 Willem Staels1,2*†
Willem Staels1,2*† Jean De Schepper1†
Jean De Schepper1† Marianne Becker3†
Marianne Becker3† Philippe Lysy4†Daniel Klink5Karl Logghe6
Philippe Lysy4†Daniel Klink5Karl Logghe6 Marieken den Brinker7†
Marieken den Brinker7† Anne Rochtus8Bruno Lapauw9†
Anne Rochtus8Bruno Lapauw9† Martine Cools10,11†Orsalia Alexopoulou12†
Martine Cools10,11†Orsalia Alexopoulou12† Marie Bex13†Bernard Corvilain14
Marie Bex13†Bernard Corvilain14 Laurent Crenier14Christophe De Block15†Julian Donckier16
Laurent Crenier14Christophe De Block15†Julian Donckier16 Robert Hilbrands17Michel Ponchon18Guy T'Sjoen9†
Robert Hilbrands17Michel Ponchon18Guy T'Sjoen9† Annick Van Den Bruel19Sara Vandewalle19†Brigitte Velkeniers17†
Annick Van Den Bruel19Sara Vandewalle19†Brigitte Velkeniers17†- 1Division of Pediatric Endocrinology, Department of Pediatrics, Vrije Universiteit Brussel (VUB), Universitair Ziekenhuis Brussel (UZ Brussel), Brussels, Belgium
- 2Genetics, Reproduction and Development (GRAD), Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 3Division of Pediatric Endocrinology, Department of Pediatrics, Centre Hospitalier de Luxembourg (CHL), Luxembourg, Luxembourg
- 4Division of Pediatric Endocrinology and Diabetes, Cliniques Universitaires Saint Luc, Brussels, Belgium
- 5Division of Pediatric Endocrinology and Diabetes, ZNA Queen Paola Children’s Hospital Antwerp, Antwerp, Belgium
- 6Department of Pediatrics, AZ Delta, Roeselare, Belgium
- 7Division of Pediatric Endocrinology, Department of Pediatrics, Universitair Ziekenhuis Antwerpen, Universiteit Antwerpen (UA), Antwerp, Belgium
- 8Division of Pediatric Endocrinology, Department of Pediatrics, Universitair Ziekenhuis Leuven, Leuven, Belgium
- 9Department of Endocrinology, Universitair Ziekenhuis Gent, Ghent, Belgium
- 10Department of Paediatrics, Division of Paediatric Endocrinology, Ghent University Hospital, Ghent University, Ghent, Belgium
- 11Department of Internal Medicine and Paediatrics, Ghent University, Ghent, Belgium
- 12Department of Endocrinology, Cliniques Universitaires Saint Luc, Brussels, Belgium
- 13Department of Endocrinology, UZ Leuven (Universitaire Ziekenhuizen Leuven), Leuven, Belgium
- 14Department of Endocrinology, Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium
- 15Department of Endocrinology and Diabetes, Universitair Ziekenhuis Antwerpen (UZA), Universiteit Antwerpen (UA), Antwerp, Belgium
- 16Department of Endocrinology, Université Catholique de Louvain CHU UCL Namur (Site Godinne), Yvoir, Belgium
- 17Department of Diabetes and Endocrinology, Vrije Universiteit Brussel (VUB), Universitair Ziekenhuis Brussel (UZ Brussel), Brussels, Belgium
- 18Department of Endocrinology, Cliniques Saint Jean, Brussels, Belgium
- 19Department of Endocrinology, AZ Sint Jan Brugge AV, Brugge, Belgium
Growth hormone (GH) deficiency (GHD) in children and adolescents can vary in severity and origin, with GH replacement therapy proving effective in achieving genetic target height. Optimal outcomes are seen in those treated early and with higher doses. As patients approach adult height, priorities shift towards optimizing metabolic effects, maintaining body composition, and enhancing bone mass and muscle strength. Transitioning from pediatric to adult care presents challenges, including accurately identifying candidates for continued GH therapy, reevaluating persistent GHD, and preventing treatment discontinuation. Assessing readiness for transition and self-management skills is crucial. This Policy and Practice Review provides a comprehensive overview of current policies, regulations, and guidelines pertinent to managing GHD transition in Belgium. We integrate perspectives from national academic and nonacademic clinical stakeholders in pediatric and adult endocrine care to provide an updated policy framework. This framework underscores the importance of sustained GH therapy during transition, particularly for individuals with persistent GHD, with the goal of optimizing practices and improving outcomes during this critical period.
1 Introduction
Growth hormone (GH) deficiency (GHD), when diagnosed during childhood or adolescence, can manifest as an isolated or a combined pituitary hormone deficiency, and it can vary in severity, ranging from severe (often defined as a peak GH following a stimulation test of < 3 ng/mL) to partial GHD (often defined as a peak GH of 3-7 ng/mL) (1). GHD may arise from idiopathic, genetic, or acquired origins, yet in each scenario, GH replacement therapy has demonstrated its efficacy in facilitating the attainment of an adult height within the genetic target height. Notably, the most favorable growth outcomes have been documented in patients with combined GHD, particularly those who started treatment at the youngest age and/or received the highest GH doses (2–5).
As individuals approach their near-adult height, the focus of GH therapy shifts from optimizing growth towards optimizing the metabolic effects of GH, primarily directed at maintaining a healthy body composition and achieving optimal peak bone mass and muscle strength in early adulthood and a transfer from pediatric to adult care becomes imperative (6–9). However, transitioning adolescents or young adults with GHD from pediatric to adult endocrinology departments poses multifaceted challenges, including medical, psychosocial, and organizational aspects (10–13). Key clinical challenges include accurately identifying candidates suitable for seamless continuation of GH treatment or reevaluation for persistent GHD, along with precise GHD diagnosis for those who require retesting (14). Additionally, strategies to mitigate the risk of follow-up loss and/or discontinuation of GH treatment need to be developed, particularly for adolescents with isolated GHD, a group that is more vulnerable to early discontinuation of medical surveillance (15). Hence, assessing readiness for transition, disease comprehension, and self-management skills are essential before transitioning patients to adult care (16).
The prevailing guidelines set forth by scientific societies for the transition care of adolescents with GHD are based on a combination of clinical expert consensus and the growing body of evidence from clinical studies, showing the advantages of continued GH therapy during the transition phase, especially for those with persistent GHD (17, 18). Differences in national policies regarding the testing of persistent GHD and the continuation of GH treatment, in addition to the observation of non-uniform GH dosing, along with the significant drop-out rates after departing pediatric services, have driven the need to update the guidelines governing the management of GH therapy during transition (19–23).
2 Assessment and discussion of policy options and implications
2.1 Why is a Belgian policy needed?
In this Belgian policy, we adapt international guidelines to our specific national context to standardize and optimize GHD management during the transition from pediatric to adult endocrine care. Key considerations included the expanding gene panel use for diagnosing GHD, addressing disparities in clinical practice among Belgian hospitals, and the recent changes in GH reimbursement criteria for GHD during the transition phase. Adherence to national policy can result in improved cost-effectiveness. However, the pivotal step lies in adopting these proposed guidelines by national insurance policies, ensuring their effective integration into the Belgian healthcare framework.
2.2 How were the Belgian guidelines established?
A writing committee comprised of national experts from Belgian university and non-university hospitals was established. First, two in-person meetings were held between several authors (WS, JDS, MB, PL, DK, KL, CDB, MB, BV) to review and discuss current evidence. The international guidelines and recent publications formed the basis for practical recommendations adapted to the Belgian context (12, 28-31). Next, the other co-authors were consulted through consecutive e-mail rounds. Finally, the guidelines were endorsed by the Belgian and Luxemburgish Society for Pediatric Endocrinology and Diabetology (BELSPEED), the Belgian Endocrine Society (BES), and the Flemish Network for Rare Diseases (VNZZ).
2.3 What is the current management of GHD during childhood and transition in Belgium?
In Belgium, GH therapy for children with GHD is managed by pediatric endocrinologists, who convene monthly for peer-review of diagnostic assessments related to GHD, coordinated by BELSPEED. These assessments encompass clinical, auxological, hormonal, brain imaging, and bone age evaluations. The diagnostic criteria include (a) growth velocity below the 25th percentile (in prepubertal patients), (b) delayed bone age maturation, (c) serum IGF1 level below the mean value for the corresponding age/bone age, and (d) GH peak < 7 µg/L (since 2009) elicited during two GH stimulation tests. Notably, at least one of these tests should be sex steroid primed in girls aged above 9 years and boys aged above 10 years, who have not yet reached an advanced pubertal stage (Tanner B3 in girls, testicular volume ≥ 10 ml in boys). Furthermore, patients diagnosed with GHD routinely undergo a brain MRI. Annually, Belgium registers 50 to 70 cases of GHD in children.
GH dosing is body weight-based at 25-30 µg/kg/day until near-adult height. GHD diagnosis is reevaluated by the treating pediatric endocrinologist after one year, juxtaposing the growth response with national references (24). Retesting for persistent GHD during mid-puberty is not customary in Belgium except when the need to continue the GH treatment is questioned by the patient, compliance is poor, or alternative diagnoses are considered. Typically, GH treatment follows the above pediatric dosing regimen as long as the growth velocity exceeds 2 cm/year and/or bone age readings show open growth plates. When these criteria are no longer met, a short-term suspension of GH therapy is done for all patients with idiopathic GHD and most patients with organic GHD. In 2003, we reported that 33% of patients with isolated idiopathic GHD exhibited a transient form when re-evaluated based on the current GHD criteria after reaching adult height (25). Unfortunately, recent national data on retesting outcomes are not available.
In most Belgian centers, an insulin tolerance test (ITT) is performed to evaluate persistent GHD. The testing is, in general, performed between 1 and 6 months after stopping GH therapy, but different cut-offs of peak GH value (varying between < 3 and < 7 µg/L) are used to diagnose persistent GHD. Clinical practice ensuring continued GH treatment beyond pediatric care varies among centers. In some, the transfer to adult endocrine care occurs upon reaching adult height, while in others a transition near the age of 18 years is more common. The handover of care in most pediatric centers is realized through a referral letter to the adult endocrinologist, while some centers arrange joint consultations with pediatric and adult care providers.
2.4 How should the transitioning of adolescents treated for GHD be organized?
The initiation of the transfer process to adult endocrine care should be guided by the adolescent’s readiness, even if final height is not yet attained. Pediatric endocrinologists should strive to equip adolescents with the skills to proficiently manage GHD without relying on parental supervision. Employing specific questionnaires, checklists, or tool kits can aid in assessing readiness and identifying knowledge gaps before transitioning to adult care. Notable examples of validated checklists include the Transition Readiness Assessment Questionnaire (TRAQ) and the Transition Readiness and Appropriateness Measure (TRAM) (16, 26).
The seamless transition of care necessitates close collaboration between pediatric and adult endocrinologists, facilitated by joint consultations. Due to the increased need for social support, lower general well-being, and the increased risk of fertility issues in adolescents with multiple pituitary hormone deficiencies (MPHD), transitioning to a multidisciplinary clinic proves most appropriate (27, 28). Such multidisciplinary transition clinics ideally encompass access to psychologists and fertility counselors, including gynecologists and andrologists (29). A dedicated adult clinic case manager can streamline patient transitions and reduce follow-up gaps.
In Belgium, established multidisciplinary transition services for GHD patients remain scarce. These clinics must establish therapeutic education programs as patients with persistent GHD often require comprehensive and repeated explanations of the benefits of continued GH treatment after having reached adult height. These include improved body composition, bone mineralization, vascular health (including serum lipids), and quality of life (30). Implementing case managers could significantly improve transition care by coordinating care and tailoring support according to individual patient needs (31). Unfortunately, case managers are unavailable in Belgium, and governmental or private healthcare insurance do not foresee their reimbursement. Particularly for patients with cognitive challenges, commonly observed in those with GHD after brain irradiation or injury, the additional guidance by case managers is beneficial. To achieve these objectives, facilitating reimbursement for transition clinics focused on endocrine conditions and organizing dedicated personnel education is paramount.
2.5 When and why should GH therapy be interrupted or continued?
The critical moment to decide on continuing GH treatment is at the end of skeletal growth. The conclusion of longitudinal growth is typically marked by a decline in height velocity to below 2 cm/year, which can, in case of doubt, be radiologically confirmed by near-final bone maturation, predictive of about 99% of residual skeletal growth, i.e., a Greulich and Pyle scored bone age of 15 years in girls and 17 years in boys.
The estimates of persistence of childhood-diagnosed GHD into adulthood are variable, ranging from 12.5% in isolated GHD to 89% in MPHD (20, 32, 33). Key determinants of GHD persistence encompass GHD etiology and severity, age at diagnosis, and the presence of concurrent pituitary hormone deficiencies (33–35). When the likelihood of persistent GHD is high, GH therapy should not be interrupted. Such interruption periods often remain asymptomatic, and some patients, especially males, are reluctant to resume GH therapy afterwards (20). Though variable, adverse changes in body composition, such as an increase in body fat and a decrease in lean mass, alongside reduced quality of life, can arise as early as 6 months after GH interruption (36, 37). Prolonged interruptions can result in dyslipidemia and increased carotid artery intima-media thickness, which compromises cardiometabolic health (37–43), a decline in muscle force, and lower quality of life (44).
Figure 1 presents a decision flowchart outlining the considerations for either adapting the GH dose and continuing or temporarily interrupting treatment to assess for persistent GHD.

Figure 1. Flowchart for the approach to growth hormone deficiency at transition. GH, growth hormone; GHD, growth hormone deficiency; GHT, growth hormone therapy; PSIS, pituitary stalk interruption syndrome; SOD, septo-optic dysplasia.
2.6 Which patients should continue GH treatment?
The decision regarding the continuation of GH therapy relies on several key factors, encompassing five distinct patient profiles.
2.6.1 Confirmed gene defects responsible for impaired GH secretion
Continuation of GH therapy is warranted for individuals with pathogenic variants in genes governing pituitary gland development, GH synthesis, or release, encompassing both isolated GHD and MPHD cases. Confirmation of genetic GHD forms requires independent genetic analysis, irrespective of affected relatives or parental consanguinity. In the absence of a molecular diagnosis at transition, clinical cues can guide targeted gene analyses.
For early-onset severe isolated GHD with a small to normal-sized pituitary on MRI, variants in GH-1 and GHRHR should be investigated. POU1F1 and PROP-1 sequencing is recommended for combined GHD without pituitary anomalies, often accompanied by TSH and PRL deficiencies (34, 45). A short neck with restricted rotation, with or without sensorineural hearing loss, warrants LHX3 mutation analysis. Cerebral malformations such as hypoplastic corpus callosum or Chiari malformation suggest LHX4 gene variants. Bilateral microphthalmia points to variants in the SOX2, OTX2, or RAX genes. Routine testing for variants in these genes is currently available at various medical genetic centers in Belgium.
2.6.2 Major congenital midline abnormalities
Adolescents with radiologically confirmed substantial congenital midline defects affecting the hypothalamic-pituitary region, such as pituitary stalk interruption syndrome (PSIS) or septo-optic dysplasia (SOD) should continue GH therapy if they present with an additional deficiency of ≥ 2 other pituitary hormones. In children with congenital cerebral midline abnormalities, like optic nerve hypoplasia/SOD or PSIS, particularly when diagnosed in infancy, GHD tends to persist, especially in cases with MPHD (32).
2.6.3 Tumor-related GHD
GH therapy should be sustained for individuals who have undergone surgery and/or high-dose irradiation for intra/supra-sellar tumors, leading to MPHD. In young adults with childhood-onset GHD and MPHD due to brain tumors, the persistence of GHD is nearly 100% (20, 32, 46).
2.6.4 Acquired non-tumoral GHD
In cases of acquired non-tumoral GHD resulting from conditions such as severe brain trauma, lymphocytic hypophysitis, sarcoidosis, or Langerhans cell histiocytosis, continuation of GH therapy is indicated, particularly in MPHD (47, 48).
2.6.5 Idiopathic GHD
Adolescents with idiopathic GHD exhibiting ≥2 other pituitary deficiencies should continue GH treatment. Although MPHD is infrequent in children with idiopathic GHD, a persistence of the GHD is observed into adulthood in such cases (25, 49).
2.7 When, where, and how to conduct retesting for GHD?
Retesting for GHD should be done in isolated GHD or GHD combined with only one other pituitary deficiency, particularly in the following scenarios:
2.7.1 Normal genetic testing and normal brain imaging, considered idiopathic GHD
Most adolescents with idiopathic GHD, either isolated or combined with only one other pituitary deficiency, exhibit normal GH reserves upon testing in adulthood (35, 50–52).
2.7.2 Isolated anterior pituitary hypoplasia or isolated ectopic posterior pituitary findings on brain MRI
GHD in children with isolated ectopic posterior pituitary or PSIS without other pituitary hormone deficiency is not invariably permanent, necessitating retesting to avoid unnecessary treatment (21, 53–55). Normal GH peaks upon retesting have also been noted in adolescents with a small pituitary (56).
2.7.3 GHD diagnosis after brain irradiation for a tumor outside the hypothalamic-pituitary region or hematologic malignancy
Up to 45% of children initially diagnosed with GHD after brain irradiation for a distant brain tumor, mainly medulloblastoma, no longer show GHD upon retesting in adulthood (57, 58). The cranial irradiation dose received during childhood determines the GH peak upon retesting in young adulthood (59).
2.7.4 GHD diagnosed after a traumatic brain injury
Only a third of children treated for GHD after traumatic brain injury demonstrated confirmed GHD in young adulthood (60).
2.7.5 GHD related to Prader-Willi syndrome
Merely a fifth of GH-treated adolescents with Prader-Willi syndrome were GHD upon reaching adult height (61).
Retesting is ideally initiated by the pediatric endocrinologist upon approaching near-adult height (defined as a height gain ≤ 2 cm in the preceding year) and performed in the pediatric clinic. A wash-out period of 3 months is recommended to mitigate false positive results in GH stimulation tests. This recovery period allows for restoring normal GH secretion physiology, optimization of other concurrent hormone therapies, and cessation of estrogen-containing oral contraceptives. These measures ensure accurate interpretation of serum IGF1 and peak GH values during the stimulation test (32, 62–64). Notably, optimizing thyroid hormone levels is essential for reliable interpretation of the GH stimulation test. Sex steroid replacement therapy, like estrogens or androgens, should not be discontinued before retesting (65). However, in patients previously diagnosed with gonadotropin deficiency, whether partial at initial testing or linked to a family history of delayed puberty, stopping testosterone or estrogens for 3 months before GH retesting is recommended. This halt should be used to confirm permanent gonadotropin deficiency and, when confirmed, recommence sex steroid treatment for at least 3 months before GH retesting.
2.8 How to confirm the diagnosis of persistent GHD in adulthood?
We recommend that the diagnosis of GHD is confirmed through a combination of a GH stimulation test and measurement of serum IGF1 levels following the discontinuation of GH treatment. We do not recommend omitting the need for a GH stimulation test when serum IGF1 levels are either < -2 SD (indicative of GHD) or normal (dismissing GHD) after halting GH therapy (17) as opposed to others (32, 46, 50). Several studies demonstrate that a low serum IGF1 level alone, or even combined with low serum IGFBP3, does not confirm GHD in adolescents or young adults (32, 46, 50, 66).
2.8.1 Pitfalls for relying on IGF1 levels alone in diagnosing GHD
First, interpreting IGF1 results can be complicated due to the variation among different IGF1 assays, stemming from differences in antibody specificity and pre-analytical sample preparation strategies to eliminate binding protein interferences (67–69). Serum IGF1 is best measured using a total IGF1 assay employing the NIBSC (02/254) standard, which offers reliable age-dependent reference values (70). Secondly, there is a major fluctuation in IGF-1 levels. After extended GH treatment, it might take 6-12 months for serum IGF-1 levels to revert to their endogenous production levels (32). Time since GH cessation, pubertal development status, body adiposity, and associated conditions can all complicate serum IGF1 interpretation. Factors such as obesity, insulin resistance, delayed gonadal development, chronic inflammation, low-calorie intake, and use of estrogens (such as oral contraceptives) can lead to varying serum IGF1 levels. Up to 50% of adolescents initially diagnosed with idiopathic GHD during childhood exhibited low serum IGF1 levels after GH treatment cessation but were later diagnosed as having transient GHD (34, 71). Conversely, around 25% of adolescents with confirmed GHD during transition had normal serum IGF1 values (generally below the mean) (21, 32, 50, 71). Therefore, a serum IGF1 below -2 SD can be considered suggestive but not diagnostic for GHD, whereas a normal IGF-1 level can be considered suggestive of normal GH production but does not exclude persistent GHD.
2.8.2 The need for GH provocation tests in diagnosing GHD
To reliably ascertain the persistence of GHD, the primary criterion is the failure to achieve a specific peak GH cut-off level during a GH stimulation test. Based on Belgium’s experience with the insulin tolerance test (ITT) and glucagon testing (GST), these two tests are preferred during the transition phase. The ITT, unless contraindicated, e.g., in the case of epilepsy, is considered the primary choice (72). In cases where epilepsy or other contraindications preclude the ITT, a glucagon stimulation test (GST) is an alternative.
An algorithm detailing the diagnostic process for persistent GHD based on serum IGF1 and ITT results is shown in Figure 2. The criteria encompass a combination of serum IGF1 levels and peak GH responses to determine the presence of GHD. Additional considerations, such as pituitary hormone deficiencies and individual medical histories, guide further assessments and evaluations for accurate diagnosis and management. Briefly, GHD can be excluded when IGF1 levels are above the mean, and a GH peak above 6 µg/L is obtained at the ITT. The diagnosis of persistent GHD can be made when serum IGF1 levels are below the - 2 SD limit and a peak GH below 6 µg/L after insulin (or below < 3 µg/L after intramuscular glucagon administration) stimulation is found. In those adolescents with MPHD and low-normal serum IGF1 levels (between -2 and 0 SD), the presence of GH peak < 6 µg/L at the ITT is sufficient to confirm persistent GHD (73). On the other hand, adolescents with low-normal serum IGF1 (near the age-related lower limit or between -2 and 0 SD) and a peak GH < 6 µg/L but no additional or only one associated pituitary hormone deficiency require a second GH stimulation test with another secretagogue (preferentially glucagon) given the known variability of peak GH values at ITT/GST testing and basal serum IGF1 levels. If the second GH stimulation test yields a GH peak response above the threshold, irrespective of the serum IGF1 level, GHD is unlikely.
However, adolescents meeting the criteria of unlikely persistent GHD, especially those with PSIS or brain irradiation, should be reassessed after 6 to 12 months. Minimum yearly measurements of IGF1, PRL, cortisol, DHEAS, LH, FSH, testosterone or estradiol, FT4, and TSH are needed in brain-irradiated children since they are at risk to develop hypopituitarism over time (55, 58, 74, 75).
2.9 How to perform the GH stimulation tests in clinical practice?
GH stimulation tests are best performed approximately three months after GH treatment cessation. They should be performed when the patient feels well, has not performed excessive physical activity in the last 3 days, and always after an overnight fast. Adequate supplementation of other pituitary hormones is key in patients with GHD before testing GH secretion. A comprehensive evaluation of adrenal, thyroid, and gonadal status is crucial for accurate GH stimulation test interpretation. Therefore, prior measurement of morning basal cortisol, ACTH, DHEAS, LH, FSH, testosterone (males), estradiol (females), FT4, and TSH levels, in addition to serum IGF1, is essential (76). Of note, central hypocortisolism should be tested at transition as it develops in up to 44% of young adults with idiopathic GHD (74, 77, 78). Furthermore, the enhanced conversion of cortisone to cortisol during the GH-deficient state may increase basal cortisol levels at retesting.
The ITT should be the first choice and performed using an insulin dose of 0.1 U/kg if BMI ≤ 30 kg/m2 or 0.2 U/kg if BMI > 30 kg/m² and sampling at -30, 0, 30, 60, 90, and 120 minutes to determine glucose, growth hormone, and cortisol responses. The -30-minute sample should be taken immediately after intravenous line placement, as in some subjects, the GH response after hypoglycemia is weak, but line placement elicits satisfactory GH secretion (79). A glucose level below 40 mg/dl or a drop of at least 50% of the basal glucose level must be obtained after insulin injection to consider the GH results reliable.
When ITT is contraindicated, the GST can be done using a fixed intramuscular dose of 1 mg (1.5 mg when BMI > 30 kg/m²). Intramuscular stimulation is suggested to be more reliable and effective in releasing GH (86). No specific change in serum glucose after glucagon is needed, but blood sampling for GH measurements is needed at -30, 0, 90, 120, 150, and 180 minutes (80). Sampling at 240 minutes during GST is not needed (81, 82).
2.10 How to interpret the insulin tolerance and glucagon stimulation test?
A peak GH value < 6 µg/L following an ITT or GST is considered diagnostic for GHD during the transition phase (50, 56, 83, 84). This cutoff lies between the GH peak of 7 µg/L used in childhood and the 3 µg/L cut-off in adulthood and accords with the physiological decrease in GH secretion after the pubertal growth spurt. In cases of severe obesity (BMI > 30 kg/m²), a lower peak GH cut-off value should be considered (17, 85). For the ITT, the cortisol response above 15 µg/dl is considered sufficient, whereas for the GST, even a lower cut-off of 11 µg/dl can be accepted, at least when the current automated immunochemiluminometric assays are used (86). The choice of GH assay should prioritize chemiluminescence immunoassays for the 22KDa GH form using the second International standard IRP 98/574 and achieve a coefficient of variation < 20% at the lower quantifiable limit. Regular national comparisons among different centers using different GH assays are essential for accurate interpretation.
2.11 How should GH treatment be given?
When persistent GHD is diagnosed at retesting, GH treatment should be resumed promptly, even without overt GHD-related complaints, as prolonged interruption of GH therapy can reduce the motivation to resume treatment (20, 87). In adolescents with fused epiphyseal growth plates, pediatric GH doses should be avoided to prevent acromegalic manifestations or fluid retention. To recommence GH treatment within the 3 to 6-month window after an interruption, it is advisable to start with half of the GH dosage administered before the interruption (88, 89). Typically, this dose will be approximately 12.5 - 15 µg/kg/day for most adolescents. Nonetheless, a commencing treatment dose should not exceed 1.2 mg/day (90). When the patient has been without GH replacement for over 6 months, starting at a total daily GH dose of 0.6 to 0.8 mg is more suitable to prevent peripheral edema, arthralgia, myalgia, and paresthesia, related to GH-induced fluid retention (89). The use of disposable pens, although requiring refrigeration, is judged as the most user-friendly device by adolescents and active young adults and can be a suitable choice for continued GH therapy. Administering GH in the evening remains the preferred timing.
Titration of the GH dose can be done after one to two months and, subsequently, every 6 months, aimed at achieving serum IGF1 levels within the appropriate age-related range. Dose adjustments of 0.1 to 0.2 mg/day can be considered when clinical assessments and serum IGF1 levels indicate either over- or undertreatment. The goal is to maintain the IGF1 level around the mean for the given age or, at least, within -2 SD and +2SD if SD reference values for age and gender are available. IGF1 assessments should best be performed at least 6 weeks after a dosage change. Generally, women will receive higher doses than men when titrating GH doses to serum IGF1 levels around the mean. A maximum GH maintenance dose of 2 mg should not be exceeded. This upper limit is rarely needed, even in women taking estrogen-containing contraceptives.
2.12 How should GH treatment be monitored, and for how long?
Regular six-monthly visits are recommended, particularly during the first years of transition, due to the substantial risk of loss to follow-up. During these visits, blood pressure, body weight, and waist circumference should be recorded. Beyond the oversight of serum IGF1 concentrations, these visits serve to track potential adverse effects, such as fluid retention, hyperglycemia, arthralgias, and paresthesia. Additionally, they allow assessing underdosing or non-compliance, indicated by increasing waist circumference, reduced vigor, generalized fatigue, or decreased overall well-being. Disease-specific quality-of-life assessment questionnaires can be used to screen for changes in well-being related to GHD. However, whether they should be used to adapt GH dosing remains unclear. After 3-5 years of treatment and/or at the age of 25 years, bone densitometry by DXA is advised. The initial evaluation of the bone mineral density at the lumbar spine and hip is best performed at the adult clinic. This approach ensures reliable longitudinal assessments since DXA techniques may differ between hospitals. Furthermore, regular brain MRI follow-up in patients harboring residual post-surgical brain tumor remnants should be coordinated with oncologists and/or neurosurgeons.
Continuing GH therapy beyond adult height is most beneficial in young adults with more severe clinical and hormonal abnormalities. In cases without discernible clinical benefits after one year of treatment, discontinuing GH therapy may be considered. However, a potential decline in lipid profile, bone mass, muscle mass or strength, and exercise capacity should factor into the decision to resume GH therapy after one year of interruption. If GH treatment is continued, yearly assessments of fasting glucose, total cholesterol, HDL cholesterol, LDL cholesterol, triglycerides, IGF1, FT4, and cortisol are advised. Regular IGF1 monitoring is essential as patients may need higher GH doses over time due to the waning response associated with aging and shifts in body fat composition. Changes from oral to transdermal estrogen replacement or starting an oral estrogen-progesterone combination as contraception could also influence GH dosing.
Around the age of 25 years, when peak bone mass and maximal muscle strength are typically reached, the need for ongoing GH treatment should be reevaluated in those adults with isolated GHD, especially when bone mineral content is within normal limits or when compliance is low. In those patients who continued GH treatment after obtaining final height, the continuation of GH or retesting for persistent GHD should be considered on a case-by-case basis. In these cases, we recommend following international clinical guidelines established for adult-onset GHD treatment (91).
2.13 What about long-acting growth hormone?
Long-acting growth hormone (LAGH) preparations for weekly injections have recently become available for treating GHD in several countries. In Belgium, only somatogron is reimbursed for treating GHD in children aged three to 18 years. Weekly LAGH injections are as effective as daily GH injections in promoting growth. While long-term outcome data are not yet available, short-term data indicate a similar safety profile and suggest a lower treatment burden, making LAGH potentially more attractive to children and parents (92, 93). Young children expected to be on GH therapy for many years, children with needle phobia, and children starting self-injections might be good candidates for LAGH (94). However, safety concerns remain, particularly for children with severe GHD experiencing hypoglycemia, cancer predisposition syndromes, GHD after cancer therapy, or severe forms of metabolic syndrome. These children may not be suitable candidates for LAGH (95).
Some safety concerns stem from maintaining supraphysiological GH levels during the day, non-physiological tissue distribution, and different temporal relationships between peak GH and IGF1 with LAGH. While mean serum IGF1 concentrations are comparable between daily and weekly injections, they are higher in the first days after the LAGH injection and might be lower immediately before the subsequent injection compared to profiles observed during standard therapy. Calculators for monitoring serum IGF1 levels in clinical practice were developed for each LAGH preparation but need further evaluation in adolescents and young adults. Additionally, the effects of LAGH during puberty on linear growth, bone, and glucose metabolism still need further study. It is unclear if concomitant thyroxine or hydrocortisone therapies require dose adjustments when switching to LAGH. An international global registry project (GloBE-Reg) was initiated to collect “real-world” safety and efficacy data in children receiving somatogron and daily GH preparation.
LAGH is not yet approved for treating adult GHD in Belgium, but transitioning from adolescents on LAGH to adult care is an opportunity for initiating LAGH therapy in selected adult patients. In adults with childhood- as well as adult-onset GHD, clinical trials have documented a similar reduction in body fat and increase in muscle mass, as well as better treatment adherence compared to daily injections. However, there are no separate data on young adults (96) or young adults transitioning from pediatric daily GH dosing to LAGH. Clinical experience with LAGH in adults is too limited to formulate guidelines on transition (97). A longer LAGH treatment interruption (up to 6 months) could be considered before retesting for GHD at transition.
Most experience with LAGH in adults comes from clinical trials with somapacitan. Based on these trials, a starting dose of 1.5 mg/week (2mg/week for women on oral estrogens) of somapacitan has been proposed after interruption of GH therapy or when initiating GH therapy in young adults, followed by dose adjustments in steps of 0.5 to 1.5 mg at 2-4 weeks intervals (98). Serum IGF1 measurements three days after somapacitan injections can be used to titrate the LAGH dose. In young adults (up to 25 years), serum IGF1 levels are targeted at the upper end of the normal range. Higher GH dosing might improve abdominal fat accumulation but may cause myalgia and insulin resistance (99). The worsening of insulin sensitivity is a concern, especially in those with risk factors for developing diabetes mellitus (e.g., obesity or a family history of diabetes). However, this negative effect of glucose metabolism might be counterbalanced by favorable body composition changes in the long term (100). The effect of LAGH on cardiovascular risk factors and endothelial function needs further study.
3 Actionable recommendations
The actionable recommendations are presented in two flow charts: the first outlines the approach to GHD at transition (Figure 1), and the second details the process for diagnosing persistent GHD at transition (Figure 2). Table 1 summarizes the key considerations for transitioning GHD from pediatric to adult clinics.
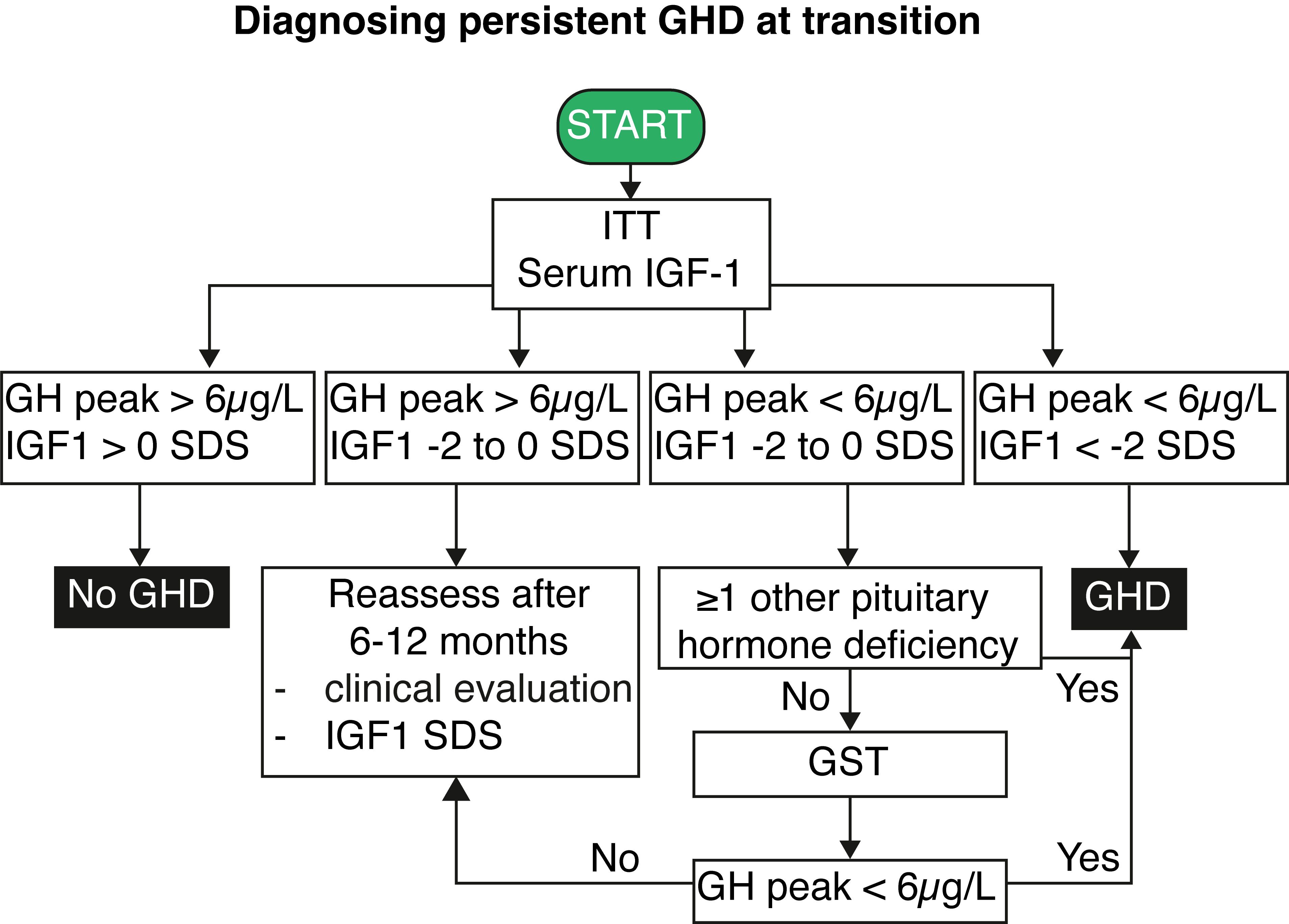
Figure 2. Flowchart for diagnosing persistent growth hormone deficiency at transition. IGF1, insulin-like growth factor 1; GH, growth hormone; GHD, growth hormone deficiency; GST, growth hormone stimulation test; ITT, insulin tolerance test; SDS, standard deviation score.
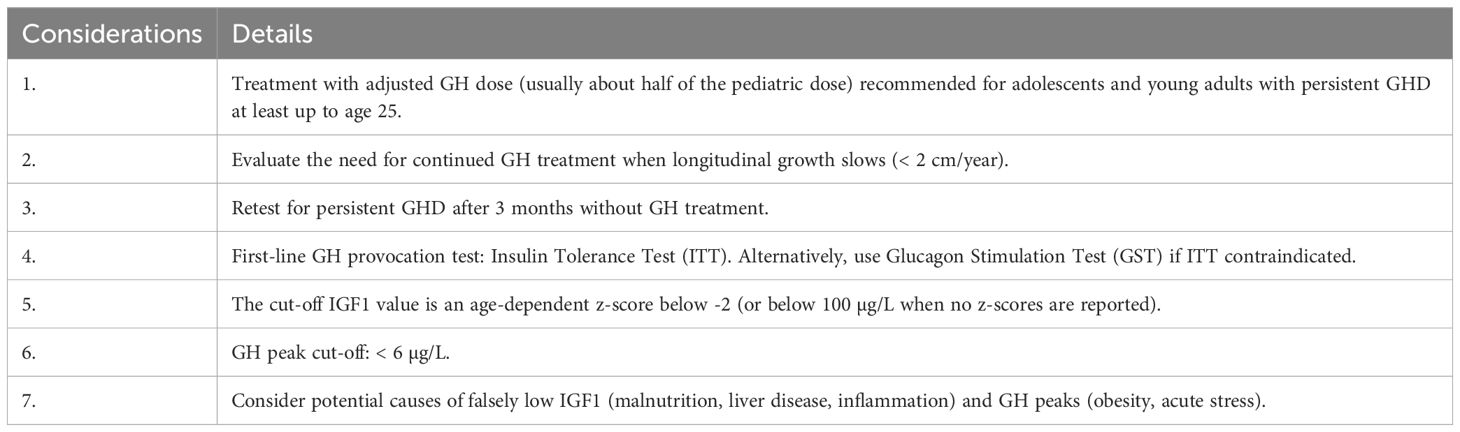
Table 1. Overview of important considerations.
Author contributions
WS: Writing – original draft, Writing – review & editing. JS: Writing – original draft, Writing – review & editing. MB: Writing – review & editing. PL: Writing – review & editing. DK: Writing – review & editing. KL: Writing – review & editing. MDB: Writing – review & editing. AR: Writing – review & editing. BL: Writing – review & editing. MC: Writing – review & editing. OA: Writing – review & editing. MAB: Writing – review & editing. BC: Writing – review & editing. LC: Writing – review & editing. CB: Writing – review & editing. JD: Writing – review & editing. RH: Writing – review & editing. MP: Writing – review & editing. GT’S: Writing – review & editing. AB: Writing – review & editing. SV: Writing – review & editing. BV: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. WS holds an FWO senior clinical investigator grant (1806421N).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Murray PG, Dattani MT, Clayton PE. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch Dis Child. (2016) 101:96–100. doi: 10.1136/archdischild-2014-307228
2. Hoybye C, Jonsson P, Monson JP, Koltowska-Haggstrom M, Hana V, Geffner M, et al. Impact of the primary aetiology upon the clinical outcome of adults with childhood-onset GH deficiency. Eur J Endocrinol. (2007) 157:589–96. doi: 10.1530/EJE-07-0364
3. Karavanaki K, Kontaxaki C, Maniati-Christidi M, Petrou V, Dacou-Voutetakis C. Growth response, pubertal growth and final height in Greek children with growth hormone (GH) deficiency on long-term GH therapy and factors affecting outcome. J Pediatr Endocrinol Metab. (2001) 14:397–405. doi: 10.1515/JPEM.2001.14.4.397
4. Sas TC, de Ridder MA, Wit JM, Rotteveel J, Oostdijk W, Reeser HM, et al. Adult height in children with growth hormone deficiency: a randomized, controlled, growth hormone dose-response trial. Horm Res Paediatr. (2010) 74:172–81. doi: 10.1159/000281323
5. Straetemans S, De Schepper J, Thomas M, Verlinde F, Rooman R. and bespeed, validation of prediction models for near adult height in children with idiopathic growth hormone deficiency treated with growth hormone: A belgian registry study. Horm Res Paediatr. (2016) 86:161–8. doi: 10.1159/000448553
6. Henry RK. When they’re done growing, don’t forget they may still need growth hormone. Metab Syndr Relat Disord. (2021) 19:257–63. doi: 10.1089/met.2020.0130
7. Leong GM, Johannsson G. Growth hormone deficiency: strategies and indications to continue growth hormone therapy in transition from adolescence to adult life. Horm Res. (2003) 60:78–85. doi: 10.1159/000071231
8. Saggese G, Baroncelli GI, Bertelloni S, Barsanti S. The effect of long-term growth hormone (GH) treatment on bone mineral density in children with GH deficiency. Role of GH in the attainment of peak bone mass. J Clin Endocrinol Metab. (1996) 81:3077–83. doi: 10.1210/jcem.81.8.8768878
9. Widdowson WM, Gibney J. The effect of growth hormone (GH) replacement on muscle strength in patients with GH-deficiency: a meta-analysis. Clin Endocrinol (Oxf). (2010) 72:787–92. 5doi: 10.1111/j.1365-2265.2009.03716.x
10. Godbout A, Tejedor I, Malivoir S, Polak M, Touraine P. Transition from pediatric to adult healthcare: assessment of specific needs of patients with chronic endocrine conditions. Horm Res Paediatr. (2012) 78:247–55. doi: 10.1159/000343818
11. Hauffa BP, Touraine P, Urquhart-Kelly T, Koledova E. Managing transition in patients treated with growth hormone. Front Endocrinol (Lausanne). (2017) 8:346. doi: 10.3389/fendo.2017.00346
12. Spaziani M, Tarantino C, Tahani N, Gianfrilli D, Sbardella E, Isidori AM, et al. Clinical, diagnostic, and therapeutic aspects of growth hormone deficiency during the transition period: review of the literature. Front Endocrinol (Lausanne). (2021) 12:634288. doi: 10.3389/fendo.2021.634288
13. Yuen KCJ, Alter CA, Miller BS, Gannon AW, Tritos NA, Samson SL, et al. Adult growth hormone deficiency: Optimizing transition of care from pediatric to adult services. Growth Horm IGF Res. (2021) 56:101375. doi: 10.1016/j.ghir.2020.101375
14. Inzaghi E, Cianfarani S. The Challenge of Growth Hormone Deficiency Diagnosis and Treatment during the Transition from Puberty into Adulthood. Front Endocrinol (Lausanne). (2013) 4:34. doi: 10.3389/fendo.2013.00034
15. Atger-Lallier L, Guilmin-Crepon S, Boizeau P, Zenaty D, Simon D, Paulsen A, et al. Factors affecting loss to follow-up in children and adolescents with chronic endocrine conditions. Horm Res Paediatr. (2019) 92:254–61. doi: 10.1159/000505517
16. van Alewijk L, Davidse K, Pellikaan K, van Eck J, Hokken-Koelega ACS, Sas TCJ, et al. Transition readiness among adolescents with rare endocrine conditions. Endocr Connect. (2021) 10:432–46. doi: 10.1530/EC-20-0304
17. Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. Drug, C. Therapeutics and S. Ethics committee of the pediatric endocrine, guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. (2016) 86:361–97. doi: 10.1159/000452150
18. Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM, et al. American association of clinical endocrinologists and american college of endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract. (2019) 25:1191–232. doi: 10.4158/GL-2019-0405
19. Aimaretti G, Attanasio R, Cannavo S, Nicoletti MC, Castello R, Di Somma C, et al. Growth hormone treatment of adolescents with growth hormone deficiency (GHD) during the transition period: results of a survey among adult and paediatric endocrinologists from Italy. Endorsed by SIEDP/ISPED, AME, SIE, SIMA. J Endocrinol Invest. (2015) 38:377–82. doi: 10.1007/s40618-014-0201-7
20. Ahmid M, Fisher V, Graveling AJ, McGeoch S, McNeil E, Roach J, et al. An audit of the management of childhood-onset growth hormone deficiency during young adulthood in Scotland. Int J Pediatr Endocrinol. (2016) 2016:6. doi: 10.1186/s13633-016-0024-8
21. Courtillot C, Baudoin R, Du Souich T, Saatdjian L, Tejedor I, Pinto G, et al. Monocentric study of 112 consecutive patients with childhood onset GH deficiency around and after transition. Eur J Endocrinol. (2013) 169:587–96. doi: 10.1530/EJE-13-0572
22. Bazarra-Castro MA, Sievers C, Schwarz HP, Pozza SB, Stalla GK. Changes in BMI and management of patients with childhood onset growth hormone deficiency in the transition phase. Exp Clin Endocrinol Diabetes. (2012) 120:507–10. doi: 10.1055/s-0032-1327599
23. Chesover AD, Dattani MT. Evaluation of growth hormone stimulation testing in children. Clin Endocrinol (Oxf). (2016) 84:708–14. doi: 10.1111/cen.2016.84.issue-5
24. Straetemans S, De Schepper J, Thomas M, Tenoutasse S, Beauloye V, Rooman R. Criteria for first-year growth response to growth hormone treatment in prepubertal children with growth hormone deficiency: do they predict poor adult height outcome? Front Endocrinol (Lausanne). (2019) 10:792. doi: 10.3389/fendo.2019.00792
25. Thomas M, Massa G, Maes M, Beckers D, Craen M, Francois I, et al. Growth hormone (GH) secretion in patients with childhood-onset GH deficiency: retesting after one year of therapy and at final height. Horm Res. (2003) 59:7–15. doi: 10.1159/000067936
26. Wood DL, Sawicki GS, Miller MD, Smotherman C, Lukens-Bull K, Livingood WC, et al. The Transition Readiness Assessment Questionnaire (TRAQ): its factor structure, reliability, and validity. Acad Pediatr. (2014) 14:415–22. doi: 10.1016/j.acap.2014.03.008
27. Lagrou K, Xhrouet-Heinrichs D, Massa G, Vandeweghe M, Bourguignon JP, De Schepper J, et al. Quality of life and retrospective perception of the effect of growth hormone treatment in adult patients with childhood growth hormone deficiency. J Pediatr Endocrinol Metab. (2001) 14 Suppl 5:1249–60.
28. Kirk J, Clayton P. Specialist services and transitional care in paediatric endocrinology in the UK and Ireland. Clin Endocrinol (Oxf). (2006) 65:59–63. doi: 10.1111/j.1365-2265.2006.02546.x
29. Savage MO, Drake WM, Carroll PV, Monson JP. Transitional care of GH deficiency: when to stop GH therapy. Eur J Endocrinol. (2004) 151 Suppl 1:S61–5. doi: 10.1530/eje.0.151s061
30. Malivoir S, Courtillot C, Bachelot A, Chakhtoura Z, Tejedor I, Touraine P. Therapeutic education programme for patients with chronic endocrine conditions: Transition from paediatric to adult services. Presse Med. (2016) 45:e119–29. doi: 10.1016/j.lpm.2015.10.025
31. Le Roux E, Menesguen F, Tejedor I, Popelier M, Halbron M, Faucher P, et al. Transition of young adults with endocrine and metabolic diseases: the ‘TRANSEND’ cohort. Endocr Connect. (2021) 10:21–8. doi: 10.1530/EC-20-0520
32. Maghnie M, Strigazzi C, Tinelli C, Autelli M, Cisternino M, Loche S, et al. Growth hormone (GH) deficiency (GHD) of childhood onset: reassessment of GH status and evaluation of the predictive criteria for permanent GHD in young adults. J Clin Endocrinol Metab. (1999) 84:1324–8. doi: 10.1210/jcem.84.4.5614
33. Rohayem J, Drechsel H, Tittel B, Hahn G, Pfaeffle R, Huebner A, et al. Genetics, and pituitary morphology in patients with isolated growth hormone deficiency and multiple pituitary hormone deficiencies: A single-centre experience of four decades of growth hormone replacement. Horm Res Paediatr. (2016) 86:106–16. doi: 10.1159/000448098
34. Quigley CA, Zagar AJ, Liu CC, Brown DM, Huseman C, Levitsky L, et al. United States multicenter study of factors predicting the persistence of GH deficiency during the transition period between childhood and adulthood. Int J Pediatr Endocrinol. (2013) 2013:6. doi: 10.1186/1687-9856-2013-6
35. Tauber M, Moulin P, Pienkowski C, Jouret B, Rochiccioli P. Growth hormone (GH) retesting and auxological data in 131 GH-deficient patients after completion of treatment. J Clin Endocrinol Metab. (1997) 82:352–6. doi: 10.1210/jcem.82.2.3726
36. Binder G, Donner J, Becker B, Bauer JL, Schweizer R. Changes in body composition in male adolescents with childhood-onset GH deficiency during transition. Clin Endocrinol (Oxf). (2019) 91:432–9. doi: 10.1111/cen.14041
37. Stouthart PJ, Deijen JB, Roffel M, Delemarre-van de Waal HA. Quality of life of growth hormone (GH) deficient young adults during discontinuation and restart of GH therapy. Psychoneuroendocrinology. (2003) 28:612–26. doi: 10.1016/S0306-4530(02)00045-8
38. Koltowska-Haggstrom M, Geffner ME, Jonsson P, Monson JP, Abs R, Hana V, et al. Discontinuation of growth hormone (GH) treatment during the transition phase is an important factor determining the phenotype of young adults with nonidiopathic childhood-onset GH deficiency. J Clin Endocrinol Metab. (2010) 95:2646–54. doi: 10.1210/jc.2009-2013
39. Johannsson G, Albertsson-Wikland K, Bengtsson BA. Discontinuation of growth hormone (GH) treatment: metabolic effects in GH-deficient and GH-sufficient adolescent patients compared with control subjects. Swedish Study Group for Growth Hormone Treatment in Children. J Clin Endocrinol Metab. (1999) 84:4516–24. doi: 10.1210/jcem.84.12.6176
40. Modesto Mde J, Amer NM, Erichsen O, Hernandez S, dos Santos CD, de Carvalho JA, et al. Muscle strength and body composition during the transition phase in patients treated with recombinant GH to final height. J Pediatr Endocrinol Metab. (2014) 27:813–20. doi: 10.1515/jpem-2013-0317
41. Colao A, Di Somma C, Rota F, Di Maio S, Salerno M, Klain A, et al. Common carotid intima-media thickness in growth hormone (GH)-deficient adolescents: a prospective study after GH withdrawal and restarting GH replacement. J Clin Endocrinol Metab. (2005) 90:2659–65. doi: 10.1210/jc.2004-1844
42. Lee YJ, Choi Y, Yoo HW, Lee YA, Shin CH, Choi HS, et al. Metabolic impacts of discontinuation and resumption of recombinant human growth hormone treatment during the transition period in patients with childhood-onset growth hormone deficiency. Endocrinol Metab (Seoul). (2022) 37:359–68. doi: 10.3803/EnM.2021.1384
43. Doknic M, Stojanovic M, Soldatovic I, Milenkovic T, Zdravkovic V, Jesic M, et al. Mapping the journey of transition: a single-center study of 170 childhood-onset GH deficiency patients. Endocr Connect. (2021) 10:935–46. doi: 10.1530/EC-21-0274
44. Wiren L, Johannsson G, Bengtsson BA. A prospective investigation of quality of life and psychological well-being after the discontinuation of GH treatment in adolescent patients who had GH deficiency during childhood. J Clin Endocrinol Metab. (2001) 86:3494–8. doi: 10.1210/jcem.86.8.7709
45. Dattani MT. DNA testing in patients with GH deficiency at the time of transition. Growth Horm IGF Res. (2003) 13 Suppl A:S122–9. doi: 10.1016/S1096-6374(03)00068-6
46. Juul A, Kastrup KW, Pedersen SA, Skakkebaek NE. Growth hormone (GH) provocative retesting of 108 young adults with childhood-onset GH deficiency and the diagnostic value of insulin-like growth factor I (IGF-I) and IGF-binding protein-3. J Clin Endocrinol Metab. (1997) 82:1195–201. doi: 10.1210/jcem.82.4.3892
47. Radojkovic D, Pesic M, Dimic D, Radjenovic Petkovic T, Radenkovic S, Velojic-Golubovic M, et al. Localised Langerhans cell histiocytosis of the hypothalamic-pituitary region: case report and literature review. Hormones (Athens). (2018) 17:119–25. doi: 10.1007/s42000-018-0024-6
48. Heinze HJ, Bercu BB. Acquired hypophysitis in adolescence. J Pediatr Endocrinol Metab. (1997) 10:315–21. doi: 10.1515/JPEM.1997.10.3.315
49. Lazar L, Dan S, Phillip M. Growth without growth hormone: growth pattern and final height of five patients with idiopathic combined pituitary hormone deficiency. Clin Endocrinol (Oxf). (2003) 59:82–8. doi: 10.1046/j.1365-2265.2003.01805.x
50. Bonfig W, Bechtold S, Bachmann S, Putzker S, Fuchs O, Pagel P, et al. Reassessment of the optimal growth hormone cut-off level in insulin tolerance testing for growth hormone secretion in patients with childhood-onset growth hormone deficiency during transition to adulthood. J Pediatr Endocrinol Metab. (2008) 21:1049–56. doi: 10.1515/JPEM.2008.21.11.1049
51. Penta L, Cofini M, Lucchetti L, Zenzeri L, Leonardi A, Lanciotti L, et al. Growth hormone (GH) therapy during the transition period: should we think about early retesting in patients with idiopathic and isolated GH deficiency? Int J Environ Res Public Health. (2019) 16:307. doi: 10.3390/ijerph16030307
52. Gelwane G, Garel C, Chevenne D, Armoogum P, Simon D, Czernichow P, et al. Subnormal serum insulin-like growth factor-I levels in young adults with childhood-onset nonacquired growth hormone (GH) deficiency who recover normal gh secretion may indicate less severe but persistent pituitary failure. J Clin Endocrinol Metab. (2007) 92:3788–95. doi: 10.1210/jc.2007-1003
53. Leger J, Danner S, Simon D, Garel C, Czernichow P. Do all patients with childhood-onset growth hormone deficiency (GHD) and ectopic neurohypophysis have persistent GHD in adulthood? J Clin Endocrinol Metab. (2005) 90:650–6. doi: 10.1210/jc.2004-1274
54. Deillon E, Hauschild M, Faouzi M, Stoppa-Vaucher S, Elowe-Gruau E, Dwyer A, et al. Natural history of growth hormone deficiency in a pediatric cohort. Horm Res Paediatr. (2015) 83:252–61. doi: 10.1159/000369392
55. di Iorgi N, Secco A, Napoli F, Tinelli C, Calcagno A, Fratangeli N, et al. Deterioration of growth hormone (GH) response and anterior pituitary function in young adults with childhood-onset GH deficiency and ectopic posterior pituitary: a two-year prospective follow-up study. J Clin Endocrinol Metab. (2007) 92:3875–84. doi: 10.1210/jc.2007-1081
56. Secco A, di Iorgi N, Napoli F, Calandra E, Calcagno A, Ghezzi M, et al. Reassessment of the growth hormone status in young adults with childhood-onset growth hormone deficiency: reappraisal of insulin tolerance testing. J Clin Endocrinol Metab. (2009) 94:4195–204. doi: 10.1210/jc.2009-0602
57. Gleeson HK, Gattamaneni HR, Smethurst L, Brennan BM, Shalet SM. Reassessment of growth hormone status is required at final height in children treated with growth hormone replacement after radiation therapy. J Clin Endocrinol Metab. (2004) 89:662–6. doi: 10.1210/jc.2003-031224
58. Nicolson A, Toogood AA, Rahim A, Shalet SM. The prevalence of severe growth hormone deficiency in adults who received growth hormone replacement in childhood [see comment. Clin Endocrinol (Oxf). (1996) 44:311–6. doi: 10.1046/j.1365-2265.1996.671492.x
59. Adan L, Trivin C, Sainte-Rose C, Zucker JM, Hartmann O, Brauner R. GH deficiency caused by cranial irradiation during childhood: factors and markers in young adults. J Clin Endocrinol Metab. (2001) 86:5245–51. doi: 10.1210/jcem.86.11.8056
60. Dassa Y, Crosnier H, Chevignard M, Viaud M, Personnier C, Flechtner I, et al. Pituitary deficiency and precocious puberty after childhood severe traumatic brain injury: a long-term follow-up prospective study. Eur J Endocrinol. (2019) 180:281–90. doi: 10.1530/EJE-19-0034
61. Grugni G, Marzullo P, Delvecchio M, Iughetti L, Licenziati MR, Osimani S, et al. Stimulated GH levels during the transition phase in Prader-Willi syndrome. J Endocrinol Invest. (2021) 44:1465–74. doi: 10.1007/s40618-020-01450-y
62. Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Shalet SM, Vance ML, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. (2006) 91:1621–34. doi: 10.1210/jc.2005-2227
63. Radovick S, DiVall S. Approach to the growth hormone-deficient child during transition to adulthood. J Clin Endocrinol Metab. (2007) 92:1195–200. doi: 10.1210/jc.2007-0167
64. Wacharasindhu S, Cotterill AM, Camacho-Hubner C, Besser GM, Savage MO. Normal growth hormone secretion in growth hormone insufficient children retested after completion of linear growth. Clin Endocrinol (Oxf). (1996) 45:553–6. doi: 10.1046/j.1365-2265.1996.00850.x
65. de Boer JA, Schoemaker J, van der Veen EA. Impaired reproductive function in women treated for growth hormone deficiency during childhood. Clin Endocrinol (Oxf). (1997) 46:681–9. doi: 10.1046/j.1365-2265.1997.1800999.x
66. Attanasio AF, Howell S, Bates PC, Blum WF, Frewer P, Quigley C, et al. Confirmation of severe GH deficiency after final height in patients diagnosed as GH deficient during childhood. Clin Endocrinol (Oxf). (2002) 56:503–7. doi: 10.1046/j.1365-2265.2002.01515.x
67. Hartman ML, Crowe BJ, Biller BM, Ho KK, Clemmons DR, Chipman JJ, et al. Which patients do not require a GH stimulation test for the diagnosis of adult GH deficiency? J Clin Endocrinol Metab. (2002) 87:477–85. doi: 10.1210/jcem.87.2.8216
68. Boguszewski CL, Lacerda CS, Lacerda Filho L, Carvalho JA, Boguszewski MC. Reappraisal of serum insulin-like growth factor-I (IGF-1) measurement in the detection of isolated and combined growth hormone deficiency (GHD) during the transition period. Arq Bras Endocrinol Metabol. (2013) 57:709–16. doi: 10.1590/S0004-27302013000900006
69. Anckaert E, Schiettecatte J, Vanbesien J, Smitz J, Velkeniers B, De Schepper J. Variability among five different commercial IGF-1 immunoassays in conditions of childhood-onset GH deficiency and GH therapy. Acta Clin Belg. (2006) 61:335–9. doi: 10.1179/acb.2006.053
70. Varewijck AJ, van der Lely AJ, Neggers S, Hofland LJ, Janssen J. Disagreement in normative IGF-I levels may lead to different clinical interpretations and GH dose adjustments in GH deficiency. Clin Endocrinol (Oxf). (2018) 88:409–14. doi: 10.1111/cen.2018.88.issue-3
71. Bechtold S, Bachmann S, Putzker S, Dalla Pozza R, Schwarz HP. Early changes in body composition after cessation of growth hormone therapy in childhood-onset growth hormone deficiency. J Clin Densitom. (2011) 14:471–7. doi: 10.1016/j.jocd.2011.05.001
72. Brabant G, Poll EM, Jonsson P, Polydorou D, Kreitschmann-Andermahr I. Etiology, baseline characteristics, and biochemical diagnosis of GH deficiency in the adult: are there regional variations? Eur J Endocrinol. (2009) 161 Suppl 1:S25–31. doi: 10.1530/EJE-09-0273
73. Donaubauer J, Kiess W, Kratzsch J, Nowak T, Steinkamp H, Willgerodt H, et al. Re-assessment of growth hormone secretion in young adult patients with childhood-onset growth hormone deficiency. Clin Endocrinol (Oxf). (2003) 58:456–63. doi: 10.1046/j.1365-2265.2003.01739.x
74. Fernandez-Rodriguez E, Quinteiro C, Barreiro J, Marazuela M, Pereiro I, Peino R, et al. Pituitary stalk dysgenesis-induced hypopituitarism in adult patients: prevalence, evolution of hormone dysfunction and genetic analysis. Neuroendocrinology. (2011) 93:181–8. doi: 10.1159/000324087
75. Marie Baunsgaard M, Sophie Lind Helligsoe A, Tram Henriksen L, Stamm Mikkelsen T, Callesen M, Weber B, et al. Growth hormone deficiency in adult survivors of childhood brain tumors treated with radiation. Endocr Connect. (2023) 12:e220365. doi: 10.1530/EC-22-0365
76. Giustina A, Wehrenberg WB. Influence of thyroid hormones on the regulation of growth hormone secretion. Eur J Endocrinol. (1995) 133:646–53. doi: 10.1530/eje.0.1330646
77. Lange M, Feldt-Rasmussen U, Svendsen OL, Kastrup KW, Juul A, Muller J. High risk of adrenal insufficiency in adults previously treated for idiopathic childhood onset growth hormone deficiency. J Clin Endocrinol Metab. (2003) 88:5784–9. doi: 10.1210/jc.2003-030529
78. Savas Erdeve S, Ocal G, Berberoglu M, Siklar Z, Hacihamdioglu B. Is adrenocorticotropic hormone deficiency really rare in patients with idiopathic growth hormone deficiency and normal thyroid function tests? Horm Res Paediatr. (2011) 75:200–5. doi: 10.1159/000320479
79. Hawkes CP, Mavinkurve M, Fallon M, Grimberg A, Cody DC. Serial GH measurement after intravenous catheter placement alone can detect levels above stimulation test thresholds in children. J Clin Endocrinol Metab. (2015) 100:4357–63. doi: 10.1210/jc.2015-3102
80. Giuffrida FM, Berger K, Monte L, Oliveira CH, Hoff AO, Maciel RM, et al. Relationship between GH response and glycemic fluctuations in the glucagon stimulation test. Growth Horm IGF Res. (2009) 19:77–81. doi: 10.1016/j.ghir.2008.06.002
81. Gomez JM, Espadero RM, Escobar-Jimenez F, Hawkins F, Pico A, Herrera-Pombo JL, et al. Growth hormone release after glucagon as a reliable test of growth hormone assessment in adults. Clin Endocrinol (Oxf). (2002) 56:329–34. doi: 10.1046/j.1365-2265.2002.01472.x
82. Leong KS, Walker AB, Martin I, Wile D, Wilding J, MacFarlane IA. An audit of 500 subcutaneous glucagon stimulation tests to assess growth hormone and ACTH secretion in patients with hypothalamic-pituitary disease. Clin Endocrinol (Oxf). (2001) 54:463–8. doi: 10.1046/j.1365-2265.2001.01169.x
83. Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M, et al. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. (2005) 152:165–70. doi: 10.1530/eje.1.01829
84. Fava D, Guglielmi D, Pepino C, Angelelli A, Casalini E, Varotto C, et al. Accuracy of glucagon testing across transition in young adults with childhood-onset growth hormone deficiency. J Clin Endocrinol Metab. (2024). doi: 10.1210/clinem/dgae408
85. Dichtel LE, Yuen KC, Bredella MA, Gerweck AV, Russell BM, Riccio AD, et al. Overweight/Obese adults with pituitary disorders require lower peak growth hormone cutoff values on glucagon stimulation testing to avoid overdiagnosis of growth hormone deficiency. J Clin Endocrinol Metab. (2014) 99:4712–9. doi: 10.1210/jc.2014-2830
86. Hamrahian AH, Yuen KC, Gordon MB, Pulaski-Liebert KJ, Bena J, Biller BM, et al. and cortisol cut-points for the glucagon stimulation test in the evaluation of GH and hypothalamic-pituitary-adrenal axes in adults: results from a prospective randomized multicenter study. Pituitary. (2016) 19:332–41. doi: 10.1007/s11102-016-0712-7
87. Camtosun E, Siklar Z, Berberoglu M. Prospective follow-up of children with idiopathic growth hormone deficiency after termination of growth hormone treatment: is there really need for treatment at transition to adulthood? J Clin Res Pediatr Endocrinol. (2018) 10:247–55. doi: 10.4274/jcrpe.0010
88. Cook DM, Rose SR. A review of guidelines for use of growth hormone in pediatric and transition patients. Pituitary. (2012) 15:301–10. doi: 10.1007/s11102-011-0372-6
89. Richmond E, Rogol AD. Treatment of growth hormone deficiency in children, adolescents and at the transitional age. Best Pract Res Clin Endocrinol Metab. (2016) 30:749–55. doi: 10.1016/j.beem.2016.11.005
90. Shalet SM, Shavrikova E, Cromer M, Child CJ, Keller E, Zapletalova J, et al. Effect of growth hormone (GH) treatment on bone in postpubertal GH-deficient patients: a 2-year randomized, controlled, dose-ranging study. J Clin Endocrinol Metab. (2003) 88:4124–9. doi: 10.1210/jc.2003-030126
91. Ho KK, G.H.D.C.W. Participants. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. (2007) 157:695–700. doi: 10.1530/EJE-07-0631
92. Mameli C, Orso M, Calcaterra V, Wasniewska MG, Aversa T, Granato S, et al. Efficacy, safety, quality of life, adherence and cost-effectiveness of long-acting growth hormone replacement therapy compared to daily growth hormone in children with growth hormone deficiency: A systematic review and meta-analysis. Pharmacol Res. (2023) 193:106805. doi: 10.1016/j.phrs.2023.106805
93. McNamara M, Turner-Bowker DM, Westhead H, Yaworsky A, Palladino A, Gross H, et al. Factors driving patient preferences for growth hormone deficiency (GHD) injection regimen and injection device features: A discrete choice experiment. Patient Prefer Adherence. (2020) 14:781–93. doi: 10.2147/PPA.S239196
94. Miller BS. What do we do now that the long-acting growth hormone is here? Front Endocrinol (Lausanne). (2022) 13:980979. doi: 10.3389/fendo.2022.980979
95. Boguszewski MCS, Boguszewski CL, Chemaitilly W, Cohen LE, Gebauer J, Higham C, et al. Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: a consensus statement. Eur J Endocrinol. (2022) 186:P35–52. doi: 10.1530/EJE-21-1186
96. Dutta D, Mahajan K, Kumar M, Sharma M. Efficacy and safety of long-acting growth hormone in adult growth hormone deficiency: A systematic review and meta-analysis. Diabetes Metab Syndr. (2022) 16:102421. doi: 10.1016/j.dsx.2022.102421
97. Kargi AY. Impact of long-acting growth hormone replacement therapy in adult growth hormone deficiency: Comparison between adolescent, adult, and elderly patients. Best Pract Res Clin Endocrinol Metab. (2023) 37:101825. doi: 10.1016/j.beem.2023.101825
98. Bidlingmaier M, Biller BMK, Clemmons D, Jorgensen JOL, Nishioka H, Takahashi Y. Guidance for the treatment of adult growth hormone deficiency with somapacitan, a long-acting growth hormone preparation. Front Endocrinol (Lausanne). (2022) 13:1040046. doi: 10.3389/fendo.2022.1040046
99. van Bunderen CC, Meijer RI, Lips P, Kramer MH, Serne EH, Drent ML. Titrating growth hormone dose to high-normal IGF-1 levels has beneficial effects on body fat distribution and microcirculatory function despite causing insulin resistance. Front Endocrinol (Lausanne). (2020) 11:619173. doi: 10.3389/fendo.2020.619173
Keywords: growth hormone deficiency (GHD), growth hormone therapy, transition, growth hormone stimulation tests, policy for transition care
Citation: Staels W, De Schepper J, Becker M, Lysy P, Klink D, Logghe K, den Brinker M, Rochtus A, Lapauw B, Cools M, Alexopoulou O, Bex M, Corvilain B, Crenier L, De Block C, Donckier J, Hilbrands R, Ponchon M, T'Sjoen G, Van Den Bruel A, Vandewalle S and Velkeniers B (2024) Policy for transitioning childhood-onset growth hormone deficiency from pediatric to adult endocrine care in Belgium. Front. Endocrinol. 15:1459998. doi: 10.3389/fendo.2024.1459998
Received: 05 July 2024; Accepted: 09 September 2024;
Published: 30 September 2024.
Edited by:
Antonio Balsamo, University Hospital S.Orsola Malpighi, ItalyReviewed by:
Martin Oswald Savage, Queen Mary University of London, United KingdomPaul B. Kaplowitz, Children’s National Hospital, United States
Copyright © 2024 Staels, De Schepper, Becker, Lysy, Klink, Logghe, den Brinker, Rochtus, Lapauw, Cools, Alexopoulou, Bex, Corvilain, Crenier, De Block, Donckier, Hilbrands, Ponchon, T'Sjoen, Van Den Bruel, Vandewalle and Velkeniers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Willem Staels, d2lsbGVtLnN0YWVsc0B1emJydXNzZWwuYmU=
†ORCID: Willem Staels, orcid.org/0000-0001-8259-3329
Jean De Schepper, orcid.org/0000-0002-2999-0250
Marianne Becker, orcid.org/0000-0002-1643-1935
Philippe Lysy, orcid.org/0000-0003-1598-7998
Bruno Lapauw, orcid.org/0000-0002-1584-4965
Martine Cools, orcid.org/0000-0002-9552-4899
Orsalia Alexopoulou, orcid.org/0000-0001-9933-7188
Marie Bex, orcid.org/0000-0002-4484-4200
Christophe De Block, orcid.org/0000-0002-0679-3203
Guy T’Sjoen, orcid.org/0000-0003-0457-9673
Sara Vandewalle, orcid.org/0000-0001-8422-4973
Brigitte Velkeniers, orcid.org/0000-0001-8577-9698
Marieken den Brinker, orcid.org/0000-0001-9278-8795