
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol., 06 February 2025
Sec. Neuroendocrine Science
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1481906
 Stéfanie Parisien-La Salle1
Stéfanie Parisien-La Salle1 Florence Perreault1Gilles Corbeil1
Florence Perreault1Gilles Corbeil1 Julie Morisset2Charles Poirier2Catherine Beauregard1Agnès Räkel1
Julie Morisset2Charles Poirier2Catherine Beauregard1Agnès Räkel1 Marjorie Labrecque3
Marjorie Labrecque3 Martine Tétreault3,4Christian Cohade5Pasquale Ferraro6
Martine Tétreault3,4Christian Cohade5Pasquale Ferraro6 Isabelle Bourdeau1*†
Isabelle Bourdeau1*†Background: Pheochromocytomas (PCCs) and paragangliomas (PGLs) (PPGLs) are rare tumours arising from the chromaffin cells. There is evidence suggesting a link between hypoxemia and PPGLs. Chronic hypoxia can lead to gain of function somatic variants in the EPAS1 gene that encodes for hypoxia-inducible factor 2-alpha (HIF-2α), involved in PPGL tumorigenesis.
Objective: To describe a rare case of PCC in a pulmonary transplant patient and characterize the tumour’s genetic background.
Clinical Case: A 47 year-old man underwent a lung transplant for chronic obstructive pulmonary disease associated with alpha-1 antitrypsin deficiency. He required home oxygen therapy for 3 years prior to transplant. Nineteen years after transplant, a CT-scan revealed a 5.8 cm x 3.9 cm heterogeneous right adrenal mass (HU of 7). Initial assessments indicated elevated 24-hour urinary catecholamines. Consequently, the patient underwent laparoscopic right adrenalectomy, confirming the PCC diagnosis.
Genetic studies: 1) Germline PPGL multigene panel: After consent, the patient underwent a panel of 14 susceptibility genes for PPGLs that revealed no pathogenic variants. 2) Somatic genetic analysis for EPAS1 gene found no variants. However, tumoral RNA sequencing unveiled activation of the HIF pathway.
Conclusion: We describe a rare case of PCC in a pulmonary transplant recipient, with genetic analyses showing no germline pathogenic variants and no somatic variants in the EPAS1 gene. RNA sequencing highlighted HIF pathway activation and angiogenic implications. Further research is necessary to elucidate the genetic and molecular mechanisms underlying PCCs in this specific case and determine its link with hypoxemia in the context of pulmonary disease.
Pheochromocytomas (PCCs) and paragangliomas (PGLs) (PPGLs) are rare neuroendocrine tumours arising from the adrenal medulla or along the sympathetic/parasympathetic ganglia chains (1). PPGLs are the most heritable tumours, with 30–35% of patients of European descent having a germline variant in a known susceptibility gene (2). More than 20 susceptibility genes for PPGLs have been described (germline and somatic) and are divided into 3 clusters: pseudohypoxia cluster 1 (1A and 1B), kinase-signalling cluster 2, and Wnt signalling cluster 3 (2, 3).
Several studies have shown that different hypoxemic states can be linked to the pathogenesis of PPGLs including high altitude, congenital heart anomalies or a pseudohypoxic state due to variants in cluster 1 genes (2–6). Indeed, tumours with pathogenic variants in cluster 1 genes display a stabilization and accumulation of hypoxia induced factor (HIF), leading to angiogenesis and tumour invasion (2, 6). Chronic hypoxia due to high altitude has been shown to be associated with a higher incidence of head and neck paragangliomas (HNPGLs) (4). Genetic analysis of a small group of high altitude HNPGLs yielded negative results for pathogenic variants in SDHB and SDHD genes (7). Recently, a meta-regression analysis was performed and showed a higher incidence of PPGLs in areas of higher altitude (8). However, this study was criticized due to the lack of inclusion of studies and heterogeneity within the studies (9). Another hypoxia inducing pathology with an association to PPGLs is cyanotic congenital heart disease (CCHD) (5). In a study by Opotowsky et al., CCHD hospitalised patients had an odds ratio of 6 for developing PPGLs compared to patients without CCHD (5). Moreover, gain of function somatic variants in the EPAS1 gene that encodes for hypoxia-inducible factor 2-alpha (HIF2α) were identified in CCHD associated PPGLs (10). In a study by Vaidya et al., 4/5 (80%) CCHD related PPGLs harboured a somatic variant in codon 530 or 531 in exon 12 of EPAS1. These residues code for the “oxygen degradation domain of HIF2α” (10). Out of the 5 patients for whom germline DNA was available (4/5), no pathogenic variants were identified in EPAS1 or the 10 PPGL susceptibility genes that were tested. This being said, it remains uncertain whether a hypoxemic state in other conditions, such as pulmonary diseases, could also contribute to the development of PPGLs. Here, we report a rare occurrence of PCC in a pulmonary transplant recipient, providing genetic characterization of the tumour.
We report the case of a 66 year-old man who underwent a unilateral lung transplant at the age of 47 for chronic obstructive pulmonary disease associated with alpha-1 antitrypsin deficiency. Prior to transplant, his forced expiratory volume in 1 second (FEV1) was 1.49 L (11% of his predicted value) and his forced vital capacity (FVC) was 1.45 L (25% of his predicted value). His exercise test showed a VO2 max of 0.7 L/min (21% of predicted value). At room air, his 6 min walk test results showed a total distance 150.4 m with a saturation of 89% at 73% of max heart rate. He required home oxygen therapy for 3 years prior to transplant. Following transplant, the patient was diagnosed with stage 4 chronic kidney disease, hypertension under dual therapy and paroxysmal atrial tachycardia and post-transplantation diabetes mellitus (PTDM). Patient’s family history was non-contributing. Hypoxia resolved after pulmonary transplant, with the patient achieving normal ambient air saturation and no longer requiring supplemental oxygen.
Eleven years after transplant, a follow-up chest CT scan described a right adrenal nodule of 1.9 x 0.8 cm. Two years later, on a chest CT scan, the nodule grew and was measured at 2.4 x 1.1 cm. No density measurements were described. Urinary catecholamines were ordered and urinary metanephrines were mildly elevated (norepinephrine 211 nmol/d (N <650), epinephrine <10 nmol/d (N <145), dopamine 867 nmol/d (N <4520), normetanephrines 507 nmol/d (N <600) and metanephrines 613 nmol/d (N <370)). However, the test result was lost to follow-up.
Nineteen years after transplant, a follow-up thoracic CT scan revealed the growth of the right adrenal nodule and described it as a heterogenous 5.8 cm x 3.9 cm mass with a density of 7 Hounsfield Units (HU) (Figure 1). An adrenal panel was ordered, and the diagnosis of PCC was confirmed by the 24 h urinary catecholamines (norepinephrine 713 nmol/d (N <650), epinephrine 588 nmol/d (N <145), dopamine 205 nmol/d (N <4520), normetanephrines 900 nmol/d (N <600) and metanephrines 1191 nmol/d (N <370)). Chromogranin A was also elevated (3297 ng/mL (N <104)), as were plasmatic metanephrines (6.99 nmol/L (n <0.48)) and normetanephrines (5.79 nmol/L (N <1.20)). The rest of the hormonal adrenal panel was normal (Table 1). The adrenal mass showed no uptake at 18F-FDG PET/CT imaging but fixation on MIBG scintigraphy (Figure 1). The patient reported rare episodes of diaphoresis, but no palpitations or headaches. He underwent a laparoscopic right adrenalectomy. The pathology report confirmed a 6.2 cm PCC with capsular invasion, micro foci of invasion of the periadrenal fatty tissue, pleomorphic cells, cellular monotony and large nests (PASS score 8-10). Staining for SDHB protein was intact and ki-67 proliferation index was <1%. Two years following PCC resection, patient’s diabetes and hypertension remained unchanged. Imaging and biochemical testing showed no signs of PCC recurrence.
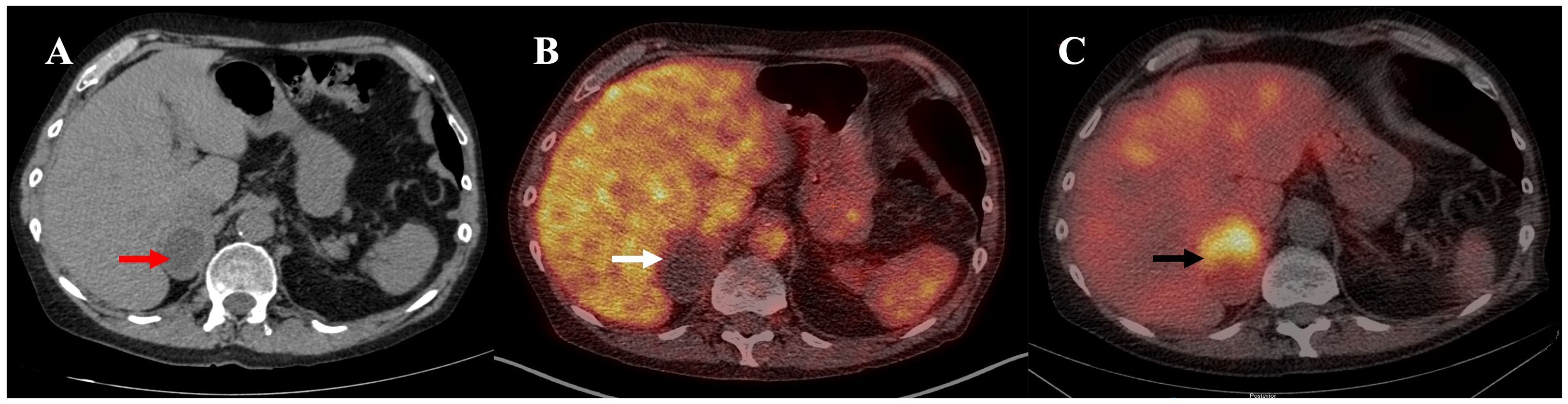
Figure 1. (A) Unenhanced CT: heterogenous 5.8 cm x 3.9 cm right adrenal mass with a density of 7 Hounsfield Units (HU) (red arrow). (B) 18F-FDG PET/CT, fused image: inactive 5.8 x 4.0 x 4.1cm right adrenal mass (white arrow). (C) 123I-MIBG SPECT/CT, fused image: right adrenal mass with intense MIBG uptake (black arrow).
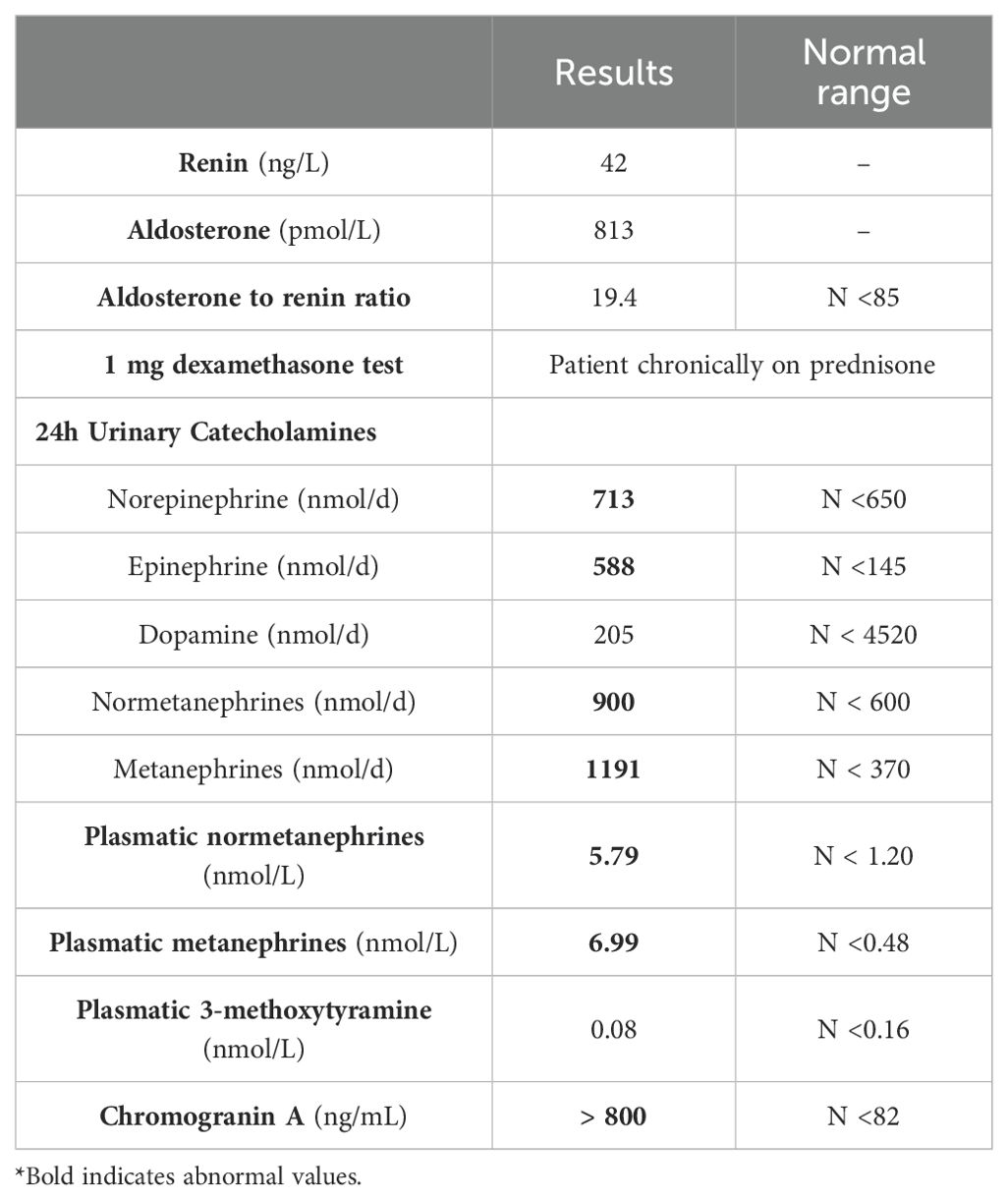
Table 1. Hormonal Adrenal panel results.
Measurement of free urinary catecholamines and metanephrines:
Samples from 24-hour urine collections, with the addition of 3,4-dihydroxybenzylamine as an internal standard, were adjusted to pH 6.5 prior to off-line solid phase extraction on ion-exchange mini-columns (Bio-Rad). Catecholamines and free metanephrines were separated on a Kinetex LC C18 100A column (150 x 4.6 mm, 5 µm; Phenomenex) using isocratic elution on an Ultimate 3000 HPLC system (Thermo Scientific) coupled to a Coulochem III electrochemical detector (ESA).
Plasma samples, with the addition of deuterated internal standards, were subjected to on-line SPE extraction (CHRO SPE Polymer WCX cartridges), followed by HPLC separation of free metanephrines on an Atlantis HILIC silica column (100 A, 2.1 X 50 mm, 3 µm; Waters) and detection with tandem mass spectrometry on a Chronect Symbiosis-TSQ Quantis LC-MSMS system (Spark Holland/Thermo).
The patient underwent genetic counselling, and a multigene panel was ordered that included 14 susceptibility genes: SDHA, SDHAF2, SDHB, SDHC, SDHD, RET, VHL, FH, NF1, MAX, TMEM127, EGLN1, KIF1B, MEN1 genes (Invitae, San Francisco, CA). Gene sequencing and deletion/duplication analysis were performed using NGS technology for all genes except for SDHA, which was not evaluated for deletion/duplication. Written consent form was obtained from the patient before genetic analysis. Leucocyte DNA was extracted from whole blood cells.
Tumoral DNA was extracted from FFPE tissues as described previously (11). Exons 9, 12 and 16 of the EPAS1 gene were studied by Sanger Sequencing (Supplementary Table S1). These 3 exons were chosen as they are sites for the three regulatory hydroxylation domains (10). The amplicons were directly sequenced using the Applied Biosystems 3730xl DNA Analyzer (The Centre d’expertise et de services Génome Québec CHU-Sainte-Justine).
RNA sequencing of patient leukocyte and tumoral RNA was performed (Research Center CHU Québec) and a commercially available pool of human adrenal total RNA (Clontech, Mountain View, CA) was used as a reference sample for tumoral RNA. Five additional patients with PPGLs, who were not exposed to chronic hypoxia and tested negative on the multigene panel for PPGLs, were also included as a combined control group (RNA and tumoral RNA).
No pathogenic variants or variants of unknown significance were detected in the multigene panel, which comprised 14 susceptibility genes for PPGLs.
No pathogenic variants were identified in all three tested exons of the EPAS1 gene.
Compared to leucocyte RNA of a pool of five patients with PPGL who did not undergo chronic hypoxia and had a negative multigene panel, our patient’s leucocyte RNA showed an overexpression of EPAS1 (or HIF2α) (log2 fold change: 5.69 p=1.26E-08). EPAS1 is implicated in multiple pathways, including ‘Pathways in cancer’ (hsa05200). We conducted KEGG pathway enrichment analysis using DAVID software (12). Notably, our analysis revealed that our top upregulated genes (452 genes with p < 0.05) are significantly associated with this pathway, showing a p-value of 0.0017 and a fold enrichment of 1.9. Other upregulated genes in this pathway included HIF1α (log2 fold change: 3.80 p=0.000145) and VEGF-C (log2 fold change: 2.46 p=0.0140). Interestingly, EGLN2, a HIF1α inhibitor, was significantly downregulated (log2 fold change: -2.65 p=0.00813) (Table 2A).
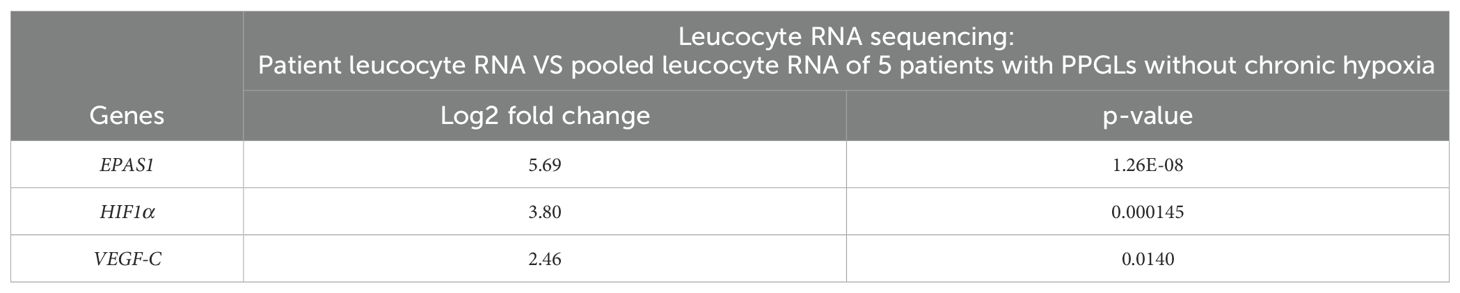
Table 2A. Leucocyte RNA sequencing.
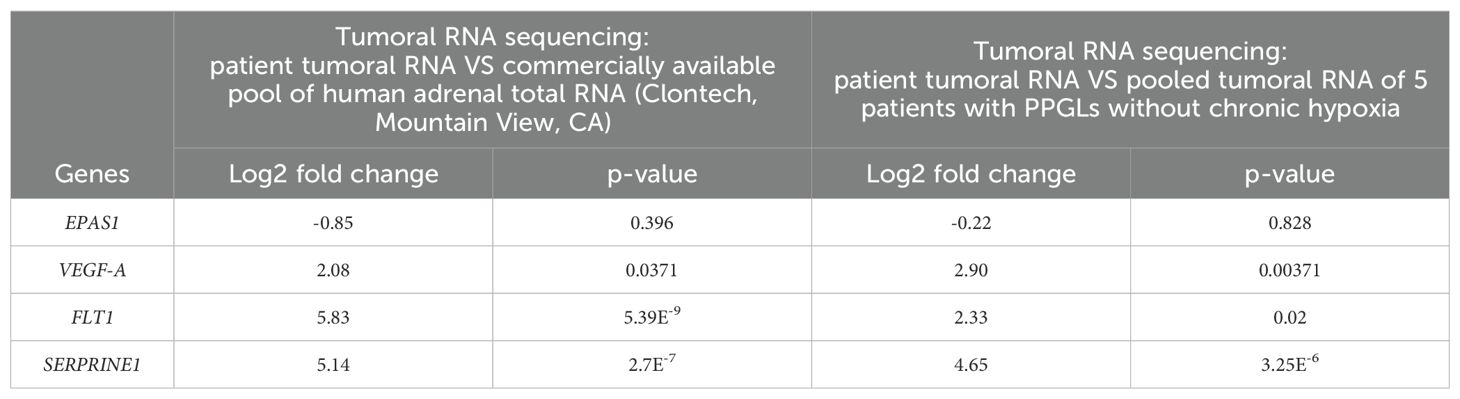
Table 2B. Tumoral RNA sequencing.
EPAS1 was not found to be significantly modified in tumour tissue comparisons in either human adrenal total RNA (log2 fold change: -0.85 p=0.396) or tumoral RNA of other PPGLs (log2 fold change: -0.22 p=0.828).
However, in the HIF-1 KEGG signalling pathway (hsa04066), three genes implicated in angiogenesis were highly upregulated when compared to human adrenal total RNA and other PPGL tumoral RNA: VEGF-A (human adrenal total RNA: log2 fold change: 2.08 p=0.0371; other PPGL tumoral RNA: log2 fold change: 2.90 p=0.00371), FLT1 (or VEGFR1) (human adrenal total RNA: log2 fold change: 5.83 p=5.39E-9; other PPGL tumoral RNA: log2 fold change: 2.33 p=0.02), SERPRINE1 (human adrenal total RNA: log2 fold change: 5.14 p=2.7E-7; other PPGL tumoral RNA: log2 fold change: 4.65 p=3.25E-6) (Table 2B).
This case illustrates the rare association between a lung transplant patient and PCC. As described above, hypoxia plays a significant role in the pathogenesis of PPGLs. Our case is of particular interest because it describes a patient with chronic hypoxia not induced by CCHD or altitude who developed a PCC.
The timing and degree of hypoxia associated with PPGLs remains unknown. In a study by Opotowsky et al., the mean duration of hypoxia for PPGLs associated with CCHD was 20 years (range 1–57 y) (5). In a review of patients with CCHD who underwent a Fontan procedure and were diagnosed with PPGLs, cyanosis duration ranged from 15 months to 13 years (13). It remains unclear whether the timing or degree of hypoxia plays a part in the pathogenesis. Interestingly, our patient did not present a known pathogenic variant in the germline PPGL multigene panel. This is often the case in patients with CCHD related PPGLs (5, 14). One patient with CCHD and a multifocal recurrent PGL harboured an SDHB pathogenic missense variant (11). Also, our patient did not present with an EPAS1 somatic variant, that is often found in CCHD associated PPGLs (10).
To explore if the patient’s chronic hypoxia played a role in the pathogenesis of his PCC, we performed RNA sequencing. We found activation of HIF pathway at various levels. Leukocyte RNA showed overexpression of EPAS1, HIF1α and VEGF-C, while tumoral RNA sequencing revealed overexpression of genes in the HIF pathway implicated in angiogenesis (VEGF-A, FLT1, SERPINE1). Interestingly, an in vitro study showed that VHL knockdown PC12 cells also overexpress SERPINE1 (15). In a recent study, PC12 cells (Rat pheochromocytoma cells) were exposed to 20 recurrent hypoxia cycles (PC12 Z20) (16). RNA sequencing demonstrated an upregulation of HIF2α (Hypoxia-Inducible Factor 2 Alpha) in these cells. The authors observe that: “PC12 Z20 cells showed a higher growth rate, and the migration and adhesion capacity were significantly increased compared with control cells.” Although these findings are interesting, they do not clearly prove that hypoxia was the driving factor for the development of the PCC.
Another driving factor for the patient’s PCC could have been his transplant. Transplant patients have higher malignancy rates than the general population (17). Chronic immunosuppressive therapy following a transplant can play a role in the development of malignancies (17). Lung transplant patients receive more immunosuppression than other soft organ transplants (17). The most frequent malignancies following lung transplant are skin cancer, lung cancer and post-transplant lymphoproliferative disease (17). In the literature, we identified two cases of PCCs in lung transplant patients (18, 19). The first case is a 44-year-old patient with heart, lung and renal transplantation secondary to cystic fibrosis and diabetes. She was diagnosed with a PCC following multiple episodes of hypertension and tachycardia. No genetic analysis was done on the tumor or germline DNA and no information was given on duration of hypoxia before diagnosis of PCC or the timing of the diagnosis after the transplant (18). The second case is a 25-year-old man with double lung transplant for cystic fibrosis who was diagnosed with a PCC after investigations for resistant hypertension. Again, no genetic analysis was reported on the tumor and germline DNA results were pending and no information was given on duration of hypoxia or timing of transplant in regards to the diagnosis of the PCC (19).
Another interesting point of this case is the radiological description of the adrenal lesion. Indeed, the patient presented with a heterogeneous PCC that had an overall density of 7 HU on a CT-scan without contrast. This is a very rare finding. So rare, that some guidelines do not recommend to screen biochemically adrenal incidentalomas with ≤ 10 HU for PCCs (20, 21). However, an important nuance is that these guidelines also recommend that a heterogeneous mass should be considered for biochemical testing (20, 21). In a large retrospective multicentre study, the sensitivity of the 10 HU cut-off for identifying a PCC was 99.6% (22). The only patient who presented with a PCC of -4 HU also had an ACTH ectopic co-secretion by the PCC, which could lead to a false negative due to higher cholesterol content within the tumor (22).
Finally, from a clinical standpoint, the patient’s diabetes and hypertension showed no improvement following PCC resection. This persistence is likely due to several contributing factors: the extended duration between the initial diagnosis and the surgery, which allowed for the establishment of irreversible metabolic and vascular changes (23); the ongoing use of diabetogenic immunosuppressive therapy required for post-transplant management, perpetuating hyperglycemia (24); and the progression of advanced chronic kidney disease, which exacerbates and sustains hypertension through mechanisms such as fluid retention, altered renin-angiotensin-aldosterone system activity, and sympathetic overactivity (25).
In sum, we report a rare case of PCC in a pulmonary transplant patient. Our genetic analyses demonstrated the absence of a pathogenic germline variant in a multigene panel of 14 genes related to PPGL and the absence of a somatic variant in the hypoxemia related gene EPAS1. RNA sequencing did demonstrate activation of the HIF pathway and implication of angiogenesis; however, further work is needed to better understand the genetic and molecular events leading to PCC in this specific case and determine its relation with hypoxemia in the context of pulmonary disease.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by CRCHUM (Centre hospitalier de l’Université de Montréal) Ethics Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
SP: Data curation, Formal analysis, Investigation, Resources, Writing – original draft, Writing – review & editing. FP: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. GC: Formal analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. JM: Investigation, Writing – original draft, Writing – review & editing. CP: Investigation, Writing – original draft, Writing – review & editing. CB: Investigation, Writing – original draft, Writing – review & editing. AR: Investigation, Writing – original draft, Writing – review & editing. ML: Investigation, Resources, Writing – original draft, Writing – review & editing. MT: Investigation, Resources, Writing – original draft, Writing – review & editing. CC: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. PF: Investigation, Writing – original draft, Writing – review & editing. IB: Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Formation de recherche pour les résidents-Résidence complémentaire en recherche: phase 1 FRQS (SPL) and in part by La Fondation du Grand défi Pierre Lavoie (IB) and The Cancer Research Society (MT, IB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1481906/full#supplementary-material
PCCs, Pheochromocytomas; PGLs, paragangliomas; PPGLs, pheochromocytomas and paragangliomas.
1. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
2. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. (2021) 43(2):199–239. doi: 10.1210/endrev/bnab019
3. Mannelli M, Rapizzi E, Fucci R, Canu L, Ercolino T, Luconi M, et al. 15 YEARS OF PARAGANGLIOMA: metabolism and pheochromocytoma/paraganglioma. Endocr Relat Cancer. (2015) 22(4):T83–90. doi: 10.1530/ERC-15-0215
4. Saldana MJ, Salem LE, Travezan R. High altitude hypoxia and chemodectomas. Hum Pathol. (1973) 4:251–63. doi: 10.1016/S0046-8177(73)80012-7
5. Opotowsky AR, Moko LE, Ginns J, Rosenbaum M, Greutmann M, Aboulhosn J, et al. Pheochromocytoma and paraganglioma in cyanotic congenital heart disease. J Clin Endocrinol Metab. (2015) 100:1325–34. doi: 10.1210/jc.2014-3863
6. Alrezk R, Suarez A, Tena I, Pacak K. Update of pheochromocytoma syndromes: genetics, biochemical evaluation, and imaging. Front Endocrinology. (2018) 9:515. doi: 10.3389/fendo.2018.00515
7. Jech M, Alvarado-Cabrero I, Albores-Saavedra J, Dahia PL, Tischler AS. Genetic analysis of high altitude paragangliomas. Endocr Pathol. (2006) 17:201–2. doi: 10.1385/EP:17:2:201
8. Leung AA, Hyrcza MD, Pasieka JL, Kline GA. Incidence of pheochromocytoma and paraganglioma varies according to altitude: meta-regression analysis. Eur J Endocrinol. (2021) 184:L21–l23. doi: 10.1530/EJE-21-0258
9. Ebbehoj A, Poulsen PL, Søndergaard E. Incidence of PPGL according to altitude – Calendar time is of the essence. Eur J Endocrinology. (2022) 186:L1–2. doi: 10.1530/EJE-21-0680
10. Vaidya A, Flores SK, Cheng Z-M, Nicolas M, Deng Y, Opotowsky AR, et al. EPAS1 mutations and paragangliomas in cyanotic congenital heart disease. New Engl J Med. (2018) 378:1259–61. doi: 10.1056/NEJMc1716652
11. Gagnon N, Cáceres-Gorriti KY, Corbeil G, El Ghoyareb N, Ludwig N, Latour M, et al. Genetic characterization of gnRH/LH-responsive primary aldosteronism. J Clin Endocrinol Metab. (2018) 103:2926–35. doi: 10.1210/jc.2018-00087
12. Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. (2022) 50:W216–21. doi: 10.1093/nar/gkac194
13. Song MK, Kim GB, Bae EJ, Lee YA, Kim HY, Min SK, et al. Pheochromocytoma and paraganglioma in Fontan patients: Common more than expected. Congenit Heart Dis. (2018) 13:608–16. doi: 10.1111/chd.12625
14. Yamamoto K, Namba N, Kubota T, Usui T, Takahashi K, Kitaoka T, et al. Pheochromocytoma complicated by cyanotic congenital heart disease: a case report. Clin Pediatr Endocrinol. (2016) 25:59–65. doi: 10.1297/cpe.25.59
15. Gao S, Liu L, Li Z, Pang Y, Shi J, Zhu F. Seven novel genes related to cell proliferation and migration of VHL-mutated pheochromocytoma. Front Endocrinol (Lausanne). (2021) 12:598656. doi: 10.3389/fendo.2021.598656
16. Helm J, Drukewitz S, Poser I, Richter S, Friedemann M, William D, et al. Treatment of pheochromocytoma cells with recurrent cycles of hypoxia: A new pseudohypoxic. In Vitro Model Cells. (2022) 11(3). doi: 10.3390/cells11030560
17. Shtraichman O, Ahya VN. Malignancy after lung transplantation. Ann Transl Med. (2020) 8:416. doi: 10.21037/atm.2020.02.126
18. Hope DC, Palan JM. Unusual presentation of phaeochromocytoma. BMJ Case Rep. (2016) 2016. doi: 10.1136/bcr-2016-214719
19. Tansey DJ, Egan JJ, Murray M, Padfield K, Conneely J, Hatunic M. Treatment-resistant hypertension in a post-transplant patient with cystic fibrosis: a rare case of phaeochromocytoma. Endocrinol Diabetes Metab Case Rep. (2021) 2021. doi: 10.1530/EDM-21-0026
20. Bancos I, Prete A. Approach to the patient with adrenal incidentaloma. J Clin Endocrinol Metab. (2021) 106:3331–53. doi: 10.1210/clinem/dgab512
21. Sherlock M, Scarsbrook A, Abbas A, Fraser S, Limumpornpetch P, Dineen R, et al. Adrenal incidentaloma. Endocrine Rev. (2020) 41:775–820. doi: 10.1210/endrev/bnaa008
22. Buitenwerf E, Korteweg T, Visser A, Haag C, Feelders RA, Timmers H, et al. Unenhanced CT imaging is highly sensitive to exclude pheochromocytoma: a multicenter study. Eur J Endocrinol. (2018) 178:431–7. doi: 10.1530/EJE-18-0006
23. Lopez C, Bima C, Bollati M, Bioletto F, Procopio M, Arata S, et al. Pathophysiology and Management of Glycemic Alterations before and after Surgery for Pheochromocytoma and Paraganglioma. Int J Mol Sci. (2023) 24:5153. doi: 10.3390/ijms24065153
24. Penfornis A, Kury-Paulin S. Immunosuppressive drug-induced diabetes. Diabetes Metab. (2006) 32:539–46. doi: 10.1016/S1262-3636(06)72809-9
Keywords: pheochromocytoma, hypoxia, genetics, RNA-sequencing, transplant
Citation: Parisien-La Salle S, Perreault F, Corbeil G, Morisset J, Poirier C, Beauregard C, Räkel A, Labrecque M, Tétreault M, Cohade C, Ferraro P and Bourdeau I (2025) Genetic characterization of a rare case of pheochromocytoma in a pulmonary transplant patient. Front. Endocrinol. 15:1481906. doi: 10.3389/fendo.2024.1481906
Received: 16 August 2024; Accepted: 20 December 2024;
Published: 06 February 2025.
Edited by:
Giuseppe Reimondo, University of Turin, ItalyReviewed by:
Sergei Tevosian, University of Florida, United StatesCopyright © 2025 Parisien-La Salle, Perreault, Corbeil, Morisset, Poirier, Beauregard, Räkel, Labrecque, Tétreault, Cohade, Ferraro and Bourdeau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Bourdeau, aXNhYmVsbGUuYm91cmRlYXVAdW1vbnRyZWFsLmNh
†ORCID: Isabelle Bourdeau, orcid.org/0000-0002-8000-4313
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.