
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 18 February 2025
Sec. Pediatric Endocrinology
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1514916
 Sarah E. Flanagan1*†‡
Sarah E. Flanagan1*†‡ Isabella-Anna Lazaridi1†
Isabella-Anna Lazaridi1† Jonna M. E. Männistö1,2
Jonna M. E. Männistö1,2 Jasmin J. Bennett1
Jasmin J. Bennett1 Oguzhan Kalyon1
Oguzhan Kalyon1 Matthew B. Johnson1Matthew N. Wakeling1Jayne A. L. Houghton3
Matthew B. Johnson1Matthew N. Wakeling1Jayne A. L. Houghton3 Thomas W. Laver1*‡
Thomas W. Laver1*‡Introduction: Congenital hyperinsulinism (HI) is characterized by inappropriate insulin secretion from the pancreatic beta-cells which causes severe hypoglycemia. Copy number variants (CNVs) encompassing multiple genes (contiguous gene CNVs) can cause syndromic forms of HI although they are not typically screened for during routine genetic testing for this condition. We aimed to assess the prevalence of disease-causing contiguous gene CNVs in a cohort of individuals referred for HI genetic testing.
Methods: Our cohort consisted of 3,763 individuals, of which 1,916 had received a genetic diagnosis for their HI and 1,847 were genetically unsolved following routine testing. We screened for 6 different contiguous gene CNVs using next-generation sequencing data from all individuals in the genetically unsolved cohort and searched for patients in our solved cohort who had already been found to have one of these CNVs.
Results: We identified a contiguous gene CNV affecting 5 of the 6 genomic loci in 53 probands; 28 from the solved cohort and 25 from the genetically unsolved cohort. Variants on the X chromosome were most common, being detected in 24/53 children. Overall, these variants represented 2.7% (53/1,941) of genetic diagnoses, which is similar to the prevalence of variants in other commonly screened HI genes.
Discussion: These results confirm that contiguous gene CNVs are an important cause of HI which should be included in standard gene panel testing processes as this will improve pick-up rates for genetic diagnoses in HI.
Congenital Hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in infants and children. In most cases, it results from the disruption of genes that control insulin secretion from the pancreatic beta-cell (1, 2).
Over 30 genetic forms of HI have been described which cause an isolated or syndromic form of the condition (3, 4). Many different types of genetic variation which disrupt the known HI genes/genetic loci are reported. These include single nucleotide variants (SNVs), indels, methylation defects, aneuploidies and large deletions or duplications (referred to as copy number variants; CNVs) (5).
Partial or whole gene deletions and/or duplications have been reported in at least 5 HI disease genes (6–10). Deletions and duplications which encompass multiple genes have also been described in individuals with HI, who typically have a syndromic condition (11–18). These variants are referred to as contiguous gene CNVs.
The molecular mechanism of HI is apparent when the contiguous gene CNV disrupts a known HI gene. For example, in Usher syndrome type 1C, the 122-kb homozygous deletion on chromosome 11p15.1 encompasses ABCC8 (13, 19), and in the chr20p deletion syndrome, HI results from heterozygous loss of the FOXA2 gene or its regulatory elements (14). For other contiguous gene CNVs, the critical region for the HI may not have been defined (12, 20) or the deletions may be coincidental or linked to phenotypic risk factors (21).
The size of the disease-causing contiguous gene CNV will vary between individuals, with their phenotype determined by the combination of genes disrupted by the copy number change. Variable penetrance and variable expressivity have also been reported (14, 22, 23). In some children, the presence of syndromic features may raise clinical suspicion of a contiguous gene CNV, prompting genome-wide copy number analyses by karyotyping, microarray or whole genome sequencing (WGS). In contrast, children who present with HI may undergo HI genetic testing as a first-line investigation.
Many genomic laboratories perform targeted analysis of the coding regions of the known HI disease genes during routine screening. While most will employ the necessary bioinformatic pipelines to detect large duplications or deletions affecting a gene on the HI panel, they often do not screen for contiguous gene CNVs where the critical region has not been determined, as these are harder to target and analyze using conventional methods. Consequently, the overall contribution of these variants to the etiology of HI has not been established although some studies have reported the prevalence of individual contiguous gene CNVs in HI cohorts (12, 14, 20).
In this study, we assessed the overall contribution of disease-causing contiguous gene CNVs to the etiology of HI. To do this we performed copy number analysis in a large cohort of patients referred to our laboratory for routine genetic testing for HI.
We studied an international cohort of 3,763 probands referred to the Exeter Genomics Laboratory for HI genetic testing over a 20-year period (2003-2023). Clinical information was provided at referral using standardized request forms. This included extra-pancreatic features that manifest as part of a syndrome, neurodevelopmental abnormalities, structural or functional organ abnormalities, growth delay, and facial dysmorphism. Seizures were not included as an additional feature in this study as they could be due to hypoglycemia. The study complied with the Declaration of Helsinki with informed consent obtained from all patients or their parents/guardians. Ethical approval for genetic testing was received from the Genetic Beta-Cell Research Bank (517/WA/0327).
The 3,763 patients were grouped into the solved cohort and unsolved cohort. The solved cohort consisted of 1,916 individuals where previous genetic testing by Sanger sequencing, targeted next generation sequencing (tNGS) and/or karyotype/microarray analysis had identified the cause of their HI. The unsolved cohort included 1,847 individuals where disease-causing SNVs and partial/whole gene deletions of a minimum of 11 HI genes (ABCC8, CACNA1D, GCK, GLUD1, HADH, HNF1A, HNF4A, INSR, KCNJ11, SLC16A1, and TRMT10A) had been excluded by tNGS (24).
We identified 6 different contiguous gene CNVs reported to be causative of HI within the literature which had sufficient evidence to support pathogenicity according to the ACMG/ClinGen CNV scoring matrix (25) (Table 1). These included deletions at chr5q35.1-q53.3 causing Sotos syndrome, deletions/duplications at chr11p15 causing Beckwith Wiedemann syndrome, chr11p14-p15 deletions causing Usher syndrome type 1c, anomalies on the X chromosome causing Turner syndrome and chr20p11.2 and chr9p24.3-p24.1 deletions. To establish the region of interest for each contiguous gene CNV we took the widest reported boundaries or the minimal critical region when defined (Table 1).
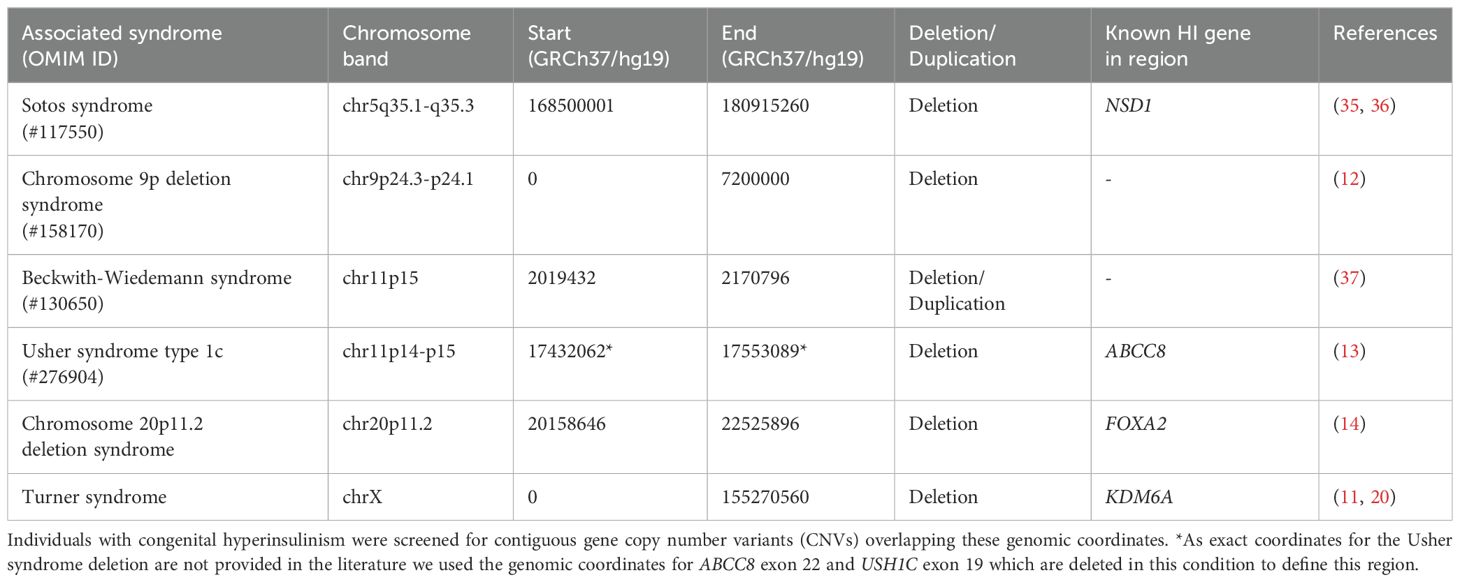
Table 1. Table detailing the six syndromes screened and their associated genomic regions.
We screened all 6 contiguous gene CNVs in the 1,847 individuals from the unsolved cohort using read-depth analysis of next-generation sequencing data. When available, WGS data was used in preference to tNGS which relied on the analysis of low-level off-target reads using our in-house software SavvyCNV (methods previously described (26)) (WGS n=178, tNGS n=1,669). To improve specificity, we ran the pipeline in non-mosaic mode. We called variants that extended across the region of interest of the reported variant, except for those on the X chromosome. For this analysis we screened the X chromosomes in all females for deletions >1Mb which encompassed the KDM6A gene or where a ring chromosome was predicted.
We undertook SNP array genotyping on samples from 8 individuals in the solved cohort with a homozygous deletion of ABCC8 exons 1-22, which had raised the possibility of an extended deletion and a diagnosis of Usher syndrome type 1C (13, 27, 28). The analysis was performed on an Illumina GSAMD-v3 array by Erasmus MC (Rotterdam, Netherlands). We used PennCNV to call deletions (29). Deletions were excluded if they had a confidence score <20 or were in a low coverage region (<10 probes covering the CNV).
When a contiguous gene CNV was identified which extended across the differentially methylated locus at chr11p15.5, we performed methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) to determine the parental origin of the affected chromosome (MRC-Holland kit ME030-C3).
Contiguous gene CNVs affecting 5 of the 6 genomic loci were identified in 53 probands: 28 were from the genetically solved cohort and 25 were from the genetically unsolved cohort (Figure 1).
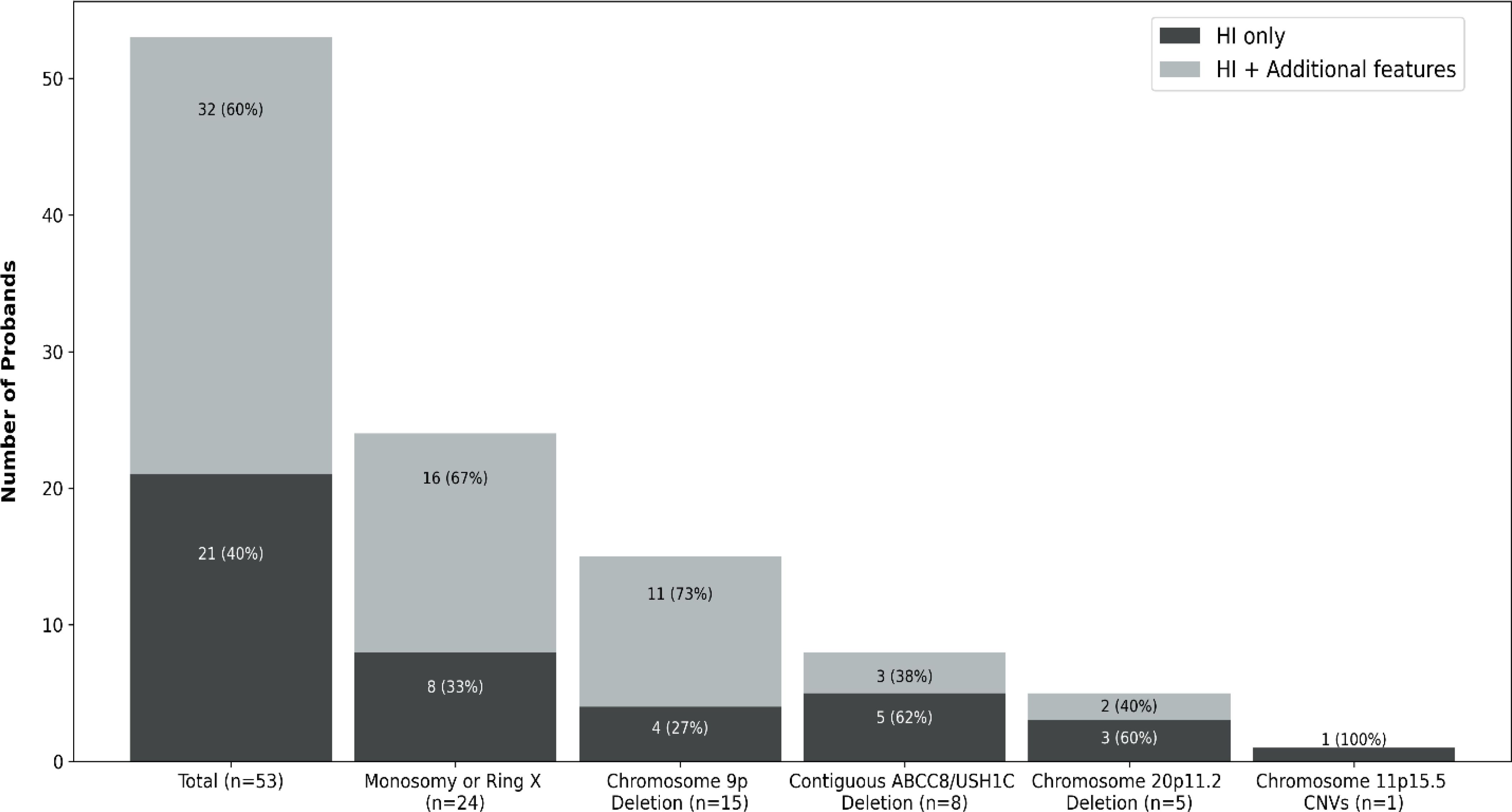
Figure 1. Bar chart representing the number of probands with HI resulting from contiguous gene copy number variants (CNVs) in our cohort. Each bar is separated according to reported additional clinical features at the time of referral for genetic testing for HI. Dark grey represents the number of children where HI was the only reported feature, light grey denotes the number of children where extra-pancreatic feature(s) were reported at referral.
We detected a contiguous gene CNV affecting the X chromosome in 24 females (24/53; 45%). Microarray analysis or karyotyping had already established a diagnosis of Turner syndrome in 14 of these children prior to referral for HI genetic testing. At least 11 individuals had a deletion that encompassed KDM6A (the exact coordinates of the variants were not available for 13 individuals). We identified a terminal chr9p deletion in 15 individuals which ranged in size from ~7.2Mb to 19.4Mb. Microarray analysis had detected the deletion in 6 of these individuals prior to referral for HI genetic testing. Five patients had a chr20p11.2 deletion ranging in size from ~3Mb to 8Mb. In one child, we identified a 4.8Mb duplication on the paternal allele at the chr11p.15.5 differentially methylated region, confirming a diagnosis of Beckwith-Wiedemann syndrome (15). SNP array analysis of all 8 individuals in the solved cohort with a deletion of ABCC8 exons 1-22, demonstrated the deletion extended over exons 1-19 of the adjacent USH1C gene, confirming a diagnosis of Usher syndrome type 1c. We did not detect any CNVs encompassing the NSD1 gene at chr5q35, which would have been consistent with Sotos syndrome (15). We previously reported clinical and genetic data for 9 of the 15 individuals with a chr9p deletion and the 5 individuals with a chr20p11.2 deletion (12, 14).
Identifying 25 contiguous gene CNVs in the genetically unsolved HI cohort increased the pick-up rate for disease-causing variants from 50.9% (95% CI: 49.3-52.5%, n=1,916/3,763) to 51.6% (95% CI: 50.0-53.2%, n=1,941/3,763). These variants had an overall prevalence of 2.7% (95% CI: 2.1-3.6%, n=53/1,941) in the solved cases, which is similar to the prevalence of variants in other routinely tested HI genes (Figure 2).
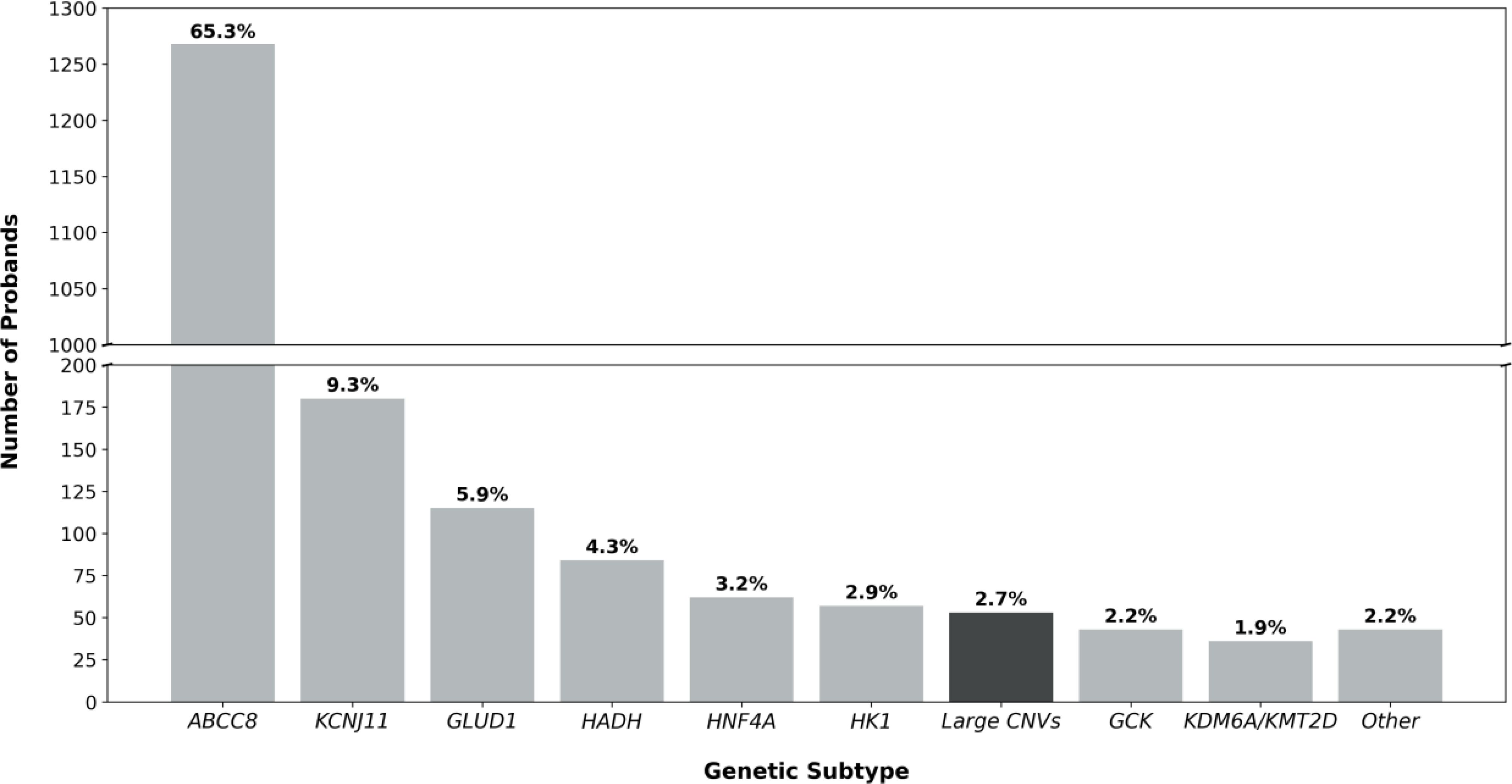
Figure 2. Bar chart detailing the genetic etiology of the 1941 probands in our genetically solved cohort. When the prevalence of a genetic etiology was less than 1%, genetic causes were grouped under ‘Other’ (EP300, NSD1, FOXA2, CACNA1D, PMM2, HNF1A, INSR). The bar highlighted in black represents the prevalence of contiguous gene copy number variants (CNVs) in the HI probands reported in this study (2.7%, 53/1941).
The median age at diagnosis of HI in the 53 individuals with a contiguous gene CNV was 1 day (range, 0 days – 6 years) and their median birthweight was +0.41 SDS (range, -2.3 to +3.1 SDS). We obtained treatment data for 43 individuals at referral for genetic testing. Among them, 35 children were treated with diazoxide, 3 with octreotide and 1 with a combination of diazoxide and octreotide. Four children with Turner syndrome were not receiving treatment at referral because their HI was in remission, and two children, both with Usher syndrome type 1C, had undergone near total pancreatectomy.
Clinicians reported additional features in 32 of the 53 individuals (60%) at the time of referral for genetic testing (Figure 1). In all but two cases, at least some of the additional features reported were consistent with the genetic diagnosis, though not necessarily pathognomonic for the condition (Figure 3).
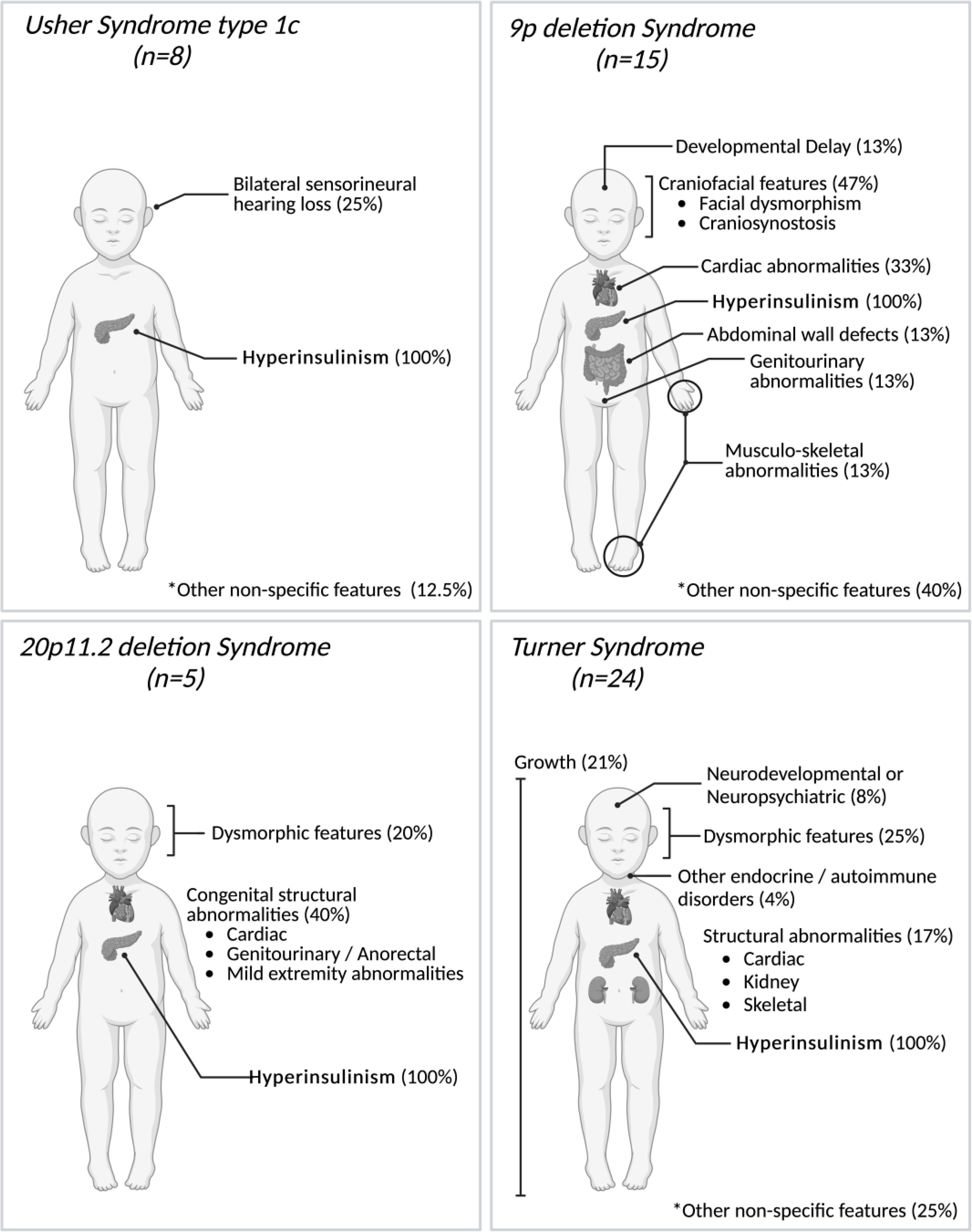
Figure 3. Phenotypic features reported in 52 individuals with contiguous gene copy number variants (CNVs) at the time of referral for genetic testing for HI. All reported features are summarized under generalized categories. Non-specific features refer to features that are rarely reported to be associated with these specific syndromes and/or that could be more common in the general population. Percentages are based on the number of individuals with these features with the possibility that one individual may have had several and/or non-specific features. The single case with a large duplication at the Beckwith-Wiedemann syndrome locus has not been depicted as no additional features were listed at the time of referral for genetic testing. Created in BioRender. Lazaridi, I. (2025) https://BioRender.com/e55o581.
The median age at referral of individuals within the solved cohort (i.e. those where the contiguous gene CNV that had already been detected prior to referral for HI testing) was 409 days (range, 11–4262 days). This was higher but not significantly different to those who underwent HI genetic testing as a first-line test (median 191 days, range 12–4065 days) (p > 0.05, Mann Whitney U).
We comprehensively screened for contiguous gene CNVs in a large cohort of patients referred for routine HI genetic testing and identified variants in 53 individuals across five distinct genetic loci. Collectively, these variants accounted for 2.7% of genetic diagnoses, a prevalence comparable to that of variants in HNF4A, HK1, and GCK (3.2%, 2.9% and 2.2% respectively; Figure 2); genes which are routinely included on gene panels for HI.
Twenty-one probands (40%) did not have extra-pancreatic features listed on the genetic testing request form, suggesting that HI was the presenting feature of their condition (30, 31). This finding highlights the need to screen for contiguous gene CNVs in all individuals with HI, regardless of their phenotype. Detecting these variants early is important because it informs prognosis, allowing for proper monitoring and the prompt diagnosis and medical management of additional clinical features if/when they develop.
The most common contiguous gene CNVs we identified were deletions on the X chromosome. These were identified in 24 females. In at least 11 patients, the deletion extended over the HI and Kabuki syndrome gene (KDM6A). Haploinsufficiency of this gene has been proposed as a mechanism for the increased prevalence of HI observed in girls with Turner syndrome (20). In 14 children, clinicians noted additional features prior to referral for HI genetic testing. These had prompted karyotyping or microarray analysis as a first line genetic test which confirmed Turner syndrome. Six of the 15 probands with a chr9p deletion had also undergone microarray analysis prior to referral for HI genetic testing which detected the deletion.
There are several reasons why patients who had already received a genetic diagnosis might have been sent for additional HI genetic testing. While the first papers describing the association between HI and Turner syndrome and HI and the 9p deletion syndrome were published in 1979 and 1996 respectively (32, 33), it has only been in the last few years that the role of these contiguous gene CNVs in the etiology of HI has been firmly established (12, 20). In keeping with this, 13 of the 14 individuals with a prior diagnosis of Turner syndrome were referred for HI genetic testing before the 2018 publication by Gibson et al., which described 12 girls with HI and X chromosome abnormalities (20). Similarly, all 6 patients with previously confirmed chr9p deletions were referred prior to the 2019 publication by Banerjee et al. (12),. In addition, it was possible that some children may have had a contiguous gene CNV and co-incidental HI (digenic disease). Because of this, clinicians may have considered comprehensive HI genetic testing necessary, particularly for those children with persistent disease that responded poorly to treatment as finding a pathogenic variant in a K-ATP channel gene for example would inform medical management (34).
We identified contiguous gene CNVs in 1.4% (95% CI: 1.1%-1.8%, n=53/3,763) of our whole HI cohort. This figure likely reflects a minimum prevalence due to several referral biases. For example, our cohort has an underrepresentation of children with Beckwith-Wiedemann syndrome as this condition is often diagnosed clinically prior to confirmatory methylation testing. We also did not search for mosaic CNVs, as these would have been called with less confidence potentially resulting in false positive calls. Therefore, it is possible that we missed some clinically relevant variants especially those causing Turner syndrome, where mosaicism has been observed (20). Finally, we limited our search to include CNVs with strong evidence for their causative role in HI, other HI disease-causing CNVs will therefore not have been detected.
In this study, we screened for contiguous gene CNVs using WGS data or off-target reads generated from routine tNGS analysis. Until recently, many laboratories have employed more cost-effective targeted gene panels or exome sequencing for screening the HI genes due to the higher costs of WGS (5). While both methods can be adapted to target these contiguous gene CNVs, their large size makes them difficult to screen for using these approaches. As the cost of WGS continues to decrease, laboratories will likely adopt it as the gold standard first-line genetic test for HI, making the detection of these contiguous gene CNVs easier. Until then, we recommend considering a CNV screen when a large deletion is detected in the ABCC8, KDM6A (females only), or FOXA2 genes, as these might indicate Usher, Turner and 20p11.2 deletion syndromes respectively.
While we have shown that our in-house software, SavvyCNV can call off-target CNVs from small tNGS panels with high precision and recall, we recognize that this methodology does not meet diagnostic standards for CNV calling (26). We used this approach in this study as it allowed for CNV analysis using existing data at no additional cost. As such, when we identified a novel contiguous gene CNV, we informed the clinicians and advised them to request orthogonal testing, which confirmed our findings in all cases.
In conclusion, we have shown that contiguous gene CNVs are an important cause of HI that should be considered by laboratories and clinicians managing children with this condition.
The datasets presented in this article are not readily available to preserve patient confidentiality. Sequencing data is available through collaboration to experienced teams working on approved studies examining the mechanisms, diagnosis and treatment of diabetes and other beta cell disorders. Requests for collaboration will be considered by a steering committee following an application to the Genetic Beta Cell Research Bank (https://www.diabetesgenes.org/current-research/genetic-beta-cell-research-bank/). Contact by email should be directed to Sarah Flanagan (s.flanagan@exeter.ac.uk).
The studies involving humans were approved by Ethical approval for genetic testing was received from the Genetic Beta-Cell Research Bank (517/WA/0327). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
SF: Resources, Conceptualization, Data curation, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing. I-AL: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. JM: Writing – original draft, Writing – review & editing, Formal analysis, Investigation, Methodology. JB: Formal analysis, Methodology, Writing – review & editing. OK: Methodology, Writing – review & editing. MJ: Methodology, Supervision, Writing – review & editing. MW: Formal analysis, Methodology, Writing – review & editing. JH: Methodology, Writing – review & editing. TL: Methodology, Supervision, Writing – original draft, Writing – review & editing, Funding acquisition.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded in whole, or in part, by the Wellcome Trust (223187/Z/21/Z). For the purpose of open access, the author has applied a CC BY public copyright license to any Author accepted Manuscript version arising from this submission.
SF has a Wellcome Trust Senior Research Fellowship [223187/Z/21/Z]. This project was supported by the Academy of Medical Sciences/the Wellcome Trust/the Government Department of Science Innovation and Technology/the British Heart Foundation/Diabetes UK Springboard Award [SBF009\1135]. JM is the recipient of a European Society for Pediatric Endocrinology (ESPE) Research Fellowship and the Foundation for Pediatric Research Postdoctoral Fellowship. MJ has a Diabetes UK/Breakthrough T1D (JDRF) funded RD Lawrence Fellowship (23/0006516). This study was supported by the National Institute for Health and Care Research Exeter Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. We are grateful to Congenital Hyperinsulinism International (a501(c)3 organization) who funded genetic testing for many of the individuals reported in this study through the Open Hyperinsulinism Genes Project. We thank Amber Luckett for providing bioinformatic support with the microarray analysis. We are grateful to the clinicians who referred families to Exeter for genetic testing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. De Leon DD, Arnoux JB, Banerjee I, Bergada I, Bhatti T, Conwell LS, et al. International guidelines for the diagnosis and management of hyperinsulinism. Horm Res Paediatr. (2024) 97:279–98. doi: 10.1159/000531766
2. Gϋemes M, Rahman SA, Kapoor RR, Flanagan S, Houghton JAL, Misra S, et al. Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management. Rev Endocrine Metab Disord. (2020) 21:577–97. doi: 10.1007/s11154-020-09548-7
3. Zenker M, Mohnike K, Palm K. Syndromic forms of congenital hyperinsulinism. Front Endocrinol (Lausanne). (2023) 14:1013874. doi: 10.3389/fendo.2023.1013874
4. Kostopoulou E, Dastamani A, Guemes M, Clement E, Caiulo S, Shanmugananda P, et al. Syndromic forms of hyperinsulinaemic hypoglycaemia-A 15-year follow-up study. Clin Endocrinol (Oxf). (2021) 94:399–412. doi: 10.1111/cen.14393
5. Hewat TI, Johnson MB, Flanagan SE. Congenital hyperinsulinism: current laboratory-based approaches to the genetic diagnosis of a heterogeneous disease. Front Endocrinol (Lausanne). (2022) 13:873254. doi: 10.3389/fendo.2022.873254
6. Flanagan S, Damhuis A, Banerjee I, Rokicki D, Jefferies C, Kapoor R, et al. Partial ABCC8 gene deletion mutations causing diazoxide-unresponsive hyperinsulinaemic hypoglycaemia. Pediatr Diabetes. (2012) 13:285–9. doi: 10.1111/j.1399-5448.2011.00821.x
7. Flanagan SE, Patch AM, Locke JM, Akcay T, Simsek E, Alaei M, et al. Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metab. (2011) 96:E498–502. doi: 10.1210/jc.2010-1906
8. Wakeling MN, Owens NDL, Hopkinson JR, Johnson MB, Houghton JAL, Dastamani A, et al. Non-coding variants disrupting a tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. Nat Genet. (2022) 54:1615–20. doi: 10.1038/s41588-022-01204-x
9. Tung JY, Boodhansingh K, Stanley CA, De Leon DD. Clinical heterogeneity of hyperinsulinism due to HNF1A and HNF4A mutations. Pediatr Diabetes. (2018) 19:910–6. doi: 10.1111/pedi.2018.19.issue-5
10. Mannisto JME, Hopkins JJ, Hewat TI, Nasser F, Burrage J, Dastamani A, et al. Congenital hyperinsulinism and novel KDM6A duplications -resolving pathogenicity with genome and epigenetic analyses. J Clin Endocrinol Metab. (2024) 30:dgae524. doi: 10.1210/clinem/dgae524
11. Alkhayyat H, Christesen HB, Steer J, Stewart H, Brusgaard K, Hussain K. Mosaic Turner syndrome and hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab. (2006) 19:1451–7. doi: 10.1515/JPEM.2006.19.12.1451
12. Banerjee I, Senniappan S, Laver TW, Caswell R, Zenker M, Mohnike K, et al. Refinement of the critical genomic region for congenital hyperinsulinism in the Chromosome 9p deletion syndrome. Wellcome Open Res. (2019) 4:149. doi: 10.12688/wellcomeopenres
13. Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet. (2000) 26:56–60. doi: 10.1038/79178
14. Laver TW, Wakeling MN, Caswell RC, Bunce B, Yau D, Mannisto JME, et al. Chromosome 20p11.2 deletions cause congenital hyperinsulinism via the loss of FOXA2 or its regulatory elements. Eur J Hum Genet. (2024) 32:813–8. doi: 10.1038/s41431-024-01593-z
15. Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. (2018) 14:229–49. doi: 10.1038/nrendo.2017.166
16. Kostopoulou E, Dastamani A, Caiulo S, Antell H, Flanagan SE, Shah P. Hyperinsulinaemic hypoglycaemia: A new presentation of 16p11.2 deletion syndrome. Clin Endocrinol (Oxf). (2019) 90:766–9. doi: 10.1111/cen.2019.90.issue-5
17. Tamame T, Hori N, Homma H, Yoshida R, Inokuchi M, Kosaki K, et al. Hyperinsulinemic hypoglycemia in a newborn infant with trisomy 13. Am J Med Genet A. (2004) 129A:321–2. doi: 10.1002/ajmg.a.v129a:3
18. Giri D, Patil P, Hart R, Didi M, Senniappan S. Congenital hyperinsulinism and Poland syndrome in association with 10p13-14 duplication. Endocrinol Diabetes Metab Case Rep. (2017) 2017:16-0125. doi: 10.1530/EDM-16-0125
19. Al Mutair AN, Brusgaard K, Bin-Abbas B, Hussain K, Felimban N, Al Shaikh A, et al. Heterogeneity in phenotype of usher-congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care. (2013) 36:557–61. doi: 10.2337/dc12-1174
20. Gibson CE, Boodhansingh KE, Li C, Conlin L, Chen P, Becker SA, et al. Congenital hyperinsulinism in infants with turner syndrome: possible association with monosomy X and KDM6A haploinsufficiency. Horm Res Paediatr. (2018) 89:413–22. doi: 10.1159/000488347
21. Hewat TI, Laver TW, Houghton JAL, Mannisto JME, Alvi S, Brearey SP, et al. Increased referrals for congenital hyperinsulinism genetic testing in children with trisomy 21 reflects the high burden of non-genetic risk factors in this group. Pediatr Diabetes. (2022) 23(4):457–461. doi: 10.1111/pedi.13333
22. Kaygusuz SB, Arslan Ates E, Vignola ML, Volkan B, Geckinli BB, Turan S, et al. Dysgenesis and dysfunction of the pancreas and pituitary due to FOXA2 gene defects. J Clin Endocrinol Metab. (2021) 106:e4142–54. doi: 10.1210/clinem/dgab352
23. Tuke MA, Ruth KS, Wood AR, Beaumont RN, Tyrrell J, Jones SE, et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet Med. (2019) 21:877–86. doi: 10.1038/s41436-018-0271-6
24. Ellard S, Lango Allen H, De Franco E, Flanagan SE, Hysenaj G, Colclough K, et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia. (2013) 56:1958–63. doi: 10.1007/s00125-013-2962-5
25. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
26. Laver TW, De Franco E, Johnson MB, Patel KA, Ellard S, Weedon MN, et al. SavvyCNV: Genome-wide CNV calling from off-target reads. PloS Comput Biol. (2022) 18:e1009940. doi: 10.1371/journal.pcbi.1009940
27. Adi A, Abbas BB, Hamed MA, Tassan NA, Bakheet D. Screening for mutations in ABCC8 and KCNJ11 genes in saudi persistent hyperinsulinemic hypoglycemia of infancy (PHHI) patients. Genes (Basel). (2015) 6:206–15. doi: 10.3390/genes6020206
28. Al-Agha AE, Ahmad IA. Characterization of the ABCC8 gene mutation and phenotype in patients with congenital hyperinsulinism in western Saudi Arabia. Saudi Med J. (2013) 34(10):1002–6.
29. Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. (2007) 17:1665–74. doi: 10.1101/gr.6861907
30. Laver TW, Wakeling MN, Hua JHY, Houghton JAL, Hussain K, Ellard S, et al. Comprehensive screening shows that mutations in the known syndromic genes are rare in infants presenting with hyperinsulinaemic hypoglycaemia. Clin Endocrinol (Oxf). (2018) 89:621–7. doi: 10.1111/cen.2018.89.issue-5
31. Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. (2011) 6:63. doi: 10.1186/1750-1172-6-63
32. McClure RJ, Telford N, Newell SJ. A mild phenotype associated with der(9)t(3;9) (p25;p23). J Med Genet. (1996) 33:625–7. doi: 10.1136/jmg.33.7.625
33. Glassman MS, Schultz RM, MacGillivray MH. Gonadal dysgenesis and leucine-sensitive hypoglycemia. J Pediatr. (1979) 94:930–1. doi: 10.1016/S0022-3476(79)80221-8
34. Banerjee I, Skae M, Flanagan SE, Rigby L, Patel L, Didi M, et al. The contribution of rapid KATP channel gene mutation analysis to the clinical management of children with congenital hyperinsulinism. Eur J Endocrinol. (2011) 164:733–40. doi: 10.1530/EJE-10-1136
35. Nakamura Y, Takagi M, Yoshihashi H, Miura M, Narumi S, Hasegawa T, et al. A case with neonatal hyperinsulinemic hypoglycemia: It is a characteristic complication of Sotos syndrome. Am J Med Genet A. (2015) 167A:1171–4. doi: 10.1002/ajmg.a.v167.5
36. Matsuo T, Ihara K, Ochiai M, Kinjo T, Yoshikawa Y, Kojima-Ishii K, et al. Hyperinsulinemic hypoglycemia of infancy in Sotos syndrome. Am J Med Genet A. (2013) 161A:34–7. doi: 10.1002/ajmg.a.v161.1
Keywords: hyperinsulinism, hypoglycemia, copy number variants, deletion, duplication, genetics
Citation: Flanagan SE, Lazaridi I-A, Männistö JME, Bennett JJ, Kalyon O, Johnson MB, Wakeling MN, Houghton JAL and Laver TW (2025) Large copy number variants are an important cause of congenital hyperinsulinism that should be screened for during routine testing. Front. Endocrinol. 16:1514916. doi: 10.3389/fendo.2025.1514916
Received: 21 October 2024; Accepted: 28 January 2025;
Published: 18 February 2025.
Edited by:
Klaus Brusgaard, Odense University Hospital, DenmarkReviewed by:
Maria Melikyan, Yerevan State Medical University, ArmeniaCopyright © 2025 Flanagan, Lazaridi, Männistö, Bennett, Kalyon, Johnson, Wakeling, Houghton and Laver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah E. Flanagan, Uy5GbGFuYWdhbkBleGV0ZXIuYWMudWs=; Thomas W. Laver, dHdsMjA3QGV4ZXRlci5hYy51aw==
†These authors have contributed equally to this work
‡ORCID: Sarah E. Flanagan, orcid.org/0000-0002-8670-6340
Thomas W. Laver, orcid.org/0000-0001-6399-0089
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.