 Lukas Plachy
Lukas Plachy Petra Dusatkova
Petra Dusatkova Shenali Anne Amaratunga
Shenali Anne Amaratunga Vit Neuman
Vit Neuman Zdenek Sumnik
Zdenek Sumnik Jan Lebl
Jan Lebl Stepanka Pruhova
Stepanka Pruhova
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 19 December 2024
Sec. Pediatric Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1506323
Genetic factors play a crucial role in determining human height. Short stature commonly affects multiple family members and therefore, familial short stature (FSS) represents a significant proportion of growth disorders. Traditionally, FSS was considered a benign polygenic condition representing a subcategory of idiopathic short stature (ISS). However, advancements in genetic research have revealed that FSS can also be monogenic, inherited in an autosomal dominant manner and can result from different mechanisms including primary growth plate disorders, growth hormone deficiency/insensitivity or by the disruption of fundamental intracellular pathways. These discoveries have highlighted a broader phenotypic spectrum for monogenic forms of short stature, which may exhibit mild manifestations indistinguishable from ISS. Given the overlapping features and the difficulty in differentiating polygenic from monogenic FSS without genetic testing, some researchers redefine FSS as a descriptive term that encompasses any familial occurrence of short stature, regardless of the underlying cause. This shift emphasizes the complexity of diagnosing and managing short stature within families, reflecting the diverse genetic landscape that influences human growth.
Genetic factors play a crucial role in determining human height with heritability exceeding 80% (1, 2). Consequently, growth failure is frequently present in multiple members of a family making familial short stature (FSS) one of the most common growth disorders (3). However, despite the widespread use of the term of FSS in routine clinical practice, it does not have a universally accepted definition.
Traditionally, FSS was considered a benign condition representing a subcategory of idiopathic short stature (ISS) – a child’s height less than 2 SD of the mean for a given age, sex and population but in concordance with midparental height (4). Polygenic inheritance is typically presumed. Moreover, socioeconomic situation may also contribute to FSS – short stature of a child and his/her parents corresponds with the situation of a population in a specific locality (5). Importantly, this definition presumes normal birth parameters and an absence of a secondary cause of short stature (e.g. systemic disease, endocrine disorder), chromosomal abnormality, monogenic condition or dysmorphic features including clinical signs of bone dysplasia (4).
Due to substantial progress in the genetic examination of short stature, it is now evident that monogenic short stature is more frequent than previously expected. Therefore, FSS may represent a monogenic condition inherited in an autosomal dominant (AD) manner as well (AD-FSS) (6, 7). Typically, one of the parents is substantially shorter than the other - the child’s height corresponds to the shorter parent’s height rather than the midparental height. In AD-FSS, body disproportionality, dysmorphic or other phenotypical signs corresponding to the specific genetic aetiology are assumed (8, 9). However, these clinical features might be subtle and frequently unrecognized (10). Due to these reasons and some others summarized in Table 1, AD-FSS might be very difficult to distinguish from classic FSS. Some authors therefore consider FSS only as a descriptive diagnosis of short stature occurring in multiple generations of a family regardless of the aetiology of the growth disorder or associated clinical features (11–14). This review covers the topic of FSS using this broader definition.
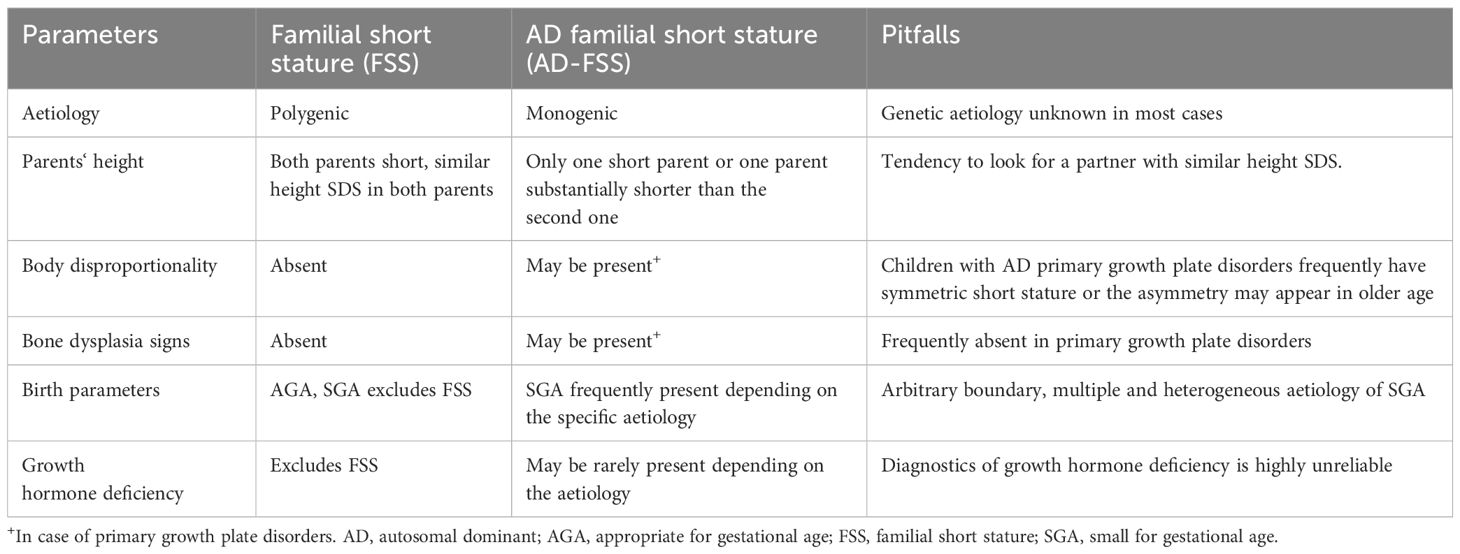
Table 1. Main differences between polygenic familial short stature and autosomal dominant short stature: the traditional approach and its pitfalls.
Polygenic inheritance has long been presumed in FSS (9). Genome wide association studies have identified hundreds of genetic variants affecting human height. These variants are relatively common (allele frequency greater than 1% in the general population), each of them alone has only a small effect on human growth (around 1-4 mm) but may cause short stature if combined (15). Interestingly, several genetic variants have been described with relatively low allele frequency (<5%) and substantially higher individual effect (more than 10 times higher than the above-mentioned common variants). As these variants are frequently located in the genes whose pathogenic variants are known to cause monogenic growth disorders, they can be thought of as variants which create interplay between polygenic and monogenic inheritance (3).
Short stature transmitted in the family in an AD manner can be caused by multiple mechanisms including primary growth plate disorders, GH deficiency/insensitivity or by the disruption of fundamental intracellular pathways (16). The sections that follow will offer insight into the specific mechanisms and phenotypic features potentially causing FSS.
Primary growth plate disorders are frequent cause of AD-FSS (10, 17). In children with pathogenic variants in one of the genes essential for the correct function of the growth plate, disproportionate short stature (with relatively shorter limbs or with relatively shorter trunk) and other signs of bone dysplasia (e.g., bone deformities, scoliosis, brachydactyly) were traditionally believed to be apparent. However, in the past years, studies have proven that the phenotype of children with primary growth plate disorders is very variable including very mild phenotypical signs frequently detectable only via detailed anthropometric examination or short stature clinically unrecognizable from polygenic ISS (16, 18–20). The genetic aetiology of FSS caused by primary growth plate disorders are summarized in a Supplementary Table.
The SHOX (Short Stature Homeobox Containing) gene located in the pseudoautosomal region of sex chromosomes encodes a nuclear transcriptional activator (SHOX protein) that promotes the differentiation of hypertrophic chondrocytes within the growth plate (21, 22). Deficiency of the SHOX protein (SHOX-D) is mostly (80%) caused by deletions involving the SHOX gene itself or its regulatory enhancer. Less frequently, SHOX-D is caused by point SHOX gene variants or by partial or complex SHOX duplication (21, 23). Heterozygous SHOX-D is a relatively frequent cause of a growth disorder with an estimated prevalence 2-15% of short children with a highly variable phenotype (21). Leri-Weil syndrome (LWS) is a condition with mesomelic shortening of the limbs (disproportionate short stature) and typical Madelung deformity of the forearm (bowing and shortening of the radius, subluxation of the distal ulna, pyramidal configuration of carpal bones). In milder forms, broadening of the forearm or its reduced ability to pronate and supinate may appear. Other clinical signs of LWS may include scoliosis, shortening of 4th and 5th metacarpals, high arched palate, micrognathia or muscular hypertrophy of the calves. Typical radiological finding in LWS is a triplet consisting of triangular shape of the distal radial epiphysis, carpal row pyramidalization and lucency of the ulnar side of the distal radius. However, auxological and radiological findings are frequently subtle or completely absent leading to a phenotype of idiopathic short stature, especially in younger children (21, 24–26). SHOX-D is one of the indications for GH therapy (27). Treatment efficacy seems to be equal compared to Turner syndrome leading to an average adult height gain of 7 cm (28).
Natriuretic peptide receptor type B encoded by the NPR2 gene plays an important role in the paracrine regulation of the growth plate. The ligand (C-type natriuretic peptide) binding to the receptor, stimulates cell proliferation, cell differentiation and extracellular matrix synthesis (29, 30). Heterozygous pathogenic variants in the NPR2 gene cause short stature (height -1.5 to -4.3 SD). In some children, clinical signs of bone dysplasia resembling LWS (e.g., disproportionate mesomelic short stature, high-arched palate, bone bowing, brachydactyly), but with no Madelung deformity present. However, most children with heterozygous pathogenic NPR2 gene variants have short stature with no associated dysmorphic features, therefore they could remain undiagnosed. Interestingly, these mutations are responsible for 2-6% of idiopathic short stature including FSS (19, 31–33). Limited data suggest a good short-term response to GH therapy (19).
The FGFR3 gene encodes fibroblast growth factor receptor type 3, a tyrosine kinase receptor that acts as a physiological negative regulator of skeletal growth by inhibiting the proliferation of chondrocytes. Gain-of-function variants in the FGFR3 gene cause multiple disorders of varying severity, which are associated with short stature and variously expressed signs of bone dysplasia (34). Achondroplasia is the most common severe skeletal dysplasia with an incidence approximately 1:20000-1:30000 live births (35). It is characterized by severe short stature (average adult height 130 cm in men and 122 cm in women) (36) with disproportionately short arms and legs, macrocephaly, frontal bossing, mid-face hypoplasia, exaggerated lumbar lordosis, genua vara and bowing of the legs. Affected individuals frequently encounter the complication of foramen magnum stenosis including sleep-breathing disorders and are at an increased risk of infant death (37). The p.Gly380Arg variant is present in 98-99% individuals with achondroplasia. The disease is transmitted in an AD manner, however, approximately 80% of variants occur de novo (34, 37).
Hypochondroplasia is a milder bone dysplasia caused by gain-of-function variants in FGFR3. The prevalence of hypochondroplasia is unknown. Skeletal features are very similar to those seen in achondroplasia but tend to be milder. Body height usually varies between -3 to -2 SD (38). In some cases, clinical signs of bone dysplasia may be very subtle and the individuals with FGFR3 gain-of-function variants have a phenotype corresponding to ISS (10, 39, 40).
Collagens are the most abundant proteins in the human body playing an important structural role. They also participate in the regulation of cell growth, differentiation and migration by interacting with cellular receptors. Collagens II, IX, X and XI are present in the extracellular matrix of the growth plate (18, 41). The defects in individual collagen molecules cause various types of growth plate disorders frequently associated with short stature (41, 42). Variants in COL2A1 gene are known to cause multiple syndromic bone dysplasias (e.g., Kniest dysplasia, Stickler syndrome) with substantial clinical heterogeneity. Bone dysplasia signs (e.g., disproportionate short stature, scoliosis, brachydactyly, metaphyseal abnormalities), distinct facial phenotype (e.g., cleft palate, mid-face hypoplasia), ocular complications (e.g., myopia, retinal detachment), sensorineural hearing loss, and joint deformities are common (18, 43, 44). Heterozygous variants in the COL11A1 gene are known to cause Stickler syndrome, Marshal syndrome and phenotypes overlapping both disorders (45). Heterozygous pathogenic variants in collagen IX genes cause multiple epiphyseal dysplasia which can be associated with proximal muscle weakness (46, 47). Heterozygous variants in the COL10A1 gene cause Schmid-type of epiphyseal chondrodysplasia characterized by short stature, widened growth plates and bowing of the long bones (48). Recently, heterozygous variants in growth plate collagen genes were shown to be a frequent cause of short stature with only subtle or absent syndromic features (18).
Aside from collagen, other proteins play an important role in the correct structure and function of the extracellular matrix of the growth plate. Among them, matrilin-3 (MATN3) and cartilage oligomeric matrix protein (COMP) are the most explored (49). Heterozygous variants in both MATN3 and COMP genes (pathological intracellular accumulation of defective proteins explain the dominant negative effect (50, 51) leading to a phenotype of multiple epiphyseal dysplasia (MED), a relatively heterogeneous condition characterized by short stature, delayed and irregular epiphyseal ossification and early onset osteoarthritis (52, 53). Besides MED, heterozygous variants in COMP gene can cause pseudoachondroplasia with typical asymmetric short limbed short stature, limitation of joint function, bone deformities, scoliosis, spinal stenosis and brachydactyly (54).
The ACAN gene encodes a proteoglycan aggrecan, one of the important components of the extracellular matrix of cartilaginous tissue (55). Pathogenic variants in the ACAN gene disrupt the correct structure and function of the growth plate and other cartilages (8). Most children with a heterozygous pathogenic variant in the ACAN gene have short stature associated with advanced bone age frequently leading to premature growth cessation. Some of the affected children may have some clinical signs of bone dysplasia including frontal bossing, midface hypoplasia, flat nasal bridge or brachydactyly. However, most children have short stature with no apparent body disproportionality or associated syndromic features (8, 55). As aggrecan is also present in the articular cartilage and intervertebral discs, early onset arthritis and intervertebral disc degenerative disease appears frequently in ACAN gene variants (8). Limited data show promising short-term response to GH therapy (8).
The clinical features of growth hormone deficiency are variable. Children with severe GHD may present in neonatal period with hypoglycemia or prolonged icterus. In older children, phenotypical signs typical for GHD (e.g., mid-face hypoplasia, truncal adiposity, thin sparce hair, high pitched voice, premature appearance) may be present. However, most children with GHD have no apparent clinical features besides short stature (56).
Autosomal dominant growth hormone deficiency (GHD) can be inherited by multiple different mechanisms. Impaired regulation of GH secretion might be caused by heterozygous variants in the GHSR gene encoding the ghrelin receptor (ghrelin is a hormone stimulating in GH release). Heterozygous variants in the GHSR gene can lead to the reduction of constitutive activity of the receptor, its intracellular retention or its binding affinity causing a variable degree of GHD (57, 58). Rarely, GHD might be caused by heterozygous mutations directly in the gene for GH. In this case, a splicing mutation in the GH1 gene causes an overproduction of the 17.5 kD isoform of GH that is subsequently retained in the endoplasmic reticulum disrupting the secretion of GH (isolated growth hormone deficiency type II) and possibly other pituitary hormones as well (59). Moreover, GHD may be caused by heterozygous variants in genes affecting pituitary development (e.g., genes SHH, PTCH1, GLI2, OTX2, SOX2, HESX1, PITX2, FGFR1, LHX4, PROKR2) (60–63).
Growth hormone mediates its effect by binding to the GH receptor (GHR). Heterozygous variants in GHR gene cause partial GH insensitivity (64). After GH is bound to GHR, the signal is transduced via a complex intracellular pathway stimulating the production of IGF-1 (insulin like growth factor type 1) (65). This intracellular signaling may be disrupted by mutations in STAT5B gene causing GH insensitivity associated with immune dysregulation that typically has an autosomal recessive inheritance. However, AD transmission has also been described (66). In blood, IGF-1 is stabilized by binding to the acid-labile subunit (ALS) and IGF binding protein type 3 (IGFBP-3) forming a ternary complex. Heterozygous variants in the IGFALS gene encoding the ALS can disrupt the ternary complex thus affecting the function of IGF-1 (65, 67). Heterozygous mutation in the IGF1R gene might lead to impaired IGF-1 sensitivity due to damage to its receptor (68).
The ubiquitous RAS/MAPK signaling pathway has a crucial role in controlling multiple functions of the human body including body height. Heterozygous variants of its components cause a heterogeneous group of disorders called RASopathies (69). In RASopathies, short stature is frequently associated with multiple syndromic features including typical facial appearance, chest deformity, cardiovascular abnormalities, psychomotor retardation or dysplasia of the lymphatic system (70). However, associated syndromic features might be mild and children with RASopathies might be concealed under the phenotype of ISS (71, 72). Besides Noonan syndrome and neurofibromatosis type 1, cardiofaciocutaneous syndrome, Costello syndrome or Legius syndrome are other examples of RASopathies. The genes whose variants are responsible for RASopathies include PTPN11, SOS1, RAF1, RIT1 or KRAS (73). Studies have proven a positive effect of GH treatment in children with Noonan syndrome (average increment of height increment 1.4 SD) (74, 75). Noonan syndrome is included among the diagnostic indications for GH treatment in both Europe and the USA (70, 76).
During the first examination of a child with FSS a possibility of AD-FSS should be considered. Apart from the detailed examination of a child, we recommend evaluating both parents’ height ideally with their body proportionality, possible dysmorphic features, orthopedic or ophthalmological problems, fracture history, presence of heart diseases or other signs that might be associated with a monogenic disorder. In some cases, a detailed pedigree with the height and phenotype of more distant relatives might also be useful. In case a child corresponds to the diagnosis of AD-FSS (see Table 1), it is advisable to indicate detailed genetic examination to determine if there is a monogenic cause for FSS. Even children whose phenotype indicates rather a polygenic aetiology of FSS might have AD-FSS. In this case, genetic examination might also be considered, especially in children with more severe growth disorder.
In the near future, facial recognition systems using artificial intelligence might be a highly effective tool for detecting even minor features typical for a specific genetic disorder associated with short stature. Multiple diseases including endocrine and metabolic disorders and rare genetic syndromes have several specific facial characteristics. Traditionally, evaluating specific facial features were a part of diagnosing genetic disorders, however, the efficacy highly depends on the experience of the physician or clinical anthropologist. Automatic recognition systems have substantial potential to save time, overcome the limited availability of experts skilled in detecting subtle phenotypic features typical for specific disorders or even detect very subtle changes hardly recognizable to the human eye (77–80).
Familial short stature is a heterogeneous entity with a substantial proportion of monogenic causes, which may have subtle phenotypic features, possible co morbidities and variable response to GH therapy. Genetic examination is frequently the only way to differentiate AD-FSS from polygenic FSS and should be considered in children with FSS. Elucidating the genetic diagnosis not only explains the cause of growth disorder in the family and enables a focus on possible hidden co morbidities associated with genetic findings, but is important from a scientific perspective as well. Studies focusing on the genetic examination of FSS have immense potential to further improve our knowledge regarding the aetiology of short stature in general. Moreover, due to the anticipated high detection rate of various genetic conditions, these results might provide crucial data for research aiming to elucidate genetic disorder-specific reaction to GH treatment.
LP: Writing – review & editing, Writing – original draft. PD: Writing – review & editing. SA: Writing – review & editing. VN: Writing – review & editing. ZS: Writing – review & editing. JL: Writing – review & editing. SP: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported by the Ministry of Health of the Czech Republic grant No. NU22J-07-00014 and conceptual development of research organization, Motol University Hospital, Prague, Czech Republic, 00064203.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1506323/full#supplementary-material
1. Jee YH, Baron J, Nilsson O. New developments in the genetic diagnosis of short stature. Curr Opin Pediatr. (2018) 30:541–7. doi: 10.1097/MOP.0000000000000653
2. Willems M, Amouroux C, Barat-Houari M, Salles J-P, Edouard T. Exploring the genetic causes of isolated short stature. What has happened to idiopathic short stature? Arch Pédiatrie. (2022) 28:28/8S27–28/8S32. doi: 10.1016/S0929-693X(22)00040-9
3. Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. (2017) 542:186–90. doi: 10.1038/nature21039
4. Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the growth hormone research society, the Lawson Wilkins pediatric endocrine society, and the European society for paediatric endocrinology workshop. J Clin Endocrinol Metab. (2008) 93:4210–7. doi: 10.1210/jc.2008-0509
5. Suh SB, Kim HS. Influences of socioeconomic status on short stature in childhood. Kosin Med J. (2020) 35:15–25. doi: 10.7180/kmj.2020.35.1.15
6. Dauber A. Genetic testing for the child with short stature - has the time come to change our diagnostic paradigm? J Clin Endocrinol Metab. (2019) 104:2766–9. doi: 10.1210/jc.2019-00019
7. Grigoletto V, Occhipinti AA, Pellegrin MC, Sirchia F, Barbi E, Tornese G. Definition and prevalence of familial short stature. Ital J Pediatr. (2021) 47:56. doi: 10.1186/s13052-021-01018-3
8. Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J Clin Endocrinol Metab. (2016) 102:460–9. doi: 10.1210/jc.2016-3313
9. Vasques GA, Andrade NLM, Jorge AAL. Genetic causes of isolated short stature. Arch Endocrinol Metab. (2019) 63:70–8. doi: 10.20945/2359-3997000000105
10. Plachy L, Petruzelkova L, Dušátková P, Maratova K, Zemkova D, Elblova L, et al. Analysis of children with familial short stature: who should be indicated for genetic testing? Endocr Connect. (2023) 12:e230238. doi: 10.1530/EC-23-0238
11. Plachy L, Strakova V, Elblova L, Obermannova B, Kolouskova S, Snajderova M, et al. High prevalence of growth plate gene variants in children with familial short stature treated with GH. J Clin Endocrinol Metab. (2019) 104:4273–81. doi: 10.1210/jc.2018-02288
12. Kim Y-M, Lim H-H, Kim E, Kim G, Kim M, So H, et al. Exploring the genetic causes for postnatal growth failure in children born non-small for gestational age. J Clin Med. (2023) 12:6508. doi: 10.3390/jcm12206508
13. Sun J, Jiang L, Liu G, Ma C, Zheng J, Niu L. Evaluation of growth hormone therapy in seven Chinese children with familial short stature caused by novel ACAN variants. Front Pediatr. (2022) 10:819074. doi: 10.3389/fped.2022.819074
14. Rani D, Shrestha R, Kanchan T, Krishan K. “Short stature.” In: StatPearls [Internet]. “Treasure Island (FL): StatPearls Publishing. (2023). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK556031/.
15. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. (2014) 46:1173–86. doi: 10.1038/ng.3097
16. Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: A new paradigm emerges. Nat Rev Endocrinol. (2015) 11:736–46. doi: 10.1038/nrendo.2015.165
17. Plachý L, Zemková D, Průhová, Lebl J. Growth plate disorders causing familiar short stature. Cesk Pediatr. (2018) 73:110–7.
18. Plachy L, Dusatkova P, Maratova K, Petruzelkova L, Elblova L, Kolouskova S, et al. Familial Short Stature - a novel phenotype of growth plate collagenopathies. J Clin Endocrinol Metab. (2021) 106:1742–9. doi: 10.1210/clinem/dgab084
19. Plachy L, Dusatkova P, Maratova K, Petruzelkova L, Zemkova D, Elblova L, et al. NPR2 variants are frequent among children with familiar short stature and respond well to growth hormone therapy. J Clin Endocrinol Metab. (2020) 105:dgaa037. doi: 10.1210/clinem/dgaa037
20. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. (2014) 99:3080–92. doi: 10.1210/jc.2014-1506
21. Binder G. Short stature due to SHOX deficiency: Genotype, phenotype, and therapy. Horm Res Paediatr. (2011) 75:81–9. doi: 10.1159/000324105
22. Marchini A, Ogata T, Rappold GA. A track record on SHOX: from basic research to complex models and therapy. Endocr Rev. (2016) 37:417–48. doi: 10.1210/er.2016-1036
23. Schneider KU, Marchini A, Sabherwal N, Röth R, Niesler B, Marttila T, et al. Alteration of DNA binding, dimerization, and nuclear translocation of SHOX homeodomain mutations identified in idiopathic short stature and Leri-Weill dyschondrosteosis. Hum Mutat. (2005) 26:44–52. doi: 10.1002/humu.20187
24. Jorge AAL, Souza SC, Nishi MY, Billerbeck AE, Libório DCC, Kim CA, et al. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf). (2007) 66:130–5. doi: 10.1111/j.1365-2265.2006.02698.x
25. Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, Quigley CA, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. (2007) 44:306–13. doi: 10.1136/jmg.2006.046581
26. Seki A, Jinno T, Suzuki E, Takayama S, Ogata T, Fukami M. Skeletal deformity associated with SHOX deficiency. Clin Pediatr Endocrinol. (2014) 23:65–72. doi: 10.1297/cpe.23.65
27. Loche S, Carta L, Ibba A, Guzzetti C. Growth hormone treatment in non-growth hormone-deficient children. Ann Pediatr Endocrinol Metab. (2014) 19:1–7. doi: 10.6065/apem.2014.19.1.1
28. Blum WF, Ross JL, Zimmermann AG, Quigley CA, Child CJ, Kalifa G, et al. GH treatment to final height produces similar height gains in patients with SHOX deficiency and turner syndrome: Results of a multicenter trial. J Clin Endocrinol Metab. (2013) 98:1383–92. doi: 10.1210/jc.2013-1222
29. Irfanullah, Zeb A, Shinwari N, Shah K, Gilani SZT, Khan S, et al. Molecular and in silico analyses validates pathogenicity of homozygous mutations in the NPR2 gene underlying variable phenotypes of Acromesomelic dysplasia, type Maroteaux. Int J Biochem Cell Biol. (2018) 102:76–86. doi: 10.1016/j.biocel.2018.07.004
30. Dickey DM, Edmund AB, Otto NM, Chaffee TS, Robinson JW, Potter LR. Catalytically active guanylyl cyclase B requires endoplasmic reticulum-mediated glycosylation, and mutations that inhibit this process cause dwarfism. J Biol Chem. (2016) 291:11385–93. doi: 10.1074/jbc.M115.704015
31. Vasques GA, Amano N, Docko AJ, Funari MFA, Quedas EPS, Nishi MY, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab. (2013) 98:1636–44. doi: 10.1210/jc.2013-2142
32. Amano N, Mukai T, Ito Y, Narumi S, Tanaka T, Yokoya S, et al. Identification and functional characterization of two novel NPR2 mutations in Japanese patients with short stature. J Clin Endocrinol Metab. (2014) 99:713–8. doi: 10.1210/jc.2013-3525
33. Yuan K, Chen J, Chen Q, Chen H, Zhu J, Fang Y, et al. NPR2 gene variants in familial short stature: A single-center study. J Pediatr Endocrinol Metab. (2022) 35:185–90. doi: 10.1515/jpem-2021-0332
34. Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: The current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat. (2012) 33:29–41. doi: 10.1002/humu.21636
35. Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, et al. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am J Med Genet A. (2008) 146A:2385–9. doi: 10.1002/ajmg.a.32485
36. Hoover-Fong JE, Schulze KJ, Alade AY, Bober MB, Gough E, Hashmi SS, et al. Growth in achondroplasia including stature, weight, weight-for-height and head circumference from CLARITY: achondroplasia natural history study—a multi-center retrospective cohort study of achondroplasia in the US. Orphanet J Rare Dis. (2021) 16:522. doi: 10.1186/s13023-021-02141-4
37. Savarirayan R, Ireland P, Irving M, Thompson D, Alves I, Baratela WAR, et al. International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia. Nat Rev Endocrinol. (2022) 18:173–89. doi: 10.1038/s41574-021-00595-x
38. Bober MB, Bellus GA, Nikkel SM, Tiller GE. Hypochondroplasia. 1999 Jul 15. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (1993–2024).
39. Heuertz S, Le Merrer M, Zabel B, Wright M, Legeai-Mallet L, Cormier-Daire V, et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur J Hum Genet. (2006) 14:1240–7. doi: 10.1038/sj.ejhg.5201700
40. Kant SG, Cervenkova I, Balek L, Trantirek L, Santen GWE, de Vries MC, et al. A novel variant of FGFR3 causes proportionate short stature. Eur J Endocrinol. (2015) 172:763–70. doi: 10.1530/EJE-14-0945
41. Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. (2011) 3:1–19. doi: 10.1101/cshperspect.a004978
42. Shen G. The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod Craniofac Res. (2005) 8:11–7. doi: 10.1111/j.1601-6343.2004.00308.x
43. Deng H, Huang X, Yuan L. Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res. (2016) 768:1–13. doi: 10.1016/j.mrrev.2016.02.003
44. Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, et al. Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum Mutat. (2016) 37:7–15. doi: 10.1002/humu.22915
45. Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am J Med Genet A. (2007) 143A:258–64. doi: 10.1002/ajmg.a.v143a:3
46. Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perälä M, Carter L, et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am J Hum Genet. (2001) 69:969–80. doi: 10.1086/324023
47. Bonnemann CG, Cox GF, Shapiro F, Wu J-J, Feener CA, Thompson TG, et al. A mutation in the alpha 3 chain of type IX collagen causes autosomal dominant multiple epiphyseal dysplasia with mild myopathy. Proc Natl Acad Sci. (2000) 97:1212–7. doi: 10.1073/pnas.97.3.1212
48. Mäkitie O, Susic M, Ward L, Barclay C, Glorieux FH, Cole WG. Schmid type of metaphyseal chondrodysplasia and COL10A1 mutations - Findings in 10 patients. Am J Med Genet. (2005) 137A:241–8. doi: 10.1002/ajmg.a.30855
49. Wagener R, Kobbe B, Aszódi A, Liu Z, Beier DR, Paulsson M. Original Contributions Structure and mapping of the mouse matrilin-3 gene (Matn3), a member of a gene family containing a U12-type AT-AC intron. Mamm Genome. (2000) 11:85–90. doi: 10.1007/s003350010018
50. Délot E, Brodie SG, King LM, Wilcox WR, Cohn DH. Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J Biol Chem. (1998) 273:26692–7. doi: 10.1074/jbc.273.41.26692
51. Cotterill SL, Jackson GC, Leighton MP, Wagener R, Mäkitie O, Cole WG, et al. Multiple epiphyseal dysplasia mutations inMATN3 cause misfolding of the A-domain and prevent secretion of mutant matrilin-3. Hum Mutat. (2005) 26:557–65. doi: 10.1002/humu.20263
52. Handa A, Grigelioniene G, Nishimura G. Skeletal dysplasia families: A stepwise approach to diagnosis. RadioGraphics. (2023) 43:e220067. doi: 10.1148/rg.220067
53. Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD. Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat Genet. (2001) 28:393–6. doi: 10.1038/ng573
54. Zhou L, Chen J, Liu Q, Yang S, Xie W, Peng Y. Case Report: Whole-exome sequencing identified two novel COMP variants causing pseudoachondroplasia. Front Endocrinol (Lausanne). (2023) 14:1267946. doi: 10.3389/fendo.2023.1267946
55. Gibson BG, Briggs MD. The aggrecanopathies; An evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis. (2016) 11:86. doi: 10.1186/s13023-016-0459-2
56. Dattani MT, Malhotra N. A review of growth hormone deficiency. Pediatr Child Health. (2019) 7:285–92. doi: 10.1016/j.paed.2019.04.001
57. Pantel J. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest. (2006) 116:760–8. doi: 10.1172/JCI25303
58. Inoue H, Kangawa N, Kinouchi A, Sakamoto Y, Kimura C, Horikawa R, et al. Identification and functional analysis of novel human growth hormone secretagogue receptor (GHSR) gene mutations in Japanese subjects with short stature. J Clin Endocrinol Metab. (2011) 96:E373–8. doi: 10.1210/jc.2010-1570
59. Miletta MC, Lochmatter D, Pektovic V, Mullis PE. Isolated growth hormone deficiency type 2: From gene to therapy. Endocr Dev. (2012) 23:109–20. doi: 10.1159/000341766
60. Alatzoglou KS, Webb EA, Le Tissier P, Dattani MT. Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev. (2014) 35:376–432. doi: 10.1210/er.2013-1067
61. Sano S, Masunaga Y, Kato F, Fujisawa Y, Saitsu H, Ogata T. Combined pituitary hormone deficiency in a patient with an FGFR1 missense variant: case report and literature review. Clin Pediatr Endocrinol. (2022) 31:2022–0020. doi: 10.1297/cpe.2022-0020
62. Kardelen AD, Najaflı A, Baş F, Karaman B, Toksoy G, Poyrazoğlu ŞChecktae, et al. PROKR2 mutations in patients with short stature who have isolated growth hormone deficiency and multiple pituitary hormone deficiency. J Clin Res Pediatr Endocrinol. (2023) 15:338–47. doi: 10.4274/jcrpe.galenos.2023.2023-4-4
63. Plachy L, Maratova K, Vesela K, Lebl J, Pruhova S. Current view on the diagnostics of growth hormone deficiency in childhood and adolescence. Cesslov Pediat. (2023) 78:5–10. doi: 10.55095/CSPediatrie2023/056
64. Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. J Endocr Soc. (2017) 1:345–58. doi: 10.1210/js.2016-1119
65. Mastromauro C, Giannini C, Chiarelli F. Short stature related to Growth Hormone Insensitivity (GHI) in childhood. Front Endocrinol (Lausanne). (2023) 14:1141039. doi: 10.3389/fendo.2023.1141039
66. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. (2018) 9:2105. doi: 10.1038/s41467-018-04521-0
67. Domené HM, Scaglia PA, Martínez AS, Keselman AC, Karabatas LM, Pipman VR, et al. Heterozygous IGFALS gene variants in idiopathic short stature and normal children: Impact on height and the IGF system. Horm Res Paediatr. (2014) 80:413–23. doi: 10.1159/000355412
68. Choi J-H, Kang M, Kim G-H, Hong M, Jin HY, Lee B-H, et al. Clinical and functional characteristics of a novel heterozygous mutation of the IGF1R gene and IGF1R haploinsufficiency due to terminal 15q26.2-<qter deletion in patients with intrauterine growth retardation and postnatal catch-up growth failure. J Clin Endocrinol Metab. (2011) 96:E130–4. doi: 10.1210/jc.2010-1789
69. Malaquias AC, Jorge AAL. Activation of the MAPK pathway (RASopathies) and partial growth hormone insensitivity. Mol Cell Endocrinol. (2021) 519:111040. doi: 10.1016/j.mce.2020.111040
70. Lebl J, Koloušková S, Toni L, Kodýtková A, Amaratunga SA, Plachý L, et al. Syndrom Noonanové a další RASopatie: Etiologie, diagnostika a terapie. Čes-slov Pediat. (2020) 75:219–26.
71. Toni L, Plachy L, Dusatkova P, Amaratunga SA, Elblova L, Sumnik Z, et al. The genetic landscape of children born small for gestational age with persistent short stature (SGA-SS). Horm Res Paediatr. (2023) 97:40–52. doi: 10.1159/000530521
72. Ferreira LV, Souza SCAL, Montenegro LR, Malaquias AC, Arnhold IJP, Mendonca BB, et al. Analysis of the PTPN11 gene in idiopathic short stature children and Noonan syndrome patients. Clin Endocrinol (Oxf). (2008) 69:426–31. doi: 10.1111/j.1365-2265.2008.03234.x
73. Hebron KE, Hernandez ER, Yohe ME. The RASopathies: from pathogenetics to therapeutics. Dis Model Mech. (2022) 15:dmm049107. doi: 10.1242/dmm.049107
74. Noonan JA, Kappelgaard A-M. The efficacy and safety of growth hormone therapy in children with Noonan syndrome: A review of the evidence. Horm Res Paediatr. (2015) 83:157–66. doi: 10.1159/000369012
75. Giacomozzi C, Deodati A, Shaikh MG, Ahmed SF, Cianfarani S. The impact of growth hormone therapy on adult height in Noonan syndrome: A systematic review. Horm Res Paediatr. (2015) 83:167–76. doi: 10.1159/000371635
76. Danowitz M, Grimberg A. Clinical indications for growth hormone therapy. Adv Pediatr. (2022) 69:203–17. doi: 10.1016/j.yapd.2022.03.005
77. Qiang J, Wu D, Du H, Zhu H, Chen S, Pan H. Review on facial-recognition-based applications in disease diagnosis. Bioengineering. (2022) 9:273. doi: 10.3390/bioengineering9070273
78. Loos HS, Wieczorek D, Würtz RP, von der Malsburg C, Horsthemke B. Computer-based recognition of dysmorphic faces. Eur J Hum Genet. (2003) 11:555–60. doi: 10.1038/sj.ejhg.5200997
79. Kosilek RP, Frohner R, Würtz RP, Berr CM, Schopohl J, Reincke M, et al. Diagnostic use of facial image analysis software in endocrine and genetic disorders: review, current results and future perspectives. Eur J Endocrinol. (2015) 173:M39–44. doi: 10.1530/EJE-15-0429
Keywords: familial short stature, autosomal dominant short stature, genetics, growth plate, short stature
Citation: Plachy L, Dusatkova P, Amaratunga SA, Neuman V, Sumnik Z, Lebl J and Pruhova S (2024) Monogenic causes of familial short stature. Front. Endocrinol. 15:1506323. doi: 10.3389/fendo.2024.1506323
Received: 04 October 2024; Accepted: 21 November 2024;
Published: 19 December 2024.
Edited by:
Huseyin Demirbilek, Hacettepe University, TürkiyeReviewed by:
Alan David Rogol, University of Virginia, United StatesCopyright © 2024 Plachy, Dusatkova, Amaratunga, Neuman, Sumnik, Lebl and Pruhova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenali Anne Amaratunga, c2hlbmFsaS5hbWFyYXR1bmdhQGZubW90b2wuY3o=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.