pycnometer and surface area analysis was carried out on a Micromeritics 3Flex gas sorption analyzer. High pressure adsorption capacities were further verified using a Hiden Isochema IGA-0002 adsorption apparatus with a pressure range of 0–10 bar.
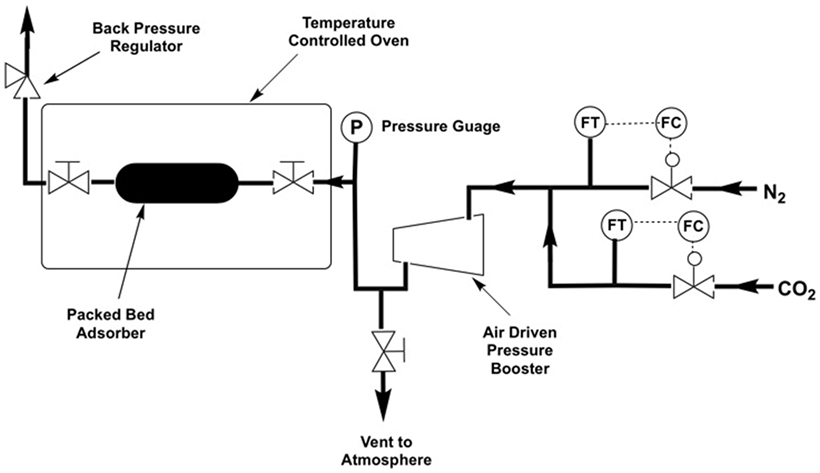
Figure 2. Simplified process flow diagram of the experimental apparatus.
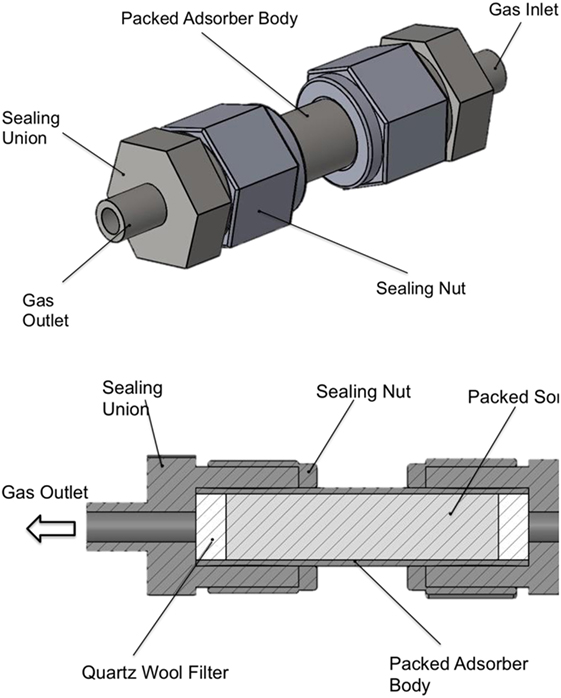
Figure 3. Schematic of the packed bed adsorber with cross-sectional view showing the internal layout and packing configuration.
Tetraethyl ammonium acetate [N4444][Ac] was synthesized by addition of glacial acetic acid (2.00 g, 33.17 mmol) to a 40 wt% aqueous solution of tetraethyl ammonium hydroxide (10.75 mL, 16.58 mmol) followed by evaporation, using heptane to assist in the removal of excess acetic acid by azeotropic distillation. Isopropanol was further used for the azeotropic removal of water. The product was isolated as a free-flowing brilliant white powder that was then further dried under high vacuum. Yield 4.87 g (97%). 1H NMR, 400 MHz, CD3OD: δ/ppm 1.05 (t, J = 7.29 Hz, 12H –CH3), 1.44 (hex, J = 7.47 Hz, 8H, –CH2–), 1.69 (br. m, 8H, –CH2–), 1.95 (s, 3 H, CH3COO), 3.26 (m, 8H, CH2-N).
Methyltrioctyl ammonium acetate [N1888][Ac] was synthesized by the addition of an excess of methyl iodide (5.00 g, 35.25 mmol) to trioctylamine (4.31 g, 11.69 mmol) in acetonitrile (50 mL). The resulting white powder precipitate [N1888][I] (5.62 g) was filtered and then dissolved in a 60:40 mixture of methanol and water (150 mL) and passed through a column of Amberlite® IRN78 Hydroxide Form anion exchange resin, which had been freshly regenerated with an aqueous solution of high purity sodium hydroxide. The resulting methyl trioctyl ammonium hydroxide was then immediately mixed with an excess of acetic acid (2.5 g, 41.6 mmol) in water (30 mL), to prevent decomposition of the unstable ammonium hydroxide species. Samples of both the column eluate and the acetic acid mixture in the receiving flask were periodically taken and shaken with silver nitrate solution, to check for cream or yellow–white precipitating AgI. As with the [N4444][Ac], this was then dried using azeotropic distillation techniques yielding a fluffy white powder. Total yield 4.11 g (82%). 1H NMR, 400 MHz, CD3OD: δ/ppm 0.93 (t, J = 7.10 Hz, 9H, –CH3), 1.38 [br. m. 30H, –(CH2)5–], 1.73 (m, 6, –CH2–), 2.18 (s, 3H, CH3COO), 3.02 (s, 3H, N-CH3), 3.27 (m, 6H, N-CH2–).
Several attempts were made to synthesize methylpyridinium acetate with anion exchange resin; however, the resulting methylpyridinium hydroxide decomposed as it was formed, leaving only a brown tarry residue on drying.
All ILs were then supported on the cellulose using wet-coating techniques. The IL was first dissolved in equal parts methanol and isopropanol before the cellulose was added at the required amount. The alcoholic solvents were then removed under vacuum using a rotary evaporator heated to 60°C and rotation set at 280 rpm, leaving the IL coated on the cellulose surface. The extent of the coating was confirmed qualitatively by SEM of the resulting free-flowing powders.
Since the gas uptake is quantified gravimetrically, it is important to know the weight of gas that is not interacting with the sorbent: this is known as the void space. The void space was calculated before each run took place. The accurate internal volume of the adsorber (empty) was found by water displacement (VA). The adsorber rig was then weighed (empty) and under vacuum. Quartz wool was used to ensure that packed ILs were not ejected from the adsorber, and this was also weighed. A portion of quartz wool was packed into one end of the adsorber and the IL to be tested was then packed on top. The second portion of quartz wool was then added at the other end to seal the IL in place and the adsorber was closed and sealed. The adsorber was then re-weighed under vacuum to give the packed sorbent weight. The volumes of the sorbent (VS) and quartz wool (VQ) were found using the density data obtained from the pycnometer measurements. These volumes were subtracted from the total internal volume to give the void space as shown in Eq. 1.
The CO2 capacity of the sorbent was calculated using a static gas pressure and was carried out using pure CO2 gas. The starting weight of the packed adsorber was taken before the gas was introduced. Pure CO2 then enters the adsorber and the total weight increase of the system was determined (MT). This was achieved by closing the valves to the reactor, removing it from the system and placing it on the balance, the mass of the empty assembly having previously been measured. The mass increase was attributed to the CO2 that had been adsorbed onto the sorbent (Mads) and CO2 in the void space (Mvoid). In order to find the mass of CO2 in the void space, the density of the gas at that specific pressure and temperature was determined. This void space mass (Mvoid) was removed from the total mass increase (MT). The remaining mass (Mads) was then attributed to the gas that had adsorbed onto the sorbent (Eq. 2).
This resulting mass of adsorbed gas (Mads) was then used to derive the sorbent capacity, which is reported as a weight percentage of the initial sorbent weight (wt%) as shown in Eq. 3.
In order to determine the rate of adsorption, the adsorber was placed under static gas pressure for set periods of time. After each time period, the detached adsorber was weighed to determine the mass of gas adsorbed. Once the adsorbed gas weight plateaued, it was assumed that the sorbent had become saturated. To test desorption rate, the adsorber was placed on the balance and opened to atmosphere. The weight was monitored and recorded at set time intervals until there was no further weight loss. At this point, it was assumed that the desorption of gas was complete.
Previous work carried out by this group (Styring et al., 2016) has demonstrated that simple tetraalkylammonium acetate SoILs are suitable low-cost sorbents for use in a pressure swing system. This is due to their rapid adsorption and desorption kinetics, their chemical and mechanical stability, and their high selectivity toward CO2.
Tetrabutylammonium acetate [N4444][Ac] was chosen for testing due to its ease of handling and low water affinity and was wet-coated onto cellulose powder at various weight loadings, the resultant sorbent powders ranging from 50 to 95% cellulose by mass. These sorbents were then measured for CO2 uptake (error ±0.01 wt%) over a range of pressures at room temperature (Figure 4). While this does not simulate the temperatures that would be expected in flue gas treatment, previous tests on unsupported SoILs have shown minimal effect of temperature on maximum CO2 capacity at a given pressure (Styring et al., 2016).
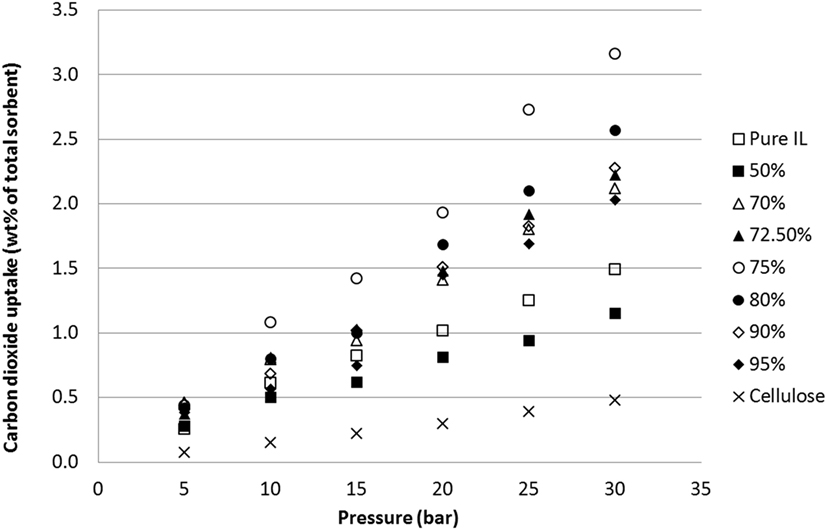
Figure 4. Uptake capacities of CO2 at various pressures on [N4444][Ac] across a range of cellulose loadings at room temperature.
In line with previous results and expectations, the linear trend of CO2 uptake with increasing pressure was observed (Figure 4). At lower pressures, the ILs have similar uptake capacities so the effect of cellulose loading is less apparent. However, as the pressure was raised to 30 bar, the differences in sorbent performance became greater. Overall, there was a clear and dramatic improvement in uptake as the sorbent trends toward a support loading of 75 wt% cellulose with CO2 total capacity at 30 bar reaching a peak of 3.16 wt% uptake. This is shown in Figure 5 in order to clarify the trend that is not immediately evident in Figure 4. If this is normalized to 1 g IL, then the adsorption scales to 12.64 wt%, 8.5 times greater than the pure IL. If the low cost of the cellulose is considered, this essentially represents an equal improvement in cost effectiveness. It should be noted that the addition of cellulose initially reduced total CO2 capacity when support loadings of 50 wt% cellulose were used. This was thought likely to be due to the formation of an aggregated matrix where the cellulose particles are bound together by the SoIL. This was borne out by the SEM images (Figure 6). Note also that despite the decrease in total sorbent uptake, when considering the uptake per unit mass IL independent of support, the uptake is still improved by a factor of 1.5 (or 54%).
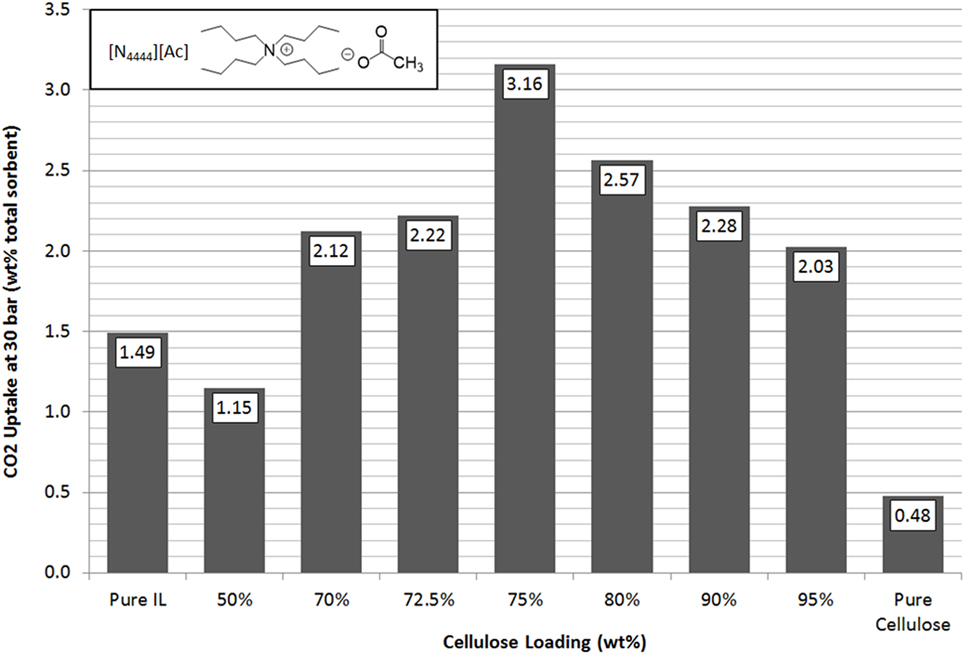
Figure 5. Trends in total sorbent CO2 uptake at 30 bar of [N4444][Ac] as a function of cellulose loading at room temperature.

Figure 6. SEM of [N4444][Ac] at 50 wt% cellulose loading showing agglomeration of cellulose particles (white) rather than surface coating.
The uptake at 75 wt% cellulose suggests that, at that loading, there is an optimum condition that allows for greater CO2 adsorption capacity. This could be due to a variety of reasons; IL layer thickness, surface morphology, and increased total surface area. This is further reinforced by the fact that total CO2 capacity is reduced as higher loadings of cellulose are used. This reduction in CO2 capacity could be due to issues with adsorption onto thinner IL layers, incomplete coverage leading to reduced IL surface area available for adsorption or simply because there is less IL in the bulk sorbent for CO2 capture. In order to help determine coverage, further SEM images of the sorbents were taken.
Figure 7 shows tested sorbents at the same magnification (×430) as each other under SEM at 20 kV. The [N4444][Ac] in its pure form shows the crystalline IL particles are very smooth and partially agglomerated, whereas cellulose exhibits a rough and very fine fibrous structure (Figures 7A,B). When supported at 75 wt% cellulose loading (image C), a coating of the smoother IL can be seen on the fibrous cellulose particle. However, when comparing Figures 7A,C, there is clear introduction of surface roughness to the IL upon loading. At the highest cellulose loading fraction of 95 wt% (Figure 7D), the smoothing effect of the IL is still visible; however, the underlying fibrous structure of the cellulose particle is more apparent, leading to areas of incomplete coverage, which would likely have selectivity implications.
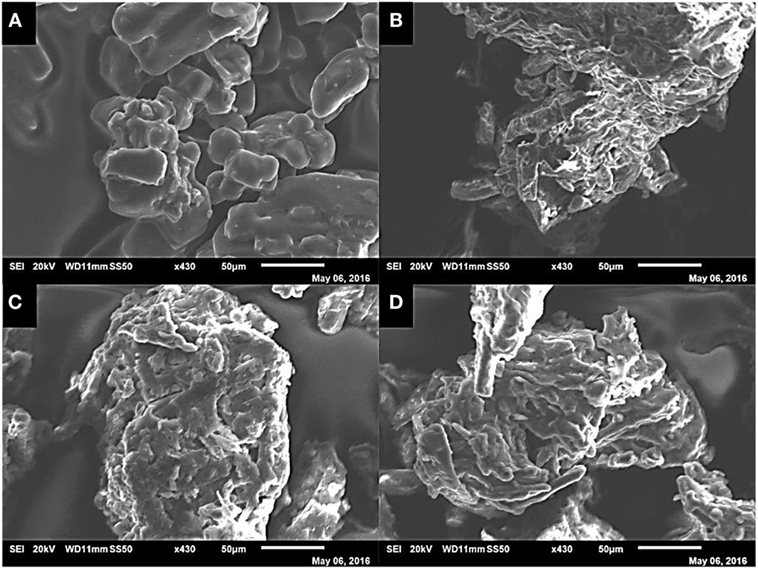
Figure 7. SEM images of unsupported [N4444][Ac] (A), powdered cellulose (B), 75 wt% cellulose [N4444][Ac] (C), and 95 wt% cellulose [N4444][Ac] (D) under the same degree of magnification (×430) at 20 kV.
The introduction of surface roughness to the IL surface through coating of the cellulose appears likely to be a cause of the observed improvement in CO2 uptake capacity. This could be explained simply by increased surface area. Alternatively, it is possible that the rough surface provides stronger binding sites that give the IL a pseudo-microporous structure. A combination of these two effects is also possible. Surface roughness causing a sorbent to behave as if microporous has been previously postulated to explain observed effects of gas adsorption onto mesoporous solids (Coasne et al., 2006).
In order to determine if this loading effect is seen with other simple ILs, a small selection of other common SoILs of both high and low capacity were tested. Based on the results of the [N4444][Ac] study, only samples of 70–80% cellulose loading by weight were considered. BET analysis of all SILs was also carried out to determine the effect of loading on surface area. This was performed in order to ascertain whether an increase in surface area was sole reason for increasing CO2 capacities.
Further testing was carried out to determine whether the trends seen for [N4444][Ac] hold true for other SoILs. [N1888][Ac], [Bmim][Br], and [N8888][Br] were chosen since they are structurally simple, low in cost, have readily available starting materials, and are solids at room temperature. Previous tests with [N8888][Br] have shown it to be particularly poor as a CO2 capture agent. It was selected in order to indicate whether the increase in CO2 capacity was proportional to the starting performance of the pure ionic liquid. If so, this would indicate that the improvement seen after cellulose support was simply down to an increase in available surface area of the IL for CO2 capture and not due to surface effects potentially suggested by SEM images.
[Bmim][Br] had a fairly poor starting capacity when using it in its pure form as it forms large crystals on synthesis and has a high affinity to water making it challenging to handle as fine powder. The resulting large crystals lead to a poor surface area to volume ratio, which was expected to be the reason for its poor uptake performance. However, once supported, [Bmim][Br] showed a similar increase in its CO2 uptake capacity at 75 wt% loading as was found for [N4444][Ac]. Following the pattern of the [N4444][Ac] (Figure 8), 70 and 80 wt% loadings showed poorer total uptake. The 75 wt% cellulose support loading gave an increase of a factor of 2.3 over pure [Bmim][Br], which amounts to an increase of CO2 adsorbed per unit mass of IL of 9.4, nearly an order of magnitude improvement in the effectiveness of the IL.
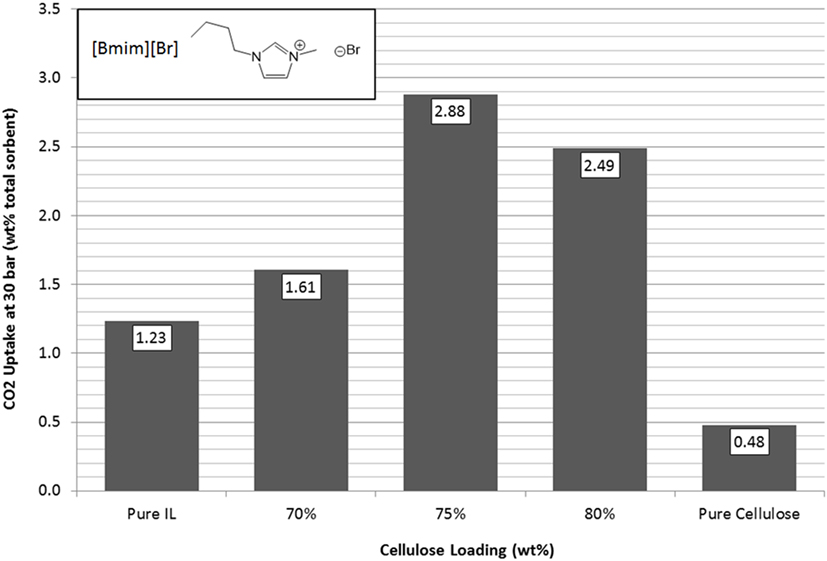
Figure 8. Trends in total sorbent CO2 uptake at 30 bar for [Bmim][Br] as a function of cellulose loading at room temperature.
Similarly, the 75 wt% cellulose sorbent shows a tripling of surface area by BET measurement (Table 2). It should also be noted that it is possible in this case that there is a sharp optimum peak loading for [Bmim][Br] in the 70–75 or 75–80 wt% range that may afford even further improvements in uptake performance. After loading, [Bmim][Br], which is highly hygroscopic, also showed a reduction in its water affinity and remained as a free-flowing powder during handling. This could also be attributed to changes in the surface properties, however, may be due to cellulose acting as a desiccant.
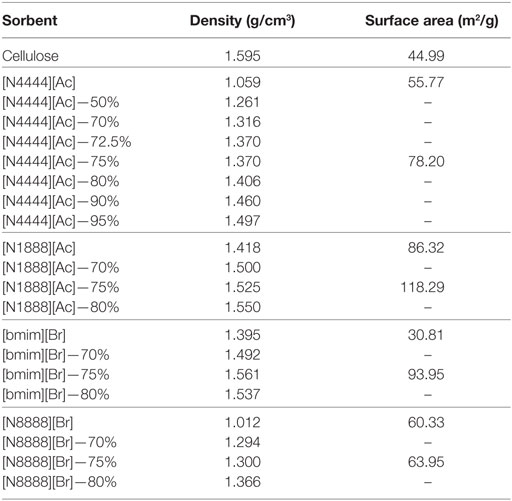
Table 2. Selected supported and unsupported sorbent physical properties.
[N1888][Ac] has the highest uptake capacity of the pure ILs that have been previously investigated by this group (Styring et al., 2016). In contrast, [N8888][Br], as previously mentioned, has one of the lowest uptake capacities measured. However, as shown in Figures 9 and 10, respectively, [N1888][Ac] showed only very modest improvement at 75 wt% loadings and loadings higher or lower than this amount actually gave a diminished performance. By contrast, [N8888][Br] showed an order of magnitude improvement in total CO2 capacity, with the highest capacity seen at 70 wt% cellulose loading. Furthermore, [N1888][Ac] exhibited a much larger increase in surface area, of around 40%, when compared to [N8888][Br] after cellulose loading (Table 2). However, since its capacity improvement was only marginal, surface area was unlikely to be the driving factor in this case. This perhaps indicates that the surface effects suggested by the SEM images on [N4444][Ac] may be important and, therefore, indicates that surface properties of [N1888][Ac] were already well-suited for CO2 adsorption. Therefore, the addition of the cellulose support had a negative or near-neutral effect at best. However, it still shows a total improvement, after the loading factor is considered, of a factor of 4.2 times better than the pure SoIL.
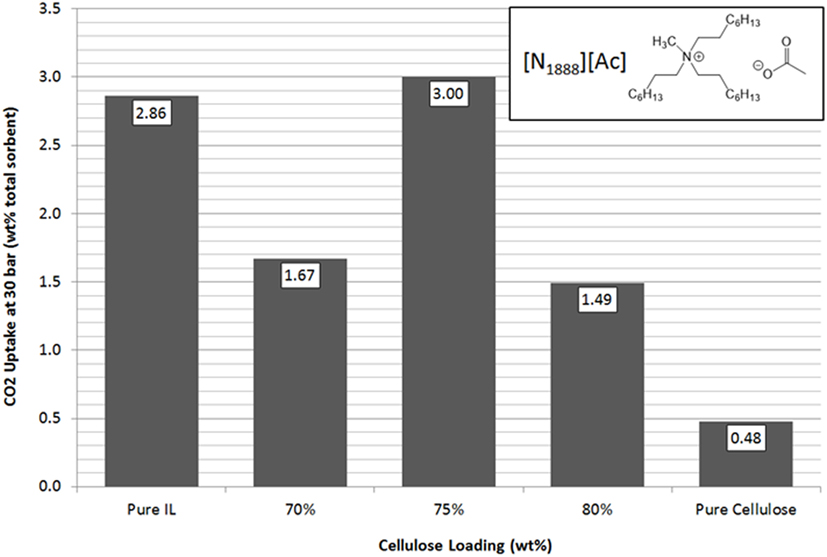
Figure 9. Trends in total sorbent CO2 uptake at 30 bar of [N1888][Ac] as a function of cellulose loading at room temperature.
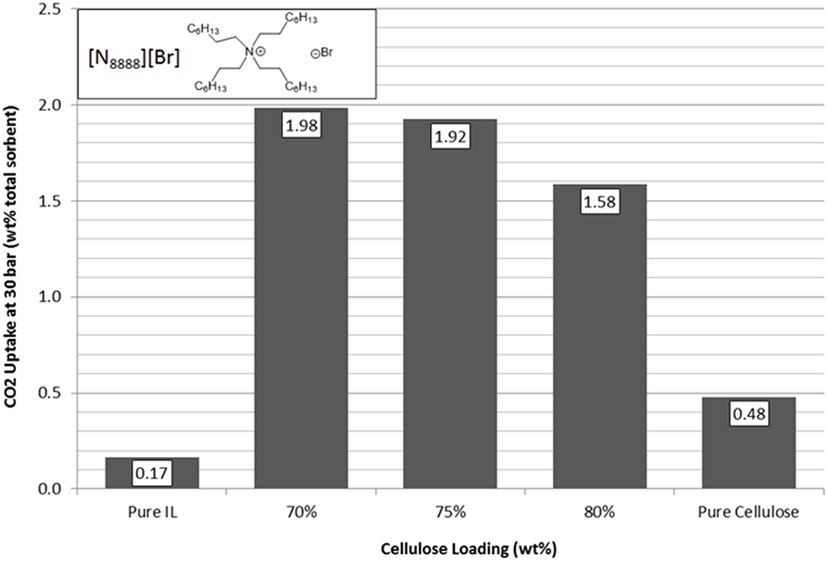
Figure 10. Trends in total sorbent CO2 uptake at 30 bar of [N8888][Br] as a function of cellulose loading at room temperature.
On the other hand, the improvement of [N8888][Br] indicates the opposite and that the unsupported salt surface morphology seems likely to be hostile to CO2 adsorption, despite its demonstrated capability to perform once supported. This is backed up by the surface area measurements shown in Table 3 where [N8888][Br] only exhibited a small increase in surface area, yet the increase in performance was large. This further suggests that surface properties play a major role in these cases.
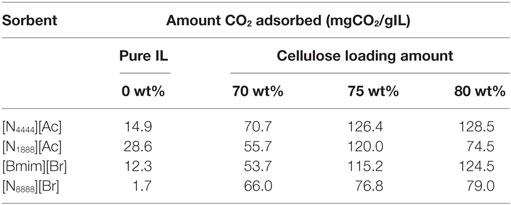
Table 3. Maximum CO2 uptake at 30 bar, as a proportion of IL present in the total sorbent.
It can be assumed that while the cellulose increases the surface area of the total sorbent, albeit with varying effect, the improvement in CO2 capacity after cellulose loading is neither proportional to the surface area increase, nor is it proportional to the pure IL uptake and is, in fact, more likely due to the increase in surface roughness imparted by the support. Overall, as cellulose loading increases, the total sorbent uses less and less ionic liquid, meaning the SoIL is used much more efficiently. The previously inaccessible centers of the SoIL particles are now instead spread over the cellulose allowing it to capture more CO2 per unit of SoIL used. In addition to what appears to be significantly improved surface effects, increased effectiveness at different cellulose loadings can be seen in Table 3: depending on the nature of the IL in question. Table 3 shows the amount CO2 captured per gram of SoIL within the total sorbent at different cellulose loadings.
In pressure swing systems, the kinetics associated with adsorption and desorption are very important. The faster these two processes can occur, the shorter the adsorb/desorb cycle times can be. This improves the efficiency by reducing adiabatic heat loss and further reduces the total sorbent inventory required to treat a given flow rate of gas. Previous work done using SoILs suggested that the kinetics for CO2 capture are very favorable (Styring et al., 2016). Pure SoILs have been shown to reach their adsorption capacity in seconds and release the captured CO2 equally as fast. The pure SoILs tested here have followed the same kinetic trends, all reaching saturation in less than 10 s with desorption being too quick to accurately measure. Cellulose loading did not have any substantial effect on the uptake kinetics, no matter the loading amount of cellulose used.
Sorbent stability was also tested and in Figure 11, the highest-capacity sorbent, N4444 acetate, loaded with 75 wt% cellulose, was tested through short 1 min adsorb/desorb cycles 40 times each at 15 and 30 bar with weight measurements taken for the first 10 and last 10 cycles to check for any overall changes. There are two scenarios presented: 30 bar adsorption—1 bar desorption and 15 bar adsorption—1 bar desorption. This test was carried out to demonstrate mechanical stability under different pressure loads and multiple cycles. Desorption was carried out at the same rate for each run, with the vessel slowly opened to atmosphere and then allowed to rest for 1 min. Sorbent stability can be shown to be very high under these conditions, with consistent uptake capacities reached. Note that a residual weight of CO2 remains at the end of each desorb cycle at both pressures. This is the CO2 that remains in the void spaces of the adsorber such as the valve assembly and piping, even after depressurization is complete. Since depressurization occurs over the same time period, the higher pressure runs have proportionately more CO2 present as less time has passed for atmospheric diffusion. Further diffusion, a flow of nitrogen or application of vacuum for a few seconds all return the starting weight of the sample to 0.
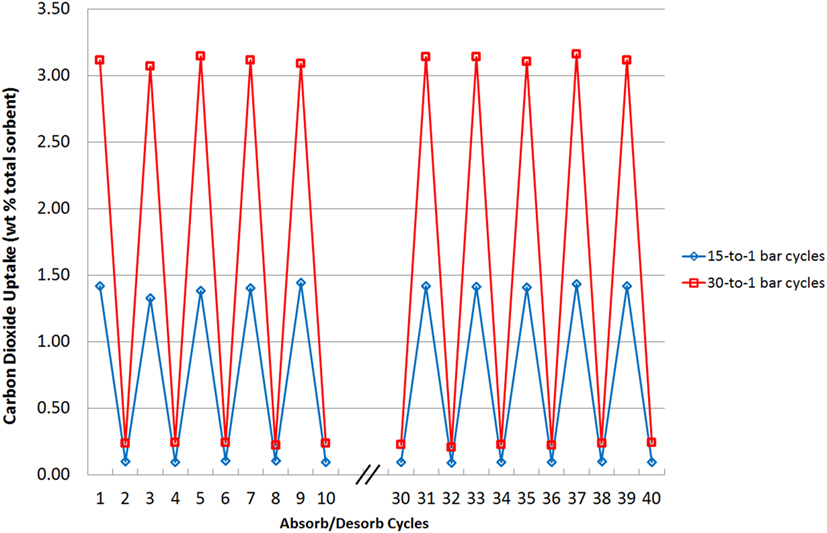
Figure 11. Sorbent stability over repeated 1-min adsorb/desorb cycles at two pressures. Desorption is carried out in each case at 1 bar pressure.
The IL component of the sorbent is also readily recovered in high yield by filtration of a slurry of the sorbent in isopropanol. The isolated IL can then readily be reused with no detectable change in performance, allowing small synthetic batches of each IL to be used to generate a variety of cellulose loading mixtures.
It is clear that further advancements in CO2 capture systems are needed in the near future to develop CCUS in order to help mitigate the potential problems caused by further carbon emissions. Existing state-of-the-art CO2 capture and separation technologies suffer from high energy costs, primarily associated with the CO2 desorption inherent in chemisorption-based TSA processes. Alternative PSA processes show many advantages in terms of both cycle speed, energy costs, and sorbent stability, due to the absence of thermal cycling. Therefore, PSA coupled with appropriate solid adsorbents would appear to be a promising alternative gas separation pathway. SoILs show good capacity and excellent adsorption rate, making them desirable for use in a PSA process. However, currently, even simple ILs have a high manufacturing cost, which reduces their attractiveness as large-scale carbon capture sorbents.
Significant reductions in amount of IL required to capture CO2 have been demonstrated by coating inexpensive cellulose particles with thin layers of IL. In addition, surface characteristics imparted to the IL by the cellulose supports have further increased CO2 uptake capacity. Together, these two effects may allow even expensive, specialized, and previously overlooked IL sorbents to be used more effectively. Fast sorption kinetics are observed, which results in large capture capacity per unit time when compared to conventional TSA systems, which exhibit significant temperature lag between adsorb and desorb cycles. Further research must be done to show their suitability as a post-combustion capture sorbents.
These studies were fundamental in nature, carried out with pure CO2 and pure nitrogen. The results of these studies will be used to monitor performance of different gas mixtures ranging from blast furnace top gases at high CO2 concentrations through to flue gas from power stations down to the parts per million concentrations of atmospheric CO2. By determining the fundamental data, we are now in a position to test these on low concentration simulated and real waste gas streams.
Each author played a major role in the development of the work and writing the manuscript. PS is the research supervisor and group leader. DGR is a postgraduate research student who undertook the work as part of his Ph.D. programme. GRMD is a postdoctoral research associate who was involved in all aspects of experimentation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the EPSRC for 4CU Programme Grant funding (EP/K001329/1) at The University of Sheffield supporting the work and GRMD, and for a DTA studentship for DGR. We also thank the ESPRC for funding of the CO2Chem Grand Challenge Network to PS (EP/K007947/1 and EP/H035702/1).
Baltus, R. E., Counce, R. M., Culbertson, B. H., Luo, H., DePaoli, D. W., Dai, S., et al. (2005). Examination of the potential of ionic liquids for gas separations. Sep. Sci. Technol. 40, 525–541. doi: 10.1081/SS-200042513
Bates, E. D., Mayton, R. D., Ntai, I., and Davis, J. H. (2002). CO2 capture by a task-specific ionic liquid. J. Am. Chem. Soc. 124, 926–927. doi:10.1021/ja017593d
Belaissaoui, B., Cabot, G., Cabot, M.-S., Willson, D., and Favre, E. (2013). CO2 capture for gas turbines: an integrated energy-efficient process combining combustion in oxygen-enriched air, flue gas recirculation, and membrane separation. Chem. Eng. Sci. 97, 256–263. doi:10.1016/j.ces.2013.04.027
Belaissaoui, B., Willson, D., and Favre, E. (2012). Membrane gas separations and post-combustion carbon dioxide capture: parametric sensitivity and process integration strategies. Chem. Eng. J. 211-212, 122–132. doi:10.1016/j.cej.2012.09.012
Brennecke, J. F., and Gurkan, B. E. (2010). Ionic liquids for CO2 capture and emission reduction. J. Phys. Chem. Lett. 1, 3459–3464. doi:10.1021/jz1014828
Chen, L., Sharifzadeh, M., and Mac Dowell, N. (2014). Inexpensive ionic liquids: [HSO4]−-based solvent production at bulk scale. Green Chem. 16, 3098–3106. doi:10.1039/c4gc00016a
Chou, C., and Chen, C. (2004). Carbon dioxide recovery by vacuum swing adsorption. Sep. Purif. Technol. 39, 51–65. doi:10.1016/j.seppur.2003.12.009
Coasne, B., Hung, F. R., Pellenq, R. J., Siperstein, F. R., and Gubbins, K. E. (2006). Adsorption of simple gases in MCM-41 materials: the role of surface roughness. Langmuir 22, 194–202. doi:10.1021/la051676g
Desideri, U., and Paolucci, A. (1999). Performance modelling of a carbon dioxide removal system for power plants. Energy Convers. Manag. 40, 1899–1915. doi:10.1016/S0196-8904(99)00074-6
Figueroa, J., Fout, T., and Plasynski, S. (2008). Advances in CO2 capture technology—the U.S. Department of Energy Carbon Sequestration Program. Int. J. Greenhouse Gas Control 2, 9–20. doi:10.1016/S1750-5836(07)00094-1
Gale, J., Bradshaw, J., Chen, Z., Garg, A., Gomez, D., Rogner, H.-H., et al. (2005). IPCC Special Report on Carbon dioxide Capture and Storage, Working Group III of the Intergovernmental Panel on Climate Change. Cambridge, UK: Cambridge University Press.
Harkin, T., Hoadley, A., and Hooper, B. (2009). A comparison of the process integration of shockwave CO2 compression with conventional turbo machinery into PCC power station design. Energy Procedia 4, 1339–1346. doi:10.1016/j.egypro.2011.01.192
Ilconich, J., Myers, C., Pennline, H., and Luebke, D. (2007). Experimental investigation of the permeability and selectivity of supported ionic liquid membranes for CO2/He separation at temperatures up to 125°C. J. Memb. Sci. 298, 41–47. doi:10.1016/j.memsci.2007.03.056
IPCC. (2014). “Climate change 2014: synthesis report,” In Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, eds R. K. Pachauri and L. A. Meyer (Geneva, Switzerland: IPCC), 1–151.
Jassim, M. S., Rochelle, G., Eimer, D., and Ramshaw, C. (2007). Carbon dioxide absorption and desorption in aqueous monoethanolamine solutions in a rotating packed bed. Ind. Eng. Chem. Res. 46, 2823–2833. doi:10.1021/ie051104r
Karadas, F., Atilhan, M., and Aparicio, S. (2010). Review on the use of ionic liquids (ILs) as alternative fluids for CO2 capture and natural gas sweetening. Energy Fuels 21, 5817–5828. doi:10.1021/ef1011337
Klein-Marcuschamer, D. (2011). Techno-economic analysis of a lignocellulosic ethanol biorefinery with ionic liquid pre-treatment. Biofuels Bioprod. Biorefin. 6, 562–569. doi:10.1002/bbb.303
Krishnamurthy, S., Rao, V., and Guntuka, S. (2014). Post combustion CO2 capture by vacuum swing adsorption: a pilot plant study. AIChE J. 60, 1830–1842. doi:10.1002/aic.14435
Kundu, P., Chakma, A., and Feng, X. (2014). Effectiveness of membranes and hybrid membrane processes in comparison with absorption using amines for post-combustion CO2 capture. Int. J. Greenhouse Gas Control 28, 248–256. doi:10.1016/j.ijggc.2014.06.031
Liu, L., Du, T., Fang, X., Che, S., and Tan, W. (2014). The 26th Chinese Control and Decision Conference. Shenyang, China: IEEE, 4038–4041.
MacDowell, N., Florin, N., Buchard, A., Hallett, J., Galindo, A., Jackson, G., et al. (2010). An overview of CO2 capture technologies. Energy Environ. Sci. 3, 1645–1669. doi:10.1039/c004106h
Mehnert, C. P. (2004). Supported ionic liquid catalysis. Chemistry 11, 50–56. doi:10.1002/chem.200400683
Moen, O. M., and Stene, H. S. (2014). Power Plant with CO2 Capture Based on PSA Cycle. Trondheim, Norway: Norwegian University of Science and Technology.
Moya, C., Palomar, J., Gonzales-Miquel, M., Bedia, J., and Rodriguez, F. (2014). Diffusion coefficients of CO2 in ionic liquids estimated by gravimetry. Ind. Eng. Chem. Res. 53, 13782–13789. doi:10.1021/ie501925d
Pershad, H., Harland, K., Stewart, A., and Slater, S. (2010). CO2 Pipeline Infrastructure: An Analysis of Global Challenges and Opportunities. Cambridge, UK: Element Energy, 1–134. Available at: www.ccsassociation.org.uk/docs/2010/IEA%20Pipeline%20final%20report%20270410.pdf
Ramdin, M., de Loos, T. W., and Vlugt, T. J. H. (2012). State-of-the-art of CO2 capture with ionic liquids. Ind. Eng. Chem. Res. 51, 8149–8177. doi:10.1021/ie3003705
Rameshni, M. (2010). Carbon Capture Overview. Monrovia, CA: WorleyParsons, 1–16. Calculated as 30% reduction in energy usage compared to MEA. Available at: http://stoppingclimatechange.com/Carbon%20Capture%20Overview%20(3).pdf
Romeo, L. M., Bolea, I., and Escosa, J. M. (2008). Integration of power plant and amine scrubbing to reduce CO2 capture costs. Appl. Therm. Eng. 28, 1039–1046. doi:10.1016/j.applthermaleng.2007.06.036
Samanta, A., and Zhao, A. (2011). Post-combustion CO2 capture using solid sorbents: a review. Ind. Eng. Chem. Res. 51, 1438–1463. doi:10.1021/ie200686q
Shiflett, M., and Yokozeki, A. (2008). Phase behavior of carbon dioxide in ionic liquids: [emim][Acetate], [emim][Trifluoroacetate], and [emim][Acetate] + [emim][Trifluoroacetate] mixtures. J. Chem. Eng. Data 54, 108–114. doi:10.1021/je800701j
Stolaroff, J., Keith, D., and Lowry, G. (2008). Carbon dioxide capture from atmospheric air using sodium hydroxide spray. Environ. Sci. Technol. 42, 2728–2735. doi:10.1021/es702607w
Styring, P., Dowson, G. R., Reed, D. G., Bellas, J. M., and Charalambous, C. (2016). Fast and selective separation of carbon dioxide from dilute streams by pressure swing adsorption using solid ionic liquids. Faraday Discuss. 192, 511–527. doi:10.1039/C6FD00035E
Swatloski, R. P., Spear, S. K., Holbrey, J. D., and Rogers, R. D. (2002). Dissolution of cellulose with ionic liquids. J. Am. Chem. Soc. 124, 4974–4975. doi:10.1021/ja025790m
United Nations. (2015). Framework Convention on Climate Change, Adoption of the Paris Agreement, A/RES/70/1, December 2015. [FCCC/CP/2015/L.9].
Wilson, G., Trusler, M., Yao, J., Lee, J. M., Graham, R., MacDowell, N., et al. (2016). End use and disposal of CO2 – storage or utilisation? General discussion. Faraday Discuss. 192, 561–579. doi:10.1039/C6FD90055K
Wong, S., and Bioletti, R. (2002). Carbon Dioxide Separation Technologies. Alberta, Canada: Carbon & Energy Management.
Xie, Y., Zhang, Y., Lu, X., and Ji, X. (2014). Energy consumption analysis for CO2 separation using imidazolium-based ionic liquids. Appl. Energy 136, 325–335. doi:10.1016/j.apenergy.2014.09.046
Keywords: carbon dioxide capture, ionic liquid, cellulose, cost reduction, pressure swing adsorption
Citation: Reed DG, Dowson GRM and Styring P (2017) Cellulose-Supported Ionic Liquids for Low-Cost Pressure Swing CO2 Capture. Front. Energy Res. 5:13. doi: 10.3389/fenrg.2017.00013
Received: 17 February 2017; Accepted: 09 June 2017;
Published: 07 July 2017
Edited by:
Angela Dibenedetto, Università degli studi di Bari Aldo Moro, ItalyReviewed by:
Hasmukh A. Patel, Northwestern University, United StatesCopyright: © 2017 Reed, Dowson and Styring. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Styring, cC5zdHlyaW5nQHNoZWZmaWVsZC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.