 Gerardo Hernán Carro1,2†
Gerardo Hernán Carro1,2† Mariano Martín1,2†
Mariano Martín1,2† Sofía Savy1,2
Sofía Savy1,2 Victoria Peyret1,2
Victoria Peyret1,2 Romina Celeste Geysels1,2
Romina Celeste Geysels1,2 Francisco Andrés Montes1,2
Francisco Andrés Montes1,2 Carlos Eduardo Bernal Barquero1,2
Carlos Eduardo Bernal Barquero1,2 Valentina Ricci3,4
Valentina Ricci3,4 María Eugenia Masnata3,4
María Eugenia Masnata3,4 Ana María Masini-Repiso1,2Patricia Papendieck3,4
Ana María Masini-Repiso1,2Patricia Papendieck3,4 Mariana Lorena Tellechea3,4
Mariana Lorena Tellechea3,4 Ana Elena Chiesa3,4
Ana Elena Chiesa3,4 Juan Pablo Nicola1,2*
Juan Pablo Nicola1,2*- 1Departamento de Bioquímica Clínica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba, Argentina
- 2Centro de Investigaciones en Bioquímica Clínica e Inmunología, Consejo Nacional de Investigaciones Científicas y Técnicas (CIBICI-CONICET), Córdoba, Argentina
- 3División de Endocrinología, Hospital de Niños Dr. Ricardo Gutiérrez, Buenos Aires, Argentina
- 4Centro de Investigaciones Endocrinológicas Dr. César Bergadá, Consejo Nacional de Investigaciones Científicas y Técnicas (CEDIE-CONICET), Buenos Aires, Argentina
Introduction: The sodium/iodide symporter (NIS) mediates active iodide accumulation in the thyroid follicular cell. Biallelic loss-of-function variants in the NIS-coding SLC5A5 gene cause congenital dyshormonogenic hypothyroidism due to a defect in the accumulation of iodide, which is required for thyroid hormonogenesis.
Objective: We aimed to identify, and if so to functionally characterize, novel pathogenic SLC5A5 gene variants in a patient diagnosed with severe congenital dyshormonogenic hypothyroidism characterized by undetectable radioiodide accumulation in a eutopic thyroid gland, as well as in the salivary glands.
Methods: The coding region of the SLC5A5 gene was sequenced using whole-exome sequencing. In silico analysis and in vitro functional characterization of missense SLC5A5 gene variants were performed.
Results: Proposita’s whole-exome sequencing revealed a novel pair of compound heterozygous missense variants in the SLC5A5 gene, c.1,627G>A (p.G543R) and c.1,684T>A (p.L562M). The parents were heterozygous carriers of the variants as determined by Sanger sequencing of the SLC5A5 gene. The p.G543R variant in the homozygous state has previously been associated with congenital hypothyroidism. The novel p.L562M variant was not reported in the Genome Aggregation Consortium dataset. In silico analysis of the pathogenic impact of the p.L562M variant yielded inconclusive results. Functional in vitro studies showed that the p.L562M variant reduces iodide accumulation due to defective expression of the mutant NIS protein at the plasma membrane. Notably, the aliphatic residue Leu at position 562 in the carboxy terminus of the protein, which is highly conserved in NIS orthologues, is required for NIS plasma membrane expression.
Conclusions: We report novel compound heterozygous missense SLC5A5 gene variants causing defective iodide accumulation, thus leading to congenital dyshormonogenic hypothyroidism.
Introduction
Congenital hypothyroidism is the most common endocrine disorder in newborns, and among the most common preventable causes of intellectual disability. In Argentina, congenital hypothyroidism occurs with an incidence of 1:2,367 based on thyrotropin-based newborn screening programs (1). Genetic defects or environmental factors affecting thyroid hormonogenesis lead to congenital dyshormonogenic hypothyroidism (2). In particular, iodide transport defect is a rare autosomal recessive disorder caused by the inability of thyroid follicular cells to accumulate iodide, resulting in congenital dyshormonogenic hypothyroidism. An iodide transport defect is suspected when normal radioiodide accumulation in a eutopic thyroid gland, as well as, in the salivary glands is reduced to absent (3–5).
Iodide accumulation, the first step in the biosynthesis of the iodine-containing thyroid hormones, is mediated by the sodium/iodide symporter (NIS) (6). NIS is an integral basolateral plasma membrane glycoprotein that mediates—with remarkable affinity—sodium-coupled active iodide accumulation into the thyroid follicular cell (7). Structurally, NIS is a 13-transmembrane segment glycoprotein with an extracellular amino-terminus and a large intracellular carboxy-terminus, which contains a conserved monoleucine-based sorting motif that is required for NIS basolateral plasma membrane expression in the thyroid follicular cell (8, 9), and involved in reduced NIS plasma membrane expression in thyroid cancer (10). Recently, the three-dimensional structure of NIS was determined at atomic resolution using single-particle cryogenic electron microscopy, providing structural information on the mechanisms underlying NIS-mediated iodide transport (11), a long-standing question in the thyroid field.
Highlighting the importance of NIS in thyroid physiology, biallelic loss-of-function variants in the NIS-coding SLC5A5 gene cause defective iodide accumulation, thus leading to congenital dyshormonogenic hypothyroidism (6). To date, more than forty pathogenic SLC5A5 variants gene have been identified in patients with congenital dyshormonogenic hypothyroidism. Detailed molecular characterization of NIS variants has provided mechanistic information on structure-function relationships, highlighting critical amino acids for substrate binding, specificity, and stoichiometry, as well as folding and plasma membrane targeting (12–17).
Here, we conducted whole-exome sequencing in a patient with severe congenital dyshormonogenic hypothyroidism characterized by undetectable radioiodide accumulation in a eutopic thyroid gland and the salivary glands. A novel pair of compound heterozygous variants in the SLC5A5 gene, c.1627G>A (p.G543R) and c.1684T>A (p.L562M), have been identified. The missense variant p.G543R in homozygous state has previously been associated with congenital hypothyroidism. Moreover, we conducted functional analysis revealing that the novel missense variant p.L562M lowers iodide accumulation due to defective NIS expression at the plasma membrane. In conclusion, we report novel compound heterozygous missense SLC5A5 gene variants causing defective iodide accumulation, thus leading to congenital dyshormonogenic hypothyroidism.
Materials and methods
Patient’s medical records
The proposita was a full-term female infant born in 1993 as the third child of non-consanguineous and healthy Caucasian parents. The proposita was diagnosed with congenital hypothyroidism (TSH 60.0 µU/ml, range 1.3-10.0 µU/ml; total T4 0.8 µg/dl, range 6.0-18.0 µg/dl) at 21 days of age, after detection of high TSH level on newborn screening (>60 µU/ml, cut off 15 µU/ml). Thyroid function analyses were performed by DELFIA system (PerkinElmer - Waltham, MA). Serum levels of thyroglobulin were not available. Clinical examination showed jaundice, lethargy, tongue protrusion, reticulated skin, and umbilical hernia. X-ray revealed lack of ossification of the distal femoral epiphysis. Levothyroxine replacement therapy was started immediately after diagnosis with a daily dose of 50 µg/kg with excellent treatment adherence over the course of 20-years follow-up period. Levothyroxine dosage was regularly adjusted based on thyroid function tests. At 2.3 years of age, thyroid function evaluation after levothyroxine withdrawal indicated permanent congenital hypothyroidism (TSH 99 µU/ml, range 0.5-6.5 µU/ml; total T4 1 µg/dl, range 4.5-12.5 µg/dl; free T4 0.1 ng/dl, range 0.8-2.2 ng/dl). Thyroid ultrasound revealed a eutopic small thyroid gland (0.23 ml, range 0.30-2.0 ml) with heterogeneous texture. Thyroid scintigraphy revealed undetectable 131I-iodide accumulation in the thyroid gland, as well as in the salivary glands, suggesting an iodide transport defect. Dosage of saliva-to-plasma radioiodide ratio was not technically available in the Division of Nuclear Medicine. At the age of 8, the Wechsler Intelligence Scale for Children (WISC-III) test showed normal neurocognitive development with lower performance in executive functions. The proposita grew normally in the 25th percentile, underwent normal pubertal development, and reached a final height of 164 cm, according to the mean parental height. Parents and older sisters were clinically euthyroid.
Ethical statement
The study was approved by the Ethics Committee of the Hospital de Niños Dr. Ricardo Gutierrez (Buenos Aires, Argentina) and was conducted with the written informed consent of the parents of the proposita’.
Whole-exome sequencing
Genomic DNA was extracted from whole blood using Wizard Genomic DNA Purification Kit (Promega – Madison, WI). Whole-exome sequencing was performed by Macrogen (Seoul, South Korea) using an Illumina platform with 150-bp paired-end reads. Exome capture and library preparation were conducted using Agilent SureSelect V6 post (Agilent Technologies, Santa Clara, CA). Raw reads were mapped to the reference human genome (GRCh38) using the Burrows-Wheeler Alignment software and processed according to the recommendations of the Genome Analysis Toolkit. Variant calling was performed using HaplotypeCaller, and variant annotation was carry out using ANNOVAR. Precomputed scores from in silico meta-predictors able to reach strong evidence for pathogenicity (18), including BayesDel, REVEL, VEST4, and MutPred2, were annotated using dbNSFP (version 4.7a) database. Variant interpretation was determined according to the American College of Medical Genetics and Genomics guidelines in a quantitative Bayesian framework (19, 20).
Sanger sequencing
The nucleotide sequence of coding exons of interest of the SLC5A5 gene was determined by Sanger sequencing by capillary electrophoresis (Macrogen) as reported previously (21).
Expression vectors and site-directed mutagenesis
The amino-terminus hemagglutinin (HA)-tagged human NIS cDNA sequence cloned into the pcDNA3.1 expression vector was kindly provided by Dr. Nancy Carrasco (Vanderbilt School of Medicine) (22).
Site-directed mutagenesis was conducted by PCR using the mutagenic oligonucleotides 5’-CCACTGTGCTGTGCAGAGCCCTCATCAGC (forward) and 5’-GCTGATGAGGGCTCTGCACAGCACAGTGG (reverse) for G543R, and 5’-CCCTGGCCCCGGGAATGTTGTGGTGG (forward) and 5’-CCACCACAACATTCCCGGGGCCAGGG (reverse) for L562M using Phusion Hot Start II DNA Polymerase (Thermo-Fisher Scientific - Waltham, MA), followed by methylated template plasmid digestion with DpnI (Promega – Madison, WI) (23). Mutagenic oligonucleotides were generated using QuickChange primer design software (Agilent Technologies - Santa Clara, CA). The fidelity of all constructs was verified by Sanger sequencing (Macrogen).
Cell culture and transfections
HeLa cells (CCL-2, American Type Culture Collection, Rockville, MD) were cultured in Dulbecco Modified Eagle’s Medium (Thermo-Fisher Scientific) supplemented with 10% fetal bovine serum (Natocor, Córdoba, Argentina). Cells were transfected with 1 µg plasmid/well in 6-well plates using TurboFect Transfection Reagent (Thermo-Fisher Scientific). All experiments were conducted two days after transfection.
125I-iodide transport assays
Transfected cells were incubated in DMEM containing 10 μM iodide supplemented with 50 μCi/μmol 125I-iodide (PerkinElmer Life Sciences - Waltham, MA) for 30 min at 37°C (24). NIS-specific iodide uptake was assessed in the presence of 40 μM perchlorate. Intracellular radioiodide was extracted with ice-cold ethanol and quantified in a Triathler Gamma Counter (Hidex - Turku, Finland). The amount of DNA was determined by the diphenylamine method after trichloroacetic acid precipitation. Results were expressed as picomoles of iodide per μg DNA, and standardized by the ratio of the percentage of mutant NIS-positive cells to the percentage of WT NIS-positive cells—both of which were determined by flow cytometry under permeabilized conditions—in order to correct for differences in transfection efficiency between samples.
Flow cytometry
Cells were fixed in 2% phosphate-buffered paraformaldehyde and stained with 0.5 μg/ml affinity-purified rabbit polyclonal anti-human NIS antibody (25) in PBS containing 0.2% human serum albumin and 0.2% Quillaja saponin (Sigma-Aldrich - St. Louis, MO) (26). After washing, cells were incubated with 1 μg/ml Alexa-488-conjugated anti-rabbit antibody (A-11008, Molecular Probes - Eugene, OR). The fluorescence of ~5x104 events per tube was assayed in a BD FACSCalibur Flow Cytometer (BD Biosciences - San Jose, CA). Data analysis was performed with FlowJo software (Tree Star - Ashland, OR).
Western blot
SDS-PAGE, electrotransference to nitrocellulose membranes, and immunoblotting were conducted as reported previously (26). Membranes were blocked and incubated with 0.2 μg/ml affinity-purified rabbit polyclonal anti-human NIS (25) and 0.2 μg/ml rabbit polyclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sc-25778, Santa Cruz Biotechnology - Santa Cruz, CA) primary antibodies. After washing, membranes were incubated with 0.07 μg/ml IRDye 680RD goat anti-rabbit (#926-68071) secondary antibody (LI-COR Biosciences - Lincoln, NE). Membranes were visualized by Odyssey Infrared Imaging System (LI-COR Biosciences). Relative band intensity was quantified using ImageJ software (National Institutes of Health - Bethesda, MD).
Immunofluorescence
Transfected cells seeded onto glass coverslips were fixed in 2% phosphate-buffered paraformaldehyde and stained with 0.5 μg/ml affinity-purified rabbit polyclonal anti-human NIS antibody (25) and 2 μg/ml mouse monoclonal anti-Calnexin (sc-23954, Santa Cruz Biotechnology) antibodies in PBS containing 0.2% human serum albumin and 0.1% Triton X-100 for permeabilized conditions (27). Alternatively, cells were stained with 1:50 mouse monoclonal anti-human NIS VJ1 antibody (28) in PBS containing 0.2% human serum albumin for non-permeabilized conditions. Secondary staining was performed with 2 μg/ml anti-rabbit Alexa-488-conjugated and anti-mouse Alexa-594-conjugated antibodies (A-11008 and A-11012, Molecular Probes). Nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes). Coverslips were mounted with FluorSave Reagent (Calbiochem - La Jolla, CA) and images were acquired on an Olympus FluoView 1200 confocal microscope (Olympus America - Center Valley, PA). Quantification of NIS expression at the plasma membrane was conducted on non-permeabilized cells using FIJI ImageJ software (National Institutes of Health).
Statistical analysis
Results are presented as the mean ± SEM of at least three independent experiments. Statistical tests were performed using Prism 8.0 software (GraphPad Software - La Jolla, CA). Multiple group analysis was conducted by one-way ANOVA and Newman-Keuls multiple-comparisons post hoc test. Differences were considered significant at p<0.05.
Results
Iodide transport defect was suspected in the proposita on the basis of severe congenital dyshormonogenic hypothyroidism characterized by undetectable radioiodide accumulation in a eutopic thyroid gland, as well as in the salivary glands. Proposita´s whole-exome sequencing revealed heterozygous missense variants in the SLC5A5 gene: c.1,627G>A, p.G543R and c.1,684T>A, p.L562M (GenBank Reference Sequence NM_000453.3, MANE Select Transcript). Pathogenic variants in other genes involved in thyroid development or physiology were not evidenced. The SLC5A5 gene variants were further confirmed by Sanger sequencing (Figure 1A). Consistent with the recessive nature of the disease, analysis of the parents showed that the father is heterozygous for p.G543R and the mother for p.L562M, while older sisters were not included in the study (Figures 1A, B).
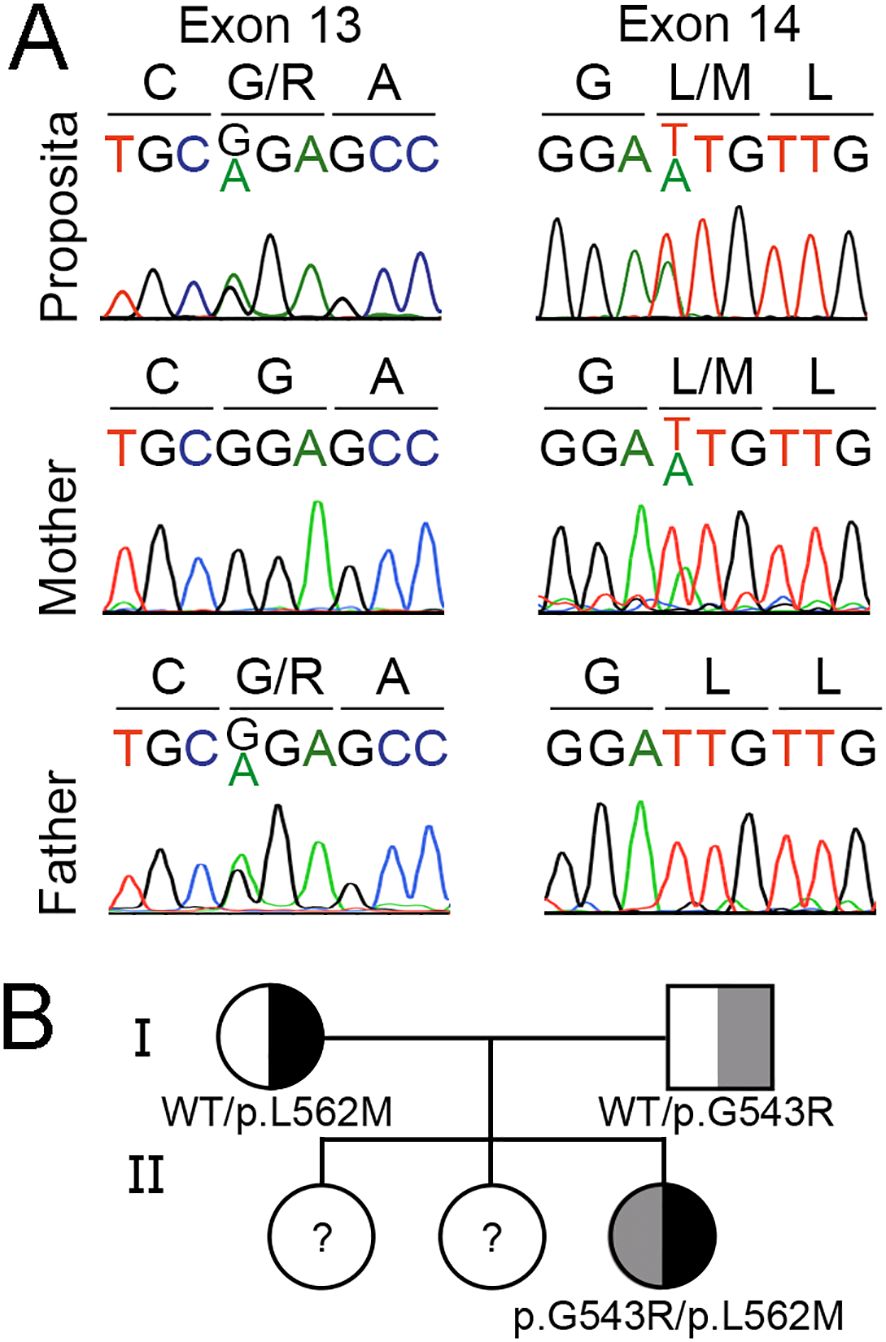
Figure 1. Identification of compound heterozygous missense SLC5A5 gene variants causing congenital dyshormonogenic hypothyroidism. (A) Sanger sequencing chromatogram showing a 9-bp fragment of SLC5A5 exon 13 (nucleotides 1,624 to 1,632) and exon 14 (nucleotides 1681 to 1689). Amino acids are indicated using the one-letter code. The proposita is compound heterozygous for the variants c.1,627G>A (p.G543R) and c.1,684T>A (p.L562M), while the mother carries the variant c.1,684T>A and the father the variant c.1,627G>A. (B) Segregation of SLC5A5 gene variants in the proposita’s family.
The variant c.1627G>A has been reported in Single Nucleotide Polymorphism database (rs776596656) in heterozygosis showing a total allele frequency of 0.00001054, according to The Genome Aggregation Database (version 4.1.0, acceded October 2024). In silico analysis predicted the p.G543R variant as pathogenic (Table 1). Significantly, the p.G543R variant was previously identified in homozygous state in a patient with goitrous congenital hypothyroidism (29), and functional characterization showed that G543R NIS lacked iodide transport activity due to impaired plasma membrane expression (30). The novel p.L562M variant was absent in The Genome Aggregation Database (version 4.1.0, acceded October 2024). In silico analysis using pathogenicity predictors revealed that the p.L562M variant did not meet the criteria to be considered either benign (BP4) or pathogenic (PP3) (Table 1) (18). According to the ACMG guidelines, the p.G543R variant was classified as pathogenic (PM2_Supporting, PM3_Supporting, PM5, PP3_Moderate, PP4, PS3), and the p.L562M variant as variant of unknown significance (PM2_supporting + PM3 + PP4).

Table 1. In silico pathogenicity prediction of variants.
According to the rat NIS three-dimensional structure (11), which shares 89% sequence identity with human NIS, the residue G543 is located on the cytoplasmic side of transmembrane segment XIII, while the residue L562 is located in the cytoplasm-facing carboxy-terminus (Figure 2A). Functional characterization was conducted to test the pathogenicity of the p.L562M variant. HeLa cells, which do not express NIS endogenously, transfected to express L562M NIS showed reduced perchlorate-sensitive iodide accumulation compared to cells expressing WT NIS (Figure 2B). Immunofluorescence confocal microscopy analysis under non-permeabilized conditions to assess plasma membrane NIS expression revealed that the levels of p.L562M NIS were significantly lower than those of WT NIS, suggesting that plasma membrane sorting of the mutant protein is severely impaired. (Figures 2C, D). Together, these findings indicate that the L562M substitution decreases NIS targeting to the plasma membrane, and consequently reduces NIS-mediated iodide transport. As previously reported (30), G543R NIS-expressing cells did not exhibit perchlorate-sensitive iodide accumulation, ultimately because G543R NIS was not targeted to the plasma membrane (Figures 2B, C).
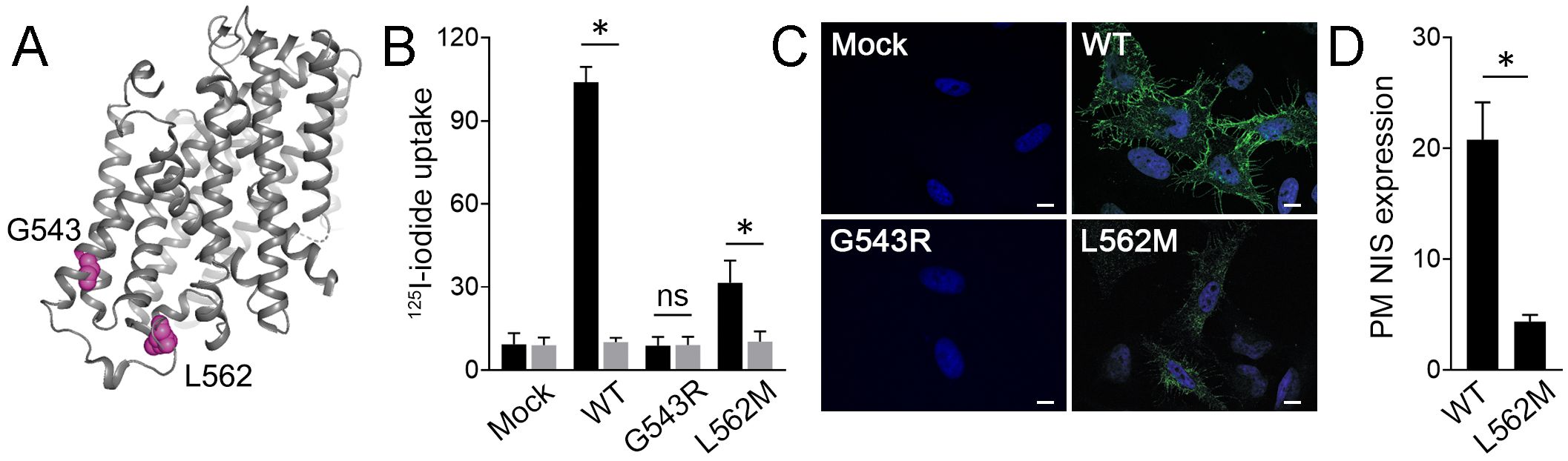
Figure 2. L652M severely reduces NIS plasma membrane expression. (A) Three-dimensional structure of rat NIS (PDB: 7UUY). The residues G543 and L562 are highlighted in pink. Image generated with The PyMOL Molecular Graphics System (Schrödinger, New York). (B) Steady-state iodide uptake in HeLa cells transiently expressing empty vector (mock) or WT, or the NIS variants G543R and L562M. Cells were incubated with iodide in the absence (black bars) or presence (gray bars) of perchlorate. Data are expressed in pmol of I−/μg of DNA ± SEM (n=3) and standardized by transfection efficiency, which was assessed by flow cytometry. *p<0.05 (ANOVA, Newman-Keuls test); ns, not significant. (C) Representative merged immunofluorescence analysis of WT or mutant NIS plasma membrane expression probed with anti-human NIS VJ1 antibody under non-permeabilized conditions. Cell nuclei were stained with DAPI (blue). Scale bars: 10 µm. (D) Mean fluorescence intensity quantification of immunofluorescence confocal microscopy images assessing plasma membrane NIS expression. Results are expressed as the mean ± SEM (n=10-12 cells). *p<0.05 (ANOVA, Student’s t-test).
On Western blots, the electrophoretic pattern of WT NIS showed a higher ratio of fully glycosylated polypeptides (~90 kDa, band B) to partially glycosylated polypeptides (~60 kDa, band A) (Figure 3A). Fully glycosylated NIS corresponds mostly to polypeptides located at the plasma membrane, whereas partially glycosylated polypeptides mostly correspond to those that have not exited the endoplasmic reticulum compartment. In contrast, the fully glycosylated NIS polypeptide (~90 kDa, band B) was minimally detected in L562M NIS-expressing cells, reinforcing the concept that only a small percentage of the mutant protein is targeted to the plasma membrane (Figure 3A). In addition, immunofluorescence confocal microscopy analysis under permeabilized conditions revealed that L562M NIS mostly colocalized with the endoplasmic reticulum-resident protein Calnexin, indicating that the mutant protein is mostly retained in the endoplasmic reticulum, whereas WT NIS was predominantly expressed at the plasma membrane (Figure 3B). As previously reported (30), G543R NIS is completely retained in the endoplasmic reticulum (Figures 3A, B). Multiple sequence alignment of NIS orthologues from different metazoan species revealed that the p.L562M variant affects a highly conserved residue (Figure 3C). Together, these findings suggest that the p.L562M variant decreases NIS targeting to the plasma membrane, and consequently reduces NIS-mediated iodide transport, thereby expanding the spectrum of congenital hypothyroidism-causing SLC5A5 gene variants.
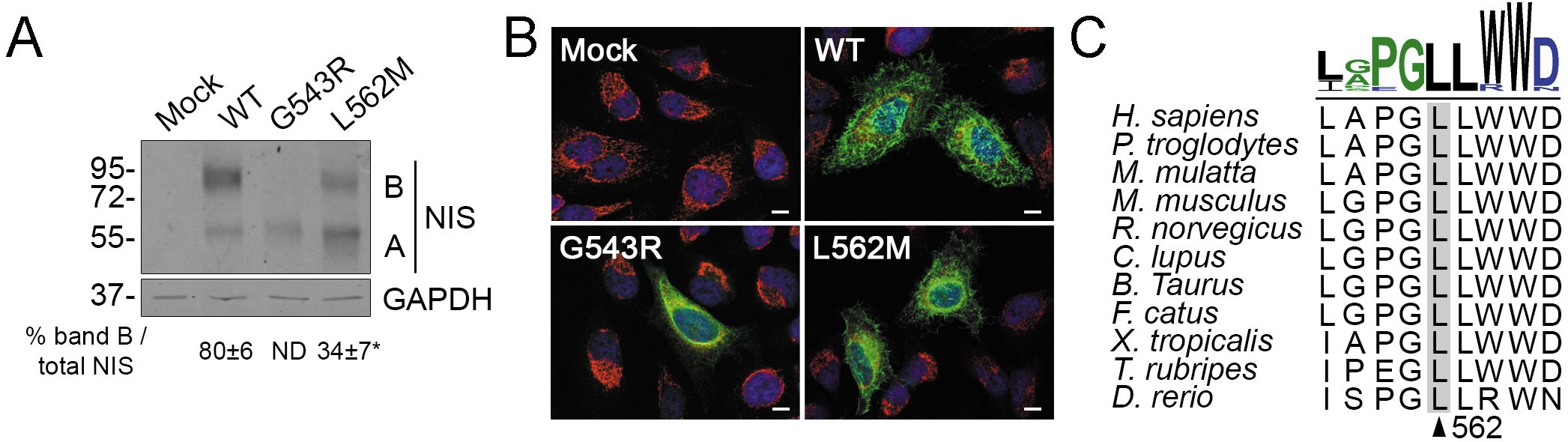
Figure 3. L562M reduces NIS exit from the endoclasmic reticulum. (A) Representative Western blot analysis of total lysates from transiently transfected HeLa cells probed with anti-human NIS and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies. Labels indicate the electrophoretic pattern of the corresponding NIS polypeptides (A: partially glycosylated; B: fully glycosylated) depending on glycosylation status. Quantification of fully glycosylated NIS polypeptides (band B) expressed as percent of total NIS is shown. Results are expressed as the mean ± SEM (n=3). *p<0.05 (ANOVA, Student’s t-test). (B) Representative merged immunofluorescence analysis of transiently transfected HeLa cells probed with anti-human NIS (green) and anti-human Calnexin (red) antibodies under permeabilized conditions. Cell nuclei were stained with DAPI (blue). Scale bars: 10 µm. (C) PSI/TM-Coffee-generated multiple amino acid sequence alignment of NIS orthologues from different metazoan species surrounding the amino acid L562 of human NIS. Sequence logo was generated using WebLogo 3 (https://weblogo.threeplusone.com).
Discussion
Next-generation sequencing has been instrumental in expanding the mutational landscape of monogenic forms of congenital hypothyroidism. In particular, whole-exome sequencing analysis has revealed pathogenic variants in novel genes involved in the pathogenesis of congenital hypothyroidism (31, 32). Here, using whole-exome sequencing, we identify a novel pair of compound heterozygous missense SLC5A5 gene variants—p.G543R and p.L562M—in a proposita with congenital dyshormonogenic hypothyroidism characterized by undetectable radioiodide accumulation in a eutopic thyroid gland, as well as in the salivary glands, suggestive of an iodide transport defect phenotype.
The p.G543R NIS variant was previously identified in homozygosity in a patient with goitrous congenital hypothyroidism (29). In addition, another variant in the same residue, p.G543E, was detected in homozygosity in two siblings with goitrous congenital hypothyroidism who showed minimal radioiodide accumulation in the thyroid gland (33). Functional characterization of the G543R/E variants revealed that they impair NIS exit from the endoplasmic reticulum, apparently due to a folding defect, thus reducing NIS-mediated iodide accumulation (30). The novel variant reported here, p.L562M, severely reduced normal iodide accumulation by repressing NIS transport to the plasma membrane. Of note, the functional evaluation supported the classification of the p.L562M NIS variant as likely pathogenic (PS3 criteria added), thus our data highlight the importance of interpreting congenital hypothyroidism-associated variants with caution according to consensus guidelines.
The residue L562 is located in the intracellularly-facing carboxy-terminus, which is required for the expression of NIS at the basolateral plasma membrane in the thyroid follicular cell (8). Underscoring the significance of the carboxy-terminal region, the missense p.S547R and p.G561E variants and the nonsense p.R636*, which generates a truncated protein missing the last eight amino acids, identified in patients with congenital dyshormonogenic hypothyroidism causes the intracellular retention of the mutant protein (34–36). Of note, the molecular characterization of the iodide transport defect-causing p.G561E NIS variant, which is adjacent to the p.L562M variant reported here, revealed the importance of a highly conserved [L/M]xW[D/E] tryptophan-acidic sorting motif involved in NIS transport to the plasma membrane (36). The p.G561E variant shifts the equilibrium of the adjacent unstructured tryptophan-acidic motif towards a structured alpha-helical conformation reducing its recognition by the kinesin-1 subunit kinesin light chain 2, thereby interfering with NIS maturation beyond the endoplasmic reticulum, and reducing iodide accumulation (36).
Considering that NIS-mediated iodide accumulation is the first step in the synthesis of iodine-containing thyroid hormones, our results suggest that the pair of compound heterozygous missense NIS variants p.G543R and p.L562M impairs normal iodide accumulation by interfering with NIS maturation and transport to the plasma membrane. Notably, the in vitro data correlate with clinical findings of impaired radioiodide accumulation in the thyroid when the patient was evaluated by thyroid scintigraphy. Therefore, the consequent lack of sufficient NIS molecules at the basolateral plasma membrane of thyroid follicular cells reveals the mechanism underlying the deficient iodide accumulation leading to congenital dyshormonogenic hypothyroidism.
Data availability statement
The raw data from whole exome sequencing remain confidential due to ethical considerations. All additional raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Hospital de Niños Dr. Ricardo Gutierrez (Buenos Aires, Argentina). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
GC: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. MM: Formal Analysis, Investigation, Writing – original draft, Writing – review & editing. SS: Formal Analysis, Investigation, Writing – review & editing. VP: Methodology, Writing – review & editing. RG: Methodology, Writing – review & editing. FM: Methodology, Writing – review & editing. CB: Methodology, Writing – review & editing. VR: Methodology, Writing – review & editing. MEM: Methodology, Writing – review & editing. AM-R: Conceptualization, Resources, Writing – review & editing. PP: Methodology, Writing – review & editing. MT: Conceptualization, Funding acquisition, Writing – review & editing. AC: Conceptualization, Funding acquisition, Resources, Writing – original draft, Writing – review & editing. JN: Conceptualization, Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Fondo para la Investigación Científica y Tecnológica - Agencia Nacional de Promoción Científica y Tecnológica (grants number PICT-2018-1596, PICT-2019-1772, PICT-2021-0005, and PICT-2021-0409 awarded to JN and PIDC-2019-0007 awarded to MT, AC, and JN), Secretaría de Políticas Universitarias - Ministerio de Educación (grant number VT12-UNCOR4153 awarded to JN), and Secretaría de Ciencia y Tecnología - Universidad Nacional de Córdoba (grant number 33620180100772CB awarded to JN).
Acknowledgments
We thank Dr. Nancy Carrasco (Vanderbilt University School of Medicine, United States) and Dr. Sabine Costagliola (Free University of Brussels, Brussels, Belgium) for kindly providing critical reagents. We also thank Dr. Pilar Crespo (Centro de Micro y Nanoscopía de Córdoba, Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Córdoba, Argentina) for imaging technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chiesa A, Prieto L, Mendez V, Papendieck P, Calcagno ML, Gruneiro-Papendieck L. Prevalence and etiology of congenital hypothyroidism detected through an argentine neonatal screening program (1997-2010). Horm Res Paediatr. (2013) 80:185–92. doi: 10.1159/000354409
2. Rose SR, Wassner AJ, Wintergerst KA, Yayah-Jones NH, Hopkin RJ, Chuang J, et al. Congenital hypothyroidism: screening and management. Pediatrics. (2023) 151:e2022060420. doi: 10.1542/peds.2022-060420
3. Zhang CX, Zhang JX, Yang L, Zhang CR, Cheng F, Zhang RJ, et al. Novel compound heterozygous pathogenic mutations of SLC5A5 in a chinese patient with congenital hypothyroidism. Front Endocrinol (Lausanne). (2021) 12:620117. doi: 10.3389/fendo.2021.620117
4. Durgia H, Nicholas AK, Schoenmakers E, Dickens JA, Halanaik D, Sahoo J, et al. Brief report: A novel sodium/iodide symporter mutation, S356F, causing congenital hypothyroidism. Thyroid. (2022) 32:215–8. doi: 10.1089/thy.2021.0478
5. Gong L, Yang N, Zhao J, Tang Y, Li L, Yang H, et al. Clinical characteristics and genetics analysis for the ITD of congenital hypothyroidism. J Pediatr Endocrinol Metab. (2022) 35:741–8. doi: 10.1515/jpem-2022-0052
6. Ravera S, Reyna-Neyra A, Ferrandino G, Amzel LM, Carrasco N. The sodium/iodide symporter (NIS): molecular physiology and preclinical and clinical applications. Annu Rev Physiol. (2017) 79:261–89. doi: 10.1146/annurev-physiol-022516-034125
7. Nicola JP, Carrasco N, Amzel LM. Physiological sodium concentrations enhance the iodide affinity of the Na+/I- symporter. Nat Commun. (2014) 5:3948. doi: 10.1038/ncomms4948
8. Martin M, Modenutti CP, Peyret V, Geysels RC, Darrouzet E, Pourcher T, et al. A carboxy-terminal monoleucine-based motif participates in the basolateral targeting of the na+/I- symporter. Endocrinology. (2019) 160:156–68. doi: 10.1210/en.2018-00603
9. Koumarianou P, Fernandez-Mendez C, Fajardo-Delgado D, Mielu LM, Santisteban P, De la Vieja A. Basolateral sorting of the sodium/iodide symporter is mediated by adaptor protein 1 clathrin adaptor complexes. Thyroid. (2022) 32:1259–70. doi: 10.1089/thy.2022.0163
10. Read ML, Brookes K, Zha L, Manivannan S, Kim J, Kocbiyik M, et al. Combined vorinostat and chloroquine inhibit sodium-iodide symporter endocytosis and enhance radionuclide uptake in vivo. Clin Cancer Res. (2024) 30:1352–66. doi: 10.1158/1078-0432.CCR-23-2043
11. Ravera S, Nicola JP, Salazar-De Simone G, Sigworth FJ, Karakas E, Amzel LM, et al. Structural insights into the mechanism of the sodium/iodide symporter (NIS). Nature. (2022) 612:795–801. doi: 10.1038/s41586-022-05530-2
12. Nicola JP, Reyna-Neyra A, Saenger P, Rodriguez-Buritica DF, Gamez Godoy JD, Muzumdar R, et al. Sodium/iodide symporter mutant V270E causes stunted growth but no cognitive deficiency. J Clin Endocrinol Metab. (2015) 100:E1353–1361. doi: 10.1210/jc.2015-1824
13. Li W, Nicola JP, Amzel LM, Carrasco N. Asn441 plays a key role in folding and function of the Na+/I- symporter (NIS). FASEB J. (2013) 27:3229–38. doi: 10.1096/fj.13-229138
14. Paroder-Belenitsky M, Maestas MJ, Dohan O, Nicola JP, Reyna-Neyra A, Follenzi A, et al. Mechanism of anion selectivity and stoichiometry of the Na+/I- symporter (NIS). Proc Natl Acad Sci U.S.A. (2011) 108:17933–8. doi: 10.1073/pnas.1108278108
15. De la Vieja A, Reed MD, Ginter CS, Carrasco N. Amino acid residues in transmembrane segment IX of the Na+/I- symporter play a role in its Na+ dependence and are critical for transport activity. J Biol Chem. (2007) 282:25290–8. doi: 10.1074/jbc.M700147200
16. Reyna-Neyra A, Jung L, Chakrabarti M, Suarez M, Amzel LM, Carrasco N. The iodide transport defect-causing Y348D mutation in the Na(+)/I(-) symporter (NIS) renders the protein intrinsically inactive and impairs its targeting to the plasma membrane. Thyroid. (2021) 31:1272–81. doi: 10.1089/thy.2020.0931
17. Bernal Barquero CE, Martín M, Geysels RC, Peyret V, Papendieck P, Masini-Repiso AM, et al. An intramolecular ionic interaction linking defective sodium/iodide symporter transport to the plasma membrane and dyshormonogenic congenital hypothyroidism. Thyroid. (2022) 32:19–27. doi: 10.1089/thy.2021.0344
18. Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/j.ajhg.2022.10.013
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
20. Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. (2018) 20:1054–60. doi: 10.1038/gim.2017.210
21. Geysels RC, Bernal Barquero CE, Martin M, Peyret V, Nocent M, Sobrero G, et al. Silent but not harmless: A synonymous SLC5A5 gene variant leading to dyshormonogenic congenital hypothyroidism. Front Endocrinol (Lausanne). (2022) 13:868891. doi: 10.3389/fendo.2022.868891
22. Ferrandino G, Nicola JP, Sanchez YE, Echeverria I, Liu Y, Amzel LM, et al. Na+ coordination at the Na2 site of the Na+/I- symporter. Proc Natl Acad Sci U.S.A. (2016) 113:E5379–5388. doi: 10.1073/pnas.1607231113
23. Rossich LE, Thomasz L, Nicola JP, Nazar M, Salvarredi LA, Pisarev M, et al. Effects of 2-iodohexadecanal in the physiology of thyroid cells. Mol Cell Endocrinol. (2016) 437:292–301. doi: 10.1016/j.mce.2016.08.036
24. Geysels RC, Peyret V, Martín M, Nazar M, Reale C, Bernal Barquero CE, et al. The transcription factor NF-κB mediates thyrotropin-stimulated expression of thyroid differentiation markers. Thyroid. (2021) 31:299–314. doi: 10.1089/thy.2020.0208
25. Tazebay UH, Wapnir IL, Levy O, Dohan O, Zuckier LS, Zhao QH, et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med. (2000) 6:871–8. doi: 10.1038/78630
26. Bernal Barquero CE, Geysels RC, Jacques V, Carro GH, Martín M, Peyret V, et al. Targeted next-generation sequencing of congenital hypothyroidism-causative genes reveals unexpected thyroglobulin gene variants in patients with iodide transport defect. Int J Mol Sci. (2022) 23:1825955. doi: 10.3390/ijms23169251
27. Peinetti N, Scalerandi MV, Cuello Rubio MM, Leimgruber C, Nicola JP, Torres AI, et al. The response of prostate smooth muscle cells to testosterone is determined by the subcellular distribution of the androgen receptor. Endocrinology. (2018) 159:945–56. doi: 10.1210/en.2017-00718
28. Pohlenz J, Duprez L, Weiss RE, Vassart G, Refetoff S, Costagliola S. Failure of membrane targeting causes the functional defect of two mutant sodium iodide symporters. J Clin Endocrinol Metab. (2000) 85:2366–9. doi: 10.1210/jc.85.7.2366
29. Li M, Li X, Wang F, Ren Y, Zhang X, Wang J, et al. Genetic analysis of iodide transporter and recycling (NIS, PDS, SLC26A7, IYD) in patients with congenital hypothyroidism. Gene. (2022) 824:146402. doi: 10.1016/j.gene.2022.146402
30. De la Vieja A, Ginter CS, Carrasco N. Molecular analysis of a congenital iodide transport defect: G543E impairs maturation and trafficking of the Na+/I- symporter. Mol Endocrinol. (2005) 19:2847–58. doi: 10.1210/me.2005-0162
31. Zhang HY, Wu FY, Zhang CX, Wu CY, Cui RJ, Liu XY, et al. Contactin 6, A novel causative gene for congenital hypothyroidism, Mediates thyroid hormone biosynthesis through notch signaling. Thyroid. (2024) 34:324–35. doi: 10.1089/thy.2023.0594
32. Sun F, Zhang RJ, Fang Y, Yan CY, Zhang CR, Wu FY, et al. Identification of eukaryotic translation initiation factor 4B as a novel candidate gene for congenital hypothyroidism. J Clin Endocrinol Metab. (2024). doi: 10.1210/clinem/dgae270
33. Kosugi S, Inoue S, Matsuda A, Jhiang SM. Novel, missense and loss-of-function mutations in the sodium/iodide symporter gene causing iodide transport defect in three Japanese patients. J Clin Endocrinol Metab. (1998) 83:3373–6. doi: 10.1210/jcem.83.9.5243
34. Martin M, Bernal Barquero CE, Geysels RC, Papendieck P, Peyret V, Masini-Repiso AM, et al. Novel sodium/iodide symporter compound heterozygous pathogenic variants causing dyshormonogenic congenital hypothyroidism. Thyroid. (2019) 29:1023–6. doi: 10.1089/thy.2019.0046
35. Martín M, Salleron L, Peyret V, Geysels RC, Darrouzet E, Lindenthal S, et al. The PDZ protein SCRIB regulates sodium/iodide symporter (NIS) expression at the basolateral plasma membrane. FASEB J. (2021) 35:e21681. doi: 10.1096/fj.202100303R
Keywords: congenital hypothyroidism, iodide transport defect, sodium iodide symporter (NIS), whole-exome sequencing, biallelic loss-of-function SLC5A5 variants
Citation: Carro GH, Martín M, Savy S, Peyret V, Geysels RC, Montes FA, Bernal Barquero CE, Ricci V, Masnata ME, Masini-Repiso AM, Papendieck P, Tellechea ML, Chiesa AE and Nicola JP (2024) Functional characterization of novel compound heterozygous missense SLC5A5 gene variants causing congenital dyshormonogenic hypothyroidism. Front. Endocrinol. 15:1465176. doi: 10.3389/fendo.2024.1465176
Received: 15 July 2024; Accepted: 04 November 2024;
Published: 19 December 2024.
Edited by:
Marian Elizabeth Ludgate, Cardiff University, United KingdomReviewed by:
Hector Manuel Targovnik, University of Buenos Aires, ArgentinaRaisa Ghosh, National Institutes of Health (NIH), United States
Copyright © 2024 Carro, Martín, Savy, Peyret, Geysels, Montes, Bernal Barquero, Ricci, Masnata, Masini-Repiso, Papendieck, Tellechea, Chiesa and Nicola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Pablo Nicola, anVhbi5uaWNvbGFAdW5jLmVkdS5hcg==
†These authors have contributed equally to this work