 Takashi Yazawa1*
Takashi Yazawa1* Yugo Watanabe1
Yugo Watanabe1 Yuko Yokohama2
Yuko Yokohama2 Yoshitaka Imamichi3
Yoshitaka Imamichi3 Kazuya Hasegawa4Ke-ichi Nakajima1
Kazuya Hasegawa4Ke-ichi Nakajima1 Takeshi Kitano5
Takeshi Kitano5 Takanori Ida6
Takanori Ida6 Takahiro Sato7Mohammad Sayful Islam1,8Akihiro Umezawa9
Takahiro Sato7Mohammad Sayful Islam1,8Akihiro Umezawa9 Satoru Takahashi10Yasuhito Kato2Sharmin Jahan11
Satoru Takahashi10Yasuhito Kato2Sharmin Jahan11 Jun-ichi Kawabe1
Jun-ichi Kawabe1- 1Department of Biochemistry, Asahikawa Medical University, Asahikawa, Japan
- 2Department of Obstetrics and Gynecology, Asahikawa Medical University, Asahikawa, Japan
- 3Department of Marine Science and Technology, Fukui Prefectural University, Obama, Japan
- 4Faculty of Health and Medical Science, Teikyo Heisei University, Tokyo, Japan
- 5Department of Biological Sciences, Graduate School of Science and Technology, Kumamoto University, Kumamoto, Japan
- 6Division of International Cooperation and Education, Center for Animal Disease Control, University of Miyazaki, Miyazaki, Japan
- 7Division of Molecular Genetics, Institute of Life Sciences, Kurume University, Kurume, Japan
- 8Department of Pharmacy, Mawlana Bhashani Science and Technology University, Tangail, Bangladesh
- 9Department of Reproduction, National Research Institute for Child Health and Development, Tokyo, Japan
- 10Department of Pediatrics, Asahikawa Medical University, Asahikawa, Japan
- 11Department of Endocrinology, Bangabandhu Sheikh Mujib Medical University (BSMMU), Dhaka, Bangladesh
3β-Hydroxysteroid dehydrogenases (3β-HSDs) catalyze the oxidative conversion of delta (5)-ene-3-beta-hydroxy steroids and ketosteroids. Human 3β-HSD type 2 (HSD3B2) is predominantly expressed in gonadal and adrenal steroidogenic cells for producing all classes of active steroid hormones. Mutations in HSD3B2 gene cause a rare form of congenital adrenal hyperplasia with varying degree of salt wasting and incomplete masculinization, resulting from reduced production of corticoids and androgens. Therefore, evaluation of the HSD3B2 enzymatic activity in both pathways for each steroid hormone production is important for accurately understanding and diagnosing this disorder. Using progesterone receptor (PR)- and androgen receptor (AR)-mediated transactivation, we adapted a method that easily evaluates enzymatic activity of HSD3B2 by quantifying the conversion from substrates [pregnenolone (P5) and dehydroepiandrosterone (DHEA)] to (progesterone and androstenedione). HEK293 cells were transduced to express human HSD3B2, and incubated medium containing P5 or DHEA. Depending on the incubation time with HSD3B2-expressing cells, the culture media progressively increased luciferase activities in CV-1 cells, transfected with the PR/AR expression vector and progesterone-/androgen-responsive reporter. Culture media from human and other mammalian HSD3B1-expressing cells also increased the luciferase activities. HEK293 cells expressing various missense mutations in the HSD3B2 gene revealed the potential of this system to evaluate the relationship between the enzymatic activities of mutant proteins and patient phenotype.
Introduction
Steroid hormones are produced from cholesterol via serial enzymatic reactions catalyzed by cytochrome P450 hydroxylases (CYPs) and hydroxysteroid dehydrogenases (HSDs) (1, 2). Among these enzymes, 3β-HSDs belong to the short-chain dehydrogenase/reductase superfamily of proteins, which requires NAD(P)+ as a co-factor in the reactions (3). With CYP11A1, a rate-limiting enzyme of steroidogenesis, they are essential for producing all classes of steroid hormones by catalyzing 3β-hydroxysteroid dehydrogenation and Δ5- to Δ4-isomerisation of the Δ5-steroid precursors. 3β-HSDs synthesize an active steroid and its precursors by converting pregnenolone (P5), 17α-pregnenolone (17-OHP5) and dehydroepiandrosterone (DHEA) into progesterone (P4), 17α-progesterone (17-OHP4) and androstenedione (A4), respectively. Although P4 itself is an active steroid hormone that strongly activates progesterone receptor (PR), it also acts as a precursor of mineralocorticoid (aldosterone) for regulating blood pressure through sodium reabsorption. 17-OHP4 and A4 are the precursors for glucocorticoid (cortisol) and androgen (testosterone) (4), respectively.
Multiple types of HSD3B/Hsd3b exist in species-specific manner (3). In human, there are two isozymes, HSD3B1 and HSD3B2 (5). Human HSD3B1 is mainly expressed in placenta and involved in the production of progesterone for maintaining the pregnancy (3, 6). It is also expressed at minor extent in other tissues, such as skin, breast and prostate. Although some single nucleotide polymorphisms (SNPs) are likely involved in prostate and breast cancers (7–13), deficiency of HSD3B1 has not been reported. In contrast, HSD3B2 is almost exclusively expressed in primary steroidogenic tissues, such as adrenal gland and gonads (testis and ovary) (3, 14). Deficiency caused by the mutations in HSD3B2 gene results in congenital adrenal hyperplasia (CAH) with salt-wasting (SW) and ambiguous genitalia, due to the reduction of glucocorticoid, mineralocorticoid and androgen (15). Although HSD3B2 deficiency is a rare autosomal recessive disorder, the clinical presentation of SW and genitalia is heterogenous among different mutations (3, 15, 16). In particular, SW phenotype ranges from no symptoms to severe forms. Therefore, the evaluation of HSD3B2 enzymatic activities is important for understanding and diagnosing this disorder. However, as mentioned above, because HSD3B2 catalyzes multiple steps in the production of various steroid hormones, evaluation of the enzymatic activities using the traditional methods like radioactive isotopes and liquid chromatography-mass spectrometry-mass spectrometry (LC–MS/MS) is complicated. We recently reported a method that easily evaluates the activities of testosterone-producing enzyme HSD17B3 using androgen receptor (AR)-mediated transactivation in cultured cells (17). Based on this methodology, we established a method in this study to evaluate HSD3B2 activities in converting substrates (P5 and DHEA) into the precursors (P4 and A4) of important steroid hormones (aldosterone and testosterone) relevant to each clinical symptom. Using this system, we evaluated the enzymatic activities of various missense mutants of HSD3B2 proteins-derived from the patients with or without SW symptom.
Materials and methods
Cell culture, transfection and luciferase assay
CV-1 and HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Cells were transfected using HilyMax (Dojindo Laboratories, Kumamoto, Japan). One day before transfection, cells were seeded on 48-well plates and cultured with DMEM supplemented with 10% Hyclone Charcoal/Dextran treated FBS (GE Healthcare UK Ltd, Buckinghamshire, England). At 24 h post-transfection, the cells were treated with vehicle (EtOH), each steroid, or supernatant of culture media for 24 h. Luciferase assays were performed as described previously (18, 19). Each data point represents the mean of at least four independent experiments.
Western blotting analysis
Extraction of total proteins from cultured cells and subsequent quantification were conducted as described previously (20, 21). Equal amounts of protein (30 μg) were resolved using 12.5% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Western blot analyses of HSD3B2, FLAG and GAPDH were performed using antibodies directed against HSD3B2 (67572-1-Ig, Proteintech Group, Inc., Rosemont, IL, USA), FLAG (Clone 871701, R&D Systems, Inc., Minneapolis, MN, USA) and GAPDH (14C10; Cell Signaling Technology, Inc.), respectively. Clarity Western ECL Substrate (Bio-Rad Laboratories Inc., Hercules, CA, USA) were used for detection.
Plasmids
The pCMV-2B expressing human HSD3B1 and HSD3B2 were generated by cloning the open reading frame (ORF) of each gene into a pCMV-2B vector (Invitrogen, Carlsbad, CA, USA). The pQCXIP expressing ovine HSD3B1 and guinea pig Hsd3b1 were generated by cloning the ORF of each gene into a pQCXIP vector. Constructs having mutations in HSD3B2 gene were prepared by the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with each primer (Supplementary Table 1). The pcDNA3 expressing human PR was generated by cloning the ORF of human PR into a pcDNA3 vector. A Slp-ARU/Luc reporter and pQCXIP/human AR were prepared as described (22).
Measurements by liquid chromatography-tandem spectrometry
P5, P4, DHEA and A4 in culture media were quantified using LC-MS/MS are based on methods as we previously described (23). As internal standards, P5, P4, DHEA and A4 were added to a medium diluted with distilled water. The steroids were extracted with methyl tert-butyl ether (MTBE). After evaporating the MTBE layer to dryness, the extract was dissolved in 0.5 mL of methanol and then diluted with 1 ml of distilled water. The sample was applied to OASIS MAX cartridge which had been successively conditioned with 3 ml of methanol and 3 ml of distilled water. After the cartridge was washed with 1 ml of distilled water, 1 ml of methanol/distilled water/acetic acid (45:55:1,v/v/v), and 1 ml of 1% pyridine solution, the steroids were eluted with 1 ml of methanol/pyridine (100:1,v/v). After evaporation, the residue was reacted with 50 μl of mixed solution (80 mg of 2-methyl-6-nitrobenzoic anhydride, 20 mg of 4-dimethylaminopyridine, 40 mg of picolinic acid and 10 μl of triethylamine in 1 ml of acetonitrile) for 30 min at room temperature. After the reaction, the sample was dissolved in 0.5 ml of ethyl acetate/hexane/acetic acid (15:35:1, v/v) and the mixture was applied to HyperSep Silica cartridge which had been successively conditioned with 3 mL of acetone and 3 ml of hexane. The cartridge was washed with 1 mL of hexane, and 2 mL of ethyl acetate/hexane (3:7, v/v). P5, P4, DHEA and A4were eluted with 2.5 ml of acetone/hexane (7:3, v/v). After evaporation, the residue was dissolved in 0.1 ml of acetonitrile/distilled water (2:3, v/v) and the solution was subjected to a LC-MS/MS.
Statistical analyses
Data are presented as the mean ± SEM or the mean ± SD. Differences between groups were assessed by the Student’s t-test or the one-way ANOVA followed by Tukey’s multiple comparison test using EZR (Easy R, Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria) (24). p-Values less than 0.05 were considered significant.
Results
Establishment of a method to evaluate enzymatic activities of HSD3B2 using cell-based assays
To establish a novel system for evaluating the enzymatic activities of HSD3B2 in multiple pathways (Figure 1), we compared the potential for PR- and AR-mediated transactivation between substrates (P5 and DHEA) and products (P4 and A4) at various concentrations in CV-1 cells (Figure 2). PR-mediated transactivation was markedly increased from 10-9 M by P4, whereas the transactivation was not increased by P5 at any concentration (Figure 2A). The ratio of P4 to P5-induced activity was also elevated from 10-9 M and the peak value was observed at 10-8 M. DHEA almost completely unaffected the AR-mediated transactivation in all the examined concentrations (Figure 2B). In contrast, A4 increased it from 10-8 M. Consistent with this, the ratio of A4 to DHEA-induced activity was significantly elevated at 10-8 and 10-7 M (Figure 2B). Based on these results, we selected 10-8 M in following experiments as optimal concentration to discriminate between substrates and products in the PR- or AR-mediated transactivation system.
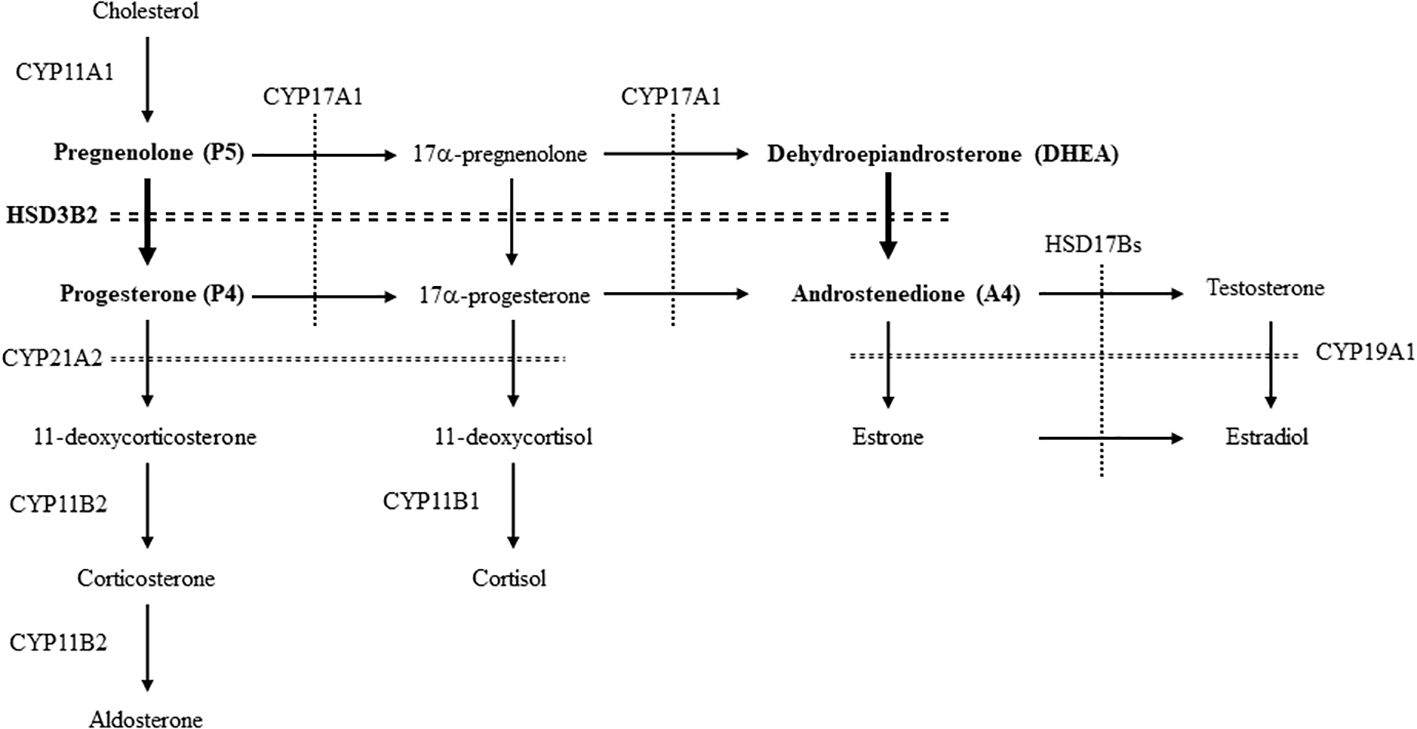
Figure 1. The enzymes and pathways for the synthesis of steroid hormones from cholesterol in human. The pathways involved in this study are showed by bold letters and arrows.
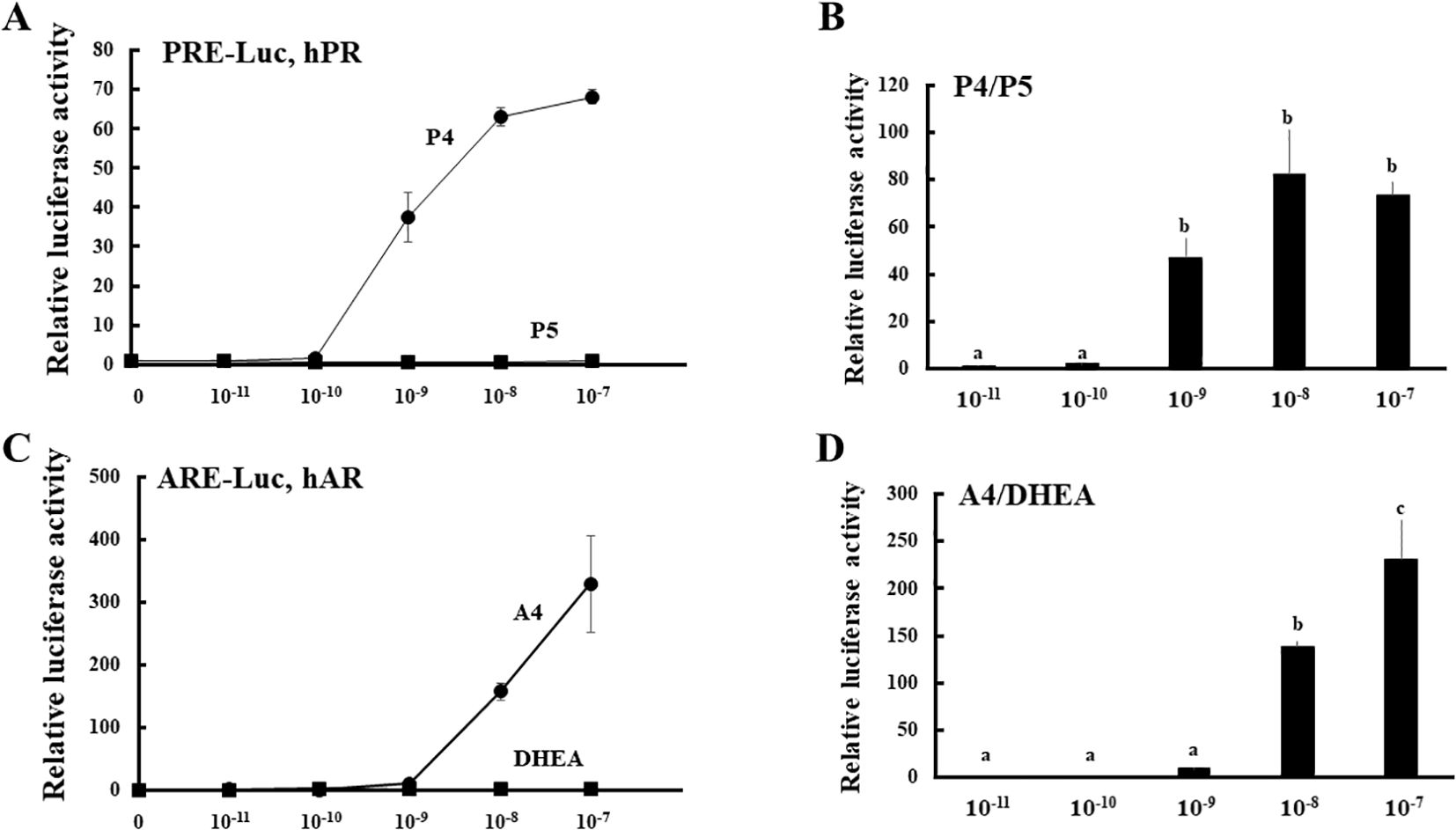
Figure 2. PR- and AR-mediated transactivation by substrates (P5 and DHEA) and products (P4 and A4) of HSD3B2 in CV-1 cells. (A) CV-1 cells were transfected with the PRE-Luc vector and the human PR-expression vector. At 24 h post-transfection, cells were incubated with or without increasing concentrations of each steroid for 24 h. Data represent the mean ± sem of at least four independent experiments. (B) Comparison for the potentials of PR-mediated transactivation between P5 and P4 at each concentration. Values of P5 were defined as 1. Values marked by the different letters (a, b, c) are significantly different with each other (P < 0.05). (C) CV-1 cells were transfected with the ARE-Luc vector and the human AR-expression vector. At 24 h post-transfection, cells were incubated with or without increasing concentrations of each steroid for 24 h. Data represent the mean ± sem of at least three independent experiments. (D) Comparison for the potentials of AR-mediated transactivation between DHEA and A4 at each concentration. Values of DHEA were defined as 1. Values marked by the different letters (a, b, c) are significantly different with each other (P < 0.05).
Next, expression vectors of GFP and human HSD3B2 genes were transfected into HEK293 cells (Figure 3A). Two days post-transfection, P5 or DHEA was added to the culture medium at 10-8 M for 3h. Then, the supernatants were collected at each time point, and transfer to culture plates of CV-1 cells that were transfected with PRE reporter/human PR expression vector or ARE reporter/human AR expression vector. Each culture medium-derived from HSD3B2-transfected HEK293 cells increased PR- and AR-mediated transactivation in CV-1 cells in a time-dependent manner after the substrate addition (Figures 3B, C). PR-mediated transactivation in CV-1 cells reached plateau in the medium cultured for 30 min with P5, whereas AR-mediated transactivation in CV-1 cells was reached plateau in the medium cultured for 2h with DHEA. Consistent with these results, P5 and DHEA were markedly converted to P4 (Figure 3D) and A4 (Figure 3E), respectively, at the end of incubation, despite these products were undetectable at the beginning of incubation (data not shown). In contrast, culture media-derived from GFP-transfected HEK293 cells did not induce the luciferase activities at all time points. These results indicate that HSD3B2-mediated conversion of P5 into P4, and DHEA into A4, in culture media are able to evaluate by PR- and AR-mediated transactivation in this system. It can also evaluate the activities of human HSD3B1 that was transiently expressed in HEK293 cells (Figure 4; Supplementary Figure 1). Activities of human HSD3B1were higher than the activities of HSD3B2, despite statistically not significant. In addition to human proteins, the activities of ovine HSD3B1 and guinea pig Hsd3b1 were detectable using this system (Supplementary Figures 2A, B).
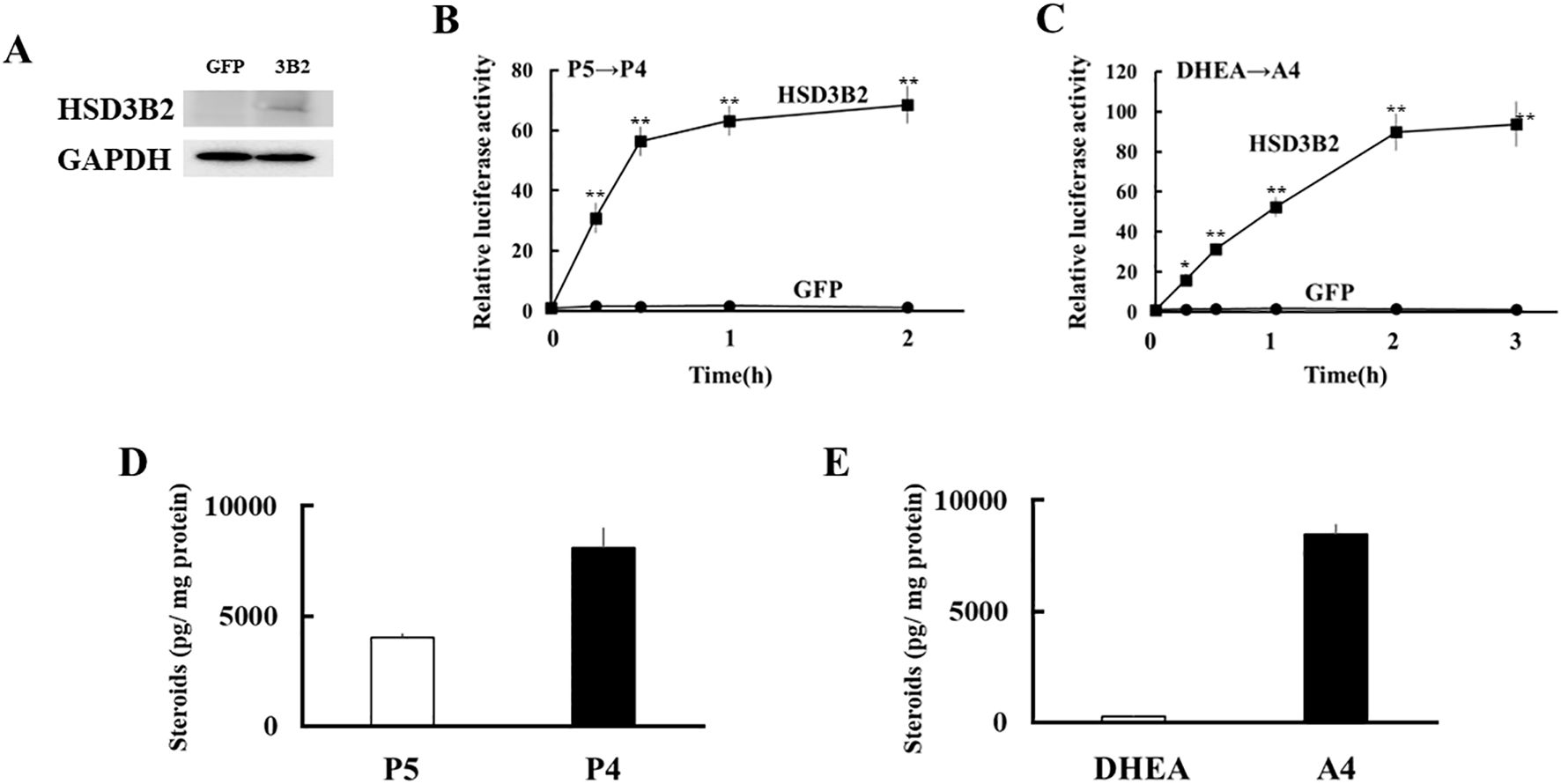
Figure 3. Development of a novel method to detect enzymatic activity of HSD3B2 by cell-based assay. (A) Western blot analyses were performed with the antibodies against HSD3B2 and GAPDH using lysates derived from GFP- or HSD3B2-expressed HEK293 cells. (B) Activation of human PR-mediated transcription by culture media from GFP- or HSD3B2-expressed HEK293 cells. CV-1 cells were transfected with PRE-Luc and human PR-expression vectors. At 24 h post-transfection, cells were incubated with each culture medium collected at indicated time after P5 (10 nM) addition for 24 h. Values for P5 (10 nM) addition in PRE-Luc vector-and human PR-expression vector-transfected CV-1 cells were defined as 1. Data represent the mean ± sem of at least four independent experiments. Differences between GFP and HSD3B2 groups in each time point are indicated by **p < 0.01. (C) Activation of human AR-mediated transcription by culture media from GFP- or HSD3B2-expressing HEK293 cells. CV-1 cells were transfected with ARE-Luc and human AR-expression vectors. At 24 h post-transfection, cells were incubated with each culture medium collected at indicated time after DHEA (10 nM) addition for 24 h. Values for DHEA (10 nM) addition in ARE-Luc vector-and human AR-expression vector-transfected CV-1 cells were defined as 1. Data represent the mean ± sem of at least four independent experiments. Differences between GFP and HSD3B2 groups in each time point are indicated by *p < 0.05 and **p < 0.01. (D, E) Concentrations of P5/P4 (D) and DHEA/A4 (E) in culture medium from HSD3B2-expressed HEK293 cells at 2 h and 3 h after addition of P5 (10 nM) and DHEA (10 nM), respectively. Each column represents the mean ± SEM of three independent experiments.
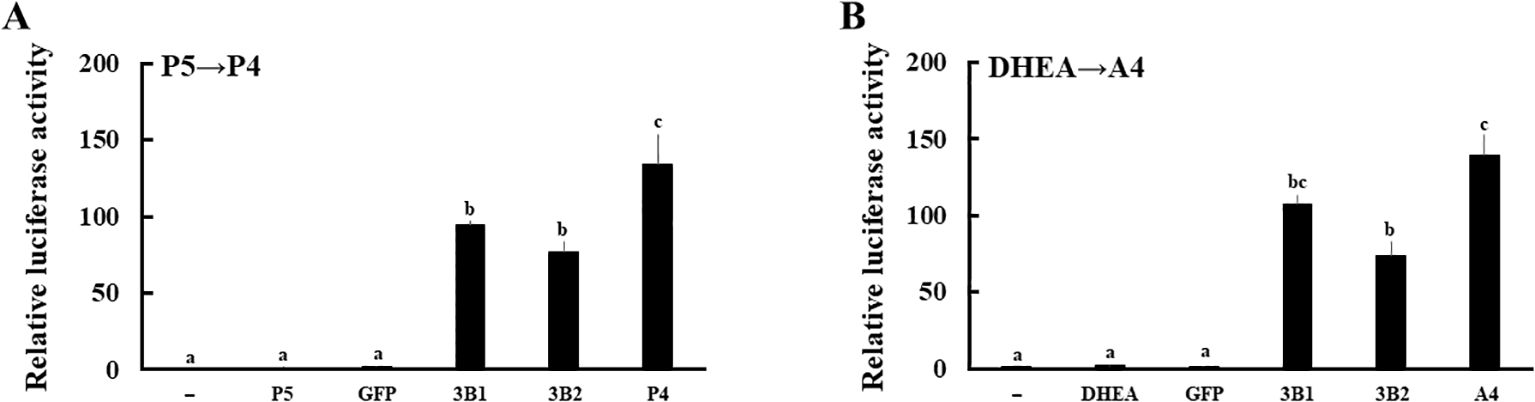
Figure 4. Evaluation of the enzymatic activities of human HSD3B1 and HSD3B2 using culture media from each gene-transfected HEK293 cells. Activation of human PR- and AR-mediated transcription by HSD3B1 and HSD3B2 using culture media from each gene-transfected HEK293 cells. CV-1 cells were transfected with PRE-Luc/human PR-expression vectors (A) and ARE-Luc/human AR-expression vectors (B). At 24 h post-transfection, cells were incubated for 24 h with vehicle (lane -), P5 (10 nM), P4 (10 nM), DHEA (10 nM),A4 (10 nM), culture medium from GFP or each HSD3B-expressing HEK293 cells collected at 2 h and 3 h after addition of P5 (10 nM) and DHEA (10 nM), respectively. Values of the vehicle were defined as 1. Data represent the mean ± sem of at least four independent experiments. Values marked by the different letters (a, b, c) are significantly different with each other (P < 0.05).
Enzymatic activities of various missense mutations in the HSD3B2 gene
We applied the above system to evaluate the enzymatic activities of four HSD3B2 mutants with or without SW (Figure 5). Although these missense mutants are identified within a decade (25–28), the enzymatic activities of mutant proteins had not been defined. Therefore, we transfected these amino acid substituted enzymes in HEK293 cells, and compared the enzymatic activities with wild type protein using the above system (Figure 6A). Consistent with patient symptoms (genitalia dysgenesis), AR-mediated transactivation by culture media adding DHEA in each mutant gene-transfected cells were markedly decreased compared with those in wild type protein transfected cells (Figure 6C). C72R, S124G and M225V completely disappeared the enzymatic activities, whereas V299I had some residual activities (19.9% versus wild type). In contrast to AR-mediated transactivation, PR-mediated transactivation by culture media in each mutant gene-transfected cells showed some difference between mutants (Figure 6B). It was never induced by culture media from C72R-, S124G- and M225V-transfected cells, whereas culture media from V299I transfected cells induced the luciferase activities despite some lesser than those from wild type gene transfected cells (67.3% versus wild type). These results indicate that heterogeneity of the clinical presentation in SW is caused by the effects of mutation on the substrate-dependent enzymatic activities.
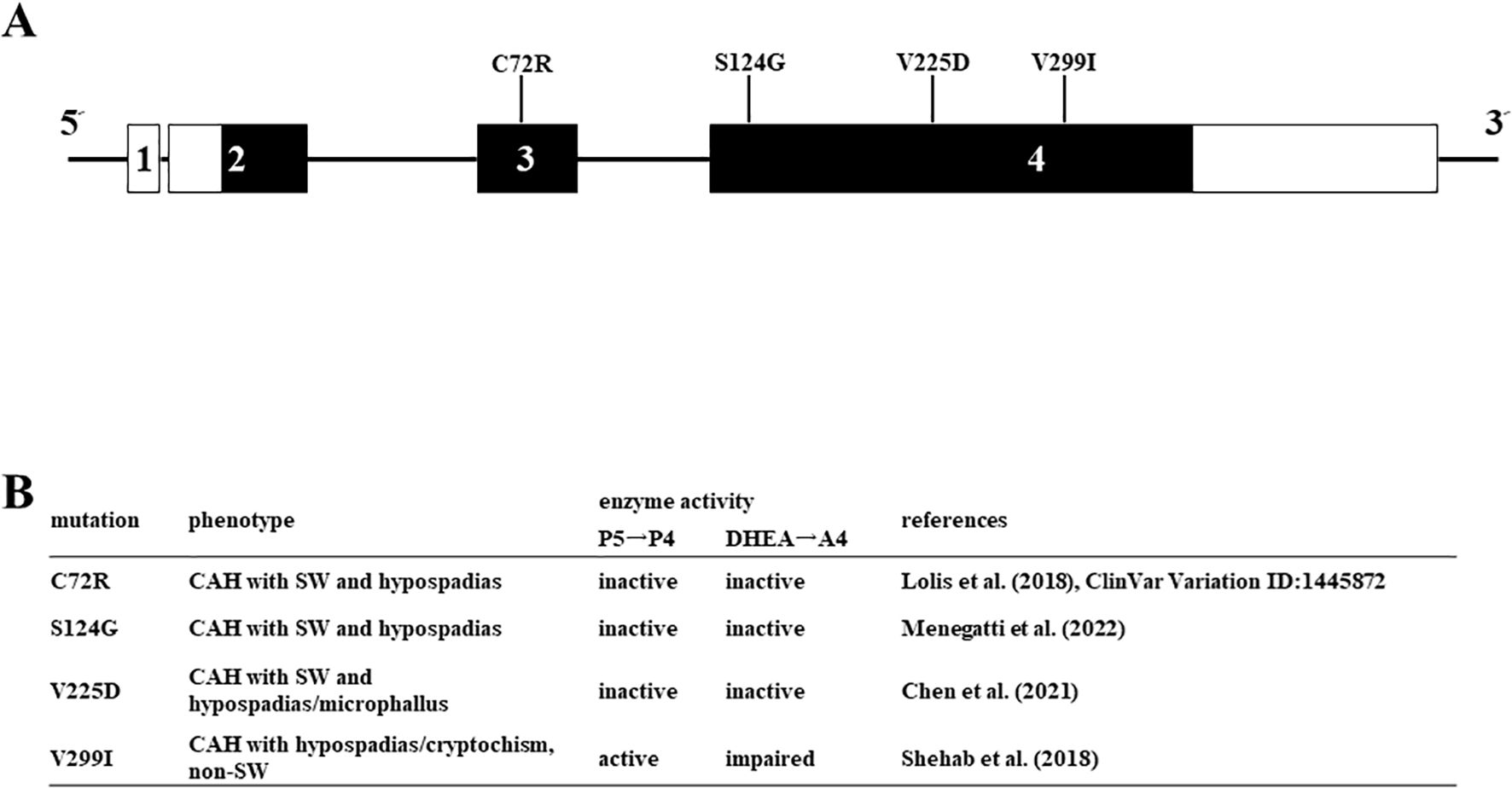
Figure 5. Missense mutations in HSD3B2 genes investigated in this study. (A) Schematic structure of the human HSD3B2 gene and substitution of amino acids. The numbered box indicates exons 5’ and 3’-untranslated regions are indicated by white bars. (B) Clinical symptoms and enzymatic activities of each mutation in the HSD3B2 gene.
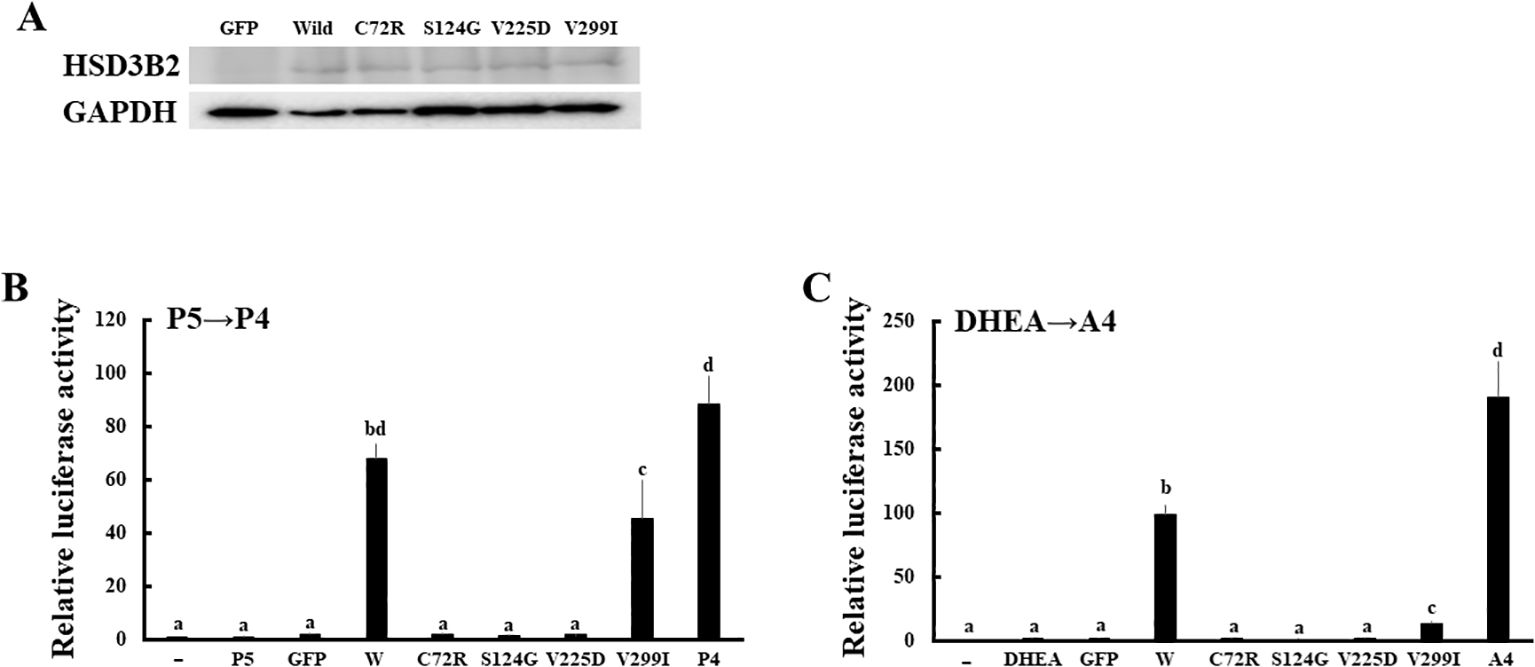
Figure 6. Enzymatic activities of missense mutations in HSD3B2 gene. (A) Expression of wild-type and mutant HSD3B2 enzymes. Western blot analyses were performed with the antibodies against HSD3B2 and GAPDH using lysates of GFP-, wild-type HSD3B2- and each mutant HSD3B2-expressed HEK293 cells. (B) Activation of human AR-mediated transcription by culture media from wild-type and mutant HSD3B2-expressing HEK293 cells. CV-1 cells were transfected with PRE-Luc and human PR-expression vectors. At 24 h post-transfection, cells were incubated with each culture medium collected at 2 h after P5 (10 nM) addition for 24 h. Values for vehicle addition in PRE-Luc vector-and human PR-expression vector-transfected CV-1 cells were defined as 1. Data represent the mean ± sem of at least four independent experiments. Values marked by the different letters (a, b, c, d) are significantly different with each other (P < 0.05). (C) Activation of human AR-mediated transcription by culture media from wild-type and mutant HSD3B2-expressing HEK293 cells. CV-1 cells were transfected with ARE-Luc and human AR-expression vectors. At 24 h post-transfection, cells were incubated with each culture medium collected at 3 h after DHEA (10 nM) addition for 24 h. Values for vehicle addition in ARE-Luc vector-and human AR-expression vector-transfected CV-1 cells were defined as 1. Data represent the mean ± sem of at least four independent experiments. Values marked by the different letters (a, b, c) are significantly different with each other (P < 0.05). Values marked by the different letters (a, b, c, d) are significantly different with each other (P < 0.05).
Discussion
We established a method to easily evaluate enzymatic activity of HSD3B2 to multiple substrates using the reporter assays in cultured cells. This system is highly sensitive and rapidly detects the conversion of substrates, due to a strong response to P4 and A4 from ectopic expression of PR and AR in CV-1 cells, which have a low background activity of C3 group nuclear receptors (29). Using this assay, we defined the effects of various missense mutations on enzymatic activities of human HSD3B2 to both P5 and DHEA.
Human PR was activated by P4 from 10-9 M. This concentration is lesser than plasma progesterone concentration in men and women at follicular phase, whereas it elevates to over 10 nM at luteal phase (30). Therefore, this concentration acts as a critical point for the actions of progesterone in menstrual cycle and early pregnancy, despite it continues to increase additional several tens-fold for maintaining the pregnancy until late phase (31–33). In contrast, A4 strongly activated human AR from 10-8 M. Because plasma concentrations of A4 is just less than this value in both men and women throughout the life (30), these facts are consistent with the concept that A4 represents as an important intermediate for sex steroids, such as testosterone and estrogens (34, 35). However, it was reported that A4 exceeds this concentration in the deficiency of some steroidogenic genes, such as HSD17B3 (36, 37) and CYP21A2 (38, 39). Although it is possible that A4 acts as an androgen in these patients, the action is insufficient for complete masculinization. This is due to the necessity of 5α-dihydrotestosterone converted from T by 5α-reductase in peripheral tissues for the male sexual differentiation (40, 41). Nevertheless, it is not ruled out that elevated A4 contributes to the partial virilization in CYP21A2-deficient female patients, even though 11-oxygenated androgens, such as 11-ketotestosterone, are likely most important (38, 42), Further detail studies are necessary to reveal the characteristics of this weak androgen in sexual development.
It is interesting that conversion of DHEA into A4 was markedly reduced in all mutants, whereas conversion of P5 into P4 was markedly reduced in three mutants (C72R, S124G and V225D). Although the conversion was also reduced in V299I, the activity was maintained at far more than 50% compared to wild type. This result is consistent with the clinical phenotypes; all the patients have ambiguous genitalia, whereas SW was observed in all the patients except for V299I (25–28). As far as we know, this is the first report revealing the different effects of amino-acid substitution of HSD3B2 on enzymatic activities by the substrates. V299 is the component of the putative membrane spanning domains that are possibly involved in the substrate-specificity (43). Therefore, it is probable that other mutation in these domains show the fluctuation of enzymatic activity dependent on the substrates as V299I. However, enzymatic activities of mutant proteins have been often evaluated using only one substrate in previous studies (44–47) Because the clinical manifestation of HSD3B2 gene mutations is heterogenous between patients, evaluation of enzymatic activities using multiple substrates is necessary for accurate diagnosis.
To resolve the problems of ambiguous genitalia by HSD3B2 deficiency, testosterone replacement therapy have been performed in the male patients, including C72R and V225D patients (27, 28, 48). This treatment attenuates microphallus by promoting the penile growth. In contrast, V299I patient suffered from premature pubarche with rapid axially/public hair and phallic enlargement at 7 years of age, despite he also showed ambiguous genitalia at birth (25). Consistent with the clinical features, plasma testosterone concentration was markedly increased. It was also reported in previous studies that parts of male patients with HSD3B2 mutations spontaneously enter puberty (45, 49–53). Although contribution of peripheral HSD3B1with aid of HSD17B5 is predicted in such patients (44), it is also possible from our results that elevation of testosterone levels is caused by the residual enzymatic activities and up-regulation of adrenal HSD3B2 expression through the elevation of ACTH in CAH (54). In fact, various mutant proteins sustain the residual activities (44). Therefore, it is conceivable again that evaluation of the enzymatic activities of mutant proteins is essential for the accurate diagnosis in this disease.
In summary, we developed a method to evaluate the enzymatic activity of 3β-HSDs to multiple substrates. It provides a useful method for comprehensive analyses of HSD3B2 mutant proteins that cause heterogenous clinical features. In addition, this system could also be useful for evaluating the enzymatic activities of HSD3B1 to develop new drugs. Although the mutations in HSD3B1 have not been linked to any genetic diseases, it was reported that its SNPs are associated with breast and prostate cancers (7–13). Therefore, it could be one of the therapeutic targets for preventing the progression of these diseases. Because the present system can easily examine the effects of added compounds through cell-based reporter assay, it could be applied to identify the compounds to suppress its activity and disease progression.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
TY: Conceptualization, Funding acquisition, Investigation, Writing – original draft. YW: Writing – original draft. YY: Investigation, Writing – original draft. YI: Funding acquisition, Writing – original draft. KH: Funding acquisition, Writing – original draft. K-iN: Writing – review & editing. TK: Writing – review & editing. TI: Funding acquisition, Writing – review & editing. TS: Funding acquisition, Writing – review & editing. MI: Investigation, Writing – review & editing. AU: Funding acquisition, Project administration, Writing – review & editing. ST: Project administration, Writing – review & editing. YK: Project administration, Writing – review & editing. SJ: Resources, Writing – review & editing. J-iK: Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by JST Japan (COI-NEXT, JPMJPF2010), Japan Sports Agency, Ministry of Education, Culture, Sports, Science and Technology-Japan, Research and development project on anti-doping science (JADA2408), the Smoking Research Foundation of Japan (2024-D002), Kobayashi Foundation (no. 181 and 215) and the Grant of National Center for Child Health and Development (2024-A2).
Acknowledgments
This study was performed under the cooperative research program of the Institute of Nature and Environmental Technology, Kanazawa University (accept no.23069) in 2023.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1480722/full#supplementary-material
References
1. Miller WL. Steroidogenesis: unanswered questions. Trends Endocrinol metabolism: TEM. (2017) 28:771–93. doi: 10.1016/j.tem.2017.09.002
2. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Rev. (2011) 32:81–151. doi: 10.1210/er.2010-0013
3. Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocrine Rev. (2005) 26:525–82. doi: 10.1210/er.2002-0050
4. Baquedano MS, Guercio G, Costanzo M, Marino R, Rivarola MA, Belgorosky A. Mutation of HSD3B2 gene and fate of dehydroepiandrosterone. Vitamins hormones. (2018) 108:75–123. doi: 10.1016/bs.vh.2018.05.002
5. McBride MW, McVie AJ, Burridge SM, Brintnell B, Craig N, Wallace AM, et al. Cloning, expression, and physical mapping of the 3beta-hydroxysteroid dehydrogenase gene cluster (HSD3BP1-HSD3BP5) in human. Genomics. (1999) 61:277–84. doi: 10.1006/geno.1999.5459
6. Luu The V, Lachance Y, Labrie C, Leblanc G, Thomas JL, Strickler RC, et al. Full length cDNA structure and deduced amino acid sequence of human 3 beta-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol (Baltimore Md.). (1989) 3:1310–2. doi: 10.1210/mend-3-8-1310
7. Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang KH, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet Oncol. (2016) 17:1435–44. doi: 10.1016/s1470-2045(16)30227-3
8. Agarwal N, Hahn AW, Gill DM, Farnham JM, Poole AI, Cannon-Albright L. Independent validation of effect of HSD3B1 genotype on response to androgen-deprivation therapy in prostate cancer. JAMA Oncol. (2017) 3:856–7. doi: 10.1001/jamaoncol.2017.0147
9. Sabharwal N, Sharifi N. HSD3B1 genotypes conferring adrenal-restrictive and adrenal-permissive phenotypes in prostate cancer and beyond. Endocrinology. (2019) 160:2180–8. doi: 10.1210/en.2019-00366
10. Flanagan MR, Doody DR, Voutsinas J, Wu Q, Banda K, Sharifi N, et al. Association of HSD3B1 genotype and clinical outcomes in postmenopausal estrogen-receptor-positive breast cancer. Ann Surg Oncol. (2022) 29:7194–201. doi: 10.1245/s10434-022-12088-w
11. Lee O, Fought AJ, Shidfar A, Heinz RE, Kmiecik TE, Gann PH, et al. Association of genetic polymorphisms with local steroid metabolism in human benign breasts. Steroids. (2022) 177:108937. doi: 10.1016/j.steroids.2021.108937
12. Zhuang Q, Huang S, Li Z. Prospective role of 3βHSD1 in prostate cancer precision medicine. Prostate. (2023) 83:619–27. doi: 10.1002/pros.24504
13. Shiota M, Endo S, Tsukahara S, Tanegashima T, Kobayashi S, Matsumoto T, et al. Importance of 3β-hydroxysteroid dehydrogenases and their clinical use in prostate cancer. Endocrine-related Cancer. (2024) 31(7):e240023. doi: 10.1530/erc-24-0023
14. Rhéaume E, Lachance Y, Zhao HF, Breton N, Dumont M, de Launoit Y, et al. Structure and expression of a new complementary DNA encoding the almost exclusive 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4-isomerase in human adrenals and gonads. Mol Endocrinol (Baltimore Md.). (1991) 5:1147–57. doi: 10.1210/mend-5-8-1147
15. Nicola AG, Carsote M, Gheorghe AM, Petrova E, Popescu AD, Staicu AN, et al. Approach of heterogeneous spectrum involving 3beta-hydroxysteroid dehydrogenase 2 deficiency. Diagnostics (Basel Switzerland). (2022) 12(9):2168. doi: 10.3390/diagnostics12092168
16. Al Alawi AM, Nordenström A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine. (2019) 63:407–21. doi: 10.1007/s12020-018-01835-3
17. Yazawa T, Imamichi Y, Uwada J, Sekiguchi T, Mikami D, Kitano T, et al. Evaluation of 17β-hydroxysteroid dehydrogenase activity using androgen receptor-mediated transactivation. J Steroid Biochem Mol Biol. (2020) 196:105493. doi: 10.1016/j.jsbmb.2019.105493
18. Yazawa T, Imamichi Y, Yuhki KI, Uwada J, Mikami D, Shimada M, et al. Cyclooxygenase-2 is acutely induced by CCAAT/enhancer-binding protein beta to produce prostaglandin E 2 and F 2alpha following gonadotropin stimulation in Leydig cells. Mol Reprod Dev. (2019) 86(7):786–97. doi: 10.1002/mrd.23163
19. Yazawa T, Imamichi Y, Kitano T, Islam MS, Khan MRI, Takahashi S, et al. Expression of Chrna9 is regulated by Tbx3 in undifferentiated pluripotent stem cells. Sci Rep. (2023) 13:1611. doi: 10.1038/s41598-023-28814-7
20. Imamichi Y, Sekiguchi T, Kitano T, Kajitani T, Okada R, Inaoka Y, et al. Diethylstilbestrol administration inhibits theca cell androgen and granulosa cell estrogen production in immature rat ovary. Sci Rep. (2017) 7:8374. doi: 10.1038/s41598-017-08780-7
21. Yazawa T, Mizutani T, Yamada K, Kawata H, Sekiguchi T, Yoshino M, et al. Differentiation of adult stem cells derived from bone marrow stroma into Leydig or adrenocortical cells. Endocrinology. (2006) 147:4104–11. doi: 10.1210/en.2006-0162
22. Yazawa T, Islam MS, Imamichi Y, Watanabe H, Yaegashi K, Ida T, et al. Comparison of placental HSD17B1 expression and its regulation in various mammalian species. Animals: an Open Access J MDPI. (2023) 13(4):622. doi: 10.3390/ani13040622
23. Yazawa T, Inaba H, Imamichi Y, Sekiguchi T, Uwada J, Islam MS, et al. Profiles of 5α-reduced androgens in humans and eels: 5α-dihydrotestosterone and 11-ketodihydrotestosterone are active androgens produced in eel gonads. Front Endocrinol. (2021) 12:657360. doi: 10.3389/fendo.2021.657360
24. Yazawa T, Sato T, Nemoto T, Nagata S, Imamichi Y, Kitano T, et al. 11-Ketotestosterone is a major androgen produced in porcine adrenal glands and testes. J Steroid Biochem Mol Biol. (2021) 210:105847. doi: 10.1016/j.jsbmb.2021.105847
25. Shehab MA, Mahmood T, Hasanat MA, Fariduddin M, Ahsan N, Hossain MS, et al. A rare variety of congenital adrenal hyperplasia with mosaic Klinefelter syndrome: a unique combination presenting with ambiguous genitalia and sexual precocity. Endocrinology Diabetes Metab Case Rep. (2018) 2018:18–0108. doi: 10.1530/edm-18-0108
26. Menegatti E, Tessaris D, Barinotti A, Matarazzo P, Einaudi S. Genetic testing for a patient with suspected 3 beta-hydroxysteroid dehydrogenase deficiency: A case of unreported genetic variants. J Clin Med. (2022) 11(19):5767. doi: 10.3390/jcm11195767
27. Chen L, Huang H, Zhang H, Zhu G, Zhu M. Three cases of 3β-hydroxysteroid dehydrogenase deficiency: Clinical analysis. Adv Clin Exp medicine: Off Organ Wroclaw Med Univ. (2021) 30:289–99. doi: 10.17219/acem/131220
28. Lolis E, Juhlin CC, Nordenström A, Falhammar H. Extensive bilateral adrenal rest testicular tumors in a patient with 3β-hydroxysteroid dehydrogenase type 2 deficiency. J Endocrine Soc. (2018) 2:513–7. doi: 10.1210/js.2018-00082
29. Campana C, Rege J, Turcu AF, Pezzi V, Gomez-Sanchez CE, Robins DM, et al. Development of a novel cell based androgen screening model. J Steroid Biochem Mol Biol. (2016) 156:17–22. doi: 10.1016/j.jsbmb.2015.11.005
30. Eisenhofer G, Peitzsch M, Kaden D, Langton K, Pamporaki C, Masjkur J, et al. Reference intervals for plasma concentrations of adrenal steroids measured by LC-MS/MS: Impact of gender, age, oral contraceptives, body mass index and blood pressure status. Clinica chimica acta; Int J Clin Chem. (2017) 470:115–24. doi: 10.1016/j.cca.2017.05.002
31. Lof M, Hilakivi-Clarke L, Sandin SS, de Assis S, Yu W, Weiderpass E. Dietary fat intake and gestational weight gain in relation to estradiol and progesterone plasma levels during pregnancy: a longitudinal study in Swedish women. BMC women’s Health. (2009) 9:10. doi: 10.1186/1472-6874-9-10
32. Zhao Y, Zhu Y, Chen X, Lin H, Qin N, Zhou Z, et al. Circulating innate lymphoid cells exhibit distinctive distribution during normal pregnancy. Reprod Sci (Thousand Oaks Calif.). (2022) 29:1124–35. doi: 10.1007/s43032-021-00834-6
33. Smith R, Smith JI, Shen X, Engel PJ, Bowman ME, McGrath SA, et al. Patterns of plasma corticotropin-releasing hormone, progesterone, estradiol, and estriol change and the onset of human labor. J Clin Endocrinol Metab. (2009) 94:2066–74. doi: 10.1210/jc.2008-2257
34. Nunes VO, Vanzellotti NC, Fraga JL, Pessoa FLP, Ferreira TF, Amaral PFF. Biotransformation of phytosterols into androstenedione-A technological prospecting study. Molecules (Basel Switzerland). (2022) 27(10):3164. doi: 10.3390/molecules27103164
35. Pang Q, Jia A, Al Masri MK, Varkaneh HK, Abu-Zaid A, Gao X. The effect of androstenedione supplementation on testosterone, estradiol, body composition, and lipid profile: a systematic review and meta-analysis of randomized controlled trials. Hormones (Athens Greece). (2022) 21:545–54. doi: 10.1007/s42000-022-00385-8
36. Fujisawa Y, Masunaga Y, Tanikawa W, Nakashima S, Ueda D, Sano S, et al. Serum steroid metabolite profiling by LC-MS/MS in two phenotypic male patients with HSD17B3 deficiency: Implications for hormonal diagnosis. J Steroid Biochem Mol Biol. (2023) 234:106403. doi: 10.1016/j.jsbmb.2023.106403
37. Gonçalves CI, Carriço J, Bastos M, Lemos MC. Disorder of sex development due to 17-beta-hydroxysteroid dehydrogenase type 3 deficiency: A case report and review of 70 different HSD17B3 mutations reported in 239 patients. Int J Mol Sci. (2022) 23(17):10026. doi: 10.3390/ijms231710026
38. Jha S, Turcu AF, Sinaii N, Brookner B, Auchus RJ, Merke DP. 11-oxygenated androgens useful in the setting of discrepant conventional biomarkers in 21-hydroxylase deficiency. J Endocrine Soc. (2021) 5:bvaa192. doi: 10.1210/jendso/bvaa192
39. Arriba M, Ezquieta B. Molecular diagnosis of steroid 21-hydroxylase deficiency: A practical approach. Front Endocrinol. (2022) 13:834549. doi: 10.3389/fendo.2022.834549
40. Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones (Athens Greece). (2018) 17:197–204. doi: 10.1007/s42000-018-0013-9
41. Batista RL, Mendonca BB. The molecular basis of 5α-reductase type 2 deficiency. Sexual development: genetics Mol biology evolution endocrinology embryology Pathol sex determination differentiation. (2022) 16:171–83. doi: 10.1159/000525119
42. Imamichi Y, Yuhki KI, Orisaka M, Kitano T, Mukai K, Ushikubi F, et al. 11-ketotestosterone is a major androgen produced in human gonads. J Clin Endocrinol Metab. (2016) 101:3582–91. doi: 10.1210/jc.2016-2311
43. Simard J, de Launoit Y, Labrie F. Characterization of the structure-activity relationships of rat types I and II 3 beta-hydroxysteroid dehydrogenase/delta 5 -delta 4 isomerase by site-directed mutagenesis and expression in HeLa cells. J Biol Chem. (1991) 266:14842–5. doi: 10.1016/S0021-9258(18)98548-0
44. Moisan AM, Ricketts ML, Tardy V, Desrochers M, Mébarki F, Chaussain JL, et al. New insight into the molecular basis of 3beta-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3B2 gene eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. (1999) 84:4410–25. doi: 10.1210/jcem.84.12.6288
45. Alos N, Moisan AM, Ward L, Desrochers M, Legault L, Leboeuf G, et al. A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3beta-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: evaluation of gonadal function after puberty. J Clin Endocrinol Metab. (2000) 85:1968–74. doi: 10.1210/jcem.85.5.6581
46. Guran T, Kara C, Yildiz M, Bitkin EC, Haklar G, Lin JC, et al. Revisiting classical 3β-hydroxysteroid dehydrogenase 2 deficiency: lessons from 31 pediatric cases. J Clin Endocrinol Metab. (2020) 105(3):dgaa022. doi: 10.1210/clinem/dgaa022
47. Rhéaume E, Sanchez R, Mébarki F, Gagnon E, Carel JC, Chaussain JL, et al. Identification and characterization of the G15D mutation found in a male patient with 3 beta-hydroxysteroid dehydrogenase (3 beta-HSD) deficiency: alteration of the putative NAD-binding domain of type II 3 beta-HSD. Biochemistry. (1995) 34:2893–900. doi: 10.1021/bi00009a020
48. Bin-Abbas BS, Sakati NA, Al-Ashwal AA. Congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase type II deficiency in 4 Saudi children. Long term follow up. Saudi Med J. (2004) 25:1293–5.
49. Ladjouze A, Donaldson M, Plotton I, Djenane N, Mohammedi K, Tardy-Guidollet V, et al. Genotype, mortality, morbidity, and outcomes of 3β-hydroxysteroid dehydrogenase deficiency in Algeria. Front Endocrinol. (2022) 13:867073. doi: 10.3389/fendo.2022.867073
50. Donadille B, Houang M, Netchine I, Siffroi JP, Christin-Maitre S. Human 3beta-hydroxysteroid dehydrogenase deficiency associated with normal spermatic numeration despite a severe enzyme deficit. Endocrine connections. (2018) 7:395–402. doi: 10.1530/ec-17-0306
51. Rhéaume E, Simard J, Morel Y, Mebarki F, Zachmann M, Forest MG, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat Genet. (1992) 1:239–45. doi: 10.1038/ng0792-239
52. Yoshimoto M, Kawaguchi T, Mori R, Kinoshita E, Baba T, Tajima T, et al. Pubertal changes in testicular 3 beta-hydroxysteroid dehydrogenase activity in a male with classical 3 beta-hydroxysteroid dehydrogenase deficiency showing spontaneous secondary sexual maturation. Hormone Res. (1997) 48:83–7. doi: 10.1159/000185492
53. Burckhardt MA, Udhane SS, Marti N, Schnyder I, Tapia C, Nielsen JE, et al. Human 3β-hydroxysteroid dehydrogenase deficiency seems to affect fertility but may not harbor a tumor risk: lesson from an experiment of nature. Eur J Endocrinol. (2015) 173:K1–k12. doi: 10.1530/eje-15-0599
Keywords: HSD3B2, CAH, progesterone, androstenedione, DSD
Citation: Yazawa T, Watanabe Y, Yokohama Y, Imamichi Y, Hasegawa K, Nakajima K-i, Kitano T, Ida T, Sato T, Islam MS, Umezawa A, Takahashi S, Kato Y, Jahan S and Kawabe J-i (2024) Evaluation of 3β-hydroxysteroid dehydrogenase activity using progesterone and androgen receptors-mediated transactivation. Front. Endocrinol. 15:1480722. doi: 10.3389/fendo.2024.1480722
Received: 14 August 2024; Accepted: 28 August 2024;
Published: 02 October 2024.
Edited by:
Takahiro Nemoto, Nippon Medical School, JapanReviewed by:
Kotaro Horiguchi, Kyorin University, JapanSaishu Yoshida, The Jikei University School of Medicine, Japan
Copyright © 2024 Yazawa, Watanabe, Yokohama, Imamichi, Hasegawa, Nakajima, Kitano, Ida, Sato, Islam, Umezawa, Takahashi, Kato, Jahan and Kawabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Yazawa, eWF6YXdhQGFzYWhpa2F3YS1tZWQuYWMuanA=