 Erika Urbano De Lima1
Erika Urbano De Lima1 Filipe Ferreira Dos Santos2,3
Filipe Ferreira Dos Santos2,3 Igor Campos Da Silva4Cláudio Rogério Alves De Lima4
Igor Campos Da Silva4Cláudio Rogério Alves De Lima4 Vitoria Sousa Frutuoso1
Vitoria Sousa Frutuoso1 Gustavo Felisola Caso1
Gustavo Felisola Caso1 Paloma Ramos De Oliveira1Ana Karina Bezerra5
Paloma Ramos De Oliveira1Ana Karina Bezerra5 Janete Maria Cerutti6
Janete Maria Cerutti6 Rodrigo Esaki Tamura1,7
Rodrigo Esaki Tamura1,7 Helton Estrela Ramos8
Helton Estrela Ramos8 Ileana Gabriela Sanchez de Rubio1,7*†
Ileana Gabriela Sanchez de Rubio1,7*†- 1Laboratório de Ciências Moleculares da Tireoide (LCMT) e Laboratório de Biologia Molecular do Câncer (LBMC), Universidade Federal de São Paulo (UNIFESP), São Paulo, Brazil
- 2Centro de Oncologia Molecular (MOC), Hospital Sírio-Libanês - Instituto de Ensino e Pesquisa (HSL-IEP), São Paulo, Brazil
- 3Department of Biochemistry, Chemistry Institute (IQ), Universidade de São Paulo (USP), São Paulo, Brazil
- 4Departamento de Cirurgia de Cabeça e Pescoço, Monte Tabor – Hospital São Rafael, Salvador, Brazil
- 5Universidade de Fortaleza – Unifor, Fortaleza, Brazil
- 6Laboratório de Bases Genéticas dos Tumores da Tiroide, Departamento de Morfologia e Genética Universidade Federal de São Paulo (UNIFESP), São Paulo, Brazil
- 7Departamento de Ciências Biológicas, Universidade Federal de São Paulo (UNIFESP), São Paulo, Brazil
- 8Laboratório de Estudos da Tireoide, Departamento de Bioregulação, Universidade Federal da Bahia (UFBA), Salvador, Brazil
Introduction: Forkhead box E1 (FOXE1) is a transcription factor with a crucial role in thyroid morphogenesis and differentiation. Promoter hypermethylation downregulates FOXE1 expression in different tumor types; nevertheless, its expression and relationship with methylation status in differentiated thyroid cancer (DTC) remain unclear.
Methods: A total of 33 pairs of matched samples of PTC tumors and non-tumors were included. Tumor cell cultures were treated with either 5-Aza-2′-deoxycytidine demethylating agent or dimethyl sulfoxide (DMSO). A real-time polymerase chain reaction (RT-PCR) and Western blotting were performed to assess FOXE1 expression. The methylation status was quantified using bisulfite sequencing. A luciferase gene assay was used to determine CpG-island functionality. Gene expression and promoter methylation of FOXE1 and FOXE1-regulated genes were also analyzed with data from The Cancer Genome Atlas (TCGA) thyroid samples.
Results: After demethylating treatment, increased FOXE1 mRNA was observed concomitantly with reduced promoter methylation of CpGisland2. A negative correlation between mRNA downregulation and an increased methylation level of CpGisland2 was observed in tumors. Diminished protein expression was also detected in some DTC cell lines and in some tumor samples, suggesting the involvement of post-transcriptional regulatory mechanisms. CPGisland2 was proved to be an enhancer. TCGA data analysis showed low FOXE1 mRNA expression in tumors with a negative correlation with methylation status and a positive correlation with the expression of most of its target genes. Reduced FOXE1 expression, accompanied by a high methylation level, was associated with PTC aggressiveness (tall cell variant, advanced extra thyroid extension, T4 American Joint Committee on Cancer (AJCC) classification), age at diagnosis (over 45 years old), and presence of a BRAFV600E mutation.
Conclusion: FOXE1 mRNA was downregulated in DTC compared with non-tumors, followed by high CpGisland methylation. A coupling of low mRNA expression and high methylation status was related to characteristics of aggressiveness in DTC tumors.
1 Introduction
Thyroid cancer is one of the most common endocrine malignancies worldwide. Differentiated thyroid cancer (DTC) accounts for approximately 90% of all thyroid cancer cases, presenting with a slow disease course and a favorable prognosis in most cases. However, an increase in the incidence and mortality rates of advanced-stage papillary thyroid cancer (PTC) has been observed in recent years (1). A large number of genetic and epigenetic events have already been described in the pathogenesis of the disease, such as the aberrant activation of the metabolic pathways associated with Mitogen-activated protein kinase (MAPK), Phosphoinositide 3-kinases/Protein kinase B (PI3K/AKT) and Isocitrate dehydrogenase 1 (IDH1) (2). Nevertheless, the etiology of DTC is still not fully understood.
The FOXE1 gene (HGNC 3806) encodes a specific thyroid transcription factor that plays a crucial role in thyroid morphogenesis, development, growth, and differentiation (3). FOXE1 regulates the transcription of thyroglobulin (TG), thyroperoxidase (TPO), NKX2.1, PAX8, Sodium/Iodine Symporter (NIS), and DUOX2, genes required for hormone synthesis (4, 5), and also regulates PDGFA (6) and ZEB1 in thyroid cancer (7). Germline FOXE1 mutations were associated with Bamforth–Lazarus syndrome (8), whereas rare somatic mutations were associated with PTC and goiter (9). Functional analysis of a novel mutation identified in one sporadic and one familial PTC suggested that the variant promoted cell proliferation and migration and may be involved in tumorigenesis (10). Previous reports and data from population databanks have shown that the presence of polymorphisms such as rs965513 and rs1867277 lead to increased susceptibility of developing DTC (11–13).
In thyroid cancer, there is no consensus on the expression of FOXE1, which has either been upregulated, downregulated, or had similar expression patterns in tumor and non-tumor samples (6, 22, 23). Aberrant methylation along with aberrant transcriptional gene expression has been identified in almost all types of cancer. Low FOXE1 expression in parallel with promoter hypermethylation was observed in colorectal, salivary gland, skin, and other types of cancer (14–17), and it was proposed that methylation of FOXE1 could be a promising marker for cancer detection (18, 19). Combined data from gene expression, polymorphisms, enhancer regions, and methylation have suggested that FOXE1 could be a novel tumor suppressor (6, 18, 20, 21). In addition, a few studies have investigated the methylation status of FOXE1 in thyroid tissue. Abu-Khudir et al. (24) observed a reduction in the methylation of two CpG sites of the FOXE1 promoter that modify its expression in non-tumor thyroid samples. Therefore, in the present study, we investigated both the expression and the promoter methylation status of the FOXE1 gene in a series of DTC tissues and thyroid tumor cell lines, and thyroid data from The Cancer Genome Atlas (TCGA) database, as well as their clinical relevance. With this approach, we contribute novel data on the reduced expression of FOXE1 in DTC, the involvement of DNA methylation in FOXE1 expression, and the association of reduced expression and high methylation with characteristics of aggressiveness.
2 Patients and methods
2.1 Clinical specimens
The series consists of 33 paired samples, either PTC tumor (T) or non-tumor (NT). Clinical data and the pathological diagnoses of all patients are shown in Supplementary Table S1 (24–27). The study was conducted under the approval of the Ethics Committee for Research Projects at the Federal University of Bahia and all patients signed an informed consent form.
2.2 Cell treatment with 5aza demethylating agent
Thyroid tumor cell lines FTC236 (FTC: follicular thyroid cancer; ECACC General collection, catalog no. 06030202), FTC238 (ECACC General collection, catalog no. 94060902), WRO (metastatic thyroid FTC), and NPA (melanoma cell line) were treated with 5aza-2′-deoxycytidine (5aza) (Sigma-Aldrich, USA). Cells (5.0 × 105) were grown at 37°C in 5% CO2 for 4 consecutive days in adequate culture medium with 15 μM 5aza diluted in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, USA) or DMSO alone as a control as previously reported (27–29). All assays were performed in triplicate. After treatment, total RNA, genomic DNA, and protein were extracted for subsequent analysis.
2.3 Quantitative real-time polymerase chain reaction
Total RNA was isolated using the Trizol® method and reverse-transcribed with a SuperScript III reverse transcriptase kit (Life Technologies, USA). Quantification was performed with a nanoespectrophotometer (KASVI, Brazil). The quantitative real-time polymerase chain reaction (qRT-PCR) was carried out using Platinum SYBR Green Master Mix (Thermo Fisher Scientific, USA) in a 7500 Real-Time PCR System (Life Technologies, USA), and normalized with S8 gene expression. PCR primers and conditions are summarized in Supplementary Table S2.
2.4 CpG island in silico identification
The gene sequence information was obtained from Ensembl Gene ID ENSG00000178919. The 10,000 bp upstream to 1000 bp downstream sequence of the FOXE1 translational start codon, which included exon 1 of the FOXE1 gene, was investigated for the detection of CpG islands using strict software, namely, the “CpG Island Search” 1.3 cpgi130.pl version (http://cpgislands.usc.edu), “CpGcluster” (http://bioinfo2.ugr.es/CpGcluster), the “RepeatMasker” open version 3.08 (http://www.repeatmasker.org), and the “CpG Island Research,” as previously described (28).
2.5 Bisulfite sequencing
DNA was purified from untreated and 5aza-treated cells as well as thyroid samples, as previously reported (24, 25, 27, 28, 31). To discriminate the methylated cytosines from the unmethylated, 1 μg of genomic DNA was converted using the EpiTect Bisulfite conversion kit (QIAgen, Germany) according to the manufacturer’s (28) protocols. Bisulfite-converted DNA (100 ng) was used as a template in 25 μL PCR reactions containing 20 pmol of specific primers: 20 mM dNTPs, 100 mM MgCl2, 20 mM Tris-HCl, 50 mM KCl, and 3U Platinum Taq DNA polymerase (Life Technologies, USA). Primer sequences and PCR conditions are described in Supplementary Table S2. EpiTect Control DNA (Qiagen) was used as a positive control. PCR products were cloned into a pCR2.1-TOPO vector using a TOPO TA Cloning kit (Life Technologies, USA) according to the manufacturer’s protocols. Individual clones from each sample were amplified and sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, USA). The degree of methylation at each CpG site in each region was expressed as a percentage, determined as the ratio of methylated cytosines to total cytosines.
2.6 Western blot analysis
Immunoblotting (10% SDS-PAGE) was performed using an anti-α TTF2 antibody (1:1,000; Affinity BioReagents, USA), Tubulin antibody (1:10,000; Sigma-Aldrich), and GAPDH antibody (1:10,000; Sigma-Aldrich). Band densitometry was performed using ImageQuant LAS 4000 (GE Healthcare Life Sciences, USA) and ImageJ software (National Institute of Health, USA).
2.7 Methylation and gene expression analysis from TCGA database
A total 564 samples (508 papillary T and 56 NT tissues) were selected for analysis of the 580 TCGA thyroid samples (THCA) (515 T, 65 NT) (http://cancergenome.nih.gov) (30). Data from samples classified as “Poorly Differentiated Thyroid Cancer” and from patients that received neoadjuvant therapy prior to resection or with incomplete clinical data were not included. The FOXE1 methylation beta values from TCGA data (level 2) were collected from the Xena Public Data Hubs (https://xena.ucsc.edu/). The CpG sites between cg15365036 and cg13696609 were considered relevant and divided into three regions named CpGisland1’ with 2 CpGs (cg15365036 and cg00282510), CpGisland2’ with 9 CpGs (from cg14270434 to cg13696609), and 2-sites-CpGisland2’ with two CpGs that surround the CpGisland2 (cg15802898 and cg13564742) (Figure 1). The methylation value of each island was calculated as the sum of its beta values and a threshold methylation value (CpGisland1’: 0.09996; CpGisland2’: 0.66276 and 2-sites-CpGisland2’: 0.2086; 1.89), which was determined by considering the distribution of the beta values for the NT and T samples as previously described (27) and used to categorize the T samples as hypermethylated. Data for FOXE1 and FOXE1 target gene expression were also collected from the Xena Public Data Hubs as RSEM values (normalized expectation maximization values) for gene expression (polyA + Illumina Hiseq) (level 3). The clinical data of these patients were downloaded from the Cbioportal for cancer genomics (level 3) (http://www.cbioportal.org/study?id=thca_tcga#clinical) (31). Clinical and pathologic characteristics are presented in Supplementary Table S3.
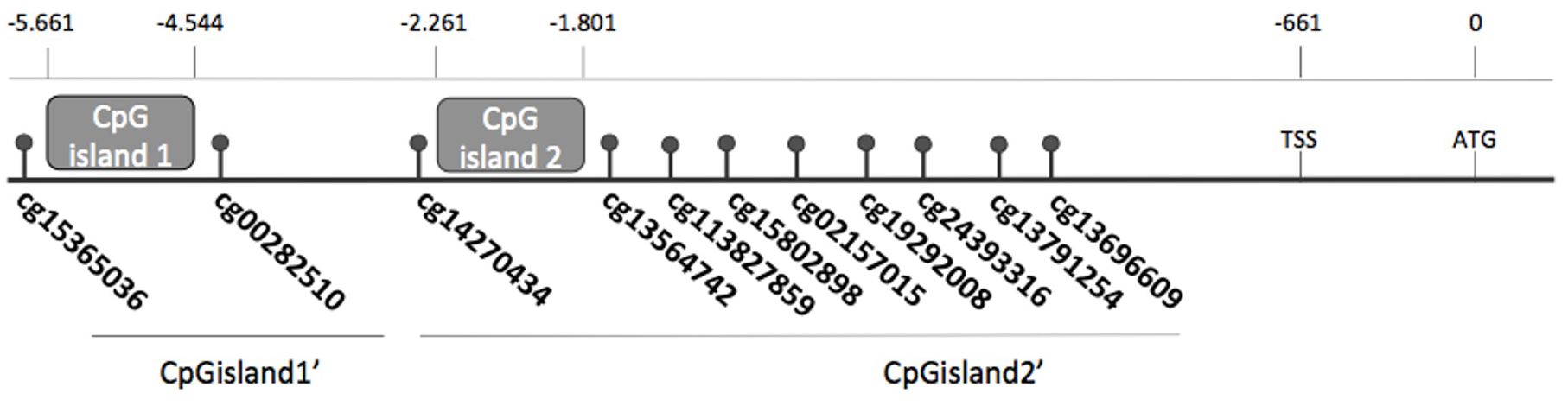
Figure 1. FOXE1 CpG (cg) sites described in TCGA database indicating the positions of CpGisland1 and CpGisland2 and TCGA sites included in CpGisland1’ and CpGisland2’.
2.8 Functional analysis of FOXE1 CpGisland2
The regulatory activity of the region containing CpGisland2 was evaluated using a luciferase reporter assay. The DNA of a thyroid control sample was used in the PCR to amplify the CpGisland2 region with the primers for cloning CpGisland2, containing Kpn1 or Xho1 restriction sites (Supplementary Table S2). The pGL3control (pGL3c) vector and the PCR product were digested with KpnI and XhoI restriction enzymes (Invitrogen), following the manufacturer’s protocol. A 10μL ligation reaction mixture containing a 1:10 insert/vector ratio, 1 μL of HiT4 DNA ligase (New England Biolabs, USA), and 1μL of 10X reaction buffer was prepared and incubated for 18 h at 16°C. Escherichia coli JM109 was transformed using electroporation and the recombinant plasmid pGL3c-CPG2 was confirmed by PCR and sequencing.
For functional analysis, HEK293 cells (1x105) were cotransfected in a 24-well plate with firefly luciferase plasmid (pGL3c-CPG2 or pGL3c) (2500 ng) and an expression vector of β-galactosidase (600 ng) to normalize the luciferase activity, with the Lipofectamine 3000 Reagent (ThermoFisher Scientific). The experiments were carried out with or without 600 ng of the vectors expressing the transcription factors PAX8 and NKX2.1 (28). Nontransfected cells were the negative control of the experiments. The cells were lysed 48 h after the transfections and 30uL of the lysates were used to measure the luciferase activity with a Luciferase Assay System kit (Promega, USA) in a Wallac Victor2 1420 Multilabel Counter (Perkin Elmer, USA). For data normalization, 80uL of ONPG (O-nitrophenyl-beta-D-galactopyranoside) (Sigma-Aldrich) was added to 30uL of the lysates and, after incubation at 37°C for 30 mins, the β-galactosidase was measured at 405 nm using the same equipment.
2.9 Statistical analysis
All data were analyzed with SPSS version 22 and GraphPad Prism 5. Results were expressed as mean ± standard error (SEM) or median (minimum-maximum). For the categorical variables, the χ2 test or the Fisher’s exact test was used. For the continuous non-parametric variables, Spearman’s correlation and the two-tailed Mann–Whitney or Kruskal–Wallis tests were performed and receiver operating characteristic (ROC) curve analysis was used to determine the cut-off value for mRNA expression. Fisher’s exact test, the χ2 test, the Kruskal–Wallis test, and multivariate logistic regression analysis with Bonferroni corrections were performed to analyze the correlation of methylation status with the clinicopathological parameters of TCGA data. For the luciferase assay, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons was used. An alpha error of 5% (p ¾ 0.05) was considered significant.
3 Results
3.1 Identification of CpG islands within the FOXE1 promoter
After in silico analysis, we identified two CpG islands. The first one was new and named CpG island1, and comprises a 200 bp sequence with 22 CpG sites and was located between positions −5,661 and −4,527 relative to the ATG site. The second one, a sequence of 460 nucleotides with 24 CpG sites, was referred to as CpGisland2 (positions -2261 to -1801 of the ATG) and has been previously described (24) (Figure 1).
3.2 FOXE1 expression and methylation analysis in thyroid carcinoma cell lines
FTC 236, FTC 238, WRO, and NPA cell cultures were treated with 5aza to investigate whether demethylation promoted increased expression of FOXE1 mRNA. The treatment resulted in a significant increase in mRNA expression in FTC 238 [9.58 (0.00031 - 175.55) AU], WRO [15.03 (2.11 - 18.32) AU], and FTC 236 [36148.2 (27.12 - 339958.7) AU] (P < 0.05) compared to untreated cells. No effect was observed in NPA [0.678 (0.2662 – 2.012) AU] (Figure 2A).
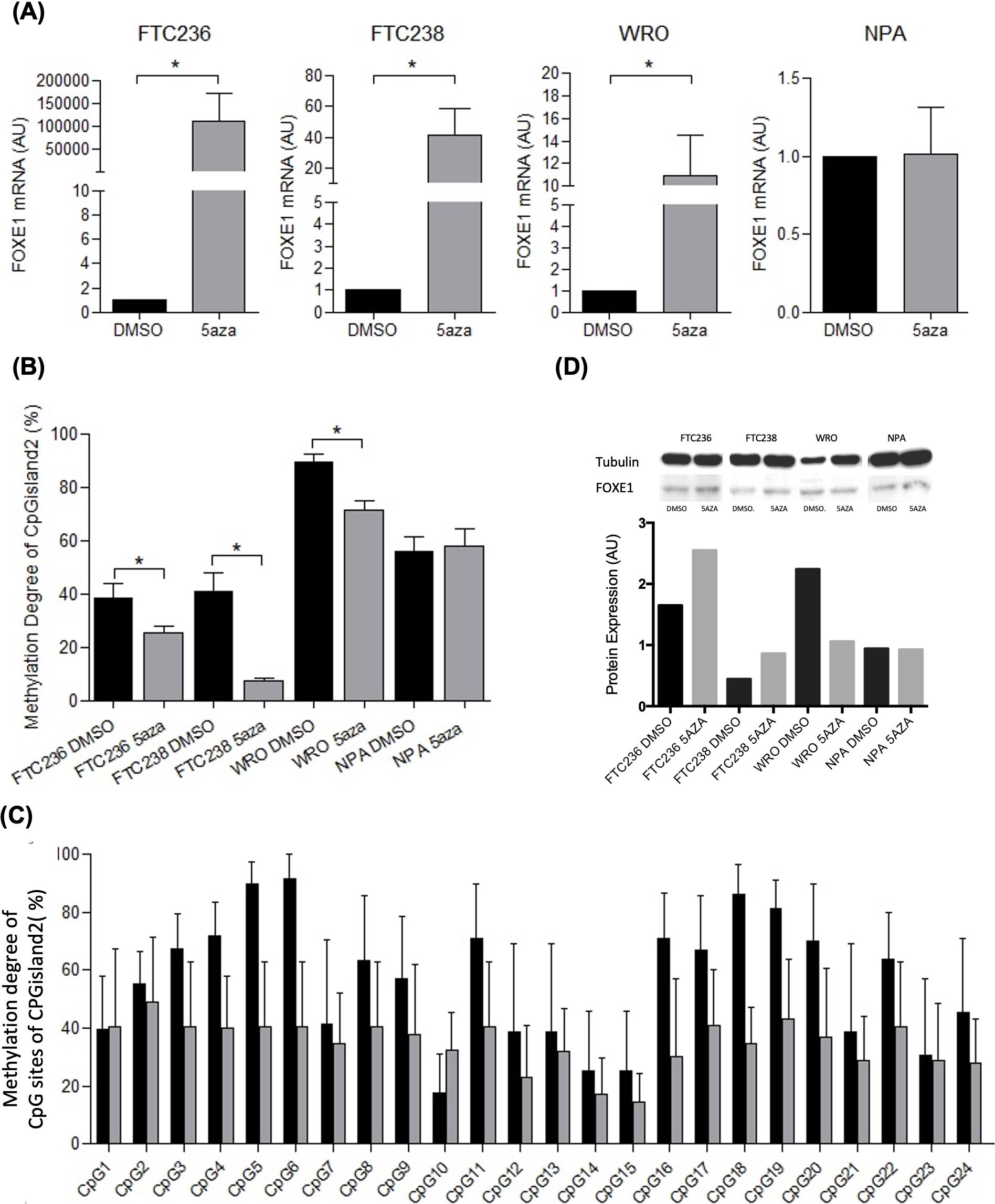
Figure 2. FOXE1 expression and methylation degree of CpGisland2 in thyroid tumor cell lines (FTC 236, FTC 238, and WRO) and melanoma (NPA) before (DMSO) and after treatment with 5aza. (A) Increased FOXE1 mRNA expression and (B) reduced methylation degree (%) in FTC 236, FTC238, and WRO cells after treatment; (C) methylation degree (%) of each CpG site in CpGisland2 in FTC 236, FTC 238, and WRO cells; and (D) increased FOXE1 protein expression after treatment in FTC236, FTC238, and WRO cells (endogenous control: tubulin). *p < 0.05 (mean ± SEM). Black bar: (DMSO) samples; gray bar: 5aza samples.
A significantly lower methylation degree of CpGisland2 was observed in FTC236, FTC238, and WRO 5aza-treated cells [30% (0%-40%), 7.69% (0%-15.38%), and 79.16% (33.33%-91.67%), respectively] compared to untreated cells [33.33% (8.33%-75%), 34.61% (0%-100%), and 94.44% (44.44%-100%), respectively] (P < 0.05) (Figure 2B). The combined analysis of the 22 CpGs in the CpGisland2 of FTC 236, FTC 238, and WRO cells showed a reduction in the methylation degree after treatment of almost all sites, but it was not statistically significant (p>0.05) (Figure 2C). In parallel, an increase in FOXE1 protein expression was observed in FTC236 and FTC238 5aza-treated cells compared to untreated cells, while increased expression was observed in the WRO cells and no difference in the NPA cells (Figure 2D). The analysis of CpGisland1 showed that the CpGs were methylated to different degrees, but there was no difference in the overall methylation levels between the treated and untreated cell lines (Supplementary Figure S1).
3.3 FOXE1 expression and promoter methylation in papillary thyroid carcinoma
We compared FOXE1 mRNA levels in T and NT matched samples from 33 patients with PTC. The T samples showed reduced expression [2.89 (0.044 - 107.86) AU] compared with their adjacent NT samples [4.06 (0.072 - 121.75) AU] (P = 0.0096) (Figure 3A). In total, 22 of the 33 pairs (69.7%) presented downregulated FOXE1 mRNA expression in the T samples compared to the corresponding NT samples (Figure 3B). An empirical mRNA expression cut-off value of 5 AU allowed us to associate higher Bethesda classifications (Bethesda IV, V and VI) with lower mRNA expression (P = 0.006).
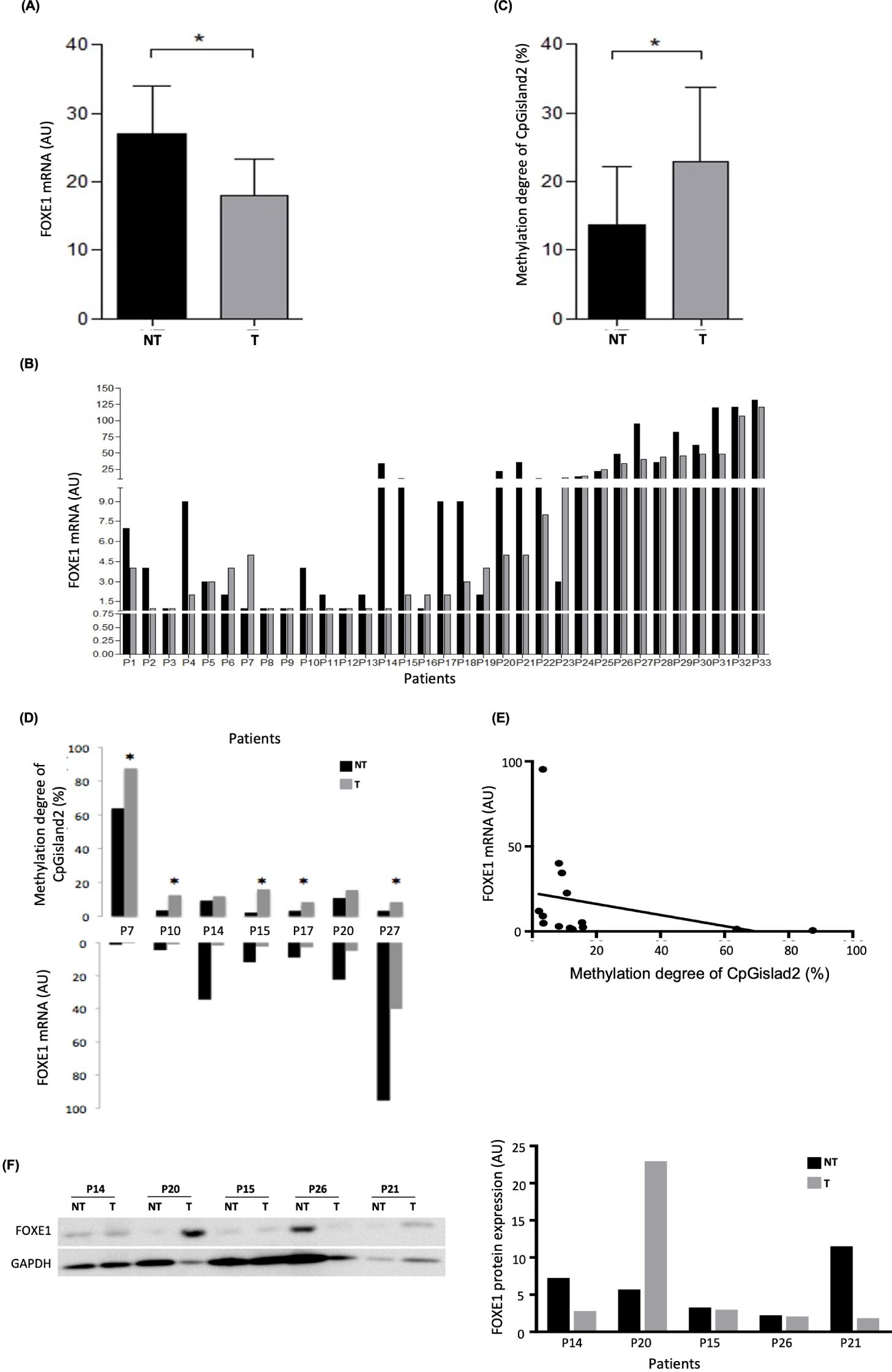
Figure 3. FOXE1 expression and methylation analysis of the 33 PTC samples pairs (T and NT). (A) Reduced FOXE1 mRNA expression in T compared with NT matched samples; (B) 69.7% of the sample pairs showing reduced FOXE1 expression in T compared to NT; (C) high methylation degree (%) of CpGisland2 in T compared to NT matched samples; (D) high methylation degree (%) coupled with reduced expression of seven pairs of T and NT matched samples. (E) Negative Spearman’s correlation between methylation and FOXE1 expression (r = -0.698, p = 0.003). (F) Relative FOXE1 protein expression in five pairs of samples with reduced mRNA expression of T compared to NT (endogenous control: β-actin). *p < 0.05 (mean ± SEM). Black bar: NT samples; gray bar: T samples.
The methylation analysis of CpGisland1 and CpGisland2 was performed in nine (P7, P9, P10, P14, P15, P17, P20, P22, and P27) and in seven pairs of samples (P7, P10, P14, P15, P17, P20, and P27), respectively. Bisulfite sequencing of the 22 CpG sites of CpGisland2 revealed a higher methylation degree in the T samples [12.5 (8.33 - 87.5) %] compared to the NT samples [3.47 (2.08 - 63.78) %] (P = 0.0177) (Figure 3C). Figure 3D details the results of the individual samples, in which all pairs presented reduced T mRNA expression with increased T methylation levels, and was significant in 71.43% of the cases (P < 0.05). There was a negative correlation between the methylation degree of CpGisland2 and mRNA expression when all samples (T and NT samples) were analyzed together (r = -0.698, P = 0.003) (Figure 3E). No difference in the methylation degree between the T and NT samples was observed for CpGisland1 (Supplementary Figure S2) and in only 44% of the pairs of samples was there an increase in the T methylation degree. There was no correlation between mRNA expression and the methylation degree of CpGisland1. Reduced relative protein expression was observed in the T samples compared to the NT samples in four of 5 pairs (two with very slight reduction) (Figure 3F).
3.4 FOXE1 expression and methylation analysis using TCGA data
TCGA data analysis showed FOXE1 mRNA expression (123511,54 ± 7912,788 AU) was downregulated in the T samples compared to the NT samples (127742.39 ± 8214.723 AU) (P < 0.001) (Figure 4). Among the PTC subtypes, the tall cell variant presented lower mRNA expression (120030.35 ± 4658.109 AU) compared to the classical (122634.57 ± 7618.959 AU) and follicular variants (127604.16 ± 8023.145 AU) (P < 0.001). When considering the clinical characteristics, the T samples with advanced extra thyroid extension exhibited lower FOXE1 mRNA expression (106590 ± 0) than those with minimal extension (118671.25 ± 5450.599) or without extension (121654.82 ± 6788,95) (P = 0.001), and the T samples with a more advanced classification presented lower FOXE1 mRNA expression (T4 < T3 < T2 < T1 and N1 < N0) (P < 0.001).
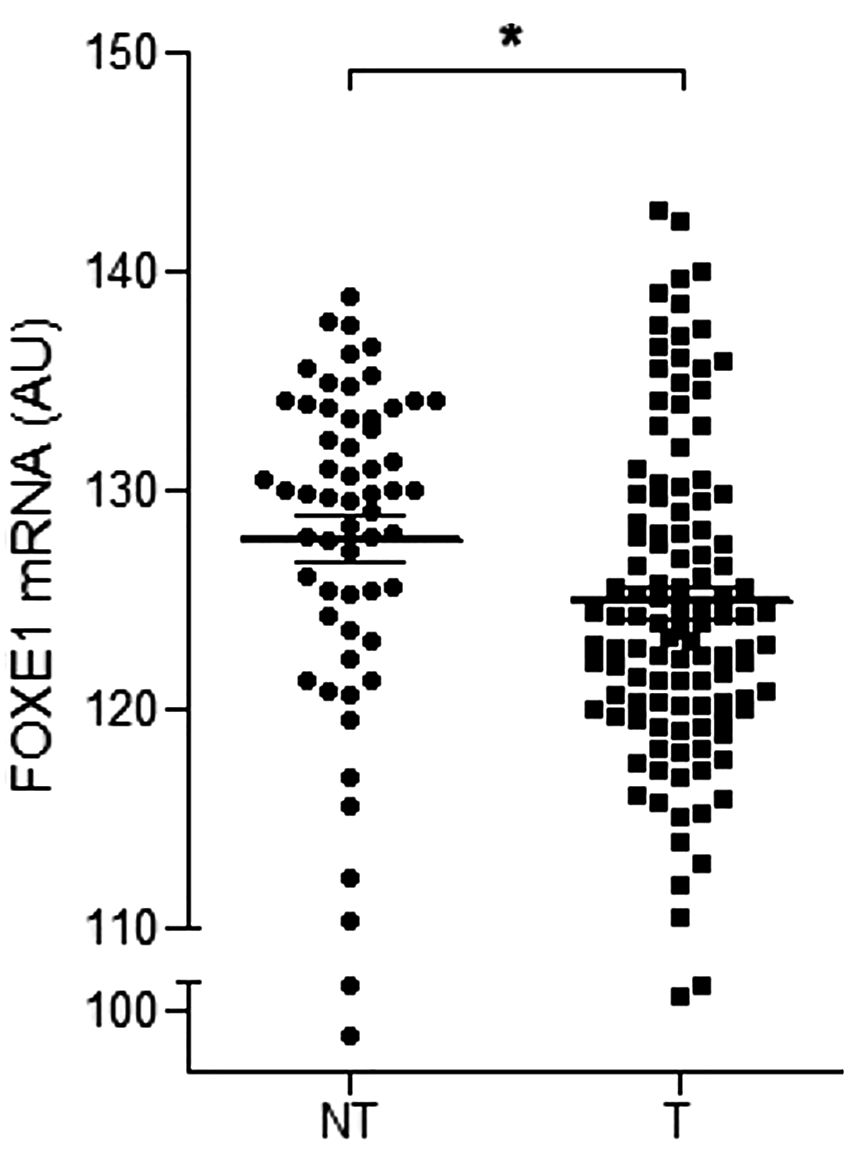
Figure 4. Downregulation of FOXE1 mRNA expression in thyroid T samples from TCGA dataset compared to NT samples. *p < 0.05 (mean ± SEM).
No difference in the methylation of CpGisland1’, CpGisland2’, and 2-sites-CpGisland2’ was detected between the T and NT samples. However, in the tumor samples, significant negative correlations between FOXE1 mRNA and the methylation values of the three regions were observed (CpGisland2’ r = -0.164; 2-sites-CpGisland2’ r = 0.218; CpGisland1’ r = -0.095, P < 0.04), while no correlation was observed in the non-tumor samples. Considering the clinical characteristics, among the PTC subtypes, the tall cell variant showed a higher methylation of CpGisland2’ (0.673637 ± 0.2435934 AU) than the classical (0.608464 ± 0.2789129 AU) (P < 0.001) and follicular variants (0.537720 ± 0.2357689 AU) (P < 0.001). Samples from older patients at diagnosis exhibited a high methylation degree in CpGisland2’ (>45 years old: 0.615680 ± 0.2916553 AU; <45 years old: 0.577995 ± 0.2398365 AU; P = 0.016). We observed a decrease in the methylation levels of CpGisland1’ and 2-sites-CpGisland2’ in the primary tumor samples (0.112480 ± 0.0416035 AU and 0.1848450 ± 0.0988664) compared to metastasis (0.1507000 ± 0.0533801 AU and 0.253413 ± 0.1108841 AU) (P = 0.042 and P = 0.046, respectively). When applying the threshold cut-off value, hypermethylation of CpGisland1’ was associated with bilateral tumors (56/87, 64.4%) (unilateral: 214/416, 51.4%, P = 0.033) and hypermethylation of CpGisland2’ and 2-sites-CpGisland2’ was associated with the tall cell PTC variant (P = 0.014 and P = 0.039, respectively).
An association between the presence of the BRAFV600E mutation, mRNA FOXE1 expression, and methylation was detected, where the BRAFV600E+ tumors had lower expression (121727.54 ± 5419.814 AU) and higher methylation (0.615242 ± 0.26681367 AU) of CpGisland2’ compared to the BRAFV600E- tumor samples (mRNA expression: 124985.91 ± 9281.498 AU and CpGisland2’ methylation: 0.584073 ± 0.2705702 AU) (P < 0.05).
3.5 Expression of FOXE1 target genes
To understand the relevance of reduced mRNA FOXE1 expression in PTC, the expression of several FOXE1 target genes, i.e., PAX8, TG, TPO, DUOX2, NIS, NKX2.1, PDGFA, and ZEB1 (6, 7, 32) and their correlation with FOXE1 mRNA expression was investigated. The analysis of the TCGA data showed reduced expression of PAX8, TG, TPO, NIS and PDGFA in T compared to NT samples (P < 0.05 each), while no differences of the expression of NKX2.1, DUOX2 and ZEB1 were detected (Supplementary Figure S3). Moreover, in the T samples, positive correlations between FOXE1 expression and PAX8, TG, TPO, DUOX2, NIS, and ZEB1 were observed, but PDGFA showed a negative correlation (P < 0.01 each). In the NT samples, only positive correlations were observed with PAX8, TG, TPO, DUOX2, and NKX2.1. Interestingly, in the T samples, a negative correlation was observed between the methylation state of the FOXE1 CPGisland 2’ and PAX8, TG, DUOX2, and NKX2.1, while a positive correlation was found with NIS and PDGFA. In the NT samples, a negative correlation was detected with PAX8, DUOX2, and NKX2.1 and a positive correlation with PDGFA (Supplementary Table S4). In the patient samples, the TG and PAX8 expression was reduced in the T samples compared with the NT samples (Supplementary Figure S4), without significance, and no correlation with FOXE1 expression was observed, probably due to the reduced number of samples.
3.6 Functional analysis of FOXE1 CpGisland2
To investigate whether the CPGisland2 was able to regulate the activity of a promoter and whether the CPGisland2 activity could be modulated by the transcription factors PAX8 and NKX2.1, a luciferase reporter assay with pGL3control-derived vectors was conducted. Cells transfected with pGL3c-CpG2 showed a significantly higher expression of luciferase compared with the pGL3c-transfected cells (P < 0.05), confirming the regulatory activity of the CPGisland2. The expression of PAX8 and NKX2.1 did not increase CPGisland2 activity. Interestingly, the expression of NKX2.1 notably reduced luciferase activity, suggesting repressor activity through the pGL3c promoter which was not overcome by the presence of CPGisland2. In contrast, CPG2island2 was able to restore luciferase activity in the presence of PAX8 (Figure 5).
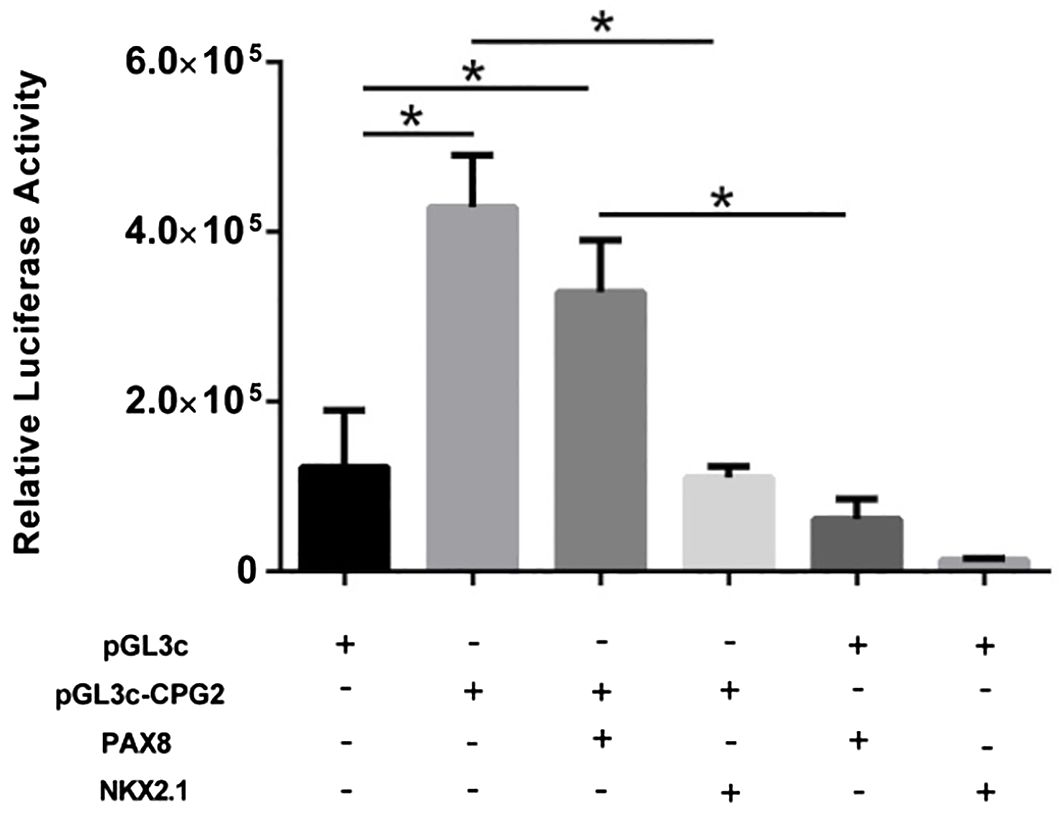
Figure 5. Relative luciferase expression in HEK293 cells transfected with pGL3c or pGL3c-CPG2 in the presence or absence of plasmids expressing the transcription factors NKX2.1 and PAX8 was evaluated to assess the regulatory activity of FOXE1 CpG-island2 (*p < 0.005).
4 Discussion
In the last few decades, studies have attempted to understand the role of the transcription factor FOXE1 in DTC by identifying mutations in sporadic or familial cases (9, 10) and polymorphisms associated with increased predisposition (11, 12), but no consensus has been reached on FOXE1 gene expression in DTC (6, 22, 23). However, a few authors have investigated FOXE1 expression and its epigenetic regulation in thyroid cancer. Thus, in this study, we sought to better understand FOXE1 gene expression in DTC in parallel with the involvement of DNA methylation and the clinical relevance of both parameters.
Initially, we performed an in silico analysis in a large sequence upstream FOXE1 gene with strict criteria and identified the new GpGisland1 and the known CpGisland2 (24) (Figure 1). Next, we investigated FOXE1 expression in cell lines and the methylation status of both CpG islands. To our knowledge, this is the first study investigating these two CPG islands in thyroid cancer. Previously, CPG2 was only investigated in ectopic thyroids and leucocytes (24). We observed increased mRNA expression in the three DTC cell lines (FTC 236, FTC 238, and WRO) concomitantly with a low methylation degree of CpGisland2 and an increased protein expression in two of them after the demethylation treatment, indicating a crosstalk between FOXE1 methylation, mRNA, and protein expression (Figure 2). The non-concordance of mRNA and protein levels in WRO suggested the involvement of post-transcriptional regulatory mechanisms (7). It has been shown that microRNAs regulate FOX protein expression (33) as miR-524-5p targets FOXE1, suppressing PTC progression (34). In addition, other events may be considered such as RNA modifications (35) and LncRNA activity (36, 37). In the melanoma (NPA) cell line, no changes were observed in FOXE1 mRNA, protein expression, and methylation degree after treatment, as we had already observed for the NIS and ABI3 genes (28, 31), probably due to the duration of the treatment or reduced activity of the 5aza demethylating agent in this cell type. We then investigated FOXE1 mRNA expression in the our patients’ samples (Figure 3). We observed reduced FOXE1 mRNA expression in tumors when compared to non-tumor samples (Figure 2), in accordance with FOXE1 expression in the TCGA dataset (Figure 3). Protein expression in the tumors was downregulated in four of the five investigated paired samples (two were slightly reduced), once more suggesting that other regulatory mechanisms play a role in FOXE1 protein synthesis (33) and need further investigation.
FOXE1 expression has been studied in different tumor types: FOXE1 downregulation was observed in salivary gland, breast, colon, and skin tumors (15, 16, 18), upregulation was described in basal cell carcinoma, liver, and airway tumors (32–34), and, in pancreatic tumors, both increased and decreased FOXE1 expression was reported (17, 35, 40). In the thyroid, previous studies suggested oncogene-like behavior for FOXE1, a correlation between the level of FOXE1 expression and thyroid tumor differentiation status (41, 42) and interference with the p53 pathway was observed. However, suppression activity of FOXE1 in the early stages of PTC has also been proposed (6). Besides that, transcriptional FOXE1 expression was significantly downregulated in poorly differentiated thyroid carcinoma and absent in anaplastic thyroid cancer (43–46). Similar results were obtained in a series of thyroid cell lines, where the expression was higher in non-tumor control and PTC than in FTC cells, with the lowest values in the ATC cell lines (7, 9). High FOXE1 expression was detected in follicular adenomas compared to FTC, as well as in Graves’ disease samples compared to PTC cases (38, 39, 47). Likewise, the overexpression of cytoplasmatic and the downexpression of nuclear FOXE1 were observed in PTC compared with surrounding NT regions (15). Yet the same group showed that FOXE1 overexpression in mouse thyroids developed multinodular goiter but not cancer (48). It has to be mentioned that FOXE1 participates in a complex transcriptional network of regulation in thyroid follicular cells (42, 49). Thus, understanding the activity of this transcription factor in thyroid cancer is challenging.
Regarding the involvement of methylation in FOXE1 expression, in our cohort of DTC samples, we observed low FOXE1 expression in tumors compared to non-tumor matched samples along with a high FOXE1 methylation degree of CpGisland2 as well as a significant negative correlation between mRNA expression and methylation. This data suggests that FOXE1 mRNA expression is likely to be modulated by methylation, particularly of CpGisland2. Interestingly, a previous study associated the high methylation of two specific sites (CpG5 and CpG6) in CpGisland2 in leukocytes, when comparing eutopic and ectopic thyroid tissues, with FOXE1 expression (24); however, our data did not confirm this association in DTC. Limitations in our study, as well as the other (24), were how to overcome technical constraints in analyzing the CG-rich region of the CpGisland2 of the FOXE1 gene in all the patient samples in the bisulfite methylation investigation and that TCGA methylation data do not include the CpG sites of the CpG-islands investigated in this study. Even so, the involvement of methylation in FOXE expression was corroborated in TCGA data, which showed a significant negative correlation between methylation and FOXE1 mRNA expression among all the samples (tumor and non-tumor) and in the tumor samples but not in the non-tumor samples. Furthermore, considering a linear regression model, the contribution of the methylation levels in gene expression of the 2-sites-CpGisland2’, which includes the two sites surrounding CpGisland2, was higher (6.3%) than the contribution of the other CpG regions (3.9% and 0.5%), suggesting that the methylation of the region that comprises CpGisland2 may be involved in mRNA regulation. These results may explain the fact that no difference in the methylation status of TCGA regions was observed between the T and NT samples. Nonetheless, the involvement of the methylation of other regions, such as regions downstream of the transcription start site, first exon, intragenic exons and first intron could not be excluded (50), as well as other genetic mechanisms that regulate FOXE1 expression, such as distant enhancers (51), FOXE1 mutations, and functional polymorphisms (22).
To advance the understanding of high methylation and reduced mRNA expression in thyroid tumors, the expression of FOXE1 target genes (TG, TPO, PAX8, NIS, DUOX2, NKX2.1, PDGFA, and ZEB1) was investigated. The analysis of TCGA data showed a moderate positive correlation between the expression of most of the FOXE1 target genes with mRNA FOXE1 expression and a negative correlation with the methylation levels of CpGisland2’ (Supplementary Table S4) in the T and NT samples. An inverse correlation was observed for PDGFA in the T samples. These results suggest the relevance of reduced FOXE1 expression in thyroid cancer dedifferentiation. Interestingly, conflicting data has been reported previously as Din et al. (6) showed increased FOXE1 mRNA and protein expression in the early stages of PTC, suggesting tumor suppression activity through the inhibition of proliferation and invasion, probably by negatively regulating PDGFA expression. Morrilo-Bernal et al. (7) showed that FOXE1 expression positively correlates with thyroid cancer differentiation, but the reduction of FOXE1 expression reduced migration and invasion through the regulation of ZEB1.Thus, further studies are necessary to understand the role of FOXE1 expression in thyroid cancer.
Still, we confirmed the enhancer activity of CPGisland2 using a luciferase gene reporter assay, and PAX8 and NKX2.1 did not modulate its activity, which was in accordance with the lack of these transcription factor sites in this region.
Considering the clinicopathological characteristics in our tumor samples, low mRNA values were associated with an increased risk of malignancy, and TCGA data analysis also showed an association between reduced FOXE1 mRNA expression and the aggressiveness of PTC subtypes, as previously reported (7), as well as with worse clinicopathological and genetics characteristics, such as extra thyroid invasion, AJCC and TNM classification, and the presence of BRAF mutations. Our results are in line with the observed downregulation of FOXE1 gene expression in PTC and no expression in the most advanced and aggressive form of TC (45). However, another report detected an increase of FOXE1 expression in PTC tumor tissues that correlated with a worse clinical prognosis (14).
In parallel with gene expression, methylation of CpGisland2’ was positively associated with the aggressiveness of PTC subtype, the presence of BRAF mutation, and age at diagnosis (>45 years old). Likewise, the methylation levels of CpGisland1’ and 2-sites-CpGisland2’ were higher in metastases than in primary tumor samples and hypermethylation of CpGisland2’ was associated with bilateral tumors and tall cell variants. Hypermethylation of FOXE1 was observed in 75% of pancreatic carcinomas, as well as in squamous cell cancer and in salivary gland carcinoma (15, 17). In breast cancer, hypermethylation was identified in the plasma of patients with a more aggressive disease (stage IV) (52) and, in bladder cancer, FOXE1 methylation was suggested as one of the high-grade markers (53). Interestingly, in colorectal cancer, it was shown that high methylation of FOXE1 contributed to the poor prognosis of the patients (16) and could be associated with the observed FOXE1 repression of tumor cell growth and glycolysis through the inhibition of HK2 (23). Besides this, other genes, mechanisms, and risk factors have been linked with DTC aggressiveness (54). We have previously shown that the hypermethylation of the HOPX promoter was associated with a worse prognosis in DTC (27) while the low DNA methylation of three SNP markers was related with recurrent or persistent disease (55). The deregulation of key signal pathways such as the hedgehog pathway was also associated with aggressive PTC (56).
In summary, these results provide novel evidence that FOXE1 mRNA expression is reduced in DTC, methylation contributes to the regulation of FOXE1 expression, and the coupling of low mRNA expression and high methylation status can be associated with some characteristics of aggressiveness in DTC tumors. Further investigations involving the methylation status of other FOXE1 regions or other genetic or epigenetic mechanisms that regulate gene expression, including microRNA, lncRNA, and RNA modifications, are warranted.
Data availability statement
Data supporting the findings of this study are available in cBIOPORTAL at doi: 10.1158/2159-8290.CD-12-0095 and doi: 10.1126/scisignal.2004088 and UCSC XENA at https://doi.org/10.1038/s41587-020-0546-8. The datasets presented in this study can also be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.cancer.gov/ccg/research/genome-sequencing/tcga, phs00017. Further information is available form the corresponding author on request.
Ethics statement
The studies involving humans were approved by Federal University of Bahia–Ethical Committee for Research Projects. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
EL: Conceptualization, Investigation, Methodology, Project administration, Writing – original draft. FF: Investigation, Methodology, Writing – original draft. IS: Methodology, Writing – original draft. CL: Methodology, Writing – original draft. VF: Investigation, Writing – original draft. GC: Investigation, Writing – original draft. PO: Investigation, Writing – original draft. AB: Methodology, Writing – original draft. JC: Resources, Writing – original draft. RT: Conceptualization, Writing – review & editing, Resources. HR: Conceptualization, Funding acquisition, Methodology, Writing – original draft. IR: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by research grants from FAPESB (Fundação de Amparo à Pesquisa no Estado da Bahia, Edital 011/2013; TOU RED010/2013; FAPESP, 2014/24549-4, 2019/15619-2 and 2023/02690-6. Fellowships were received from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for VF, PO, and GC and from FAPESP for EUL (2013/19598-3), RT (2020/08271-7) and FF (2020/14158-9).
Acknowledgments
The authors would like to thank all the participants who contributed samples to this study and Dr Alfredo Fusco for the NPA and WRO cells.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1454349/full#supplementary-material
References
1. Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. (2017) 317:1338. doi: 10.1001/jama.2017.2719
2. Zaballos MA, Santisteban P. Key signaling pathways in thyroid cancer. J Endocrinol. (2017) 235:R43–61. doi: 10.1530/JOE-17-0266
3. Felice MDE, Lauro RDI, Zoologica S, Dohrn A, Biology M. Thyroid development and its disorders : genetics and. Endocr Rev. (2004) 25:722–46. doi: 10.1210/er.2003-0028
4. Zannini M, Avantaggiato V, Biffali E, Arnone MI, Sato K, Pischetola M, et al. TTF-2, a new forkhead protein, shows a temporal expression in the developing thyroid which is consistent with a role in controlling the onset of differentiation. Embo. (1997) 16:3185–97. doi: 10.1093/emboj/16.11.3185
5. Redondo-blanco S, Fernández J, Gutiérrez-del-río I, Villar CJ, Lombó F. New insights toward colorectal cancer chemotherapy using natural bioactive compounds. Front Pharmacol (2017) 8:1–22. doi: 10.3389/fphar.2017.00109
6. Ding Z, Ke R, Zhang Y, Fan Y, Fan J. Molecular and Cellular Endocrinology FOXE1 inhibits cell proliferation, migration and invasion of papillary thyroid cancer by regulating PDGFA. Mol Cell Endocrinol. (2019) 483:110420. doi: 10.1016/j.mce.2019.03.010
7. Morillo-bernal J, Fernández LP, Santisteban P. FOXE1 regulates migration and invasion in thyroid cancer cells and targets ZEB1. Endocr Rev. (2020) 27:137–51. doi: 10.1530/ERC-19-0156
8. Clifton-bligh RJ, Wentworth JM, Heinz P, Crisp MS, John R, Lazarus JH, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. (1998) 19:399–401. doi: 10.1038/1294
9. Mond M, Bullock M, Yao Y, Clifton-bligh RJ, Gilfillan C, Fuller PJ. Somatic mutations of FOXE1 in papillary thyroid cancer. Thyroid. (2015) 25:904–10. doi: 10.1089/thy.2015.0030
10. Pereira JS, Gomes J, Alexandra R, Maria P, Cavaco BM. Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine. (2015) 49:204–14. doi: 10.1007/s12020-014-0470-0
11. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet. (2009) 41:460–4. doi: 10.1038/ng.339
12. Landa I, Ruiz-llorente S, Montero-conde C, Jara-albarra A, Rodrı P, Castello R, et al. The Variant rs1867277 in FOXE1 Gene Confers Thyroid Cancer Susceptibility through the Recruitment of USF1/USF2 Transcription Factors. PloS Genet. (2009) 5:5–10. doi: 10.1371/journal.pgen.1000637
13. Kang J, Deng XZ, Fan YB WB. Relationships of FOXE1 and ATM genetic polymorphisms with papillary thyroid carcinoma risk: A meta-analysis. Tumor Biol. (2014) 35:7085–96. doi: 10.1007/s13277-014-1865-5
14. Fan Y, Ding Z, Yang Z, Deng X, Kang JIE, Wu BO, et al. Expression and clinical significance of FOXE1 in papillary thyroid carcinoma. Mol Med Rep. (2013) 8:123–7. doi: 10.3892/mmr.2013.1494
15. Bychkov A, Saenko V, Nakashima M, Mitsutake N, Rogounovitch T, Nikitski A, et al. Patterns of FOXE1 expression in papillary thyroid carcinoma by immunohistochemistry. Thyroid. (2013) 23:271–7. doi: 10.1089/thy.2012.0466
16. Sugimachi K, Matsumura T, Shimamura T, Hirata H, Uchi R, Ueda M, et al. Aberrant methylation of FOXE1 contributes to a poor prognosis for patients with colorectal cancer. Ann Surg Oncol. (2016) 23:3948–55. doi: 10.1245/s10434-016-5289-x
17. Bell A, Bell D, Weber RS, El-naggar AK. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer. (2011) 117:2898–909. doi: 10.1002/cncr.25818
18. Venza I, Visalli M, Tripodo B, De Grazia G, Loddo S, Teti D, et al. FOXE 1 is a target for aberrant methylation in cutaneous squamous cell carcinoma. Br J Dermatol. (2010) 162:1093–7. doi: 10.1111/j.1365-2133.2009.09560.x
19. Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron JL, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. (2003) 63:3735–42.
20. Melotte V, Yi JM, Lentjes MHFM, Smits KM, Van Neste L, Niessen HEC, et al. Spectrin repeat containing nuclear envelope 1 and forkhead box protein E1 are promising markers for the detection of colorectal cancer in blood. Cancer Prev Res. (2015) 8:157–65. doi: 10.1158/1940-6207.CAPR-14-0198
21. Papadia C, Louwagie J, Del Rio P, Grooteclaes M, Coruzzi A, Montana C, et al. FOXE1 and SYNE1 genes hypermethylation panel as promising biomarker in colitis associated colorectal neoplasia. Inflammation Bowel Dis. (2016) 20:1–2. doi: 10.1097/01.MIB.0000435443.07237.ed.Full
22. Lidral AC, Liu H, Bullard SA, Bonde G, Machida J, Visel A, et al. A single nucleotide polymorphism associated with isolated cleft lip and palate, thyroid cancer and hypothyroidism alters the activity of an oral epithelium and thyroid enhancer near FOXE1. Hum Mol Genet. (2015) 24:3895–907. doi: 10.1093/hmg/ddv047
23. Dai W, Meng X, Mo S, Xiang W, Xu Y, Zhang L, et al. FOXE1 represses cell proliferation and Warburg effect by inhibiting HK2 in colorectal cancer. Cell Commun Signal. (2020) 8:1–13. doi: 10.1186/s12964-019-0502-8
24. Abu-khudir R, Magne F, Chanoine J, Deal C, Van Vliet G, Deladoëy J. Role for tissue-dependent methylation differences in the expression of FOXE1 in nontumoral thyroid glands. J Clin Endocrinol Metab. (2014) 99:1120–9. doi: 10.1210/jc.2013-4414
25. Galrão AL, Sodré AK, Camargo RY, Friguglietti CU, Kulcsar MA, Lima EU, et al. Methylation levels of sodium-iodide symporter (NIS) promoter in benign and Malignant thyroid tumors with reduced NIS expression. Endocrine. (2013) 43: 225–9. doi: 10.1007/s12020-012-9779-8
26. Sodré AKMB, Rubio IGS, Galrão ALR, Knobel M, Tomimori EK, Alves VAF, et al. Association of low sodium-iodide symporter messenger ribonucleic acid expression in Malignant thyroid nodules with increased intracellular protein staining. J Clin Endocrinol Metab. (2008) 93: 4141–5. doi: 10.1210/jc.2007-0353
27. Lima EU, Rubio IGS, Da Silva JC, Galrão AL, Pêssoa D, Oliveira TC, et al. HOPX homeobox methylation in differentiated thyroid cancer and its clinical relevance. Endocr Connect. (2018) 7: 1333–42. doi: 10.1530/EC-18-0380
28. Galrão AL, Camargo RY, Friguglietti CU, Moraes L, Cerutti JM, Serrano-Nascimento C, et al. Hypermethylation of a new distal sodium/iodide symporter (NIS) enhancer (NDE) Is associated with reduced nis expression in thyroid tumors. J Clin Endocrinol Metab. (2014) 99: E944–52. doi: 10.1210/jc.2013-1450
29. Moraes L, Galrão ALR, Rubió I, Cerutti JM. Transcriptional regulation of the potential tumor suppressor ABI3 gene in thyroid carcinomas: Interplay between methylation and NKX2-1 availability. Oncotarget. (2016) 7: 25960–70. doi: 10.18632/oncotarget.8416
30. Network TCGAR. Integrated genomic characterization of papillary thyroid carcinoma. Cell. (2014) 159:678–90. doi: 10.1016/j.cell.2014.09.050
31. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Arman B, et al. The cBio cancer genomics portal : an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
32. Fernández LP, López-Márquez A, Martínez ÁM, Gómez-López G, Santisteban P. New insights into foxE1 functions: identification of direct foxE1 targets in thyroid cells. PloS One. (2013) 8: e62849. doi: 10.1371/journal.pone.0062849
33. Li C, Zhang K, Chen J, Chen L, Wang R, Chu X. MicroRNAs as regulators and mediators of forkhead box transcription factors function in human cancers. Oncotarget. (2017) 8:12433–50. doi: 10.18632/oncotarget.14015
34. Liu H, Chen X, Lin T, Chen X, Yan J, Jiang S. MicroRNA-524-5p suppresses the progression of papillary thyroid carcinoma cells via targeting on FOXE1 and ITGA3 in cell autophagy and cycling pathways. J Cell Physiol. (2019) 234:18382–91. doi: 10.1002/jcp.28472
35. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. BioMed Pharmacother. (2019) 112:108613. doi: 10.1016/j.biopha.2019.108613
36. Mancheng AD, Ossas U. How does lncrna regulation impact cancer metastasis. Cancer Insight. (2023) 1:15–34. doi: 10.58567/ci01010002
37. Brancaccio A, Minichiello A, Grachtchouk M, Antonini D, Sheng H, Parlato R, et al. Requirement of the forkhead gene Foxe1, a target of sonic hedgehog signaling, in hair follicle morphogenesis. Hum Mol Genet. (2004) 13:2595–606. doi: 10.1093/hmg/ddh292
38. Eichberger T, Regl ÃG, Ikram ÃMS, Neill GW, Philpott MP, Aberger F, et al. FOXE1, A new transcriptional target of GLI2, is expressed in human epidermis and basal cell carcinoma. J Investig Dermatotl. (2004) 122:1180–7. doi: 10.1111/j.0022-202X.2004.22505.x
39. Zhang J, Yang Y, Yang T, Yuan S, Wang R, Pan Z, et al. Double-negative feedback loop between microRNA-422a and forkhead box (FOX)G1/Q1/E1 regulates hepatocellular carcinoma tumor growth and metastasis. Hepatology. (2015) 61:561–73. doi: 10.1002/hep.27491
40. Xu Y, Chang R, Peng Z, Wang Y, Ji W, Guo J, et al. Loss of polarity protein AF6 promotes pancreatic cancer metastasis by inducing Snail expression. Nat Commun. (2015) 6:7184. doi: 10.1038/ncomms8184
41. Credendino SC, Moccia C, Amendola E, D’avino G, Di Guida L, Clery E, et al. Foxe1 gene dosage affects thyroid cancer histology and differentiation. vivo Int J Mol Sci. (2021) 22:1–9. doi: 10.3390/ijms22010025
42. Li S, Zhang Z, Peng H, Xiao X. YY1-induced up-regulation of FOXE1 is negatively regulated by miR-129-5p and contributes to the progression of papillary thyroid microcarcinoma. Pathol Res Pract. (2021) 221:153337. doi: 10.1016/j.prp.2020.153337
43. Nonaka D, Tang Y, Chiriboga L, Rivera M, Ghossein R. Diagnostic utility of thyroid transcription factors Pax8 and TTF-2 (FoxE1) in thyroid epithelial neoplasms. Mod Pathol. (2008) 2:192–200. doi: 10.1038/modpathol.3801002
44. Sequeira MJ, Morgan JM, Fuhrer D, Wheeler MH, Jasani B, Ludgate M. Thyroid transcription factor-2 gene expression in benign and Malignant thyroid lesions. Thyroid. (2001) 11:995–1001. doi: 10.1089/105072501753271662
45. Hajian R, Javadirad SM, Kolahdouzan M. FOXE1 gene is a probable tumor suppressor gene with decreased expression as papillary thyroid cancers grow, and is absent in anaplastic thyroid cancers. Biochem Genet. (2024). doi: 10.1007/s10528-023-10642-z
46. Zhang P, Zuo H, Nakamura Y, Nakamura M, Wakasa T, Kakudo K. Immunohistochemical analysis of thyroid-specific transcription factors in thyroid tumors. Pathol Int. (2006) 56:240–5. doi: 10.1111/j.1440-1827.2006.01959.x
47. Kallel R, Belguith-maalej S, Akdi A, Mnif M, Charfeddine I, Vel A. Genetic investigation of FOXE1 p olyalanine tract in thyroid diseases : New insight on the role of FOXE1 in th yroi d c arcinoma. Cancer Biomarkers. (2011) 8:43–51. doi: 10.3233/DMA-2011-0824
48. Nikitski A, Saenko V, Shimamura M, Nakashima M, Matsuse M, Suzuki K, et al. Targeted foxe1 overexpression in mouse thyroid causes the development of multinodular goiter but does not promote carcinogenesis. Endocrinology. (2016) 157:2182–95. doi: 10.1210/en.2015-2066
49. López-Márquez A, Fernández-Méndez C, Recacha P, Santisteban P. Regulation of foxe1 by thyrotropin and transforming growth factor beta depends on the interplay between thyroid-specific, CREB and SMAD transcription factors. Thyroid. (2019) 29:714–25. doi: 10.1089/thy.2018.0136
50. Li S, Zhang J, Huang S, He X. Genome-wide analysis reveals that exon methylation facilitates its selective usage in the human transcriptome. Brief Bioinform. (2018) 19:754–64. doi: 10.1093/bib/bbx019
51. He H, Li W, Liyanarachchi S, Srinivas M, Wang Y, Akagi K, et al. Multiple functional variants in long-range enhancer elements contribute to the risk of SNP rs965513 in thyroid cancer. Proc Natl Acad Sci. (2015) 112: 6128–33. doi: 10.1073/pnas.1506255112
52. Weisenberger DJ, Trinh BN, Campan M, Sharma S, Long TI, Ananthnarayan S, et al. DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. Nucleic Acid Res. (2008) 36:4689–98. doi: 10.1093/nar/gkn455
53. Olkhov-mitsel E, Savio AJ, Kron KJ, Pethe VV, Hermanns T, Fleshner NE. Tr a n s l a t i o n a l O n c o l o g y Epigenome-Wide DNA Methylation Profiling Identifies Differential Methylation Biomarkers in High-Grade Bladder Cancer. Transl Oncol. (2017) 10:168–77. doi: 10.1016/j.tranon.2017.01.001
54. Zhang J, Xu S. High aggressiveness of papillary thyroid cancer: from clinical evidence to regulatory cellular networks. Cell Death Discovery. (2024) 10: 378. doi: 10.1038/s41420-024-02157-2
55. Park JL, Jeon S, Seo EH, Bae DH, Jeong YM, Kim Y, et al. Comprehensive DNA methylation profiling identifies novel diagnostic biomarkers for thyroid cancer. Thyroid. (2020) 30:192–203. doi: 10.1089/thy.2019.0011
Keywords: FOXE1, expression, DNA methylation, differentiated thyroid cancer, aggressiveness
Citation: De Lima EU, Dos Santos FF, Da Silva IC, De Lima CRA, Frutuoso VS, Caso GF, De Oliveira PR, Bezerra AK, Cerutti JM, Tamura RE, Ramos HE and Rubio IGSd (2024) Reduced expression of FOXE1 in differentiated thyroid cancer, the contribution of CPG methylation, and their clinical relevance. Front. Endocrinol. 15:1454349. doi: 10.3389/fendo.2024.1454349
Received: 25 June 2024; Accepted: 15 October 2024;
Published: 11 November 2024.
Edited by:
Maria Del Mar Montesinos, CONICET Centre for Research in Clinical Biochemistry and Immunology (CIBICI), ArgentinaCopyright © 2024 De Lima, Dos Santos, Da Silva, De Lima, Frutuoso, Caso, De Oliveira, Bezerra, Cerutti, Tamura, Ramos and Rubio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ileana Gabriela Sanchez de Rubio, aWxlcnViaW9AZ21haWwuY29t; aWxlYW5hLnJ1YmlvQHVuaWZlc3AuYnI=
†ORCID: Ileana Gabriela Sanchez de Rubio, orcid.org/0000-0003-4779-4533