 Diba Chowdhury
Diba Chowdhury Chloe E. Jang
Chloe E. Jang Patrick Lajoie
Patrick Lajoie Stephen J. Renaud
Stephen J. Renaud- 1Department of Anatomy and Cell Biology, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 2Department of Biochemistry, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 3Children’s Health Research Institute, Lawson Health Research Institute, London, ON, Canada
The placenta is a temporary organ that forms during pregnancy and is essential for fetal development and maternal health. As an endocrine organ, proper placental function requires continual production, folding, and transport of proteins and lipids. Central to these processes is the endoplasmic reticulum (ER), a dynamic organelle responsible for maintaining cellular protein and lipid synthesis and processing. ER stress occurs when there is an accumulation of unfolded or misfolded proteins, which triggers the activation of cellular pathways collectively called the unfolded protein response. Unfolded protein response pathways act to alleviate the misfolded protein burden and restore ER homeostasis, or if unresolved, initiate cell death. While prolonged ER stress has been linked to deficient placental function and adverse pregnancy outcomes, basal activation of unfolded protein response pathways is required for placental development and function. This review explores the importance of ER homeostasis in placental development and function, examining how disruptions in ER stress responses may contribute to adverse pregnancy outcomes.
1 Introduction
The endoplasmic reticulum (ER) is a membrane-bound organelle that plays a central role in the synthesis, folding, and transport of proteins, as well as lipid production and calcium storage. It is a dynamic structure, adapting and remodeling itself in response to cellular demands. This adaptability is critical for maintaining cellular proteostasis, thereby balancing protein synthesis, folding, and degradation. ER stress occurs when the capacity of the ER to properly fold proteins is overwhelmed, leading to an accumulation of misfolded or unfolded proteins in the ER. ER stress triggers the activation of cellular signaling pathways designed to take corrective actions and restore proteostasis, or to initiate cell death pathways if the burden is prolonged or severe. While ER stress and downstream signaling can occur in any cell, those with a high demand for protein synthesis such as hormone-producing cells are particularly susceptible. The placenta is one such organ tasked with producing numerous hormones and other proteins critical for fetal development and pregnancy success, where disruptions in proteostasis can have severe consequences for maternal and fetal health. In this review, we will present the importance of ER homeostasis for placental development and function, and discuss evidence linking ER stress with deficient placentation and adverse pregnancy outcomes.
2 The placenta
The placenta is a transient yet highly sophisticated organ that intimately connects the gestational parent and fetus. It forms the primary interface separating maternal and fetal tissue and facilitates metabolic exchange, endocrine functions, and fetal protection. The placenta regulates the exchange of nutrients, oxygen, carbon dioxide, and other substances between maternal and fetal circulations, while restricting the transfer of pathogens, xenobiotics, maternal immune cells and many other potentially harmful substances from accessing fetal blood (1, 2). The placenta is also an adaptive organ that can integrate information on maternal nutrient availability and fetal demands, and undergo dynamic morphological and molecular changes to support fetal development (3). As an endocrine organ, the placenta produces a plethora of hormones and other factors that regulate maternal adaptations to pregnancy along with fetal growth and development (4, 5).
3 An overview of placental structure
The human placenta is arranged into highly branched tree-like villi containing an inner core of macrophages, fibroblasts, pericytes, connective tissue, and capillaries that connect to the fetal circulation through the umbilical vessels. The villous core is separated from maternal blood by a trophoblast bilayer: an inner portion of cytotrophoblasts (CTBs) resting on a basement membrane, and an outer syncytiotrophoblast (STB) layer (Figure 1). CTBs are self-renewing progenitor cells that initially form a continuous layer during early pregnancy, and replenish the STB layer by fusing into it. In late pregnancy, the proportion of CTBs dwindles and they form a disjointed layer beneath the STB. The STB layer, on the other hand, is a multinucleated entity with a vast interconnected cytoplasm that lines the placental villi and bathes in maternal blood. It forms the primary boundary between maternal blood and fetal tissue, and is responsible for numerous placental functions including maternal-fetal gas and nutrient exchange (6, 7). Given its proximity to maternal blood, STB is responsible for the production and metabolism of numerous hormones, such as human chorionic gonadotropin (hCG). These factors are deposited into maternal circulation to alter maternal physiology and metabolism for fetal benefit. Furthermore, since the STB does not contain intercellular junctions, it forms a semi-exclusive barrier that restricts the passage of many substances from accessing fetal circulation (8).
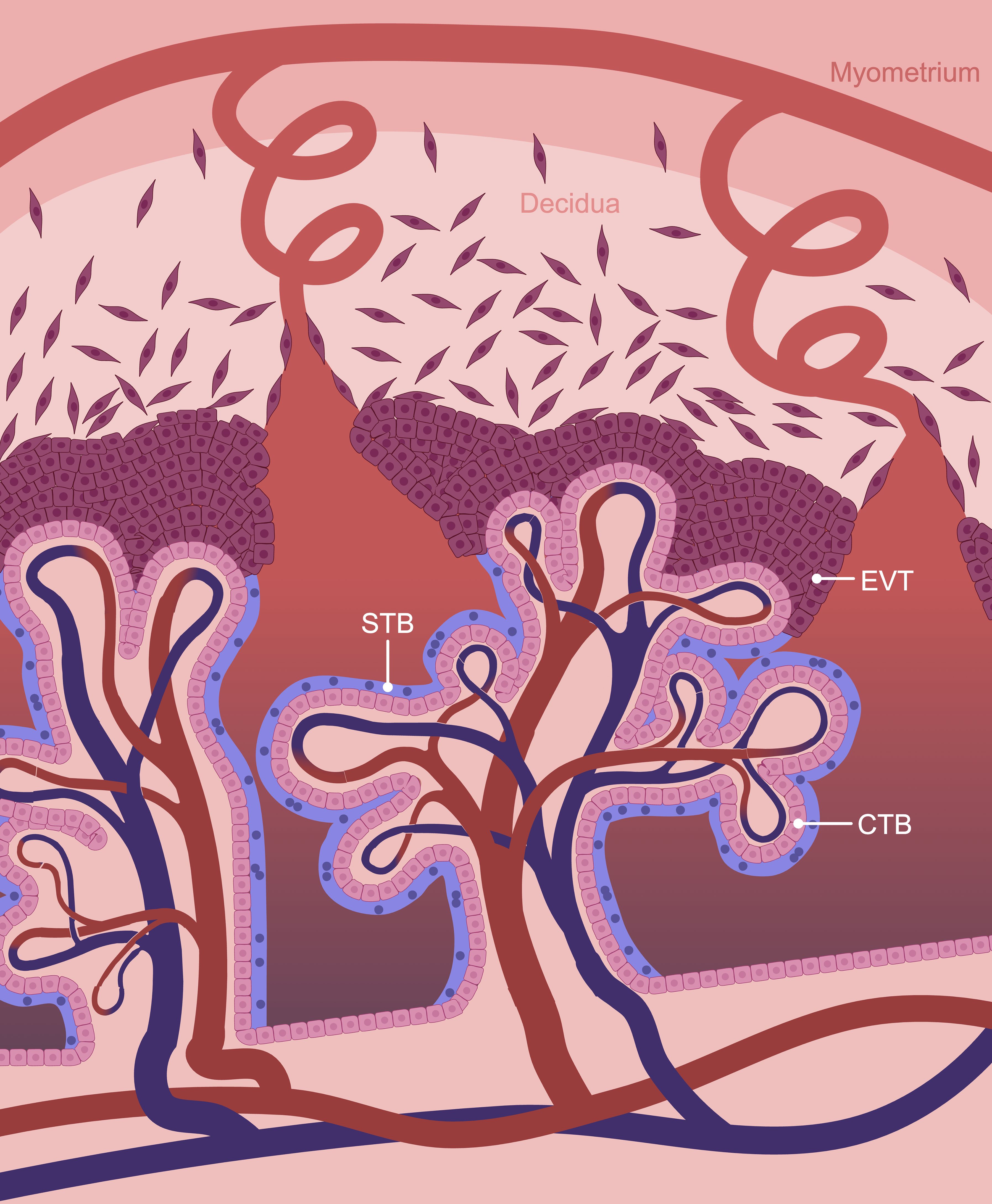
Figure 1. Schematic representation of the maternal-fetal interface. The human placenta is composed of chorionic villi which are lined by a trophoblast bilayer containing an inner cytotrophoblast (CTB) and outer syncytiotrophoblast (STB) layer. Extravillous trophoblasts (EVTs) form the cell columns at the tips of anchoring villi adjacent to the decidua and are the source of invasive trophoblasts. Created in BioRender. Chowdhury, D. (2024) https://BioRender.com/f22l744.
At sites where large anchoring villi contact the decidua basalis (the specialized endometrial tissue adjacent to where the placenta forms), CTBs differentiate into a distinct cell-type: extravillous trophoblasts (EVTs). Proximally, EVTs proliferate into stratified cellular columns. At the distal tips of these columns, cells stop replicating and gain invasive properties. Invasive EVTs infiltrate into the decidua basalis and inner third of the myometrium. EVTs are versatile cells that affix the placenta to the decidua basalis, interact with decidual stromal cells and immune cells to support immunological tolerance, and remodel uterine blood vessels and glands to ensure that a consistent supply of nutrients and oxygen are delivered to the placenta to support fetal sustenance (9).
The decidua basalis is a dynamic tissue derived from the endometrium adjacent to where the placenta forms. Transformation of the endometrium into the decidua begins during the secretory phase of the menstrual cycle and accelerates upon fertilization. Decidualization involves the differentiation of fibroblast-like endometrial stromal cells (ESCs) into polygonal decidual stromal cells (DSCs) along with extensive development of uterine glands and blood vessels, processes that are tightly regulated by estrogen and progesterone (10). Transformation of ESCs into DSCs is associated with a substantial increase in the metabolic demand and secretory activity of the cells that serve essential roles in early embryo nutrition and maternal-fetal communication (11).
Placental structure exhibits marked diversity between species. In humans and closely-related primates, the placenta is termed “hemochorial” since maternal blood directly contacts trophoblasts. Other species with hemochorial placentation include common laboratory rodents like mice, rats, and guinea pigs. Although these species exhibit notable differences in placental anatomy and physiology compared to humans, there are also many similarities. For example, placentas of mice, rats, and guinea pigs have a labyrinth zone containing syncytialized trophoblasts that specialize in nutrient and gas exchange (akin to the placental villi in humans), and a junctional zone adjacent to the decidua basalis that anchors the placenta to the decidua and is the site where invasive trophoblasts emanate (analogous to human EVTs). Consequently, rodents are often used as laboratory models to gain deeper mechanistic insight into placental development and maternal-placental-fetal interactions (12). Although this review will focus mostly on placentation in humans, much knowledge has been derived from studies using rodent models and these studies will be mentioned when appropriate.
4 Placenta-associated pregnancy complications
Deficient placental development and function is a major culprit underlying severe pregnancy complications that compromise maternal and fetal well-being and survival. For example, inadequate placental development resulting in placental insufficiency (inadequate maternal blood supply to the placenta) can lead to preeclampsia—a common and dangerous pregnancy disorder characterized by sudden-onset maternal hypertension, endothelial dysfunction, and organ damage. In many cases, in preeclampsia the fetus does not obtain adequate oxygen and nutrients, resulting in fetal growth restriction (FGR) and the potential for long-term health deficiencies. Placentas from preeclampsia and FGR often exhibit evidence of hypoxia, oxidative stress and inflammation—conditions that can adversely affect ER homeostasis and result in ER stress (13–15). In this review, we will discuss ER homeostatic mechanisms required for placental development and function, and describe how these mechanisms are perturbed following cellular exposure to stress during placenta-associated pregnancy complications.
5 ER stress
Protein turnover in the placenta increases throughout pregnancy, requiring efficient quality control mechanisms to ensure that the protein folding capacity of the ER meets protein synthesis demands (16, 17). Key components of this quality control machinery include ER-resident chaperones such as binding immunoglobulin protein (BiP; also called glucose regulated protein 78, GRP78), calreticulin, calnexin, and protein disulfide isomerases (PDIs). These chaperones assist proteins in achieving their native conformation (18). However, if chaperones cannot keep pace with protein folding demands, an accumulation of unfolded or misfolded proteins in the ER lumen can result, leading to ER stress and the activation of the unfolded protein response (UPR) (19). The role of ER-associated chaperones for placentation and decidualization are summarized in Supplementary Table 1.
ER stress can occur due to an accumulation of abnormal lipids or misfolded proteins in the ER (20, 21). The accumulation and aggregation of misfolded proteins can occur due to increased rates of protein synthesis, deficiency in autophagy, nutrient deprivation, and dysregulated calcium levels (22, 23). Many cell types experience a high protein load during various stages of differentiation and maturation, resulting in ER stress. Induction of ER stress can have morphological and biochemical consequences to cells. Morphologically, ER stress can result in dilation of ER cisternae to increase the lumen size and accommodate the sudden increase in protein accumulation (24, 25). Many cells that experience ER stress undergo a degree of epithelial-to-mesenchymal transition, including increased N-cadherin levels and decreased E-cadherin levels (26). Biochemically, ER stress can activate several signaling pathways including the UPR, in an effort to resolve the stress and restore proteostasis (27).
Recent studies have characterized early-onset preeclampsia as a proteinopathy due to presence of misfolded proteins in maternal urine and serum (28–30). Misfolded oligomeric proteins and amyloids, arising from defective chaperone function or impaired autophagy, may either deposit in the placenta from maternal circulation or be produced directly by placental cells (31). Several amyloidogenic proteins, including amyloid-β, transthyretin, and α-1-antitrypsin, have been identified in the preeclampsia-associated misfoldome and proposed as potential diagnostic markers for preeclampsia (31–33). Deposition of protein aggregates in the placenta may induce cellular toxicity, impair nutrient and gas exchange, and lead to placental insufficiency. Exposure of placental explants to serum from preeclamptic pregnancies induces ER stress and the formation of syncytial knots, suggesting that maternal serum in preeclampsia may contain ER stress-inducing factors that promote protein aggregation in placental cells (34, 35). While the placenta can efflux misfolded proteins into maternal blood, the accumulation of protein aggregates in maternal blood may pose risks for maternal organs (31). Therefore, targeting of misfolded proteins and resolution of ER stress could be promising therapeutic strategies to restore cellular homeostasis and improve pregnancy outcomes (36–38).
6 The UPR in placentation
The UPR is a conserved cellular response to ER stress. In principle, the UPR seeks to correct the accumulation of unfolded and misfolded proteins in the ER, thereby restoring ER homeostasis. Basal levels of UPR activation promote cellular homeostasis, but in cases of prolonged or severe ER stress, unmitigated UPR activation can lead to cell death (39, 40). The UPR pathway consists of three ER-resident sensor proteins: inositol-requiring enzyme 1 (lRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6), all of which trigger a transcriptional response to adapt the ER folding environment by upregulating genes involved in protein folding and degradation (Figure 2) (41). The following sections will summarize the role of IRE1, PERK, and ATF6 signaling in the context of placentation and decidualization.
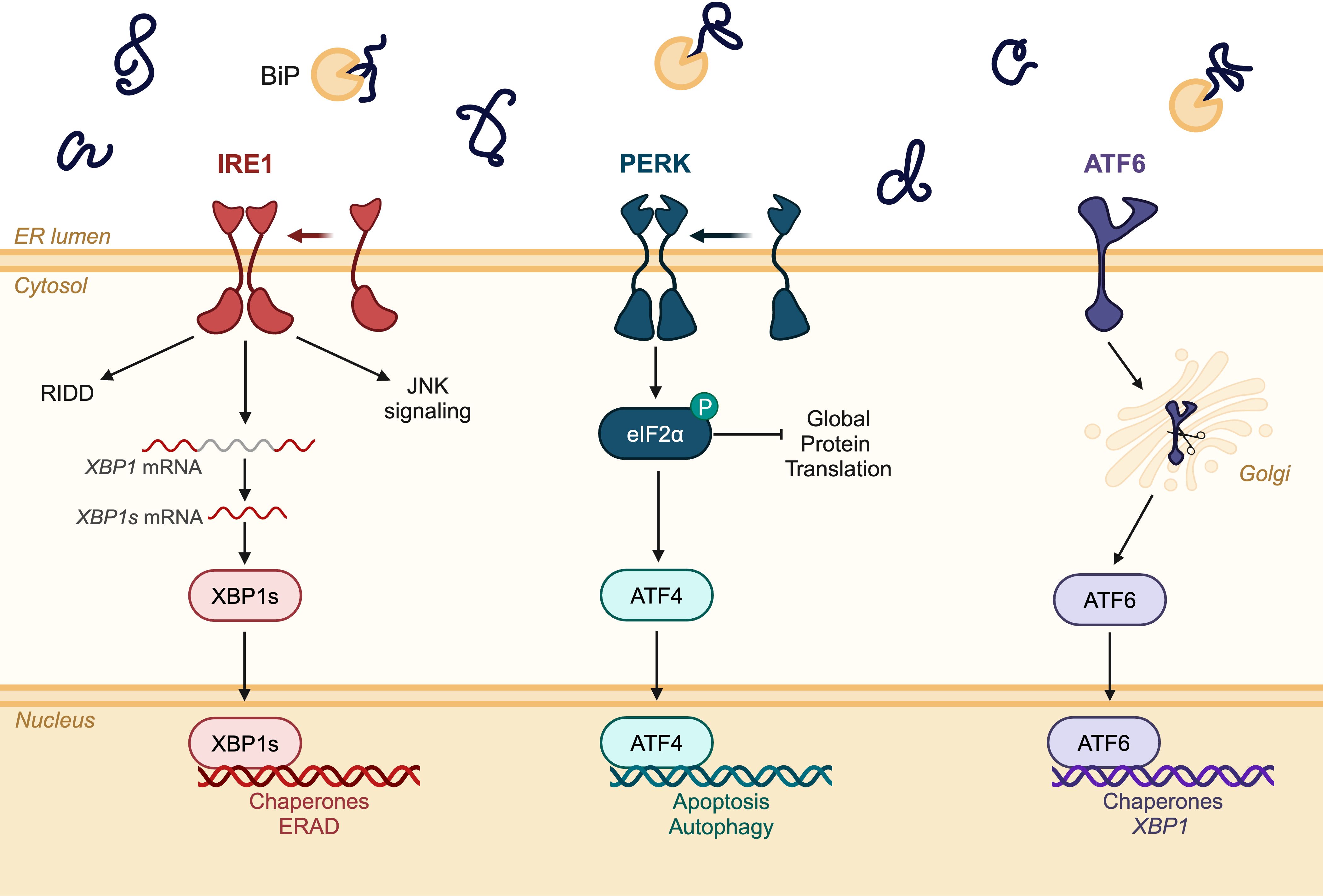
Figure 2. Schematic diagram of the three signaling branches of the unfolded protein response (UPR). Inositol-requiring enzyme 1 (lRE1); protein kinase R-like ER kinase (PERK); activating transcription factor 6 (ATF6); regulated IRE1-dependent decay (RIDD); jun N-terminal kinase (JNK). Created in BioRender. Chowdhury, D. (2024) https://BioRender.com/r03w626.
7 IRE1
IRE1 is a transmembrane protein that is evolutionarily conserved from yeast to humans (42, 43). It contains a serine/threonine kinase domain and a cytosol-facing domain with endoribonuclease activity. There are two isoforms of IRE1 in mammals – IRE1α and IRE1β – with IRE1α being the ubiquitously expressed isoform (44). Upon detection of misfolded or unfolded proteins, IRE1 homodimerizes and activates its kinase function through trans-autophosphorylation of Ser724, Ser726, and Ser729 in the kinase activation loop (45). Once activated, IRE1 splices out a 26-nucleotide intron from the X-box binding protein 1 (XBP1) mRNA sequence, encoding for the spliced XBP1 (XBP1s) transcription factor (46). Thus, IRE1 activation is typically measured through the phosphorylation of IRE1, splicing of XBP1 mRNA, or the accumulation and nuclear localization of the XBP1s transcription factor. XBP1s transcriptionally regulates genes encoding various chaperones, ER-associated degradation (ERAD) components, and proteins involved in lipid biosynthesis to relieve ER stress (47). The endoribonuclease activity of IRE1 can also degrade other mRNA sequences through regulated IRE1-dependent decay to reduce protein synthesis in the ER lumen (48, 49). While the activation of IRE1 is associated with cell survival mechanisms to relieve ER stress, persistent IRE1 activity can induce apoptosis by activating tumor necrosis factor receptor-associated factor 2, apoptosis signal-regulating kinase 1, and mitogen-activated protein kinases (26, 50).
7.1 IRE1 is required for placental development in mice
IRE1α (herein referred to as IRE1) is essential for placental development in mice. Embryos and placentas with a deletion in Ern1, the gene that encodes IRE1, are smaller than wild-type and heterozygous siblings, and die around embryonic day 12.5 (51–53). Ern1-/- placentas have reduced blood spaces in the labyrinth zone compared to wild-type mice, accompanied by decreased expression of vascular endothelial growth factor, suggesting that IRE1 contributes to placental vascular development (52–54). There does not appear to be an effect on decidualization in these mice (52). Interestingly, activation of IRE1 signaling, based on detecting IRE1 phosphorylation and Xbp1 splicing, occurs in the placenta throughout early and midgestation but not in the embryo (52), suggesting that IRE1 signaling is particularly important for placental development. In line with this observation, midgestation lethality is avoided in mice lacking Ern1 in embryonic tissue but not trophoblasts (using a Mox2+/Cre transgenic mouse), demonstrating that the cause of embryonic demise in Ern1-/- mice is due to defective placental development (52). Therefore, IRE1 appears to have a critical role for placental development in mice.
In line with an essential role of IRE1 signaling for placentation, high levels of XBP1s are detectable in the mouse placenta, but not in other embryonic organs (52). XBP1s is also spatiotemporally expressed in the mouse uterus where it is mainly detected in epithelial cells during early pregnancy and then subsequently in DSCs (55, 56). To investigate whether canonical IRE1-XBP1s signaling is involved in placental development, Xbp1-/- mice have been generated. Interestingly, embryos and placentas with a deletion in Xbp1 exhibit embryonic lethality between embryonic day 12.5 and 14.5; however, unlike Ern1-/- mice, the cause of lethality is attributed to liver hypoplasia. There does not seem to be a notable difference in placental morphology or vascular endothelial growth factor levels between Xbp1-/- mice and wild-type mice (52, 57). Therefore, the essential role of IRE1 in mouse placental development may be through a noncanonical signaling pathway independent of XBP1s.
7.2 IRE1 in human STB formation
There is strong evidence that IRE1 signaling is active and required during human STB formation. Activation of IRE1 signaling may be downstream of cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling, a well-established inducer of trophoblast syncytialization (58, 59). PKA phosphorylates IRE1 on Ser724 (58, 60), and also activates cAMP-response element binding protein, which is a transcriptional activator of ERN1 (61). STB formation in vitro using primary CTBs from term placenta is associated with IRE1 activation. Similar results are observed using BeWo cells, a CTB-like choriocarcinoma line that fuses to form STB-like cells after exposure to forskolin (62). Interestingly, the addition of small molecule inhibitors targeting IRE1, or RNA-interference targeting all three UPR branches, reduces CTB fusion, hCG secretion, and autophagy (62). Therefore, similar to its important role in mouse placentation, IRE1 appears to be required for human STB formation.
Why is IRE1 signaling needed for STB formation? While this answer remains unclear, it may be related to the increased synthesis and secretion of numerous proteins involved in the differentiation of CTBs and their fusion into STBs. Increased demand for protein synthesis during STB formation can exceed the ER’s folding capacity, resulting in the activation of the UPR to adapt to the new protein burden. Since IRE1-XBP1s signaling promotes the production of chaperones and ERAD components, its activation during STB formation may promote protein folding and clearance to ensure proper proteostasis in the ER, thereby enabling STB function. IRE1 activation may also promote the expression of genes involved in phospholipid biogenesis to remodel the ER membrane and expand the ER lumen during STB formation (63, 64).
Apart from responding to ER stress signals, XBP1s transcriptionally regulates the expression of differentiation-associated genes in other cells (65, 66). It is therefore possible that XBP1s may exert transcriptional control of genes needed for STB formation and function, such as ER-resident proteins needed for protein processing of STB-related hormones. As one example, the prototypical STB hormone, hCG, is a glycoprotein containing 11 disulfide bonds. These disulfide bonds are essential for hCG to fold into its proper protein configuration (67–69). XBP1s transcriptionally regulates the expression of PDIs, which are needed in the ER for disulfide bond formation and stability of this protein (70). Furthermore, XBP1s also transcriptionally regulates secretory pathway components, including those involved in protein glycosylation and vesicle trafficking needed for maturation and secretion of hCG. Thus, IRE1-XBP1s signaling may be required for the synthesis, processing, and secretion of hCG, along with other STB-associated proteins (71).
CTB fusion involves dynamic changes in membrane composition and membrane-membrane interactions facilitated by syncytins, resulting in the formation of fusion pores, merging of membranes, and intermixing of cytoplasm between adjacent cells (72). While there is no evidence to-date that IRE1-XBP1s signaling transcriptionally regulates the expression of syncytins or their receptors, a role for IRE1-XBP1s signaling has been identified in other models of cell fusion. For example, deletion of IRE1 in mouse muscle satellite cells inhibits myoblast fusion and impairs skeletal muscle regeneration. IRE1-XBP1s signaling directly controls the expression of several genes involved in myoblast fusion, including Mymk (encoding Myomaker) (73). IRE1-XBP1s may similarly regulate the expression of genes involved in STB formation; or at minimum, may be needed to produce chaperones and other proteins that facilitate the synthesis, transport, and membrane presentation of fusogens.
Aside from direct transcriptional control of fusogens, IRE1 may play a role in cell fusion during STB formation by interacting with filamin A, an actin binding protein that regulates cytoskeletal organization. IRE1 acts as a scaffold by recruiting kinases, such as protein kinase C alpha, to promote phosphorylation of filamin A and induce cytoskeletal remodeling, an essential process that occurs during cell fusion (74–76). Furthermore, IRE1 may facilitate cell fusion by initiating contact sites between the ER and plasma membrane through store-operated calcium channels, which regulate intracellular calcium levels and maintain lipid homeostasis. IRE1 interacts with stromal interacting molecule 1 (STIM1), a sensor of calcium levels in the ER lumen (77). When calcium levels in the ER lumen are low, STIM1 undergoes a conformational change to promote calcium entry from the cytosol to the ER lumen (77, 78). Therefore, by interacting with STIM1, IRE1 may regulate cytosolic calcium levels, which along with increased cAMP, is a requirement for STB formation (79).
7.3 IRE1 in human EVT formation and function
IRE1 signaling is enriched in first trimester EVTs compared to CTBs, and gene set enrichment analysis of EVTs derived from human trophoblast stem cells shows enrichment of XBP1s-induced chaperones (80). Furthermore, the same study showed that inhibition of IRE1 using a small molecule inhibitor, 4µ8c, reduces cell surface HLA-G expression, suggesting that IRE1-XBP1s signaling is active during EVT formation and may be required for the synthesis and/or transport of cell surface antigens.
While the specific role of IRE1-mediated signaling in EVT formation and invasiveness is not fully understood, interactions with other factors deemed important for EVT function have been noted. For instance, Krüppel-like factor 6 (KLF6) is a transcription factor highly expressed in the placenta that modulates EVT formation and invasiveness. Using HTR-8/SVneo cells, an immortalized cell-line derived from first trimester placental outgrowths and often used as a model of invasive EVTs, silencing KLF6 reduces IRE1 and XBP1s levels, suggesting that KLF6 may be upstream of IRE1 signaling. Interestingly, both KLF6 and IRE1 deficient mouse models are embryonic lethal at embryonic day 12.5, suggesting that they may be part of a similar developmental pathway (52, 81). IRE1 may also promote the expression of high-temperature requirement A serine peptidase 1 (HTRA1) following exposure to ER stress conditions, which promotes invasion of HTR-8/SVneo cells (82, 83). Notably, in other cells, IRE1-XBP1s signaling promotes the transcription of genes encoding factors associated with epithelial-to-mesenchymal transition, such as SNAI1, SNAI2, ZEB2 and TCF3 (84). EVTs undergo epithelial-to-mesenchymal transition as they gain invasive properties (85), but further studies are needed to determine whether IRE1-XBP1s signaling may contribute to this aspect of EVT development.
7.4 IRE1 signaling during human decidualization
There is evidence that IRE1-XBP1 signaling is active during decidualization. For example, cytoplasmic and nuclear reactivity for unspliced and spliced variants of XBP1 are detected in both DSCs and glandular epithelium (86). Decidualization of ESCs is associated with increased levels of IRE1 and its downstream targets, XBP1s and CCAAT/enhancer-binding protein homologous protein (CHOP). Treatment of DSCs with the IRE1 inhibitor, STF-083010, downregulates the expression of CHOP and prevents the secretion of interleukin-1β (IL-1β) associated with decidualization (87). Furthermore, in a co-culture model designed to mimic implantation, inhibition of IRE1 reduced invasion of trophoblast spheroids (derived from Swan-71 immortalized trophoblasts) into DSCs, suggesting that IRE1 may be required for this process (87). While IRE1-mediated signaling may be required for decidualization, hyperactivation may induce autophagy and disrupt decidualization. For example, the IRE1-XBP1s pathway is negatively regulated by the heat shock cognate 71 kDa (Hsc70). Knockdown of Hsc70 increases XBP1 protein expression, triggers autophagy, and impairs decidualization of ESCs (88). These results suggest that moderate activation of IRE1 supports successful decidualization, whereas too little or too much activation can interfere with this process.
7.5 IRE1 signaling in the placenta during pregnancy complications
Aberrant ER stress and hyperactivation of IRE1 signaling is associated with dysregulated placental development and adverse pregnancy outcomes. This has been most frequently documented in pregnancies complicated by preeclampsia and FGR. Compared to placentas from normotensive pregnancies, placentas from preeclamptic pregnancies (particularly early-onset preeclampsia) exhibit increased IRE1 phosphorylation, XBP1 splicing, and ER luminal dilation (89, 90). Likewise, expression of XBP1 is increased in decidual tissues from preeclampsia with and without FGR (86). Endometrial biopsies from patients with recurrent pregnancy loss and implantation failure have elevated expression of IRE1 compared to controls, but surprisingly lower expression of XBP1s, suggesting that dysregulated IRE1 activation may contribute to these pregnancy complications (87). However, phosphorylated IRE1 was not measured in the samples from recurrent pregnancy loss and implantation failure, and should be evaluated in future studies to determine whether IRE1 activity may be altered in these pregnancy complications.
IRE1 phosphorylation, along with other markers of ER stress and apoptosis, is also increased when subjecting trophoblasts to hypoxia-reoxygenation in vitro to mimic the conditions that placental cells would experience in early-onset preeclampsia (91–93). Therefore, IRE1 signaling likely contributes to the cellular stress response during pathological conditions. Other pregnancy complications associated with ER stress and IRE1 activation include gestational cholestasis, a condition in which bile acids build up in the liver and enter the bloodstream. In a mouse model of gestational cholestasis, increased IRE1 activation is apparent in placental tissue, and this was associated with apoptosis and FGR. Inhibition of IRE1 signaling using 4µ8c prevents trophoblast apoptosis and rescues FGR in mice. Likewise, inhibiting IRE1 signaling prevents cell death in the HTR-8/SVneo cell-line following exposure to deoxycholic acid (94).
Physiological and environmental stressors that increase the risk of pregnancy complications can induce ER stress and activate IRE1 signaling. For example, maternal obesity is a risk factor for various pregnancy complications. Compared to placentas from normal weight pregnancies, obese pregnancies exhibit increased levels of phosphorylated IRE1 and XBP1s in placental tissue (95, 96). Palmitate is the most common saturated fatty acid in the body and is detectable at higher levels in the circulation of obese persons. Increased levels of palmitate alter ER morphology, impair invasiveness, and induce ER stress and apoptotic signaling in various human trophoblast cell-lines (97, 98). Other environmental stressors associated with ER stress and IRE1 hyperactivation in trophoblasts include viral infections (e.g., ZIKV) and exposure to toxins such as nicotine and ethanol (99–102). In the latter case, only total IRE1 protein levels were assessed, and therefore the contribution of active IRE1-XBP1s signaling to placental function following exposure to toxins remains elusive.
8 PERK
PERK detects and responds to ER stress by reducing the amount of protein in the ER lumen through the attenuation of translation (103). When there is an overaccumulation of misfolded proteins in the ER lumen, PERK homodimerizes and trans-autophosphorylates at Thr980, resulting in the activation of its kinase function (104). PERK will subsequently phosphorylate eukaryotic initiation factor 2α (eIF2α), preventing the assembly of ribosomes at the initiator codon of mRNA transcripts and blocking protein synthesis (105). As part of the PERK signaling branch, phosphorylated eIF2α promotes mRNA translation of the activating transcription factor 4 (ATF4), which can modulate transcription depending on its binding partners. The classical transcriptional target of ATF4 during ER stress is DDIT3, which encodes for CHOP, a transcription factor that regulates apoptosis (106). ATF4 can also transcriptionally regulate genes involved in amino acid metabolism and autophagy (107).
8.1 PERK regulates proper protein folding in the mouse placenta
Mice lacking PERK are viable and do not initially differ in weight compared to their wild-type counterparts, suggesting that PERK activity is not required (or is redundant) for placental and embryo development (108). However, other organs with secretory activity are severely impacted by PERK deficiency. For example, mice lacking PERK exhibit progressive loss of pancreatic β cells, neonatal development of diabetes mellitus, and exocrine pancreatic atrophy. Pancreatic cells in these mice have ER lumen dilation and accumulation of electron-dense material in the ER. Mice lacking PERK also have severe skeletal dysplasias and dwarfism associated with reduced hepatic secretion of insulin-like growth factor 1 (108–110). Since the placenta is also a secretory organ, PERK deficiency may affect aspects of placental endocrine function.
To determine a potential contribution of PERK signaling to placental endocrine activity, a conditional mouse model with PERK depletion specifically in the junctional zone has been investigated. The junctional zone secretes numerous hormones and growth factors, and is therefore considered as the endocrine component of the murine placenta (111). There are no differences in litter size, placental weight or fetal weight between wild-type mice and those with junctional zone-specific PERK depletion (as expected, since the global PERK knockout also does not show these differences). However, PERK deficiency exacerbates ER stress in the junctional zone when mice are housed in a reduced oxygen atmosphere, which was done to induce tissue hypoxia and ER stress. When exposed to hypoxia, PERK-deficient junctional zone trophoblasts exhibit dilation of the ER cisternae and accumulation of protein aggregates, indicating possible loss of ER homeostasis through aggregation of misglycosylated secretory proteins in the ER (111, 112). Furthermore, these trophoblasts have a reduced capacity to stimulate maternal physiological adaptions, such as the induction of glycogenolysis in the liver. Therefore, in the placenta, PERK may promote proper protein folding and processing in the ER, whereas PERK-deficient mice may be more susceptible to proteinopathies under ER stress-inducing conditions.
Activation of PERK attenuates translation through the phosphorylation of eIF2α on Ser51 (113). A mouse model has been generated in which this serine is mutated to alanine, preventing the ability of eIF2α to reduce translation (114). These mice die shortly after birth due to hypoglycemia, resulting from a deficiency in gluconeogenesis and loss of pancreatic β cells. In the placenta, eIF2α mutant mice exhibit increased basal translation, correlating with reduced placental and fetal growth compared to mice possessing at least one wild-type copy (112). Additionally, these placentas have an accumulation of glycoproteins in the junctional zone and reduced labyrinth zone volume, suggesting that eIF2α dysfunction may disrupt placental endocrine function (112).
8.2 PERK signaling in human placental and decidual development
As a key regulator of protein translation, PERK signaling may contribute to the control of trophoblast differentiation. For example, PERK signaling is activated in BeWo cells during forskolin-induced differentiation as shown through increased levels of phosphorylated eIF2α, ATF4, and CHOP. Exposing BeWo cells to a PERK inhibitor, GSK2656157, inhibits cell fusion, but interestingly there is no effect on secreted hCG levels. Similar results are obtained using primary CTBs from term placentas (62). Like IRE1, PERK may promote trophoblast fusion through its interaction with filamin A to regulate cytoskeletal remodeling and ER calcium levels (115). In HTR-8/SVneo cells and JEG-3 cells, another transformed trophoblast cell-line, induction of ER stress using tunicamycin, thapsigargin, or pro-inflammatory cytokines reduces matrix metalloproteinase 2 (MMP2) levels and cell invasion. However, inhibition of PERK restores MMP2 levels (116). Given the importance of PERK signaling for ER homeostasis in other secretory cells, PERK may maintain cellular integrity in the placenta, particularly under ER stress-inducing conditions.
Within the decidua, DSCs from pregnancies deemed healthy express high levels of phosphorylated PERK. Treatment of ESCs and glandular cells with progesterone activates the PERK pathway, as shown by the increased levels of phosphorylated eIF2α, ATF4, and CHOP (87, 90, 117). Progesterone-driven CHOP expression induces several apoptosis markers (e.g., BAX and cleaved caspase-3) in ESCs and glandular cells. Furthermore, BiP/GRP78, CHOP, and cleaved caspase-3 are upregulated during the secretory phase in stromal and glandular regions of healthy human endometrium, confirming that UPR activation and CHOP-mediated apoptosis increase in response to higher progesterone levels (117). Altogether, these findings indicate that progesterone-driven activation of the PERK/eIF2α/ATF4 pathway, and subsequent CHOP-mediated apoptosis, may regulate endometrial remodeling during the late secretory phase.
8.3 PERK signaling in the placenta during pregnancy complications
PERK signaling may have important roles in determining cell function and cell fate during placental pathologies. For example, in placentas from preeclamptic pregnancies, increased levels of phosphorylated PERK, phosphorylated eIF2α, ATF4 and CHOP are evident, and these proteins appear to localize predominantly to the STB layer (118, 119). Increased levels of phosphorylated PERK are also evident in the decidua (90). Decidual tissue from preeclamptic pregnancies with FGR have increased phosphorylation of eIF2α and ATF4 levels compared to control pregnancies (86, 120). These findings suggest that dysregulation of PERK signaling in the decidua may impair decidualization and contribute to the development of pregnancy complications.
Maternal serum from preeclamptic pregnancies activates the PERK pathway in placental explants and HTR-8/SVneo cells, as shown through increased eIF2α phosphorylation and CHOP levels. Cell death was also apparent after explants and cells were exposed to serum from preeclamptic pregnancies, but the specific contribution of PERK signaling to cytotoxicity was not assessed (34). It is possible that the PERK-mediated cell death observed in placentas from preeclamptic pregnancies occurs as a result of histone deacetylase (HDAC) deficiency (121). HDACs are essential for trophoblast differentiation and are often downregulated in placentas from preeclamptic pregnancies (121–124). HDAC2 silencing in HTR-8/SVneo cells increases pyroptosis, which was prevented when PERK was knocked down (121). Therefore, PERK activation may underlie the increased incidence of cell death in placental pathologies.
To recapitulate the ER stress-inducing conditions that may induce PERK signaling and affect trophoblast function and viability, several in vitro models of cell stress have been devised. For example, exposure of BeWo cells to hypoxia/reoxygenation (fluctuating between room air and 1% O2) leads to increased levels of phosphorylated eIF2α levels and reduced cell number (39). IL-1β, a pro-inflammatory cytokine that is increased in preeclampsia, induces apoptosis in BeWo cells through the PERK pathway, which is inhibited by progesterone (118). PERK inhibition can also reduce apoptosis in BeWo cells treated with endocannabinoid 2-arachidonoylglycerol (125). Cadmium, an environmental pollutant and carcinogen, reduces 11β-HSD2 expression in JEG-3 cells, which is restored either by knocking down PERK expression or by treating cells with antioxidants such as melatonin or N-acetylcysteine (126, 127). Thus, signaling through PERK may regulate trophoblast survival under various ER stress-inducing conditions.
9 ATF6
ATF6 is the third ER transmembrane sensor that is activated in response to ER stress (128). There are two isoforms of ATF6: ATF6α and ATF6β. Signaling through ATF6α generally results in a more potent but transient transcriptional response, whereas ATF6β is a comparatively weaker transcriptional modulator (129). During ER stress, the cytosolic domain of ATF6 translocates to the Golgi apparatus to be cleaved by site 1 and site 2 proteases (130). These proteases remove the luminal and transmembrane portions of ATF6, yielding the ATF6 transcription factor. ATF6 binds to the ER stress response element motif to transcriptionally regulate chaperones and ERAD components that enhance protein folding and contribute to clearance of misfolded proteins, respectively (131, 132).
9.1 ATF6 in mouse placental and decidual development
While various aspects of the UPR have been studied in the context of placental development, the role of ATF6 remains largely unexplored, highlighting a gap in understanding its potential contributions to trophoblast function and placental health. Knockout of either ATF6α or ATF6β in mice produces viable progeny, but double ATF6α/β knockout mice die early during development (around the time of implantation), suggesting that ATF6α and ATF6β may share an overlapping function that is essential for decidualization and early embryonic development in mice (133, 134). In the mouse uterus, expression of ATF6α is primarily localized to luminal and glandular epithelial cells, specifically near the blastocyst implantation site, suggesting a potential role in uterine receptivity (135). Since ATF6α is also present in primary and secondary decidualization zones, it is plausible that ATF6α could contribute to the progression of decidualization. Indeed, artificial decidualization of pseudopregnant mice increases ATF6α expression in decidual cells (135). Altogether, increased ATF6α levels in the uterus seem to correlate with the implantation window and initial stages of decidualization in mice, but whether ATF6α directly contributes to these processes requires further exploration.
9.2 ATF6 signaling in human placental and decidual development
ATF6α is present in the human placenta, and staining is particularly strong in nuclei clustered in syncytial knots (90). Exposure of primary CTBs to an ATF6α inhibitor, AEBSF, reduces cell fusion and hCG secretion (62, 136). It is possible that ATF6α promotes STB formation by regulating expression of BiP/GRP78 (137). Additionally, since ATF6α transcriptionally regulates XBP1, there may be contributory or compensatory mechanisms through which ATF6α influences STB formation by supporting the IRE1-XBP1s axis (131). ATF6α is also detected in EVTs, suggesting its participation in the invasion of EVTs into the decidua and interactions with decidual cells (86). In cancer cells, ATF6α promotes cell invasion and metastasis (138). Therefore, it is possible that ATF6α may regulate genes involved in EVT migration and invasion, but this requires further investigation.
Decidual cells from healthy pregnancies exhibit high levels of ATF6α localized primarily to the cytoplasm, indicating the presence of primarily inactive forms of ATF6α (90). Decidualization of ESCs is associated with an upregulation of ATF6α. Inhibition of ATF6 using AEBSF reduces the decidualization-associated increase in IL-1β secretion, which was also reported when using an IRE1 inhibitor (87, 139). It is unclear whether the mechanistic control of inflammatory cytokine production by ATF6α overlaps with the IRE1 signaling pathway.
9.3 ATF6 signaling in the placenta during pregnancy complications
In early-onset preeclampsia with FGR, ATF6α protein levels are increased in placental tissue compared to placentas from pregnancies deemed healthy (39, 86). It is unclear if ATF6α activation is also increased, as the levels of active ATF6α, or nuclear localization of the protein, were not assessed. A hallmark of placentas from preeclampsia is reduced secretion of placental growth factor, which is a pro-angiogenic molecule important for maintaining endothelial integrity. Interestingly, reduced placental growth factor in preeclampsia is associated with nuclear localization of ATF4 and ATF6β (but not ATF6⍺ or XBP1) in the STB layer (140, 141). Combined ATF4 and ATF6β silencing in BeWo cells exposed to the ER stressor thapsigargin or hypoxia/reoxygenation increased expression of placental growth factor (141), suggesting that ATF4 and ATF6β negatively regulate placental growth factor expression. Therefore, interfering with ATF4 and ATF6β may be a feasible means to enhance placental growth factor production in compromised pregnancies.
10 Future applications: targeting ER stress to alleviate placental dysfunction
As highlighted in the preceding sections, placental development and function require a fine balance of ER stress and UPR activation to maintain cellular homeostasis. Too much or too little UPR activation can impair placental and decidual function and lead to adverse pregnancy outcomes. Therefore, therapeutic approaches targeting ER stress pathways may be promising avenues to restore proper placental function and improve fetal and maternal outcomes (34, 142). As one example, tauroursodeoxycholic acid (TUDCA), a bile acid derivative that is currently being evaluated for the treatment for neurodegeneration, alleviates ER stress by acting as a chemical chaperone and reducing the expression of ER stress-related proteins (143). In a rat model of advanced maternal age, placental insufficiency accompanied by increased ER stress markers (p-eIF2α and CHOP) was observed in aged, pregnant rats. TUDCA treatment administered via drinking water throughout pregnancy reduced ER stress in the placenta and improved placental blood flow and fetal growth (38, 144–146). TUDCA treatment of pre-implantation embryos can increase the rate of blastocyst formation and enhance the success of implantation and pregnancy in mice (147). Another example includes phenylbutyric acid, a fatty acid that is naturally produced through fermentation by colonic bacteria, which ameliorates ER stress by acting as a chemical chaperone (148). Phenylbutyric acid reduces high glucose-induced ER stress in BeWo cells, suggesting that this compound may be useful to reduce placental ER stress in pregnancies with gestational diabetes (149). Additionally, phenylbutyric acid can also reduce hypoxia-induced apoptosis in HTR-8/SVneo cells by assisting in protein folding and reducing PERK activation (150). Overall, the use of chemical chaperones to facilitate protein folding and prevent UPR hyperactivation may be a promising avenue to treat placental pathologies and improve fetal outcomes.
Future studies could also explore whether selectively targeting one or more UPR pathways is an effective way to improve placental function (39, 151, 152). There are several small molecule inhibitors that specifically target these pathways and are being tested in humans for their efficacy in disease settings like cancer (153–155), but it remains uncertain whether these inhibitors can be safely used during pregnancy. Unintended off-target effects and safe delivery strategies need to be considered. Conversely, since UPR pathways are essential for various aspects of placental and decidual function, it is possible that transiently stimulating one or more pathways may be beneficial to restore ER homeostasis and promote cell survival. More research is required to determine whether ER stress-related pathways can be safely modulated to fine-tune placental ER homeostasis.
11 Conclusion and perspectives
In this review, we highlighted the importance of ER homeostasis and UPR signaling for placental and decidual development and function. Although there is resounding evidence of increased ER stress and UPR hyperactivation during pregnancy pathologies, each UPR signaling branch contributes to basic processes needed for placental and decidual function, and these functions may shift depending on cellular conditions (Figure 3). Therefore, tweaking the activity of UPR branches to restore ER homeostasis holds promise as a treatment approach to improve placental function in compromised pregnancies.
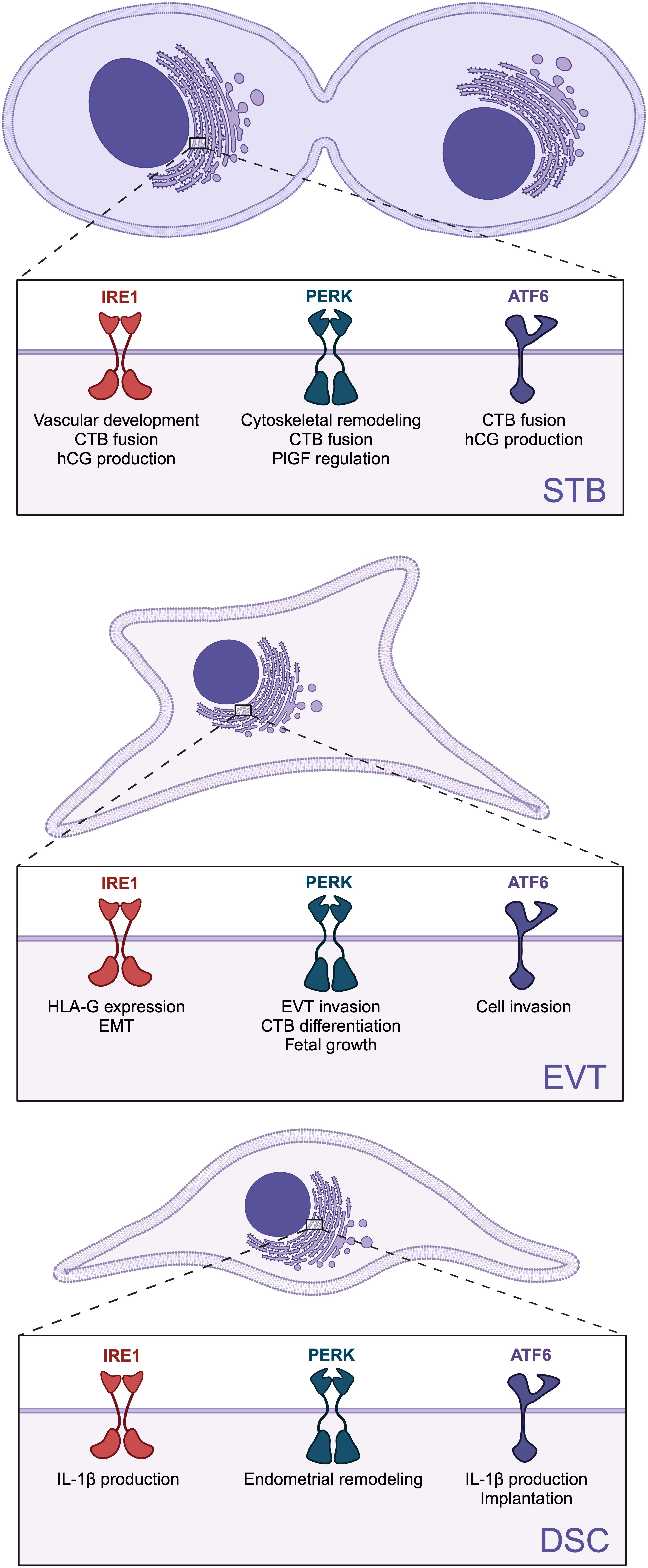
Figure 3. Role of specific unfolded protein response (UPR) branches on placental and decidual development and function. The three UPR sensors: inositol-requiring enzyme 1 (lRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) are involved in the differentiation and function of syncytiotrophoblast (STB), extravillous trophoblasts (EVTs), and decidual stromal cells (DSCs). Created in BioRender. Chowdhury, D. (2024) https://BioRender.com/x70x431.
Despite significant advances in understanding the role of UPR pathways in placental and decidual development, several gaps remain. It is apparent that UPR functions extend beyond merely responding to stress, as UPR sensors participate in homeostatic mechanisms to facilitate the high demand for protein synthesis and processing in secretory cells. However, the precise roles of each UPR branch in placental and decidual functions are still elusive. Determining their specific contributions can be challenging because there is significant overlap and compensation between the branches and other stress-related pathways. For instance, Ern1-deficient mouse placentas exhibit increased PERK and ATF6 activation, which may mask additional ways that IRE1 regulates placental development and function (52). As another example, kinases associated with the integrated stress response, which are not connected to the UPR, can activate eIF2α independently of PERK (156). Therefore, phosphorylation of eIF2α does not necessarily guarantee that the PERK branch of the UPR is active. Additionally, the source of ER stress and UPR activation during placental and decidual development remains elusive. The high demand for protein and lipid production could be contributing to ER stress and UPR activation during placental and decidual development.
Another challenge with assessing ER stress, particularly in the context of placentation, is the limited tools that are available to confirm UPR branch activation. Many studies rely on detecting a handful of changes in gene expression or protein levels of UPR markers in bulk tissue or cell-lines. As the field evolves and better cell models and tools are developed, a more comprehensive understanding of each UPR branch is likely to emerge. For instance, the use of more complex cellular models of the decidua and placenta, such as decidual and trophoblast organoid cultures or “on-a-chip” platforms, offer promising avenues for better representing cellular interactions at the maternal-fetal interface. The use of these models in combination with more precise detection of UPR activation, such as reporter plasmids to measure activation of specific UPR branches in combination with omics technologies for more comprehensive coverage of downstream UPR targets, may provide a deeper understanding of the cellular response to stress at the maternal-fetal interface. The use of CRISPR-mediated gene targeting to disrupt each ER sensor in trophoblast and decidual models will also be enlightening. Collectively, uncovering the role of UPR branches holds potential for a greater understanding of ER homeostasis during normal and pathological placentation.
Author contributions
DC: Writing – original draft, Writing – review & editing. CJ: Writing – original draft, Writing – review & editing. PL: Writing – original draft, Writing – review & editing. SR: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Our research is funded by the Natural Sciences and Engineering Research Council of Canada (04543: SR, 05267: PL) and Canadian Institutes of Health Research (PJT180483: SR, PJT168882 and ARB192062: PL).
Acknowledgments
Figures were designed using BioRender (www.biorender.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1525189/full#supplementary-material
References
1. Zeldovich VB, Clausen CH, Bradford E, Fletcher DA, Maltepe E, Robbins JR, et al. Placental syncytium forms a biophysical barrier against pathogen invasion. PloS Pathog. (2013) 9:e1003821. doi: 10.1371/journal.ppat.1003821
2. Deval G, Boland S, Fournier T, Ferecatu I. On placental toxicology studies and cerium dioxide nanoparticles. IJMS. (2021) 22:12266. doi: 10.3390/ijms222212266
3. Fowden AL, Sferruzzi-Perri AN, Coan PM, Constancia M, Burton GJ. Placental efficiency and adaptation: endocrine regulation. J Physiol. (2009) 587:3459–72. doi: 10.1113/tjp.2009.587.issue-14
4. Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocrine Rev. (2006) 27:141–69. doi: 10.1210/er.2005-0011
5. Costa MA. The endocrine function of human placenta: an overview. Reprod BioMedicine Online. (2016) 32:14–43. doi: 10.1016/j.rbmo.2015.10.005
6. Kojima J, Ono M, Kuji N, Nishi H. Human chorionic villous differentiation and placental development. IJMS. (2022) 23:8003. doi: 10.3390/ijms23148003
7. Kim SM, Kim JS. A review of mechanisms of implantation. Dev Reprod. (2017) 21:351–9. doi: 10.12717/DR.2017.21.4.351
8. Jaremek A, Jeyarajah MJ, Jaju Bhattad G, Renaud SJ. Omics approaches to study formation and function of human placental syncytiotrophoblast. Front Cell Dev Biol. (2021) 9:674162. doi: 10.3389/fcell.2021.674162
9. Velicky P, Meinhardt G, Plessl K, Vondra S, Weiss T, Haslinger P, et al. Genome amplification and cellular senescence are hallmarks of human placenta development. PloS Genet. (2018) 14:e1007698. doi: 10.1371/journal.pgen.1007698
10. Gellersen B, Brosens JJ. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocrine Rev. (2014) 35:851–905. doi: 10.1210/er.2014-1045
11. Giudice LC, Martina NA, Crystal RA, Tazuke S, Druzin M. Insulin-like growth factor binding protein-1 at the maternal-fetal interface and insulin-like growth factor-I, insulin-like growth factor-II, and insulin-like growth factor binding protein-1 in the circulation of women with severe preeclampsia. Am J Obstetrics Gynecology. (1997) 176:751–8. doi: 10.1016/S0002-9378(97)70598-2
12. Soares MJ, Varberg KM, Iqbal K. Hemochorial placentation: development, function, and adaptations†. Biol Reproduction. (2018) 99:196–211. doi: 10.1093/biolre/ioy049
13. Burton GJ, Redman CW, Roberts JM, Moffett A. Pre-eclampsia: pathophysiology and clinical implications. BMJ. (2019) 5:l2381. doi: 10.1136/bmj.l2381
14. Ornaghi S, Tyurmorezova A, Giardini V, Algeri P, Vergani P. 797: The role of inflammation in late-onset preeclampsia. Am J Obstetrics Gynecology. (2012) 206:S350–1. doi: 10.1016/j.ajog.2011.10.815
15. Robillard PY, Dekker G, Scioscia M, Bonsante F, Iacobelli S, Boukerrou M, et al. Increased BMI has a linear association with late-onset preeclampsia: A population-based study. PloS One. (2019) 14:e0223888. doi: 10.1371/journal.pone.0223888
16. Jackson A, Duggleby S, Grove G. Whole body protein turnover can be measured non-invasively in women using the end product method with [15N]glycine to show changes with the menstrual cycle and pregnancy. Eur J Clin Nutr. (2000) 54:329–36. doi: 10.1038/sj.ejcn.1600958
17. Khorami Sarvestani S, Shojaeian S, Vanaki N, Ghresi-Fard B, Amini M, Gilany K, et al. Proteome profiling of human placenta reveals developmental stage-dependent alterations in protein signature. Clin Proteom. (2021) 18:18. doi: 10.1186/s12014-021-09324-y
18. Sun H, Wu M, Wang M, Zhang X, Zhu J. The regulatory role of endoplasmic reticulum chaperone proteins in neurodevelopment. Front Neurosci. (2022) 16:1032607. doi: 10.3389/fnins.2022.1032607
19. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
20. Lajoie P, Moir RD, Willis IM, Snapp EL. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol Biol Cell. (2012) 23:955–64. doi: 10.1091/mbc.e11-12-0995
21. Fun XH, Thibault G. Lipid bilayer stress and proteotoxic stress-induced unfolded protein response deploy divergent transcriptional and non-transcriptional programmes. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158449. doi: 10.1016/j.bbalip.2019.04.009
22. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. (2016) 529:326–35. doi: 10.1038/nature17041
23. Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hänelt I, et al. Activation of the unfolded protein response by lipid bilayer stress. Mol Cell. (2017) 67:673–684.e8. doi: 10.1016/j.molcel.2017.06.012
24. Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J Cell Biol. (1990) 110:1501–11. doi: 10.1083/jcb.110.5.1501
25. Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. (2009) 187:525–36. doi: 10.1083/jcb.200907074
26. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS J. (2019) 286:241–78. doi: 10.1111/febs.2019.286.issue-2
27. Liu Y, Chang A. Heat shock response relieves ER stress. EMBO J. (2008) 27:1049–59. doi: 10.1038/emboj.2008.42
28. Burton GJ, Yung HW. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertension: Int J Women’s Cardiovasc Health. (2011) 1:72–8. doi: 10.1016/j.preghy.2010.12.002
29. Buhimschi I, Nayeri U, Zhao G, Shook L, Pensalfini A, Funai E, et al. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci Trans Med. (2014) 6:245ra92–245ra92. doi: 10.1126/scitranslmed.3008808
30. Cheng S, Banerjee S, Daiello L, Nakashima A, Jash S, Huang Z, et al. Novel blood test for early biomarkers of preeclampsia and Alzheimer’s disease. Sci Rep. (2021) 11:null. doi: 10.1038/s41598-021-95611-5
31. Medegan Fagla B, Buhimschi IA. Protein misfolding in pregnancy: current insights, potential mechanisms, and implications for the pathogenesis of preeclampsia. Molecules. (2024) 29:610. doi: 10.3390/molecules29030610
32. Gerasimova EM, Fedotov SA, Kachkin DV, Vashukova ES, Glotov AS, Chernoff YO, et al. Protein misfolding during pregnancy: new approaches to preeclampsia diagnostics. IJMS. (2019) 20:6183. doi: 10.3390/ijms20246183
33. Lederer W, Schaffenrath H, Alomar-Dominguez C, Thaler J, Fantin R, Dostal L, et al. Cerebrospinal beta-amyloid peptides(1-40) and (1-42) in severe preeclampsia and HELLP syndrome – a pilot study. Sci Rep. (2020) 10:5783. doi: 10.1038/s41598-020-62805-2
34. Castro KR, Prado KM, Lorenzon AR, Hoshida MS, Alves EA, Francisco RPV, et al. Serum from preeclamptic women triggers endoplasmic reticulum stress pathway and expression of angiogenic factors in trophoblast cells. Front Physiol. (2022) 12:799653. doi: 10.3389/fphys.2021.799653
35. Capatina N, Burton GJ, Yung HW. Elevated homocysteine activates unfolded protein responses and causes aberrant trophoblast differentiation and mouse blastocyst development. Physiol Rep. (2022) 10:15467. doi: 10.14814/phy2.15467
36. Chen X, Shi C, He M, Xiong S, Xia X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Sig Transduct Target Ther. (2023) 8:352. doi: 10.1038/s41392-023-01570-w
37. Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. (2008) 105:18525–30. doi: 10.1073/pnas.0809677105
38. Pasha M, Kirschenman R, Wooldridge A, Spaans F, Cooke CLM, Davidge ST. The effect of tauroursodeoxycholic acid (TUDCA) treatment on pregnancy outcomes and vascular function in a rat model of advanced maternal age. Antioxidants. (2022) 11:1275. doi: 10.3390/antiox11071275
39. Yung HW, Atkinson D, Campion-Smith T, Olovsson M, Charnock-Jones DS, Burton GJ. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J Pathol. (2014) 234:262–76. doi: 10.1002/path.2014.234.issue-2
40. Read A, Schröder M. The unfolded protein response: an overview. Biol (Basel). (2021) 10:384. doi: 10.3390/biology10050384
41. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. (2018) 69:169–81. doi: 10.1016/j.molcel.2017.06.017
42. Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. (2009) 6:743–50. doi: 10.1093/jb/mvp166
43. Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. (1993) 73:1197–206. doi: 10.1016/0092-8674(93)90648-A
44. Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. (2013) 23:547–55. doi: 10.1016/j.tcb.2013.06.005
45. Prischi F, Nowak PR, Carrara M, Ali MMU. Phosphoregulation of Ire1 RNase splicing activity. Nat Commun. (2014) 5:3554. doi: 10.1038/ncomms4554
46. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. (2002) 415:92–6. doi: 10.1038/415092a
47. Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. (1988) 54:209–20. doi: 10.1016/0092-8674(88)90553-3
48. Hollien J, Weissman J. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. (2006) 313:104–7. doi: 10.1126/science.1129631
49. Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. (2014) 39:245–54. doi: 10.1016/j.tibs.2014.02.008
50. Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int J Biochem Cell Biol. (2001) 33:1047–63. doi: 10.1016/S1357-2725(01)00093-0
51. Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman R. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. (2005) 2:268–81. doi: 10.1172/JCI200521848
52. Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. (2009) 106:16657–62. doi: 10.1073/pnas.0903775106
53. Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PloS One. (2010) 5:e9575. doi: 10.1371/journal.pone.0009575
54. Woods L, Perez-Garcia V, Hemberger M. Regulation of placental development and its impact on fetal growth—New insights from mouse models. Front Endocrinol. (2018) 9:570. doi: 10.3389/fendo.2018.00570
55. Guo SW, Yu W, Wang X, Liu JH, Zhang XY, He CQ, et al. Expression and regulation of XBP1 in mouse uterus during early pregnancy. Sheng Li Xue Bao. (2021) 73:208–16. doi: 10.13294/j.aps.2020.0091
56. Gu XW, Yan JQ, Dou HT, Liu J, Liu L, Zhao ML, et al. Endoplasmic reticulum stress in mouse decidua during early pregnancy. Mol Cell Endocrinology. (2016) 434:48–56. doi: 10.1016/j.mce.2016.06.012
57. Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. (2000) 14:152–7. doi: 10.1101/gad.14.2.152
58. Mao T, Shao M, Qiu Y, Huang J, Zhang Y, Song B, et al. PKA phosphorylation couples hepatic inositol-requiring enzyme 1α to glucagon signaling in glucose metabolism. Proc Natl Acad Sci USA. (2011) 108:15852–7. doi: 10.1073/pnas.1107394108
59. Keryer G, Alsat E, Taskén K, Evain-Brion D. Cyclic AMP-dependent protein kinases and human trophoblast cell differentiation in vitro. J Cell Sci. (1998) 111:995–1004. doi: 10.1242/jcs.111.7.995
60. Knerr I, Schubert SW, Wich C, Amann K, Aigner T, Vogler T, et al. Stimulation of GCMa and syncytin via cAMP mediated PKA signaling in human trophoblastic cells under normoxic and hypoxic conditions. FEBS Letters. (2005) 579:3991–8. doi: 10.1016/j.febslet.2005.06.029
61. Kikuchi D, Tanimoto K, Nakayama K. CREB is activated by ER stress and modulates the unfolded protein response by regulating the expression of IRE1α and PERK. Biochem Biophys Res Commun. (2016) 469:243–50. doi: 10.1016/j.bbrc.2015.11.113
62. Bastida-Ruiz D, Yart L, Wuillemin C, Ribaux P, Morris N, Epiney M, et al. The fine-tuning of endoplasmic reticulum stress response and autophagy activation during trophoblast syncytialization. Cell Death Dis. (2019) 10:651. doi: 10.1038/s41419-019-1905-6
63. Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, et al. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. (2007) 282:7024–34. doi: 10.1074/jbc.M609490200
64. Cox JS, Chapman RE, Walter P. The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. MBoC. (1997) 8:1805–14. doi: 10.1091/mbc.8.9.1805
65. Iwakoshi N, Lee AH, Vallabhajosyula P, Otipoby K, Rajewsky K, Glimcher L. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. (2003) 4:321–9. doi: 10.1038/ni907
66. Hu CCA, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. (2009) 28:1624–36. doi: 10.1038/emboj.2009.117
67. Singh V, Merz WE. Disulfide bond formation is not required for human chorionic gonadotropin subunit association. J Biol Chem. (2000) 275:11765–70. doi: 10.1074/jbc.275.16.11765
68. Mise T, Bahl OP. Assignment of disulfide bonds in the beta subunit of human chorionic gonadotropin. J Biol Chem. (1981) 256:6587–92. doi: 10.1016/S0021-9258(19)69030-7
69. Huth JR, Perini F, Lockridge O, Bedows E, Ruddon RW. Protein folding and assembly in vitro parallel intracellular folding and assembly. Catalysis of folding and assembly of the human chorionic gonadotropin alpha beta dimer by protein disulfide isomerase. J Biol Chem. (1993) 268:16472–82. doi: 10.1016/S0021-9258(19)85444-3
70. Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, et al. IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. (2012) 16:473–86. doi: 10.1016/j.cmet.2012.09.003
71. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. (2004) 21:81–93. doi: 10.1016/j.immuni.2004.06.010
72. Lu X, Wang R, Zhu C, Wang H, Lin HY, Gu Y, et al. Fine-tuned and cell-cycle-restricted expression of fusogenic protein syncytin-2 maintains functional placental syncytia. Cell Rep. (2017) 21:1150–9. doi: 10.1016/j.celrep.2017.10.019
73. Joshi AS, Tomaz Da Silva M, Roy A, Koike TE, Wu M, Castillo MB, et al. The IRE1α/XBP1 signaling axis drives myoblast fusion in adult skeletal muscle. EMBO Rep. (2024) 25:3627–50. doi: 10.1038/s44319-024-00197-4
74. Tigges U, Koch B, Wissing J, Jockusch BM, Ziegler WH. The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase Cα. J Biol Chem. (2003) 278:23561–9. doi: 10.1074/jbc.M302302200
75. Urra H, Henriquez DR, Cánovas J, Villarroel-Campos D, Carreras-Sureda A, Pulgar E, et al. IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat Cell Biol. (2018) 20:942–53. doi: 10.1038/s41556-018-0141-0
76. Ishikawa A, Omata W, Ackerman WE, Takeshita T, Vandré DD, Robinson JM. Cell fusion mediates dramatic alterations in the actin cytoskeleton, focal adhesions, and E-cadherin in trophoblastic cells. Cytoskeleton. (2014) 71:241–56. doi: 10.1002/cm.21165
77. Carreras-Sureda A, Zhang X, Laubry L, Brunetti J, Koenig S, Wang X, et al. The ER stress sensor IRE1 interacts with STIM1 to promote store-operated calcium entry, T cell activation, and muscular differentiation. Cell Rep. (2023) 42:113540. doi: 10.1016/j.celrep.2023.113540
78. Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. (2005) 437:902–5. doi: 10.1038/nature04147
79. Vatish M, Tesfa L, Grammatopoulos D, Yamada E, Bastie CC, Pessin JE. Inhibition of akt activity and calcium channel function coordinately drive cell-cell fusion in the beWO choriocarcinoma placental cell line. PloS One. (2012) 7:e29353. doi: 10.1371/journal.pone.0029353
80. Morey R, Farah O, Kallol S, Requena DF, Meads M, Moretto-Zita M, et al. Transcriptomic drivers of differentiation, maturation, and polyploidy in human extravillous trophoblast. Front Cell Dev Biol. (2021) 9:702046. doi: 10.3389/fcell.2021.702046
81. Matsumoto N, Kubo A, Liu H, Akita K, Laub F, Ramirez F, et al. Developmental regulation of yolk sac hematopoiesis by Krüppel-like factor 6. Blood. (2006) 107:1357–65. doi: 10.1182/blood-2005-05-1916
82. Yoshida K, Kusama K, Fukushima Y, Ohmaru-Nakanishi T, Kato K, Tamura K. Alpha-1 antitrypsin-induced endoplasmic reticulum stress promotes invasion by extravillous trophoblasts. IJMS. (2021) 22:3683. doi: 10.3390/ijms22073683
83. Yoshida K, Kusama K, Azumi M, Yoshie M, Kato K, Tamura K. Endoplasmic reticulum stress-regulated high temperature requirement A1 (HTRA1) modulates invasion and angiogenesis-related genes in human trophoblasts. J Pharmacol Sci. (2022) 150:267–74. doi: 10.1016/j.jphs.2022.10.003
84. Cuevas EP, Eraso P, Mazón MJ, Santos V, Moreno-Bueno G, Cano A, et al. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci Rep. (2017) 7:44988. doi: 10.1038/srep44988
85. Davies J E, Pollheimer J, Yong HEJ, Kokkinos MI, Kalionis B, Knöfler M, et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adhesion Migration. (2016) 10:310–21. doi: 10.1080/19336918.2016.1170258
86. Lian IA, Løset M, Mundal SB, Fenstad MH, Johnson MP, Eide IP, et al. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta. (2011) 32:823–9. doi: 10.1016/j.placenta.2011.08.005
87. Grasso E, Gori S, Soczewski E, Fernández L, Gallino L, Vota D, et al. Impact of the Reticular Stress and Unfolded Protein Response on the inflammatory response in endometrial stromal cells. Sci Rep. (2018) 8:12274. doi: 10.1038/s41598-018-29779-8
88. Brosens JJ, Salker MS, Teklenburg G, Nautiyal J, Salter S, Lucas ES, et al. Uterine selection of human embryos at implantation. Sci Rep. (2014) 4:3894. doi: 10.1038/srep03894
89. Du L, He F, Kuang L, Tang W, Li Y, Chen D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J Hum Hypertens. (2017) 31:49–55. doi: 10.1038/jhh.2016.17
90. Thomson S, Waters KA, Hennessy A, Machaalani R. The unfolded protein response and apoptotic regulation in the human placenta due to maternal cigarette smoking and pre-eclampsia. Reprod Toxicology. (2021) 105:120–7. doi: 10.1016/j.reprotox.2021.09.001
91. Cheng S, Huang Z, Jash S, Wu K, Saito S, Nakashima A, et al. Hypoxia-reoxygenation impairs autophagy-lysosomal machinery in primary human trophoblasts mimicking placental pathology of early-onset preeclampsia. Int J Mol Sci. (2022) 23:null. doi: 10.3390/ijms23105644
92. Li H, Zhao W, Wang L, Luo Q, Yin N, Lu X, et al. Human placenta-derived mesenchymal stem cells inhibit apoptosis of granulosa cells induced by IRE1α pathway in autoimmune POF mice. Cell Biol Int. (2019) 43:899–909. doi: 10.1002/cbin.11165
93. Fu J, Zhao L, Wang L, Zhu X. Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwanese J Obstetrics Gynecology. (2015) 54:19–23. doi: 10.1016/j.tjog.2014.11.002
94. Gao X, Lin S, Jiang P, Ye M, Chen W, Hu C, et al. Gestational cholestasis induced intrauterine growth restriction through triggering IRE1α-mediated apoptosis of placental trophoblast cells. FASEB J. (2022) 36:e22388. doi: 10.1096/fj.202101844RR
95. Liong S, Lappas M. Endoplasmic reticulum stress is increased in adipose tissue of women with gestational diabetes. PloS One. (2015) 10:e0122633. doi: 10.1371/journal.pone.0122633
96. Shen WB, Wang B, Yao R, Goetzinger KR, Wu S, Gao H, et al. Obesity impacts placental function through activation of p-IRE1a-XBP1s signaling. Front Cell Dev Biol. (2023) 11:1023327. doi: 10.3389/fcell.2023.1023327
97. Sahoo PK, Krishnamoorthy C, Wood JR, Hanson C, Anderson-Berry A, Mott JL, et al. Palmitate induces integrated stress response and lipoapoptosis in trophoblasts. Cell Death Dis. (2024) 15:31. doi: 10.1038/s41419-023-06415-6
98. Rampersaud AM, Dunk CE, Lye SJ, Renaud SJ. Palmitic acid induces inflammation in placental trophoblasts and impairs their migration toward smooth muscle cells through plasminogen activator inhibitor-1. Mol Hum Reproduction. (2020) 26:850–65. doi: 10.1093/molehr/gaaa061
99. Huovinen M, Ietta F, Repo JK, Paulesu L, Vähäkangas KH. The effect of ethanol and nicotine on ER stress in human placental villous explants. Curr Res Toxicology. (2022) 3:100081. doi: 10.1016/j.crtox.2022.100081
100. Repo J K, Pesonen M, Mannelli C, Vähäkangas K, Loikkanen J. Exposure to ethanol and nicotine induces stress responses in human placental BeWo cells. Toxicol Letters. (2014) 224:264–71. doi: 10.1016/j.toxlet.2013.10.032
101. Wang X, Lin P, Li Y, Xiang C, Yin Y, Chen Z, et al. Brucella suis Vaccine Strain 2 Induces Endoplasmic Reticulum Stress that Affects Intracellular Replication in Goat Trophoblast Cells In vitro. Front Cell Infect Microbiol. (2016) 6:19/abstract. doi: 10.3389/fcimb.2016.00019/abstract
102. Muthuraj PG, Sahoo PK, Kraus M, Bruett T, Annamalai AS, Pattnaik A, et al. Zika virus infection induces endoplasmic reticulum stress and apoptosis in placental trophoblasts. Cell Death Discovery. (2021) 7:24. doi: 10.1038/s41420-020-00379-8
103. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. (2000) 5:897–904. doi: 10.1016/S1097-2765(00)80330-5
104. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. (1999) 397:271–4. doi: 10.1038/16729
105. Liu Z, Lv Y, Zhao N, Guan G, Wang J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. (2015) 6:e1822–2. doi: 10.1038/cddis.2015.183
106. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. (2000) 6:1099–108. doi: 10.1016/S1097-2765(00)00108-8
107. Neill G, Masson GR. A stay of execution: ATF4 regulation and potential outcomes for the integrated stress response. Front Mol Neurosci. (2023) 16:1112253. doi: 10.3389/fnmol.2023.1112253
108. Li Y, Iida K, O’Neil J, Zhang P, Li S, Frank A, et al. PERK eIF2α Kinase regulates neonatal growth by controlling the expression of circulating insulin-like growth factor-I derived from the liver. Endocrinology. (2003) 144:3505–13. doi: 10.1210/en.2003-0236
109. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk–/– mice reveals a role for translational control in secretory cell survival. Mol Cell. (2001) 7:1153–63. doi: 10.1016/S1097-2765(01)00264-7
110. Jones CJP, Fox H. An ultrastructural and ultrahistochemical study of the human placenta in maternal pre-eclampsia. Placenta. (1980) 1:61–76. doi: 10.1016/S0143-4004(80)80016-6
111. Yung HW, Zhao X, Glover L, Burrin C, Pang PC, Jones CJP, et al. Perturbation of placental protein glycosylation by endoplasmic reticulum stress promotes maladaptation of maternal hepatic glucose metabolism. iScience. (2023) 26:105911. doi: 10.1016/j.isci.2022.105911
112. Yung HW, Hemberger M, Watson ED, Senner CE, Jones CP, Kaufman RJ, et al. Endoplasmic reticulum stress disrupts placental morphogenesis: implications for human intrauterine growth restriction. J Pathol. (2012) 228:554–64. doi: 10.1002/path.2012.228.issue-4
113. Sudhakar A, Ramachandran A, Ghosh S, Hasnain SE, Kaufman RJ, Ramaiah KVA. Phosphorylation of Serine 51 in Initiation Factor 2α (eIF2α) Promotes Complex Formation between eIF2α(P) and eIF2B and Causes Inhibition in the Guanine Nucleotide Exchange Activity of eIF2B. Biochemistry. (2000) 39:12929–38. doi: 10.1021/bi0008682
114. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. (2001) 7:1165–76. doi: 10.1016/S1097-2765(01)00265-9
115. Van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell. (2017) 65:885–899.e6. doi: 10.1016/j.molcel.2017.01.020
116. Lee CL, Veerbeek JHW, Rana TK, Van Rijn BB, Burton GJ, Yung HW. Role of endoplasmic reticulum stress in proinflammatory cytokine–mediated inhibition of trophoblast invasion in placenta-related complications of pregnancy. Am J Pathology. (2019) 189:467–78. doi: 10.1016/j.ajpath.2018.10.015
117. Choi JY, Jo MW, Lee EY, Lee DY, Choi DS. Ovarian steroid dependence of endoplasmic reticulum stress involvement in endometrial cell apoptosis during the human endometrial cycle(2018). Available online at: https://rep.bioscientifica.com/view/journals/rep/155/6/REP-17-0713.xml (accessed November 6, 2024).
118. Liu L, Zhang Y, Wang Y, Peng W, Zhang N, Ye Y. Progesterone inhibited endoplasmic reticulum stress associated apoptosis induced by interleukin-1β via the GRP78/PERK/CHOP pathway in BeWo cells. J Obstet Gynaecol. (2018) 44:463–73. doi: 10.1111/jog.2018.44.issue-3
119. Yung H, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathology. (2008) 173:451–62. doi: 10.2353/ajpath.2008.071193
120. Gupta MB, Abu Shehab M, Nygard K, Biggar K, Singal SS, Santoro N, et al. IUGR is associated with marked hyperphosphorylation of decidual and maternal plasma IGFBP-1. J Clin Endocrinol Metab. (2019) 104:408–22. doi: 10.1210/jc.2018-00820
121. Liu J, Yang W. Mechanism of histone deacetylase HDAC2 in FOXO3-mediated trophoblast pyroptosis in preeclampsia. Funct Integr Genomics. (2023) 23:152. doi: 10.1007/s10142-023-01077-1
122. Xie D, Zhu J, Liu Q, Li J, Song M, Wang K, et al. Dysregulation of HDAC9 represses trophoblast cell migration and invasion through TIMP3 activation in preeclampsia. Am J Hypertension. (2019) 32:515–23. doi: 10.1093/ajh/hpz006
123. Wang P, Zhao C, Zhou H, Huang X, Ying H, Zhang S, et al. Dysregulation of Histone Deacetylases Inhibits Trophoblast Growth during Early Placental Development Partially through TFEB-Dependent Autophagy-Lysosomal Pathway. IJMS. (2023) 24:11899. doi: 10.3390/ijms241511899
124. Jaju Bhattad G, Jeyarajah MJ, McGill MG, Dumeaux V, Okae H, Arima T, et al. Histone deacetylase 1 and 2 drive differentiation and fusion of progenitor cells in human placental trophoblasts. Cell Death Dis. (2020) 11:311. doi: 10.1038/s41419-020-2500-6
125. Almada M, Costa L, Fonseca B, Alves P, Braga J, Gonçalves D, et al. The endocannabinoid 2-arachidonoylglycerol promotes endoplasmic reticulum stress in placental cells. Reproduction. (2020) 160:171–80. doi: 10.1530/REP-19-0539
126. Shi XT, Zhu HL, Xiong YW, Liu WB, Zhou GX, Cao XL, et al. Cadmium down-regulates 11β-HSD2 expression and elevates active glucocorticoid level via PERK/p-eIF2α pathway in placental trophoblasts. Chemosphere. (2020) 254:126785. doi: 10.1016/j.chemosphere.2020.126785
127. Shi XT, Zhu HL, Xu XF, Xiong YW, Dai LM, Zhou GX, et al. Gestational cadmium exposure impairs placental angiogenesis via activating GC/GR signaling. Ecotoxicology Environ Safety. (2021) 224:112632. doi: 10.1016/j.ecoenv.2021.112632
128. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. MBoC. (1999) 10:3787–99. doi: 10.1091/mbc.10.11.3787
129. Thuerauf DJ, Marcinko M, Belmont PJ, Glembotski CC. Effects of the isoform-specific characteristics of ATF6α and ATF6β on endoplasmic reticulum stress response gene expression and cell viability. J Biol Chem. (2007) 282:22865–78. doi: 10.1074/jbc.M701213200
130. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. (2000) 6:1355–64. doi: 10.1016/S1097-2765(00)00133-7
131. Lei Y, Yu H, Ding S, Liu H, Liu C, Fu R. Molecular mechanism of ATF6 in unfolded protein response and its role in disease. Heliyon. (2024) 10:e25937. doi: 10.1016/j.heliyon.2024.e25937
132. Hillary RF, FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. J BioMed Sci. (2018) 25:48. doi: 10.1186/s12929-018-0453-1
133. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. (2007) 13:365–76. doi: 10.1016/j.devcel.2007.07.018
134. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6α Optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. (2007) 13:351–64. doi: 10.1016/j.devcel.2007.07.005
135. Xiong Y, Li W, Lin P, Wang L, Wang N, Chen F, et al. Expression and regulation of ATF6α in the mouse uterus during embryo implantation. Reprod Biol Endocrinol. (2016) 14:65. doi: 10.1186/s12958-016-0199-0
136. Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, et al. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. (2003) 278:31024–32. doi: 10.1074/jbc.M300923200
137. Fradet S, Pierredon S, Ribaux P, Epiney M, Shin Ya K, Irion O, et al. Involvement of membrane GRP78 in trophoblastic cell fusion. PloS One. (2012) 7:e40596. doi: 10.1371/journal.pone.0040596
138. Sicari D, Fantuz M, Bellazzo A, Valentino E, Apollonio M, Pontisso I, et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene. (2019) 38:6184–95. doi: 10.1038/s41388-019-0878-3
139. Soczewski E, Gori S, Paparini D, Grasso E, Fernández L, Gallino L, et al. VIP conditions human endometrial receptivity by privileging endoplasmic reticulum stress through ATF6α pathway. Mol Cell Endocrinol. (2020) 516:110948. doi: 10.1016/j.mce.2020.110948
140. Chau K, Hennessy A, Makris A. Placental growth factor and pre-eclampsia. J Hum Hypertens. (2017) 31:782–6. doi: 10.1038/jhh.2017.61
141. Mizuuchi M, Cindrova-Davies T, Olovsson M, Charnock-Jones DS, Burton GJ, Yung HW. Placental endoplasmic reticulum stress negatively regulates transcription of placental growth factor via ATF4 and ATF6β: implications for the pathophysiology of human pregnancy complications. J Pathology. (2016) 238:550–61. doi: 10.1002/path.2016.238.issue-4
142. Capatina N, Hemberger M, Burton GJ, Watson ED, Yung HW. Excessive endoplasmic reticulum stress drives aberrant mouse trophoblast differentiation and placental development leading to pregnancy loss. J Physiol. (2021) 599:4153–81. doi: 10.1113/tjp.v599.17
143. Lee YY, Hong SH, Lee YJ, Chung SS, Jung HS, Park SG, et al. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem Biophys Res Commun. (2010) 397:735–9. doi: 10.1016/j.bbrc.2010.06.022
144. Pasha M, Kirschenman R, Wooldridge A, Spaans F, Cooke CLM, Davidge ST. The effect of tauroursodeoxycholic Acid (TUDCA) treatment on placental endoplasmic reticulum (ER) stress in a rat model of advanced maternal age. PloS One. (2023) 18:e0282442. doi: 10.1371/journal.pone.0282442
145. Dionísio PA, Amaral JD, Ribeiro MF, Lo AC, D’Hooge R, Rodrigues CMP. Amyloid-β pathology is attenuated by tauroursodeoxycholic acid treatment in APP/PS1 mice after disease onset. Neurobiol Aging. (2015) 36:228–40. doi: 10.1016/j.neurobiolaging.2014.08.034
146. Freitas IN, Da Silva JA Jr., Oliveira KMD, Lourençoni Alves B, Dos Reis Araújo T, Camporez JP, et al. Insights by which TUDCA is a potential therapy against adiposity. Front Endocrinol. (2023) 14:1090039. doi: 10.3389/fendo.2023.1090039
147. Lin T, Diao YF, Kang JW, Lee JE, Kim DK, Jin DI. Tauroursodeoxycholic acid improves the implantation and live-birth rates of mouse embryos. Reprod Biol. (2015) 15:101–5. doi: 10.1016/j.repbio.2015.01.004
148. Mimori S, Ohtaka H, Koshikawa Y, Kawada K, Kaneko M, Okuma Y, et al. 4-Phenylbutyric acid protects against neuronal cell death by primarily acting as a chemical chaperone rather than histone deacetylase inhibitor. Bioorganic Medicinal Chem Letters. (2013) 23:6015–8. doi: 10.1016/j.bmcl.2013.08.001
149. Yung HW, Alnæs-Katjavivi P, Jones CJP, El-Bacha T, Golic M, Staff AC, et al. Placental endoplasmic reticulum stress in gestational diabetes: the potential for therapeutic intervention with chemical chaperones and antioxidants. Diabetologia. (2016) 59:2240–50. doi: 10.1007/s00125-016-4040-2
150. Li Y, Guo Y, Wu D, Ai L, Wu R, Ping Z, et al. Phenylbutyric acid inhibits hypoxia-induced trophoblast apoptosis and autophagy in preeclampsia via the PERK/ATF-4/CHOP pathway. Mol Reprod Devel. (2024) 91:e23742. doi: 10.1002/mrd.23742
151. Mukherjee I, Dhar R, Singh S, Sharma JB, Nag TC, Mridha AR, et al. Oxidative stress-induced impairment of trophoblast function causes preeclampsia through the unfolded protein response pathway. Sci Rep. (2021) 11:18415. doi: 10.1038/s41598-021-97799-y
152. Marek-Iannucci S, Yildirim AD, Hamid SM, Ozdemir AB, Gomez AC, Kocatürk B, et al. Targeting IRE1 endoribonuclease activity alleviates cardiovascular lesions in a murine model of Kawasaki disease vasculitis. JCI Insight. (2022) 7:e157203. doi: 10.1172/jci.insight.157203
153. Li X, Bo Y, Zeng Q, Diao L, Greene S, Patterson J, et al. Population pharmacokinetic model for oral ORIN1001 in Chinese patients with advanced solid tumors. Front Pharmacol. (2024) 15:1322557. doi: 10.3389/fphar.2024.1322557
154. Gabrail NY, Hamilton EP, Elias AD, Rimawi MF, Li C, Corvez MM, et al. A phase 1/2 trial of ORIN1001, a first-in-class IRE1 inhibitor, in patients with advanced solid tumors. JCO. (2021) 39:3080–0. doi: 10.1200/JCO.2021.39.15_suppl.3080
155. Stokes ME, Calvo V, Fujisawa S, Dudgeon C, Huang S, Ballal N, et al. PERK inhibition by HC-5404 sensitizes renal cell carcinoma tumor models to antiangiogenic tyrosine kinase inhibitors. Clin Cancer Res. (2023) 29:4870–82. doi: 10.1158/1078-0432.CCR-23-1182
Keywords: ER stress, UPR, placenta, trophoblast, decidua
Citation: Chowdhury D, Jang CE, Lajoie P and Renaud SJ (2024) A stress paradox: the dual role of the unfolded protein response in the placenta. Front. Endocrinol. 15:1525189. doi: 10.3389/fendo.2024.1525189
Received: 08 November 2024; Accepted: 03 December 2024;
Published: 20 December 2024.
Edited by:
Reinaldo Marín, Instituto Venezolano de Investigaciones Científicas (IVIC), VenezuelaReviewed by:
Estela Bevilacqua, University of São Paulo, BrazilFernanda Regina Giachini, Federal University of Mato Grosso, Brazil
Copyright © 2024 Chowdhury, Jang, Lajoie and Renaud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen J. Renaud, c3JlbmF1ZDRAdXdvLmNh; Patrick Lajoie, cGxham9pZTNAdXdvLmNh