 Joanna Sobolewska
Joanna Sobolewska Wioleta Respondek2
Wioleta Respondek2 Przemyslaw Witek
Przemyslaw Witek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 11 February 2025
Sec. Adrenal Endocrinology
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1533711
This article is part of the Research TopicEnhancing Adrenal Tumor Diagnostics: Biomarkers and Molecular MechanismsView all 12 articles
The primary management in the care of patients with adrenal incidentalomas is to determine the oncologic risk, namely, the possibility of malignancy. The first place among adrenal incidentaloma lesions requiring diagnosis and treatment promptly is adrenocortical carcinoma (ACC). Similarly, in the case of pheochromocytoma, the lack of early diagnosis worsens the patient’s prognosis. Even though both ACC and pheochromocytoma are among the less frequent adrenal lesions, neither should be excluded during differential diagnostics, especially in patients with an equivocal clinical presentation and non-typical adenoma radiological features. ACC presenting as pheochromocytoma is one of the few cases described in the literature, some of which could not collect exhaustive clinical data. Herein, in this article, we would like to provide an overview of reported ACC cases clinically manifesting as pheochromocytoma, based on the clinical image of a 59-year-old female patient with unintentional weight loss, non-specific abdominal pain, a diagnosis of hypertension, and significantly elevated excretion of 3-methoxytyramine in a 24-h urine collection, histopathologically diagnosed with ACC. The case presented emphasizes how crucial a comprehensive diagnostics and individual approach to the patient would be.
Adrenal incidentalomas are adrenal masses detected on imaging studies performed for reasons other than suspected adrenal disease. The etiology of adrenal focal lesions is heterogeneous and includes both benign and malignant lesions originating from the adrenal cortex, medulla, and masses of extra-adrenal source (1). Autopsy evaluation indicates the prevalence of adrenal lesions at approximately 2%, increasing with age (1). The most relevant diagnostic tools for adrenal tumors are unenhanced computed tomography (CT), magnetic resonance imaging (MRI) with technique of chemical-shift imaging, and 18F-fluorodeoxyglucose positron emission tomography/CT (18F-FDG-PET-CT) (2). Abdominal ultrasound remains of limited importance (1, 2). In adrenal imaging, MRI is the preferred choice over CT in children, adolescents, and pregnant women (1). A special position in adrenal incidentaloma management is given to lesions of potential malignancy, which, on unenhanced CT, are manifested by the presence of heterogeneous adrenal lesions with a diameter ≥4cm and a density exceeding 10 Hounsfield units (HU), usually >20 HU (1). Two primary malignant lesions may originate from the adrenal glands—adrenocortical carcinoma (ACC) and pheochromocytoma, both of which remain infrequent lesions (1, 3). ACC is a malignant tumor with an unfavorable prognosis (2), which is estimated to occur in 0.5–2 people per million per year (3). The ACC accounts for 0.4% to 4% of all adrenal incidentalomas (1), and its peak incidence occurs in the fifth and sixth decades of life (4). In 30%–40% of ACC cases, the diagnosis is led by clinical manifestations due to an excess of cortisol, less frequently androgens, and most rarely mineralocorticosteroids (2). The characteristic features of ACC in the CT, besides density exceeding 10 HU in the native phase, comprise necrotic and hemorrhagic areas visible in the tumor (2).The majority of ACC are sporadic, but 5%–10% may arise in the course of germline mutations, such as Li–Fraumeni syndrome, Lynch syndrome, and several cancer hereditary predisposition syndromes—multiple endocrine neoplasia type 1, neurofibromatosis type 1, familial adenomatous polyposis, Gardner syndrome, Beckwith–Wiedemann syndrome, and Carney complex (2, 5). Because of this, molecular diagnostics’ role in managing patients with ACC seems particularly encouraging. The investigation should include comorbid cancers and medical family history, while clinicians must individualize screening based on the gene of interest and age of onset of related cancers (2, 6). Any suspected cancer hereditary predisposition syndromes should involve referring the patient for genetic testing, which, if Lynch syndrome is confirmed, may open up other therapeutic options for the patient, such as immunotherapy with immune checkpoint inhibitors (2, 6).
Pheochromocytoma is a neuroendocrine tumor producing catecholamines, originating in the chromaffin cells of the adrenal medulla or extra-adrenal paraganglia (3, 7). Pheochromocytoma accounts for 1%–5% of all adrenal incidentalomas (1). Pheochromocytoma and paraganglioma are described collectively as PPGL group, with an incidence of two to eight cases per million people per year (3). Pheochromocytoma should always be considered in the differential diagnosis because of the risk of causing a life-threatening catecholamine crisis and specific management before surgical treatment (3). The risk of metastatic lesions of PPGL increases with a tumor ≥50 mm in diameter, the presence of paraganglioma with extra-adrenal localization, a germline mutation of the iron sulfur B subunit of the succinate dehydrogenase complex (SDHB), and plasma 3-methoxytramine (3MT) levels more than three times the upper reference limit (URL) (3). The gene that encodes the B subunit of the SDHB complex remains a key molecular contributor to malignant PPGL, with mutations in this gene found in at least 40% of all metastatic PPGL cases (3). Vigilance should be maintained, especially when the clinical picture is ambiguous. Therefore, we would like to present the case of a patient with ACC, clinically manifested as pheochromocytoma, and a review of similar cases available in the literature. The indicated case is one of the few described, so we would like to emphasize the role of interdisciplinary cooperation, extensive diagnostics, and individual approach in the care of patients with adrenal lesions.
A 59-year-old female patient with incidentally detected left adrenal gland focal lesion previously qualified for surgical treatment was admitted to the Endocrinology Department in January 2024 for preoperative hormonal evaluation. In the patient’s history, an unintentional weight loss (approximately 15 kg over several months) and non-specific abdominal pain for a few weeks were present. An ultrasound examination performed in October 2023 for the listed symptoms visualized the aforementioned lesion of mixed echogenicity in the left adrenal gland, with dimensions of 90 × 61 mm. Outpatient abdominal MRI performed in December 2023 revealed a polycyclic nodular lesion of the left adrenal gland, measuring 85 × 83 × 83 mm, with intermediate signal in T1 and T2 images, heterogeneously enhancing after administration of a contrast agent, with areas of central necrosis—an image suggestive of ACC/pheochromocytoma. The patient has not systematically monitored her blood pressure at home. However, in the results presented by the patient from several days prior to hospitalization, systolic blood pressure (BP) values exceeded 160 mmHg, which allowed the diagnosis of hypertension. Due to suspected secondary etiology of hypertension, treatment with doxazosin was implemented. The patient denied other symptoms suggestive of a pheochromocytoma. On physical examination, she did not present typical clinical features of overt Cushing’s syndrome (CS) and other endocrine stigmatization hallmarks.
Throughout the hospital stay, the hormonal evaluation confirmed a mild autonomic cortisol secretion (MACS)—morning serum cortisol: 20.5 (URL 19.4) μg/dL, midnight serum cortisol: 6.9 (URL 5.4) μg/dL, adrenocorticotropin: 12.95 (URL 48) pg/mL, serum cortisol after 1 mg of dexamethasone: 4 (URL 1.8) μg/dL, dehydroepiandosterone sulfate: 77.1 (URL 182.2) μg/dL. In twice 24-h urine collection, a significantly elevated (4.5–6 × URL) excretion of 3MT with normal concentrations of metanephrine and normetanephrine was found. Other laboratory assays did not indicate any significant abnormalities: 17-hydroxyprogesterone: 0.87 (URL 0.9) ng/mL and total testosterone 0.72 (URL 1.24) nmol/L. The potential influence of non- and pharmacological factors on the metoxycatecholamine measurement was excluded. An unenhanced CT scan was performed to control the dimensions of the lesion and visualized a solid, heterogeneous tumor in the left adrenal gland field measuring about 87 × 64 × 107 mm with a density of approximately 35 HU (Figure 1).
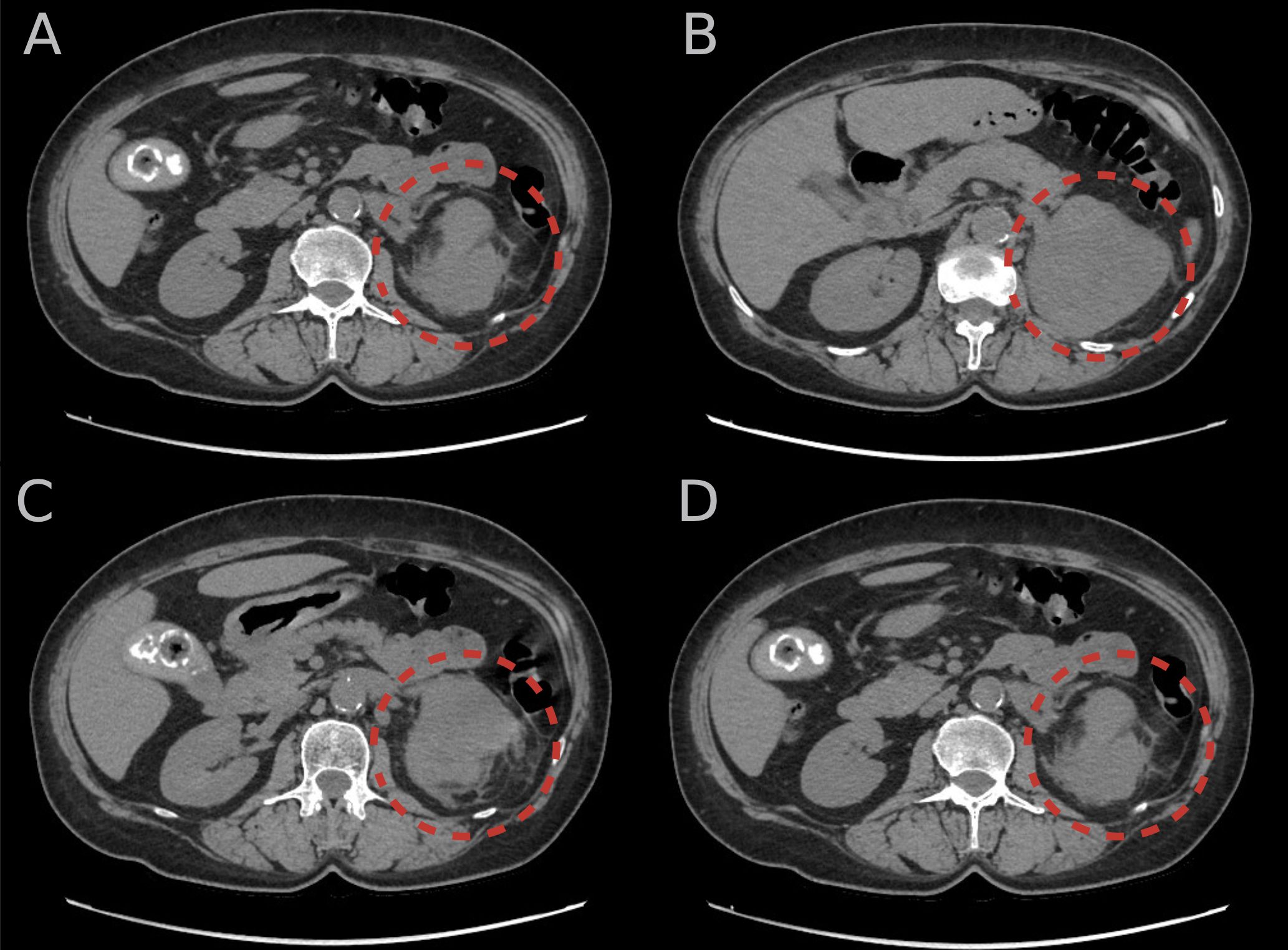
Figure 1. (A–D) An unenhanced computer tomography scans of the abdomen showing solid, heterogeneous tumor in the left adrenal gland (red circular lines) measuring approximately 87 × 64 × 107 mm with a density of approximately 35 Hounsfield units.
The patient was referred for 18F-FDG-PET-CT, which revealed a metabolically active nodular lesion measuring 93 × 79 × 94 mm with a maximal SUV of 27.3 originating from the left adrenal gland in the retroperitoneal cavity. No metabolic active metastatic lesion was found in this examination. Due to suboptimal BP control, the dose of doxazosin was escalated (4 mg, taken two times a day). At the end of January 2024, after the required pharmacological preparation, a total left adrenalectomy with simultaneous splenectomy and regional lymphadenectomy was performed. A postoperative hormonal evaluation did not show adrenal insufficiency. Clinically, after surgery, the patient required continued hypotensive treatment at a reduced doses (doxazosin, 1 mg daily).
Histopathological evaluation identified adrenocortical carcinoma pT3N0. The preparation exhibited a high-grade mitotic tumor index and a 7/7 score on the modified Weiss scale. The microscopic assessment revealed high-grade nuclear atypia of epithelioid and pleomorphic cells, forming a solid tumor, with necrosis and small hemorrhages. Tumor invasion was also found in small vessels surrounding and across the organ capsule. Immunohistochemical (IHC) staining was negative for inhibin, calretinin, chromogranin, synaptophysin, and neuron-specific enolase (NSE). The Ki-67 index was approximately 80%. No cancer metastasis was found in the sampled lymph nodes. A subsequent histopathological verification, also performed by another experienced pathomorphologist, resulted in an 8/9 Weiss score and a 7/7 score according to Aubert’s modified Weiss system. IHC evaluation yielded a positive reaction for CKPAN, Vimentin, CD10, GATA-3, and S100 (in part of the cells). Markers of adrenal cortical differentiation, or markers typical of the conventional form of ACC, were negative; however, single, diffuse atypical cells exhibited a positive reaction for calretinin, which may indicate cortical differentiation of some of the tumor cells. Although not entirely characteristic and typical, the morphological picture and immunophenotype may correspond to the “sarcomatoid” subtype of adrenocortical carcinoma. The decision about adjuvant therapy with mitotane was made by the oncology team, and the patient has undergone this treatment.
The most common clinical manifestations of ACC include symptoms resulting from excess hormones, mainly cortisol, hence plethora, muscle atrophy, or metabolic complications such as diabetes mellitus (DM) or osteoporosis (4). The next most frequently produced by ACC hormones are adrenal androgens, causing hirsutism, and virilization (4). Hyperaldosteronism remains the fewest endocrine disorder in ACC, and the hypokalemia that may occur in this group of patients is more likely to arise from severe hypercortisolemia, as excess of cortisol affects as many as 50%–70% of ACC patients (2). It is worth mentioning the possibility of combined endocrine disorders in patients with adrenal tumors, which clinically may be reflected in an increased possibility of ACC (2). However, the presented patient did not exhibit clinical signs of overt CS but rather an abdominal discomfort that may result from the dimensions of the adrenal lesion—the tumor to one extent measured above 10 cm. ACCs are usually large tumors at the time of diagnosis, usually exceeding 6 cm in diameter (4). Thus, the clinical picture of an ACC patient may also include symptoms secondary to the effect of the tumor mass, mainly mentioned abdominal pain, as well as a feeling of abdominal fullness or early satiety (4). Symptoms resulting from the tumor mass effect usually emerge in hormonally inactive ACC cases (2). Noteworthily, ACC usually occurs unilaterally, while bilateral lesions, especially in patients with active malignancy, may suggest metastatic adrenal lesions (2).
In the presented patient, MACS was diagnosed. Although MACS patients are not considered to be at high risk of developing overt CS, it is recommended to actively search for potential cortisol-related comorbidities, such as hypertension or type 2 DM (1). In our patient, glycated hemoglobin was 5.7%, and further observation was planned.
Most patients with pheochromocytoma present with paroxysmal symptoms such as palpitation, sweating, anxiety attacks, or hypertension that poorly responds to conventional treatment, even leading to hypertensive crises (7). Although most pheochromocytoma tumors secrete excess norepinephrine, it is essential to mention the rare epinephrine-secreting tumors, which are associated with hypotension and shock after alpha-blocker administration caused by the solid beta-adrenergic effects of epinephrine (7). Alpha-blockers are medications preventing hypertensive crises during surgical treatment (7). As mentioned in the presentation, the patient was diagnosed with hypertension, and due to the clinical suspicion of pheochromocytoma, pharmacotherapy with doxazosin was implemented. After adrenalectomy, hypertension persisted in the presented patient, which supports that it did not result from excess secretion of the catecholamines by the tumor. However, there are some doubts: in our patient, of the three metoxycatecholamine metabolites tested in twice 24-h urine collection, only 3MT excretion was significantly elevated (4.5–6 × URL). The 3MT is a metabolite of dopamine metabolism, which is often excreted by extra-adrenal lesions (8). Postoperative evaluation of our patient has shown a normalization of 3MT.
In a similar case to ours, described by Ni and Htet, the 28-year-old female patient also did not present symptoms suggestive of CS or virilization but reported those indicative of pheochromocytoma, namely, palpitations, excessive sweating, flushing, and cold extremities, which correlated with high vanillylmandelic acid (VMA) urine excretion (3 × URL) which had been normalized after adrenalectomy, but the histopathological examination of resected tumor showed an ACC (9).
In the absence of clinical features of steroid hormone production, it is advisable to biochemically exclude pheochromocytoma (4) by measuring plasma-free metanephrines or fractionated urinary metanephrines in all patients with adrenal lesions not presenting typical adenomas features (1). It has been indicated that in patients without clinical signs suggestive of pheochromocytoma, determining plasma fractions provides higher sensitivity than assessing urinary metanephrines. Despite considering the determination of plasma-free metanephrines as the most sensitive currently available diagnostic assay, the rate of false-negative results is estimated at 2.1%. False-positive results can be induced by either clinical conditions (heart failure, anxiety, hypoglycemia, pain, sleep apnea syndrome, renal failure), medications (sympathomimetics, tricyclic antidepressants, atypical neuroleptics), stimulants (coffee, nicotine), or dietary factors (fruits, especially bananas) (10).
Alert signs, such as night sweats or cachexia, and paraneoplastic syndromes in the course of ACC are rather rare (4). However, cases of cancer-related hypoglycemia in patients with ACC have been described (11) or hyperrenin hyperaldosteronism (12) and erythropoietin-related polycythemia (13). The presented patient, however, reported significant unintentional weight loss.
The pathomorphological diagnosis of ACC remains a diagnostic challenge due to its rarity (14) and the presence of several variants—conventional, pediatric, oncocytic, myxoid, and sarcomatoid (15, 16). Biopsy plays a limited role in the diagnosis of adrenal tumors, being contraindicated in the majority of cases because of the risk of tumor dissemination and the possibility of inducing a catecholamine crisis in the case of pheochromocytoma. However, it acquires significance in adrenal lesions with a possible metastatic manifestation of extra-adrenal tumors (3). Evaluation of Ki-67, one of the most relevant factors of proliferation, remains crucial, while its assessment in biopsy material may not reflect the index titer in the entire tumor (2). For the proper pathomorphological evaluation, several tools may be used: the Weiss system, the reticulin algorithm, the Helsinki system, and the Lin–Weiss–Bisceglia system (2, 17). In general clinical practice, the diagnosis of malignant neoplasm is based on finding at least three of nine morphological parameters on light microscopy, according to the Weiss scoring system (3, 15). In the case of our patient, histopathological verification was firmly supportive of ACC, i.e., high-grade mitotic tumor index, nuclear atypia, presence of necrosis and hemorrhages, and vascular invasion. Specific ACC markers (inhibin and calretinin) were negative in the first IHC determination, and positive, only for calretinin in some of the diffuse atypical cells in the reevaluation, so the sarcomatoid subtype of ACC was diagnosed. This subtype is the least common variant of ACC, characterized by aggressive behavior and a worse prognosis than other ACC variants (18). In a study by Papathomas et al. showing six cases of sarcomatoid ACC, obtained from five pathomorphology departments, SF-1 was the only adrenal cortex marker positive in the epithelial component in all cases, but was always negative in the sarcomatoid areas; cadherins were positive only in the epithelial component (18). In our patient’s case, SF-1 and cadherin were not determined, but as reported above, they may be negative in the sarcomatoid components of the tumors. The histologic and immunohistochemical findings also ruled out the possibility of pheochromocytoma. Conversely, the indicated ambiguous immunohistochemistry results do not oppose the diagnosis of ACC and may be helpful in determining the prognosis. In the study conducted by Zlatibor et al. involving 30 ACC cases, among subjects with negative inhibin staining, half of them died within the first 6 months, and only one person in this group survived longer than 1 year (19). In that study, a similar relationship between survival length and a worse prognosis was also observed with a Ki-67 index ≥7% (19).
Collected data highlight the predominance of diagnoses of advanced ACC cases with distant metastases (stage 4) in the past; now, it is reported that 25% to 30% of patients with ACC present with metastases (4). The most common locations for metastatic lesions include the lungs, liver, and bones (4). A complete staging evaluation, including at least a chest CT and 18F-FDG-PET-CT, is recommended before surgical treatment (1). In our patient, the staging was performed, and no metastases were found.
Reviewing the literature, we found a few similar cases besides those mentioned above, some of which have been dated distantly and thus may not contain all the clinical data. A summary of the described cases is presented in Table 1; however, several of them are worth additional emphasize. In the paper by Alsabeh et al., five patients with elevated catecholamine secretion in urine or serum were described and the histopathological evaluation confirmed two cases of ACCs and three cases of adrenal cortical adenomas (ACAs) (20). Both cases with a diagnosis of ACC involved men with previously diagnosed hypertension. In each, distant metastases were found in the months following adrenalectomy (20). In both ACC cases, as well as in one ACA case, immunohistochemical evaluation revealed positive staining for NSE and synaptophysin and negative for chromogranin (20). Another case worth emphasizing is the patient described by Fassina et al. in which, due to further diagnostics because of ongoing left-sided pain symptoms, slides obtained from a performed 7-month-earlier laparotomic excision of a pheochromocytoma in the left adrenal gland were reevaluated, which resulted in a final diagnosis of coexisting pheochromocytoma and ACC (21). The patient was presumed to be genetically predisposed to develop endocrine dysplasia, but germline mutations in VHL, RET, SDHB, SDHC, and TMEM127 were excluded, implying sporadic co-occurrence of the tumors in mentioned case (21). Since pheochromocytoma may produce adrenocorticotropin, insulin-like growth factor 2, somatostatin, growth hormone-releasing hormone, corticotropin-releasing hormone, and interleukin 6, it was considered, however, that it might also stimulate the adrenal cortex to hyperfunction and proliferation through the paracrine pathway (21).
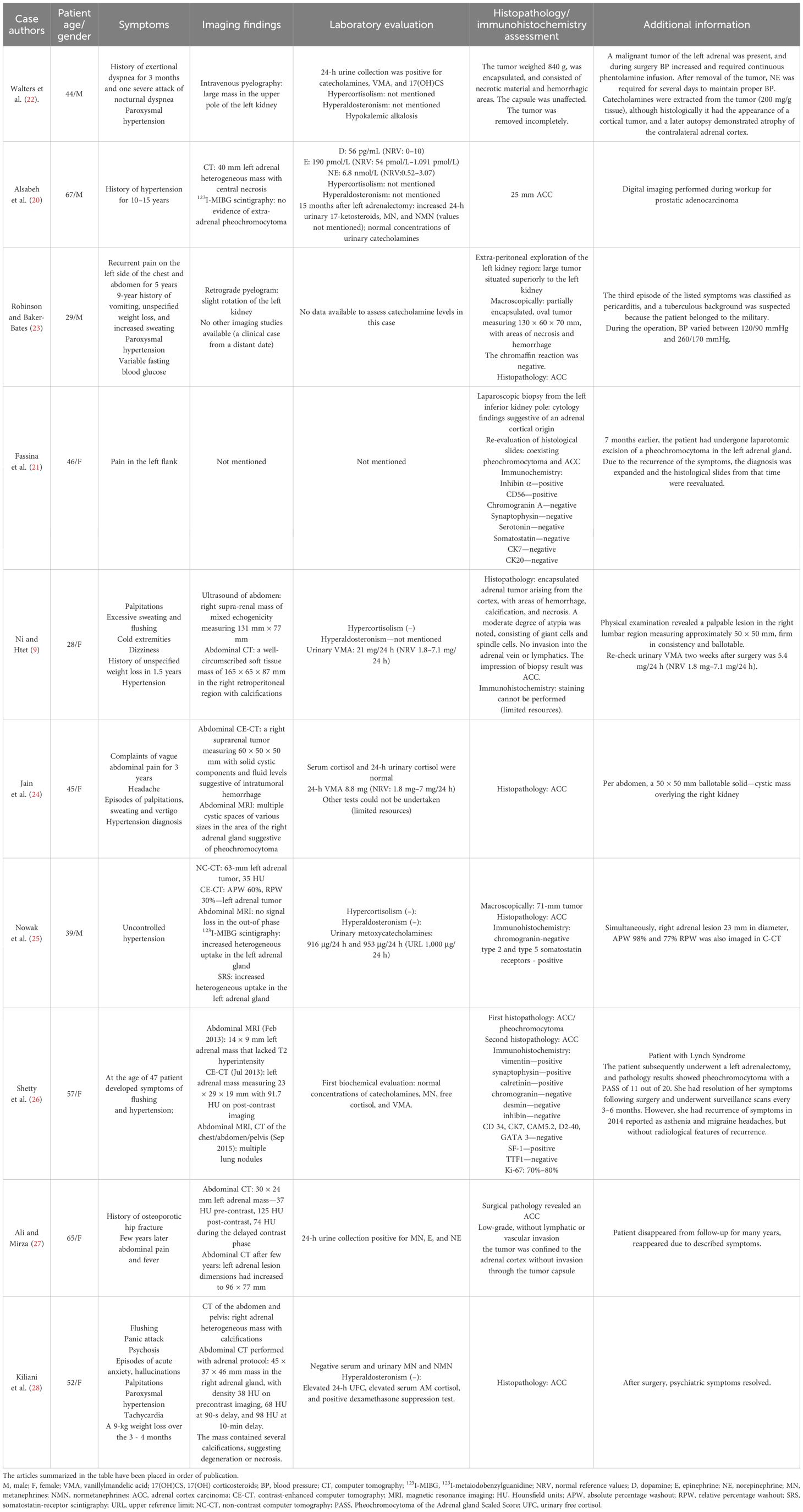
Table 1. Table presenting an overview of published clinical cases of adrenocortical carcinoma manifesting as pheochromocytoma (9, 20–28).
As we have summarized in our paper, ACC is among the rare neoplasms with challenging pathomorphological evaluation and multiple clinical manifestations, including those mimicking pheochromocytoma. Although our review may have some limitations, such as some clinical cases dating to distant years (the 1960s), when the possibilities of radiological and hormonal evaluation were limited, most provide solid clinical data supporting such rare ACC presentation. The necessity of including these distant in time cases in our review only confirms how infrequent the reported manifestation of ACC is and how extensive a literature search may be confronted by clinicians struggling with comparable concerns while diagnosing patients with adrenal tumors of equivocal clinical presentation. Based on the ambiguities in our case report and the available papers referenced from the literature, we recommend managing this group of patients with a comprehensive multidisciplinary team of clinicians using the available laboratory and imaging modalities. The case presented here emphasizes the importance of an individualized approach in caring for patients with adrenal tumors. Overall, the case we describe is one of the few in which we have obtained laboratory, imaging, histopathologic, and immunohistochemical results, which provides a further basis for advancing our knowledge of this rare presentation of ACC.
JS: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. WR: Investigation, Supervision, Writing – review & editing. PW: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The publication fee was covered by Medical University of Warsaw.
Thanks are given to the Professor Lukasz Koperski (Department of Pathology, Medical University of Warsaw) for consulting the pathomorphological result.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fassnacht M, Tsagarakis S, Terzolo M, Tabarin A, Sahdev A, Newell-Price J, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. (2023) 189:G1–42. doi: 10.1093/ejendo/lvad066
2. Handkiewicz-Junak D, Dedecjus M, Ambroziak U, Barczyński M, Bednarek-Papierska L, Chmielik E, et al. Polish diagnostic and therapeutic recommendations for adrenocortical carcinoma. Endokrynol Polska. (2024) 75:339–58. doi: 10.5603/ep.101677
3. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HR, et al. Adrenocortical carcinomas and Malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2020) 31:1476–90. doi: 10.1016/j.annonc.2020.08.2099
4. Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, et al. Adrenocortical carcinoma. Endocr Rev. (2014) 35:282–326. doi: 10.1210/er.2013-1029
5. Kamilaris CDC, Hannah-Shmouni F, Stratakis CA. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract Res Clin Endocrinol Metab. (2020) 34:101428. doi: 10.1016/j.beem.2020.101428
6. Ahuja K, Goudar R. A novel lynch syndrome kindred with hereditary adrenal cortical carcinoma. Cancer Genet. (2024) 288–289:137–40. doi: 10.1016/j.cancergen.2024.11.005
7. Walther MM, Keiser HR, Linehan WM. Pheochromocytoma: evaluation, diagnosis, and treatment. World J Urol. (1999) 17:35–9. doi: 10.1007/s003450050102
8. Eisenhofer G, Goldstein DS, Sullivan P, Csako G, Brouwers FM, Lai EW, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. (2005) 90:2068–75. doi: 10.1210/jc.2004-2025
9. Ni H, Htet A. Adrenal cortical carcinoma masquerading as pheochromocytoma: a case report. Ecancermedicalscience. (2012) 6:277. doi: 10.3332/ecancer.2012.277
10. Eisenhofer G, Pamporaki C, Lenders JWM. Biochemical assessment of pheochromocytoma and paraganglioma. Endocr Rev. (2023) 44:862–909. doi: 10.1210/endrev/bnad011
11. Ishikura K, Takamura T, Takeshita Y, Nakagawa A, Imaizumi N, Misu H, et al. Cushing’s syndrome and big IGF-II associated hypoglycaemia in a patient with adrenocortical carcinoma. BMJ Case Rep. (2010) 2010:bcr07.2009.2100. doi: 10.1136/bcr.07.2009.2100
12. Yamanaka K, Iitaka M, Inaba M, Morita T, Sasano H, Katayama S. A case of renin-producing adrenocortical cancer. Endocr J. (2000) 47:119–25. doi: 10.1507/endocrj.47.119
13. Oka T, Onoe K, Nishimura K, Tsujimura A, Sugao H, Takaha M, et al. Erythropoietin-producing adrenocortical carcinoma. Urol Int. (1996) 56:246–9. doi: 10.1159/000282852
14. Aiba M, Fujibayashi M. Histopathological diagnosis and prognostic factors in adrenocortical carcinoma. Endocr Pathol. (2005) 16:13–22. doi: 10.1385/EP:16:1:013
15. Papotti M, Libè R, Duregon E, Volante M, Bertherat J, Tissier F. The Weiss score and beyond–histopathology for adrenocortical carcinoma. Horm Cancer. (2011) 2:333–40. doi: 10.1007/s12672-011-0088-0
16. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol. (2022) 33:155–96. doi: 10.1007/s12022-022-09710-8
17. Gambella A, Volante M, Papotti M. Histopathologic features of adrenal cortical carcinoma. Adv Anat Pathol. (2023) 30:34–46. doi: 10.1097/PAP.0000000000000363
18. Papathomas TG, Duregon E, Korpershoek E, Restuccia DF, van Marion R, Cappellesso R, et al. Sarcomatoid adrenocortical carcinoma: a comprehensive pathological, immunohistochemical, and targeted next-generation sequencing analysis. Hum Pathol. (2016) 58:113–22. doi: 10.1016/j.humpath.2016.08.006
19. Zlatibor L, Paunovic I, Zivaljevic V, Dundjerovic D, Tatic S, Djukic V. Prognostic significance of immunohistochemical markers in adrenocortical carcinoma. Acta Chir Belg. (2020) 120:23–9. doi: 10.1080/00015458.2018.1543822
20. Alsabeh R, Mazoujian G, Goates J, Medeiros LJ, Weiss LM. Adrenal cortical tumors clinically mimicking pheochromocytoma. Am J Clin Pathol. (1995) 104:382–90. doi: 10.1093/ajcp/104.4.382
21. Fassina A, Cappellesso R, Schiavi F, Fassan M. Concurrent pheochromocytoma and cortical carcinoma of the adrenal gland. J Surg Oncol. (2011) 103:103–4. doi: 10.1002/jso.v103.1
22. Walters G, Wyatt GB, Kelleher J. Carcinoma of the adrenal cortex presenting as a pheochromocytoma: report of a case. J Clin Endocrinol Metab. (1962) 22:575–80. doi: 10.1210/jcem-22-6-575
23. Robinson PL, Baker-Bates ET. Adrenal cortical carcinoma simulating a phæochromocytoma. Br J Surgery. (2005) 41:399–403. doi: 10.1002/bjs.18004116818
24. Jain S, Agarwal L, Nadkarni S, Ameta A, Goyal A, Kumar R, et al. Adrenocortical carcinoma posing as a pheochromocytoma: a diagnostic dilemma. J Surg Case Rep. (2014) 2014:rju030. doi: 10.1093/jscr/rju030
25. Nowak KM, Łebek-Szatańska A, Samsel R, Roszkowska-Purska K, Ćwikła JB, Papierska L. Adrenocortical carcinoma mimicking pheochromocytoma on iodine 123-labeled metaiodobenzylguanidine scintigraphy. Pol Arch Intern Med. (2019) 129:822–3. doi: 10.20452/pamw.14958
26. Shetty I, Fuller S, Raygada M, Merino MJ, Thomas BJ, Widemann BC, et al. Adrenocortical carcinoma masquerading as pheochromocytoma: a histopathologic dilemma. Endocrinol Diabetes Metab Case Rep. (2020) 2020:19–0147, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Shetty%20I%22%5BAuthor%5D" Impana Shetty 1, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Fuller%20S%22%5BAuthor%5D" Sarah Fuller 1, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Raygada%20M%22%5BAuthor%5D" Margarita Raygada 1, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Merino%20MJ%22%5BAuthor%5D" Maria J Merino 2, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Thomas%20BJ%22%5BAuthor%5D" B J Thomas 1, HYPERLINK "https://pubmed.ncbi.nlm.nih.gov/?term=%22Widemann%20BC%22%5BAuthor%5D" Brigitte C Widemann . doi: 10.1530/EDM-19-0147
27. Ali M, Mirza L. An unusual case of Adrenocortical Adenocarcinoma with Biochemical Masquerade of Pheochromocytoma. Pak J Med Sci. (2021) 37:1241–3. doi: 10.12669/pjms.37.4.3916
Keywords: adrenocortical carcinoma, pheochromocytoma, adrenal incidentaloma, symptoms, mild autonomous cortisol secretion
Citation: Sobolewska J, Respondek W and Witek P (2025) A rare manifestation of adrenocortical carcinoma as a mimic of pheochromocytoma: a case report and literature review. Front. Endocrinol. 16:1533711. doi: 10.3389/fendo.2025.1533711
Received: 24 November 2024; Accepted: 20 January 2025;
Published: 11 February 2025.
Edited by:
Piotr Glinicki, Centre of Postgraduate Medical Education, PolandReviewed by:
Corin Badiu, Carol Davila University of Medicine and Pharmacy, RomaniaCopyright © 2025 Sobolewska, Respondek and Witek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Sobolewska, am9hbm5hLnNvYm9sZXdza2FAd3VtLmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.