 Ana-Maria Sigarteu Chindris1*
Ana-Maria Sigarteu Chindris1* Michael Rivera2
Michael Rivera2 Yaohua Ma3Asha Nair3Yi Liu3
Yaohua Ma3Asha Nair3Yi Liu3 Xue Wang3
Xue Wang3 Brian M. Necela4Jennifer M. Kachergus4John D. Casler5
Brian M. Necela4Jennifer M. Kachergus4John D. Casler5 Christopher Brett6Ana M. Rivas Mejia1Victor J. Bernet1
Christopher Brett6Ana M. Rivas Mejia1Victor J. Bernet1 John A. Copland III4
John A. Copland III4 Keith L. Knutson4,7
Keith L. Knutson4,7 E. Aubrey Thompson4Robert C. Smallridge1
E. Aubrey Thompson4Robert C. Smallridge1- 1Division of Endocrinology, Mayo Clinic, Jacksonville, FL, United States
- 2Division of Anatomic Pathology, Mayo Clinic, Rochester, MN, United States
- 3Department of Quantitative Health Sciences, Mayo Clinic, Jacksonville, FL, United States
- 4Department of Cancer Biology, Mayo Clinic, Jacksonville, FL, United States
- 5Department of Otorhinolaryngology/Audiology, Mayo Clinic, Jacksonville, FL, United States
- 6Department of Internal Medicine, Mayo Clinic, Jacksonville, FL, United States
- 7Department of Immunology, Mayo Clinic, Jacksonville, FL, United States
Introduction: BRAFV600E mutation (BRAFmut) is common in papillary thyroid cancer (PTC), and most patients have an excellent outcome. However, a TERT-promoter mutation (pTERTmut) in the presence of BRAFmut (BRAFmutpTERTmut) has been demonstrated to confer a more aggressive behavior to PTC. Lymphocytic infiltration is often present in PTC. In this study, we sought to decipher the relationship between the BRAF and pTERT mutations and immune gene dysregulation in tumor samples from a cohort of 147 samples of PTC.
Methods: The abundance of 770 immune gene transcripts was determined by multiprex capture/detection and digital counting of mRNA transcripts using the NanoString nCounter® PanCancer Immune Profiling Panel.
Results: We identified 40 immune transcripts differentially expressed in BRAFmutpTERTmut vs BRAFmutpTERT wildtype (pTERTwt) (P<0.05). Transcripts induced by BRAFmut alone were significantly repressed in BRAFmutpTERTmut samples, such as genes expressed by lymphoid cells, antigen-presenting cells, and cytotoxic cells, including chemokines, cytokines, checkpoint control proteins, interferon downstream markers, TNF superfamily proteins and BMP markers. A validation analysis using 444 samples from The Cancer Genome Atlas (TCGA) PTC dataset yielded similar results. Deconvolution analysis confirmed differences in the immune cell populations such as increased presence of M2 macrophages in the BRAFmutpTERTmut Mayo cohort and a lower abundance of M1 macrophages in the BRAFmutpTERTmut TCGA cohort compared to BRAFmutpTERTwt. Most of the immune gene pathways were enriched in the BRAFmutpTERTwt tumors in both Mayo and TCGA cohorts but not in BRAFmutpTERTmut. BRAFmutpTERTwt had higher stromal lymphocytes infiltration as compared to BRAFwtpTERTwt tumors, corroborating the transcriptomic findings.
Discussion: To our knowledge this is the first report of a potential link between TERT and the immune microenvironment, offering an explanation for the aggressive nature of BRAFmutpTERTmut PTC.
1 Introduction
Thyroid cancer has increased in incidence and mortality in the United States over the last few decades, with papillary thyroid cancer (PTC) as the most common histological subtype (1). While most PTCs are diagnosed early and highly curable, about 10% have an aggressive course, leading to multiple recurrences, distant metastatic disease, and eventually death. The molecular features responsible for tumor aggressiveness in PTC have not yet been fully deciphered.
The most common genetic alteration in PTC is BRAF V600E mutation (BRAFmut), which plays an important role in tumorigenesis (2). While most patients with PTC, including those with BRAFmut, have excellent outcomes, BRAFmut in the presence of a C228T or C250T TERT-promoter mutation (pTERTmut) appears to confer a more aggressive phenotype to PTC than BRAFmut alone (3). Interestingly, 12% to 18% of patients with PTC have BRAFmutpTERTmut, similar to the percentage of patients with PTC who experience poor outcomes (3, 4). To date, there is insufficient understanding of how the presence of BRAFmutpTERTmut results in an aggressive PTC phenotype.
BRAF is a key regulator of growth factor/MAPK signaling, and one might expect that the constitutively active BRAF mutation might be a powerful oncogenic driver, such as RAS, RAF, ERBB2, and EGFR. However, the link between BRAFmut and clinical aggressiveness of PTC has proved to be difficult to establish. In general, BRAFV600E does not appear to be an independent predictor of PTC-associated mortality (5). Despite that, it has been reported that BRAFmut is associated with a more aggressive phenotype in patients older than 55 years of age (6, 7) and may be an independent risk factor for recurrence in this population (8).
The relationship between age, BRAF mutation status, and tumor aggressiveness is further complicated by the observation that pTERTmut is associated with older age at diagnosis (9–11). Since earlier studies did not consider pTERTmut a variable, it is possible that the relationship between BRAFmut and age at diagnosis may reflect an age-associated increase in double mutant tumors, which express both BRAFmut and pTERTmut. Such tumors have been shown to be larger, have a higher degree of extrathyroidal extension and are more aggressive than PTC tumors with wild type BRAF (BRAFwt) or BRAFmut alone (3, 10).
The relationship between chronic lymphocytic thyroiditis and differentiated thyroid cancer has been the subject of ongoing debate. While up to 35% of PTC arise on a background of lymphocytic thyroiditis (BLT) (12), the effect of the immune microenvironment on the clinicopathological features of PTC is less clear, owing to the complexity of interactions between the tumor and different lymphocyte subsets (12–21). Nevertheless, the enormous impact of tumor immune microenvironment on cancer cell biology is now well established and targeted for therapeutic purposes in several malignancies, and to some extent, in advanced thyroid malignancies (22).
BRAFmut is common in PTC with a reported prevalence of 48% in a cohort of 2,099 patients with PTC (7) and about 60% in The Cancer Genome Atlas (TCGA) PTC cohort (9). Analyses of molecular features from PTC tumors in TCGA have revealed that BRAFmut tumors tend to express a lower abundance of markers of thyrocyte differentiation compared to BRAFwt PTC (12). We previously published a transcriptomic analysis of a small cohort of patients with BRAF tumors (12 BRAFmut, 8 BRAFwt) and from these data posited that BRAFmut tumors are immune-suppressive (23). Conversely, a larger TCGA study has subsequently revealed that BRAFmut tumors express higher levels of several immune cell marker transcripts, consistent with an immune-activated state (24). However, a proteomic analysis of 30 PTC tumors (15 each BRAFmut and BRAFwt) led to the conclusion that BRAFmut tumors express lower levels of major histocompatibility complex class II (MHCII) components, indicative of immune suppression (25). In that study, it was proposed that down-regulation of MHCII engendered escape from immune surveillance, which could be relieved by BRAF inhibitors in preclinical models. Thus, it appears that although there is a link between BRAFmut and the immune system, it is unclear whether BRAFmut tumors are more or less immunogenic than BRAFwt tumors.
The analyses described herein were carried out to evaluate immune gene expression in an independent cohort of patients with PTC tumors from the Mayo Clinic Thyroid Cancer Registry. An immune function gene expression platform (NanoString Technologies, Inc) was used to analyze 770 immune transcripts as a function of BRAFmutpTERTmut status in 147 formalin-fixed, paraffin-embedded (FFPE) PTC tumor samples. The results were validated using the PTC cohort in TCGA. The results of our study reveal a novel, heretofore unappreciated, link between TERT and the immune system, a link which may account for the more aggressive phenotype of BRAFmutpTERTmut PTC.
2 Materials and methods
2.1 Study approval
Our study was conducted in accordance with recognized ethical guidelines, including the US Common Rule, and Mayo Clinic Institutional Review Board approval was obtained in accordance with a filed assurance approved by the Department of Health and Human Services.
2.2 Patients and sample preparation
We reviewed the Mayo Clinic thyroid cancer database and identified patients with PTC who were treated for this diagnosis at Mayo Clinic in Jacksonville, Florida (MCF) between January 1st 2001 and December 31st 2015. Medical records of these patients were reviewed for the following clinical characteristics: demographic characteristics, pathology, presence of local or distant persistent/recurrent disease, and clinical outcome. We retrieved 147 FFPE samples from the Mayo Clinic biorepository. The original histology was reviewed and the degree of lymphocyte infiltration into the tumor stroma (TIL) was determined by a single endocrine pathologist (MR). TIL was graded as 0 (no inflammation) 1 (10% or less), 2 (11-30%) or 3 (>30%) of tumor involved by chronic inflammation. BLT in uninvolved thyroid tissue was also scored and reported as absent (0), mild (1) (scattered/lymphoid aggregates), moderate (2) (easily identifiable lymphoid aggregates), or marked (3) (large confluent lymphoid aggregates involving most of the tissue).
2.3 Mutation status determination and immune-related gene expression profiling
Ten-μm-thick sections from formalin-fixed paraffin-embedded archival tissue blocks were cut and fixed to glass microscopy slides. For each block, an additional consecutive 5-μm-thick section was stained with hematoxylin and eosin (H&E) and used as a template for identification of tumor and stroma on the corresponding unstained slides. In the Mayo cohort, the BRAF status was determined by immunohistochemistry (IHC) as previously described (26). The IHC findings were then confirmed by digital droplet polymerase chain reaction (dd PCR) (QX200 Droplet Digital™ PCR BioRad). The presence of 10% or more of mutant alleles in a tumor was used as the cutoff for classifying it as BRAFmut; those with <10% mutant alleles were deemed BRAFwt. TERT promoter status was determined by ddPCR.
Based on the corresponding H&E slide, 10 µm FFPE sections from each tumor were macrodissected and used for simultaneous purification of DNA and RNA using QIAGEN All Prep kit (catalog no. 80 204). For all samples, total cellular RNA (at a minimum concentration of 10 ng/µl) was assessed for yield (NanoDrop 2000 Thermo Scientific). Immune-related gene expression profiling was performed utilizing the nCounter® PanCancer Immune Profiling Panel comprised of 770 immune genes and associated controls (Supplementary Table S1). 200ng of each total RNA sample was prepared as per the manufacturer’s instructions. The nSolver analysis software version 4.0 was used for custom QC and normalization of the nCounter data.
For the TCGA cohort, patient characteristics and pre-processed gene expression data, which included the BRAF and TERT mutational status were downloaded from https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp.
2.4 Differential gene expression analysis
Generalized linear model (GLM) was performed to access differential expression of individual genes in binary comparisons based on NanoString PTC data. The comparisons included 1) BRAFmutpTERTwt vs BRAFwtpTERTwt; 2) BRAFmutpTERTmut vs BRAFwtpTERTwt; 3) BRAFmutpTERTmut vs BRAFmutpTERTwt; and 4) BRAFwtpTERTmut vs BRAFwtpTERTwt. (Supplementary Figure S1). Log2 fold change (FC) (ratio of geometric mean) with 95% confidence interval (CI) was estimated by GLM. Benjamini & Yekutieli (BY) adjusted P-values were calculated. For effective validation, we selected all the genes from TCGA data (if their names showed in NanoString PTC data) and ran the same differential expression analysis (Supplementary Figure S2). P values < 0.05 were considered statistically significant. All analyses were performed using R software, version 3.6.2. TIL status was compared using 2-tailed Fisher’s exact test. Chi-square test of independence was used for comparison of categorical data and Kruskal-Wallis test to compare continuous variables across groups.
2.5 Pathway analysis
In order to gain further insight to what drives the suppression of the immune genes in the BRAFmutpTERTmut samples compared to BRAFmutpTERTwt, we conducted pathway analyses of the two cohorts. The Gene Set Enrichment Analysis (GSEA) software is publicly available from the Broad Institute of MIT and Harvard University (http://software.broadinstitute.org/gsea/index.jsp). Gene counts from Mayo cohort and TCGA cohort were utilized for Hallmark, chemical and genetic perturbations (CGP), and Gene Ontology Biological Process (GOBP) analysis (version v2024.1). GSEA was performed using the gene permutation option and gene sets smaller than 15 or larger than 500 were excluded. The false discovery rate (FDR) with less than 0.25 (FDR < 0.25) was considered as significantly enriched. Additionally, GSEA was conducted using gene set signatures from MsigDB database, with enrichment scores (NES) calculated to elucidate biological pathways associated with BRAFmutpTERTwt and BRAFmutpTERTmut tumors.
2.6 Immune cell deconvolution analysis
The relative abundance of immune cell levels were estimated from the gene expression profiles using CIBERSORTx (CIBERSORTx (stanford.edu) We used the leukocyte signature matrix 22 (LM22) from CIBERSORTx to deconvolute and quantify the abundance of 22 immune cell types from the NanoString gene expression data. Quantile normalization was applied, and the data was run in absolute mode using 100 permutations. The analysis was repeated utilizing the TCGA PTC data. The resulting absolute abundance values for each immune cell type was compared between BRAFmutpTERTwt and BRAFmutpTERTmut using linear regression analysis. Immune cells with p-value <0.05 were reported statistically significant.
3 Results
3.1 Patient characteristics
A total of 147 patients treated for PTC at MCF were identified using our thyroid cancer registry. There were 107 females and 40 males with a median (range) age at diagnosis of 50.3 years (18.1-88.3) (Supplementary Table S2). Two patients with recurrent disease had their initial surgery elsewhere and were operated at MCF for locally recurrent disease. One patient had a thyroid lobectomy elsewhere demonstrating a 9.5 cm PTC and completion thyroidectomy at MCF 15 years later, with a 1.7 cm PTC found in the remaining lobe. The other 144 patients had their initial surgery at MCF. One hundred and eighteen (78%) pathology specimens were PTC classic variant, 23 (16%) encapsulated variant, 4 (3%) oncocytic variant, and 2 (1%) tall cell variant. The cohort of 147 samples included two specimens of cribriform morular thyroid carcinoma which was considered a histological subtype of PTC according to the 2017 WHO classification of tumors, and a distinct histological category per the 2022 WHO classification (Supplementary Table S2). Twenty patients did not have their subsequent follow-up at MCF after the initial diagnosis and treatment. The remaining 127 patients were followed over a median (range) of 163.5 months (2-325 months). For the 145 primary tumor samples, median (range) tumor size was 1.5 cm (0.6-6.5). Primary tumor size was unknown for one patient who was operated at MCF for recurrent disease. One hundred and twenty two patients were TNM stage I, 17 were stage II, five were stage III, and three were stage IVB. Fourteen patients died during follow-up, all from causes other than thyroid cancer. At the last follow-up, 109 patients did not have any evidence of disease, six had persistent, anatomically detectable local disease, and one had detectable distant disease.
3.2 BRAF and TERT mutational status and lymphocytic infiltrate profiles
Supplementary Table S3 displays the BRAF and TERT mutational status of the samples comprised in the Mayo cohort. BRAFmut was identified in 93 samples (63.3%), and 54 (36.7%) were BRAFwt. Ten of the BRAFmut samples also harbored pTERTmut (6 C228T and 4 C250T). The remaining 83 samples were BRAFmutpTERTwt. In 49 samples, neither BRAFmut nor pTERTmut were identified. pTERTmut was detected in five BRAFwt samples (2 C228T and 3 C250T).
TIL and BLT status are presented in Supplementary Tables S2–S4. BRAFmutpTERTwt tumors had TIL scores significantly elevated: (>0, Fisher exact two-tailed P=.001) and (>1, P=.042) compared to BRAFwtpTERTwt. In contrast none of the pTERTmut tumors had a TIL score of ≥ 2, irrespective of the BRAF status. There was no statistically significant difference in the BLT profile across groups (p=0.63) (Supplementary Table S2).
3.3 Differential gene expression
We initially evaluated differential expression of immune function genes between BRAFmutpTERTwt and BRAFwtpTERTwt tumors. Since this was our largest set of samples, we used BY.adj.P to identify immune function transcripts that were differentially expressed in BRAFmutpTERTwt with a high degree of confidence. We identified 345 transcripts that were differentially expressed at P<0.05. After adjustment, 148 transcripts remained significantly differentially expressed Benjamini-Yekutieli adjusted P values (BY.adj.P ≤ 0.05) (Supplementary Table S5). We found that most of these transcripts were more abundant in BRAFmutpTERTwt tumors (127 induced vs 21 repressed) than in BRAFwtpTERTwt tumor Supplementary Figure S3 lists the 127 genes induced in the BRAFmut tumors, which include the T-cell marker, CD4; myeloid marker, ITGAX/CD11c; macrophage marker, CHIT1; check point transcripts CD274/B7-H3 which were robustly overexpressed (BY.adj.P<0.05). In general, data indicates that BRAFmutpTERTwt tumors are more immunogenic than BRAFwtpTERTwt tumors as also demonstrated in Supplementary Figure S1A.
3.3.1 BRAFmut-induced immune gene expression is generally reversed by pTERTmut
While BRAFmut did not significantly affect the abundance of BRAF mRNA, pTERTmut, TERT mRNA abundance was significantly higher in BRAFmutpTERTmut cells than in BRAFmutpTERTwt (Figure 1A). Among the 127 immune transcripts induced by BRAFmut in pTERTwt tumors (log2 FC BRAFmutpTERTwt vs BRAFwtpTERTwt > 0, BY.adj.P<0.05), 87 transcripts were expressed at approximately the same level with pTERTmut (BRAFmutpTERTmut vs BRAFmutpTERTwt, P>0.05), while the other 40 transcripts were differentially expressed in BRAFmutpTERTmut vs BRAFmutpTERTwt, either higher or lower (critical P<0.05) (Supplementary Table S6).
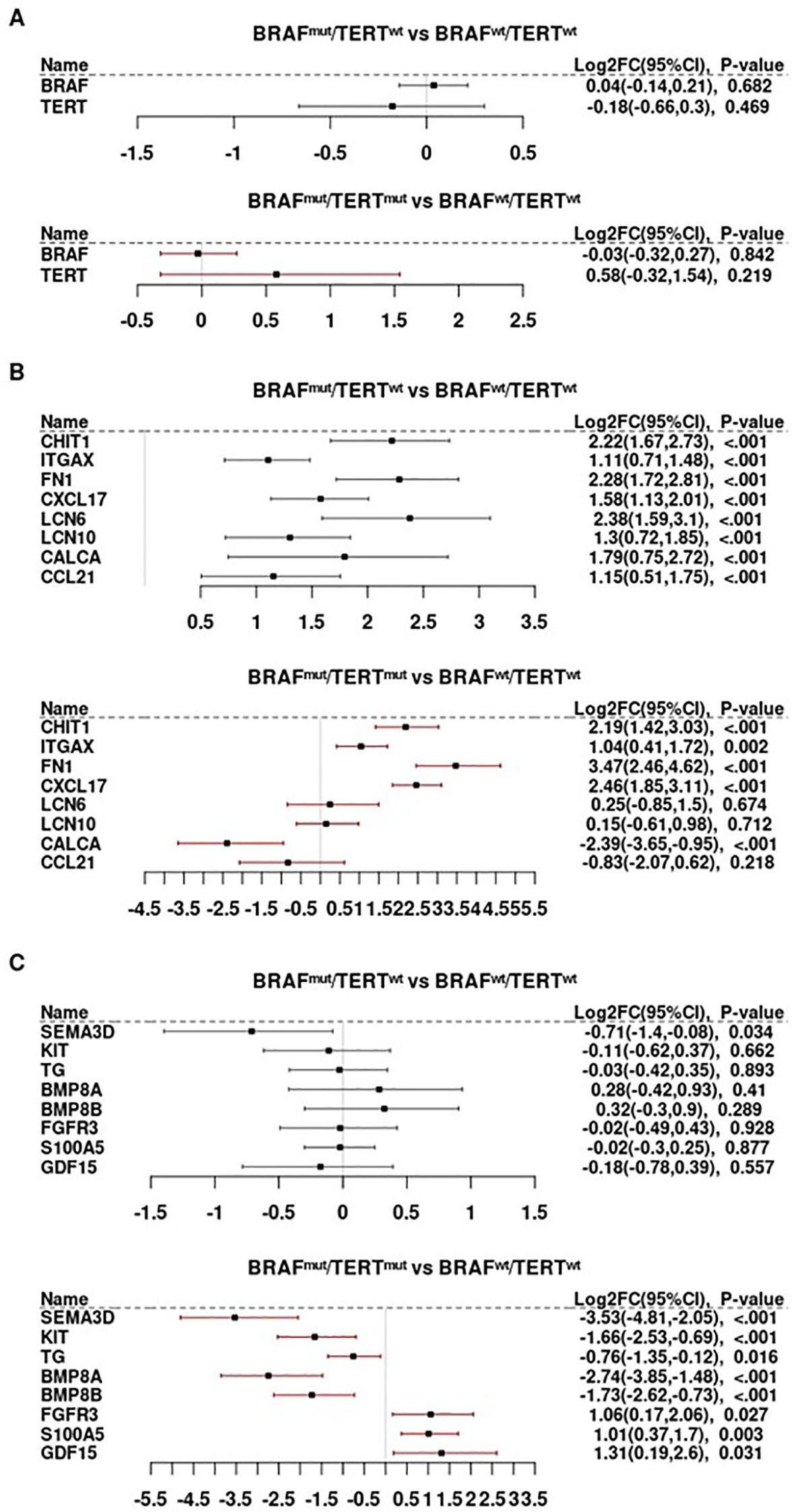
Figure 1. Differential gene expression as a function of BRAF and TERT mutational status. (A) BRAF and TERT mRNA abundance in relationship to the presence of BRAFV600E and TERT promoter mutations. (B) The TERT promoter mutations are associated with repression of CALCA, CCL21, strong induction of FN1, CXCL17, LCN6 and LCN10, and seems to have no effect on CHIT1 and ITGAX expression in BRAFV600E mutated tumors. (C) Immune genes whose expression is repressed in the presence of TERT promoter mutation but appears unaltered by the presence of BRAFV600E.
Most of the transcripts that were strongly induced by BRAFmut in the absence of pTERTmut were repressed in double mutant tumors (BRAFmutpTERTwt > BRAFmutpTERTmut), as summarized in Supplementary Figure S3. However, the effect of BRAFmut on some genes was unaltered by the addition of pTERTmut, as illustrated by the macrophage marker, CHIT1, and myeloid marker, IGTAX/CD11c (Figure 1B). The well-known BRAFmut target, FN1/fibronectin, and monocyte/dendritic cell chemokine, CXCL17, were more potently induced in the double mutant tumors; however, some BRAFmut-induced genes (LCN6, LCN10) were not induced in BRAFmutpTERTmut tumors and others (eg, CALCA/calcitonin and T-cell chemokine, CCL21) were suppressed in presence of pTERTmut to levels below those observed in BRAFwtpTERTwt tumors (Figure 1B). Moreover, several genes not significantly regulated by BRAFmut were either repressed or induced in double mutant tumors as a function of TERT (Figure 1C).
Lymphoid cell markers were generally induced in BRAFmutpTERTwt but repressed in BRAFmutpTERTmut tumors (Figure 2A). Notably the pan-leukocyte marker, PTPRC/CD45, and CD45 effector kinase, LCK, were less abundant in BRAFmutpTERTmut tumors. The T-cell markers, CD247/CD3Z, CD4, and CD8B; B-cell marker, CD19; and putative natural killer (NK) cell marker, NCAM1/CD56, were repressed by pTERTmut. The Treg marker, FOXP3, was largely unaltered by pTERTmut. Consistent with the decreased CD8B and NCAM1/CD56 markers, we also noted decreased cytotoxicity markers in BRAFmutpTERTmut tumors, including GZMB and NK cell markers, KLRB1, KLRC1 and KLRD1 (Figure 2B).
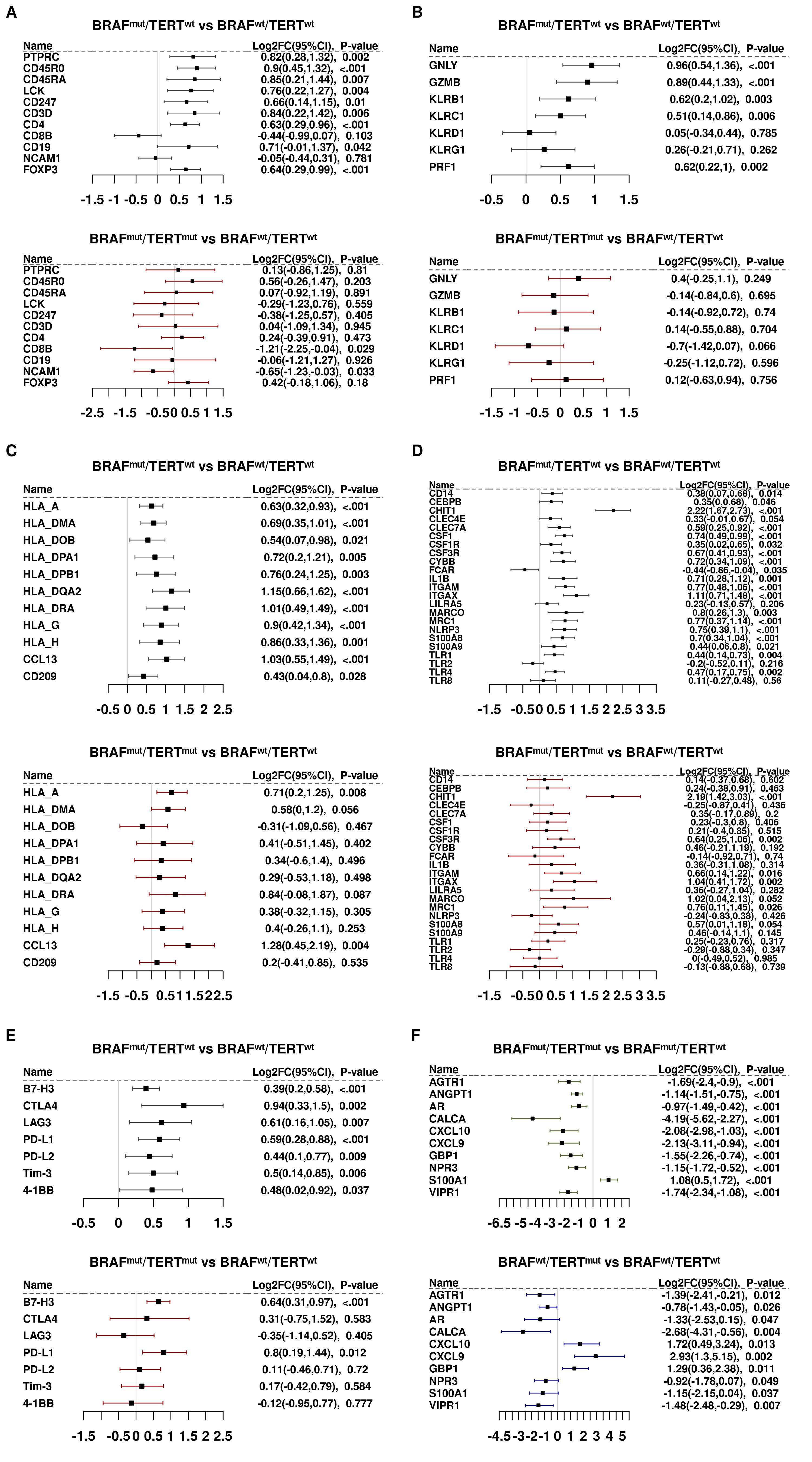
Figure 2. Panels (A-E). Differential gene expression of immune gene subsets. (A) Lymphoid markers. (B) Cytotoxic cell markers. (C) Antigen presentation cell markers. (D) Myeloid markers. (E) Checkpoint Control I/IO drug targets. Panel (F). Differentially expressed transcripts in 5 BRAFwt samples harboring the TERT promoter mutation vs BRAFwtpTERTwt.
Genes associated with antigen presenting cells and myeloid cells did not appear to be significantly repressed by pTERT with the notable exceptions of the macrophage-activating marker, TLR4; monocyte activator, CSF1; and inflammasome subunit, NLRP3 (Figures 2C, D).
Figure 2E summarizes the effects of pTERTmut on immune checkpoint genes and immuno-oncology targets. Expression of transcripts that encode two of the more actively studied checkpoint proteins, CD274/PD-L1 and CD276/B7-H3, were induced in BRAFmut tumors and were not significantly affected by the addition of pTERTmut. Similarly, CTLA4 was induced by BRAF and was not significantly repressed by pTERTmut. Conversely, LAG3, which encodes a cell surface molecule expressed on activated T cells, NK cells, B cells and plasmacytoid cells and has been recently reported to drive T cell exhaustion, was markedly repressed in double mutant tumors (27, 28).
Given the observation that pTERTmut significantly alters the BRAFmut-induced immune landscape, the question arises: is this the additive effect of two oncogenic mutations, or does the interaction of the two mutant genes lead to a molecular phenotype that is different from that imposed by either mutation alone? To address this question, we interrogated the immune landscape of a very small cohort of BRAFwtpTERTmut PTC tumors (n=5). To this end, we compared transcripts that were differentially expressed (BY.adj.P<0.05) in BRAFmutpTERTmut vs BRAFmutpTERTwt with transcripts that were differentially expressed (critical P<0.05) in BRAFwtpTERTmut vs BRAFwtpTERTwt. Only 10 genes were identified (Figure 2F), but the results suggest that pTERTmut may directly repress some genes that were differentially expressed in BRAFmutpTERTmut vs BRAFmutpTERTwt (eg, CALCA/calcitonin, which is strongly induced in BRAFmutpTERTwt and strongly repressed in BRAFmutpTERTmut). On the other hand, some genes appeared to be expressed in the double mutant tumors in a manner opposed to the effects of pTERTmut alone (eg, interferon target genes, CXCL9, CXCL10, and GBP1). Given the very small sample cohort available for analysis, any conclusions must be advanced with caution. Nevertheless, there is some suggestion that BRAFmut and pTERTmut may interact in a fashion that results in an immune landscape that is different from that imposed by either mutation acting alone.
3.3.2 Signaling activity
Several interferon target genes were differentially expressed, generally induced in BRAFmutpTERTwt and repressed in BRAFmutpTERTmut samples (Figure 3A). Notable among these transcripts are CXCL9,10, and 11, which are ligands for the tumor-suppressive CXCR3 receptor. CXCR3 is also induced by BRAFmut (Supplementary Table S5) but is not induced in double mutant tumors. A similar pattern prevailed with TNF superfamily transcripts, as well as AP1 subunits, FOS and JUN (Figure 3B). BMP markers were strongly repressed by pTERTmut; whereas TGFB1, TGFB2, and TGFBR1 were induced by BRAFmut and unaffected by pTERTmut. However, the catalytic subunit of the TGFB receptor, TGFBR2, was significantly repressed by pTERTmut (Figure 3C).
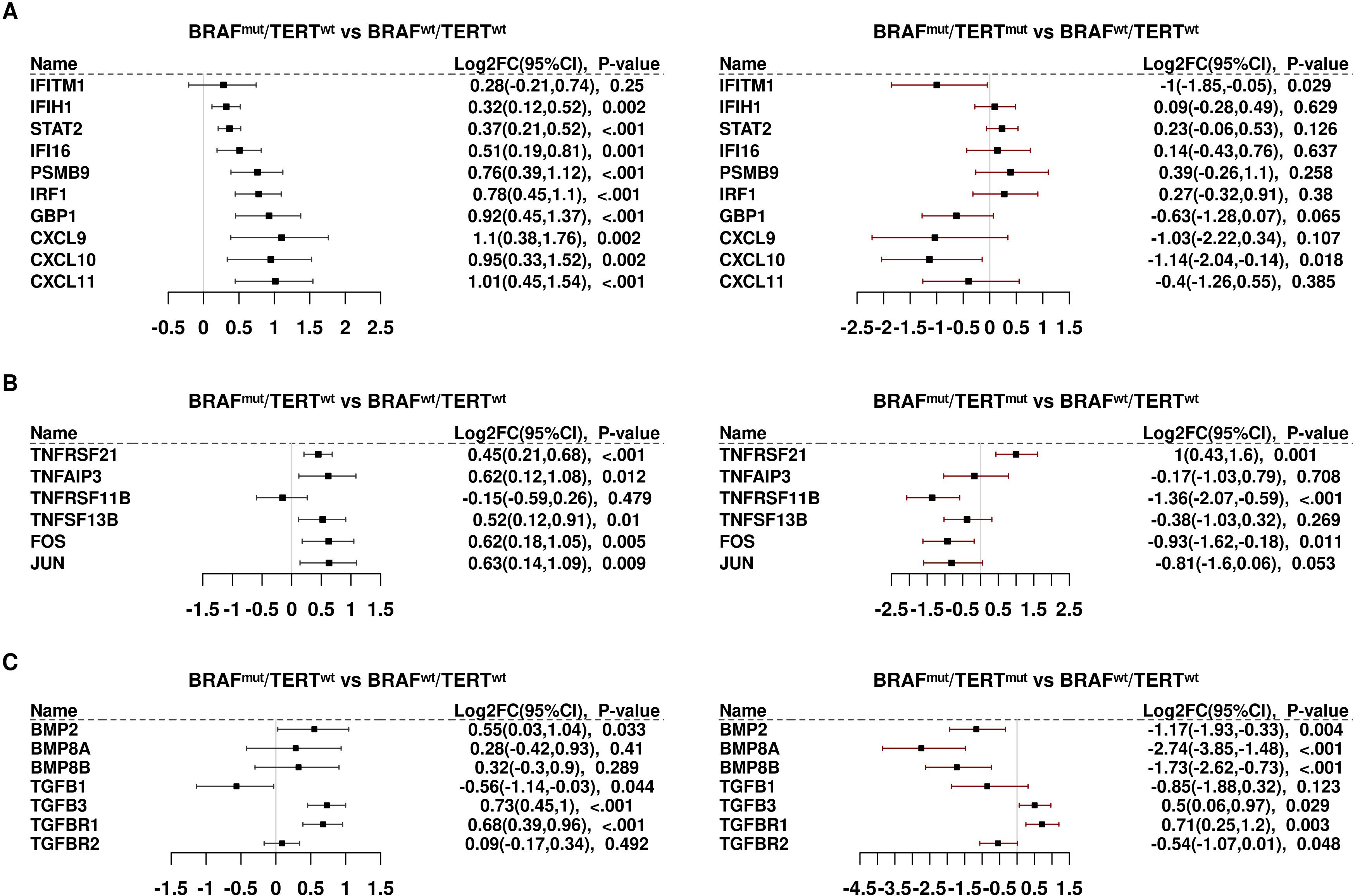
Figure 3. Differential expression of genes involved in signaling activity: (A) Interferon (IFN) downstream markers. (B) Tumor necrosis factor/TNF/AP1 signaling. (C) Bone morphogenetic proteins/transforming growth factor beta (BMP/TGFB).
3.3.3 Analysis of TCGA RNAseq data
Of the 496 patients with PTC in TCGA, the 444 with known BRAF and TERT status were included as our validation cohort (Supplementary Tables S7, S8). While BRAFmut is relatively common in PTC, present is approximately half of all PTC tumors, pTERTmut is rare. Thus, the number of BRAFmutpTERTmut tumors available for analysis in our Mayo Clinic cohort was small, only 10 samples. We sought to validate our major findings using RNAseq data from our TCGA PTC cohort (Supplementary Table S9). Distribution of the various genotypes in TCGA samples is shown in Supplementary Table S8, noting that genotypes in this cohort were defined by sequence analysis of DNA extracted from fresh-frozen tumors; whereas, genotypes in the Mayo Clinic cohort were determined by dd PCR and IHC for BRAFmut and dd PCR for pTERTmut using DNA from FFPE samples. Transcript abundance in TCGA was determined by RNAseq analysis of fresh frozen RNA; while transcript abundance in Mayo Clinic samples was determined by NanoString analysis of RNA from FFPE samples. With these potentially confounding variables in mind, we sought to determine if the general pattern of immunosuppression observed in the double mutant samples from Mayo Clinic was recapitulated in the TCGA cohort. Supplementary Figure S2 displays comparisons between the differential gene expression in BRAFmutpTERTwt vs BRAFwtpTERTwt (A), BRAFmutpTERTmut vs BRAFwtpTERTwt (B), BRAFmutpTERTmut vs BRAFmutpTERTwt (C) and BRAFwtpTERTmut vs BRAFwtpTERTwt (D) cohorts.
Figure 4A shows the 40 transcripts that were strongly suppressed by pTERTmut on the BRAFmut background (log2 FC TCGA BRAFmutpTERTmut vs BRAFmutpTERTwt< −0.5, P<0.05) in the Mayo Clinic samples being also suppressed in the TCGA cohort. As shown in Figure 4A, essentially all these transcripts were repressed in both TCGA and Mayo Clinic samples. Likewise, the T-cell receptor subunits, CD247/CD3Z, CD3D, and CD3E, were repressed by pTERTmut in both datasets (Figure 4B). CD4 was slightly less abundant, albeit not significantly in Mayo Clinic patients, and CD4 was essentially unchanged in TCGA samples. Strikingly, markers of cytotoxic CD8+ T-cells (CD8A and CD8B) were strongly downregulated by pTERTmut in both Mayo Clinic and TCGA samples (BRAFmutpTERTmut vs BRAFmutpTERTwt) (Figure 4B). The NK cell markers, KLRB1 and KLRC1, as well as cytotoxic cell markers, GZMA, GZMB, and PRF1, exhibited similar patterns of regulation in the two sample cohorts (Figure 4C). A puzzling exception was NCAM1/CD56, which was downregulated in the Mayo Clinic pTERTmut samples but not significantly altered in TCGA samples. It should be noted that, in addition to being an NK cell marker, CD56 is a marker of neuroendocrine differentiation, and therefore, not interpreted strictly as an immune cell marker in PTC tumors (29).
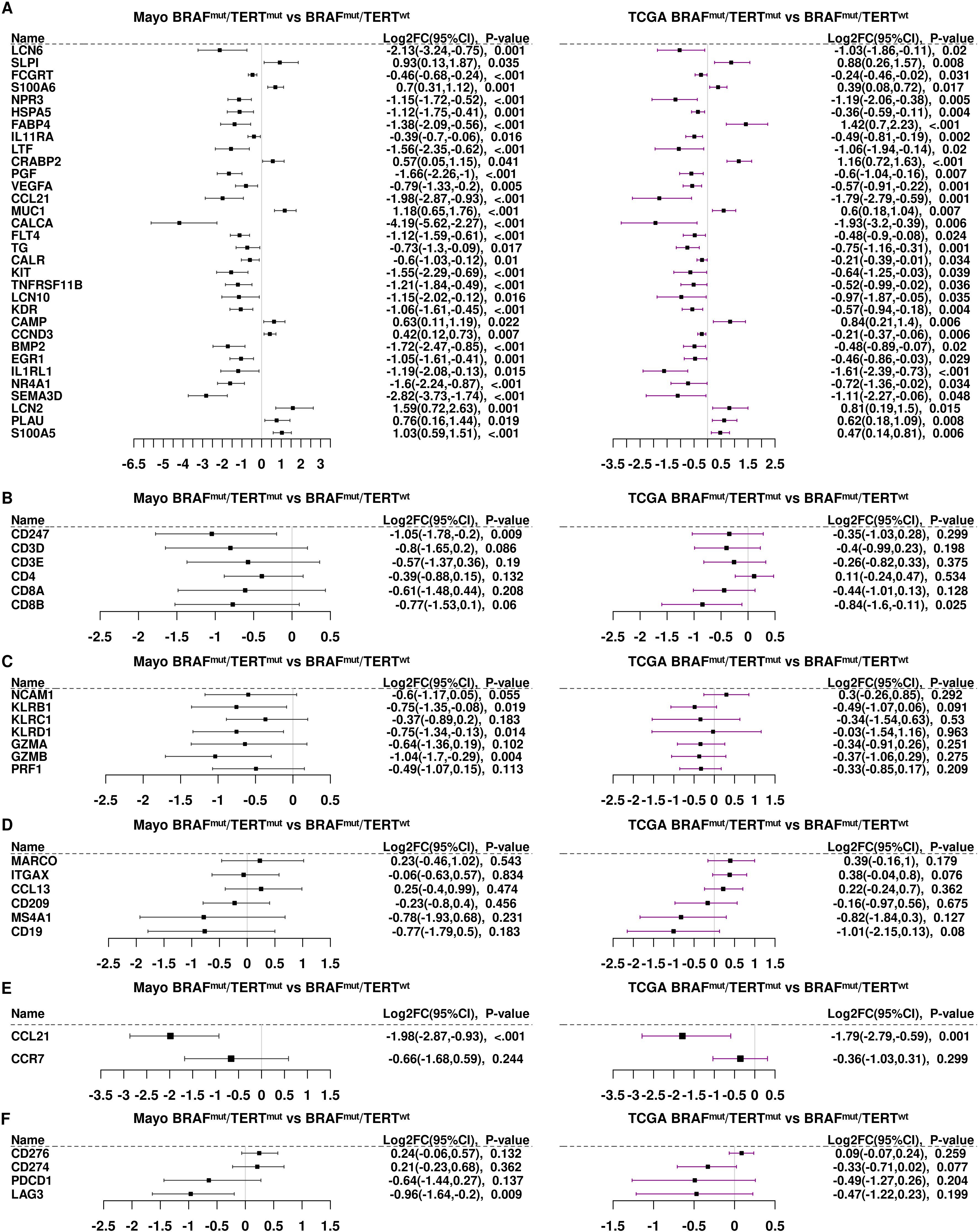
Figure 4. Differential gene expression of immune genes in the TCGA samples compared to the Mayo Clinic cohort. (A) We identified 40 immune genes significantly differentially expressed (p<0.05) in both the Mayo Clinic cohort (black bars) and in the TCGA samples (purple bars). Panels (B-F). Differential gene expression of immune gene subsets: (B) T cell markers; (C) NK/cytotoxic cell markers; (D) antigen presenting cell markers; (E) CCL21 and CCR7 and (F) checkpoint genes.
Similarities in the patterns of expression were observed regarding macrophage and myeloid cell markers, MARCO and ITGAX/CD11c, genes expressed by antigen presenting cells such as CCL13 and CD209. B-cell markers, MS4A1/CD20 and CD19, were strongly repressed in BRAFmutpTERTmut tumors compared to BRAFmutpTERTwt in both sample cohorts (Figure 4D).
CCL21, the second most highly repressed gene in TCGA BRAFmutpTERTmut samples, was also strongly repressed in the Mayo Clinic cohort (Figure 4E). CCR7, the receptor for CCL21, was somewhat less abundant in BRAFmutpTERTmut vs BRAFmutpTERTwt in both sample cohorts. The CCL21/CCR7 axis is known to function in recruitment of cytotoxic cells (30) and downregulation of this pathway in double mutant cells may account, at least in part, for the observed decrease in cytotoxic markers in double mutant tumors. As shown in Figure 4E, the checkpoint factors, CD276/B7-H3 and CD274/PD-L1, were also equally abundant in both Mayo Clinic and TCGA samples. PCDC1/PD-1 and LAG3 were significantly repressed by pTERTmut on the BRAFmut background in both Mayo Clinic and TCGA samples (Figure 4F). Thus, the general patterns of regulation: pTERTmut repression of CD8+ and NK cytotoxic cells with or dendritic cells; and relatively high levels of CD276/B7-H3 and CD274/PD-L1 were exhibited by both tumor sample cohorts.
3.4 Pathway analysis
To evaluate the immune landscape function of the BRAF and TERT mutations, we performed pathway analysis of both Mayo and TCGA sample cohorts. (Figure 5, Supplementary Table S10). The overwhelming majority of immune related pathways were enriched in both BRAFmutpTERTwt cohorts compared to the BRAFmutpTERTmut cohorts. Four pathways were enriched in the BRAFmutpTERTmut TCGA cohort: Positive regulation of IL-4 production, chronic inflammatory response, neutrophil chemotaxis and leukocyte tethering and rolling.
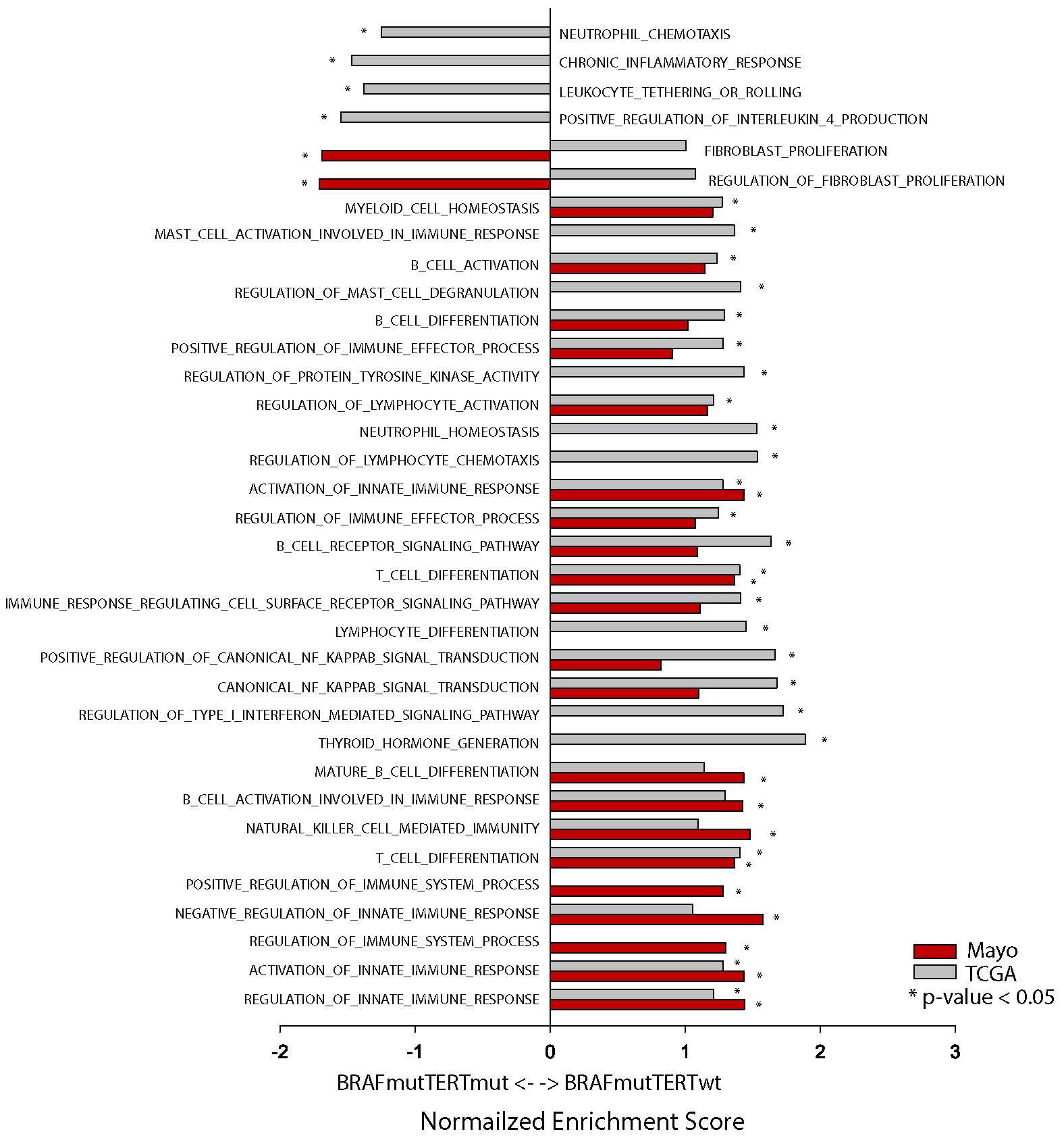
Figure 5. Pathway analysis of Mayo (red bars) and TCGA (gray bars) BRAFmutpTERTwt vs BRAFmutpTERTmut samples. * indicates p-value <0.05.
IL-4 is known to induce M2 macrophages which have immunosuppressive functions and have been linked to tumor progression (31–33). The TERT mutational status did not appear to influence the fibroblast proliferation and regulation of fibroblast proliferation pathways.
Despite the differences regarding sample preparation (fresh frozen vs FFPE), genotyping (DNA sequencing vs IHC and ddPCR), and transcriptome analysis (RNAseq vs NanoString), the data from both cohorts indicates that BRAFmutpTERTmut tumors display a suppressed immune environment compared to BRAFmutpTERTwt.
3.5 Immune cell profiles
The differences between the immune cell composition between BRAFmutpTERTwt and BRAFmutpTERTmut was achieved by using CIBERSORTx, following the method proposed by Newman et al. (34), and summarized in Figure 6. In the Mayo cohort, the most notable observation was an increased proportion of the M2 macrophage in the BRAFmutpTERTmut compared to the BRAFmutpTERTwt (p<0.05). Additionally, the BRAFmutpTERTmut demonstrated a reduced population of NK resting cells and increased neutrophils and activated dendritic cell populations. As this analysis was carried out with a limited set of 143 genes due to incomplete overlap of the two platforms (NanoString and CIBERSORTx) certain immune cell types may be underrepresented, therefore these results must be interpreted with caution. Absolute immune cell fractions in the individual pTERTmut samples are listed in Supplementary Figure S4 We repeated the same analysis utilizing the TCGA BRAFmutpTERTwt and BRAFmutpTERTmut sample set. While we did not observe a significant difference in the M2 macrophage population in the BRAFmutpTERTmut cohort compared to the BRAFmutpTERTwt, the double mutant cohort exhibited a decreased proportion of the proinflammatory M1 macrophages. (Figure 6B) Absolute immune cell fractions in the individual pTERTmut samples are presented in Supplementary Figure S5.
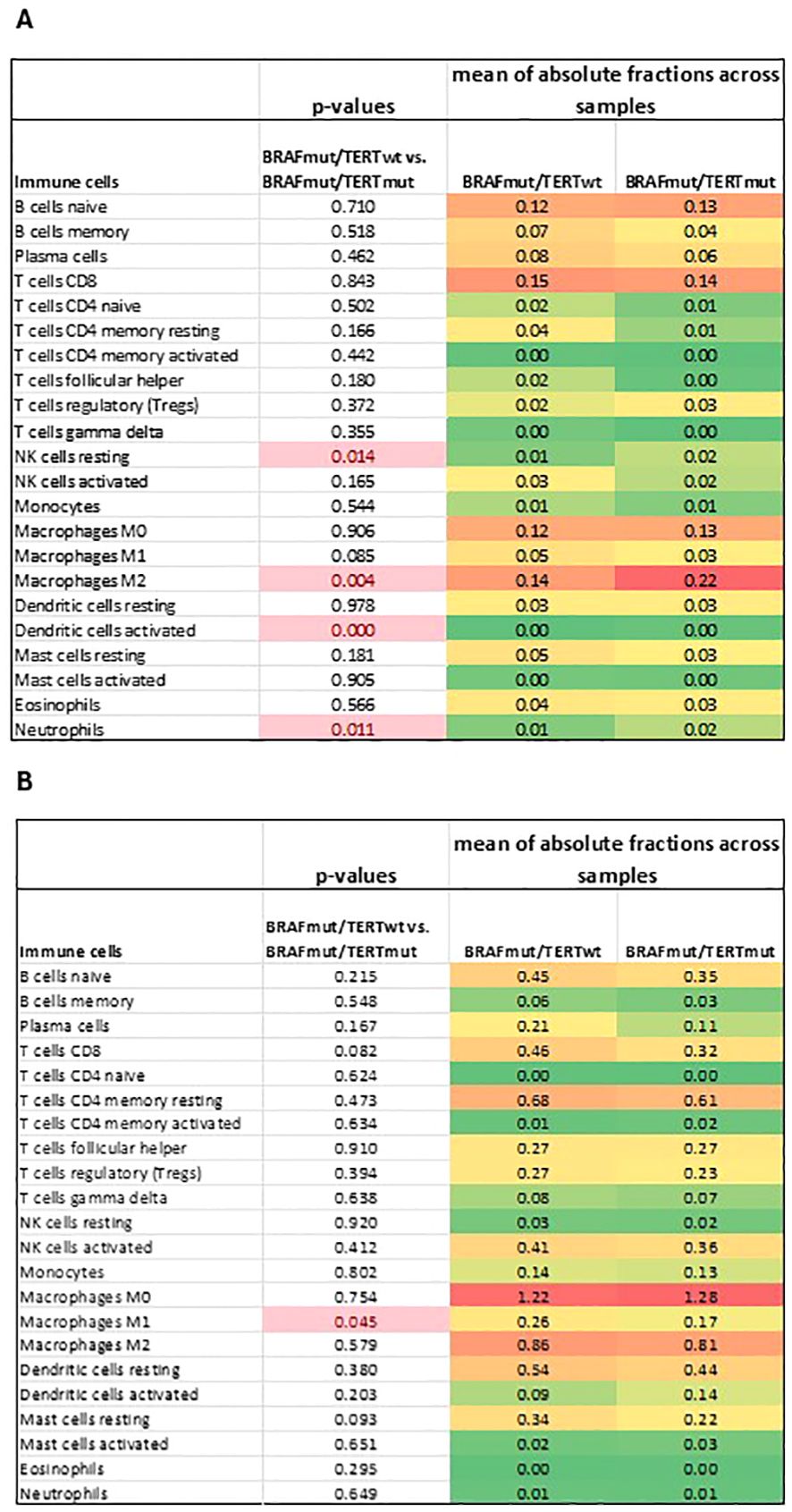
Figure 6. Deconvolution analysis of (A) Mayo and (B) TCGA BRAFmutpTERTwt vs BRAFmutpTERTmut sample cohorts. p-value <0.05 was considered statistically significant. ¥, mean of absolute fraction of activated dendritic cells in BRAFmutpTERTwt, 0.00006; &, mean of absolute fraction of activated dendritic cells in BRAFmutpTERTmut, 0.00137.
4 Discussion
To our knowledge, this is the first report of a potential link between TERT and the immune system in thyroid cancer. The TERT gene encodes the catalytic subunit of telomerase, which, in combination with the RNA subunit encoded by TERC, plays an important role in immortalization of cells. In thyroid cancer, TERT reactivation appears to be a late event in tumorigenesis, primarily the result of activating pTERTmut and clinically associated with more aggressive disease (35). It is unclear whether the effects we observed are secondary to telomere elongation and immortalization, or whether there is some unknown function of TERT related to the immune system. In this context, it should be emphasized that the effects we observe in pTERTmut tumors do not necessarily reflect direct actions of TERT but might be secondary consequences of telomere lengthening on the BRAFmut background.
In an initial analysis of a small cohort of BRAFmut PTC tumors (with no knowledge of pTERT status), we advanced the hypothesis that BRAFmut suppressed the immune landscape of PTC tumors (23). A subsequent report from TCGA analysis, however, was inconsistent with this conclusion, reporting that myeloid cells, B-cells, and regulatory T-cells were significantly higher in the BRAFmut group (12). The analysis reported herein is generally consistent with that TCGA publication. BRAFmutpTERTwt PTC express significantly higher levels of immune markers linked to T-cells, B-cells, macrophage and dendritic cells. CD247 has been previously reported as downregulated in PTC tissue and postulated to attenuate NK cell–mediated toxicity (36). Downstream markers of interferon signaling were also markedly induced (GBP1, CXCL9, CXCL10, CXCL11), as were checkpoint control genes (CD276/B7-H3, CD274/PD-L1, CTLA4, and LAG3). Thus, the molecular data strongly suggest that BRAFmutpTERTwt PTC tumors are associated with an immune-enriched microenvironment compared to BRAFwt tumors. Further, analysis of stromal tumor-infiltrating lymphocytes is consistent with this conclusion.
BRAFmut is common in PTC; most PTC sample cohorts contain about half BRAFmut tumors. Conversely, pTERTmut is rare in PTC; only 10 of 147 Mayo Clinic samples and 26 of 444 TCGA samples were BRAFmutpTERTmut. Thus, one must acknowledge from the outset that statistical power is limited for analysis of the double mutation genotype. This cautionary note applies even more to any attempt to analyze the exceedingly rare BRAFwtpTERTmut PTC tumors. Nevertheless, the molecular landscapes of BRAFmutpTERTwt and BRAFmutpTERTmut tumors were strikingly different, allowing for interpretation of at least the more robust differences between these two genotypes.
pTERTmut is believed to activate TERT transcription, and we observed a significant increase in TERT mRNA abundance in BRAFmutpTERTmut cells. The abundance of BRAF mRNA did not appear to be affected by either BRAF or TERT mutation status. In general, pTERTmut appeared to suppress the immune enriching effects of BRAFmut, although in some cases the gene expression did not seem influenced by TERT while in other cases the stimulatory effects of BRAFmut were enhanced. Some genes were unaffected by BRAFmut alone but were differentially expressed (compared to BRAFwtpTERTwt) in double mutant tumors. The effects of pTERTmut on the BRAFmut background were numerous and complex. To the extent that one may rely upon data from analysis of the extremely rare BRAFwtpTERTmut tumors, it may be that some of the observed effects are simply additive; however, there is at least a suggestion that pTERTmut and BRAFmut may interact together to induce a molecular phenotype that is distinct from that imposed by either mutation alone. We noted that while TERT is strongly induced in BRAFmutpTERTmut tumors, this gene is not differentially expressed in BRAFwtpTERTmut tumors (compared to BRAFwtpTERTwt tumors). Although the very small number of samples engenders caution, our data are consistent with a previous report that BRAFmut induces TERT in pTERTmut but not pTERTwt tumors (37).
Despite the complexity of the observed difference in gene expression in BRAFmutpTERTwt and BRAFmutpTERTmut tumors, several important features clearly emerge. First, and perhaps the most important observation was the effect of pTERTmut on the tumor associated macrophage (TAM) population. TAMs are generally categorized into classical activated macrophages, M1, and alternatively activated macrophages, M2, both able to be converted into each other in response to changes in the tumor microenvironment (38). While M1 macrophages have an anti-tumor effects, either by direct cytotoxic effect or by antibody-mediated cytotoxicity, M2 macrophages are immunosuppressive, promoting tumor progression, angiogenesis and invasion (32, 33, 38).
In our study, BRAFmutpTERTmut tumors in the Mayo cohort displayed an increased proportion of M2 macrophages compared to the BRAFmutpTERTwt group. This finding is supported by the down-regulation of TLR4. TLR4 deficiency has been associated with attenuation of adipose tissue inflammation, and promoting M2 macrophage polarization (39). In addition, the positive regulation of IL-4 pathway was enriched in BRAFmutpTERTmut samples, IL-4 being a known activator of M2 macrophages (31, 40).
As the sample processing and methodology used to determine the genotype and transcript abundance Mayo and TCGA sample cohorts were different, our expectation was not to obtain the exact same results but to observe a similarly suppressed immune environment in the BRAFmut pTERTmut compared to the BRAFmut pTERTwt. As such, while there was no significant difference in the M2 abundance in the TCGA cohort, the M1 macrophages were significantly less abundant in the BRAFmutpTERTmut samples, indicative of a less immunogenic environment. A potential explanation may lay in the knowledge that activation of Pi3K signaling in TLR4 activated cells augments NF-κB activation, promoting M1 macrophage response (41). In our study, positive regulation of NF-κB pathway was suppressed in BRAFmutpTERTmut compared to BRAFmutpTERTwt.
Second, profound effects of pTERTmut were observed with B-cell markers (MS4A1/CD20 and particularly CD19) and markers of cytotoxic cells (CD8+ T-cells and NK cells). Key markers of these cell types are robustly and reproducibly (in Mayo Clinic and TCGA samples) repressed in double mutant tumors. It is plausible that these effects are linked to downregulation of interferon signaling, albeit effects on BMP and TNFSF signaling may also play a role (37). Third, the most striking effect of pTERTmut on the BRAFmut background was on the calcitonin gene, CALCA. This gene is strongly induced by BRAFmut in Mayo Clinic samples and log2FC=-1.93 (critical p<0.006) in TCGA samples. CALCA encodes the neuropeptide αCGRP, which has been extensively studied as an anti-inflammatory signal (42). Thus, downregulation of CALCA should lead to inflammation, but this was not observed in BRAFmutpTERTmut PTC tumors.
It is important to acknowledge that, despite seeing several markers being significantly downregulated, we did not observe corresponding major shifts in the immune cell populations. This could be explained by a decrease in the markers at individual level without driving a change in the cell populations, reflecting hyporesponsiveness of the immune effectors as seen with CD8 expression on cytotoxic T cells (43).
Our studies were motivated, at least in part, by the desire to identify features that account for the clinical observation that BRAFmutpTERTmut PTC tumors are considerably more aggressive than BRAFmutpTERTwt or BRAFwtpTERTwt tumors (10). In a sense, our data appear to answer one question: why is the double mutant more aggressive than the BRAFmutpTERTwt genotype? The answer plausibly lies within the immune microenvironment, which is clearly suppressed by pTERTmut. BRAFmutpTERTwt tumors are more immunogenic that BRAFmutpTERTmut tumors, and progression is more likely to be suppressed by the host immune system. Noncanonical functions of TERT include promoting epithelial to mesenchymal transition and cancer cell stemness (10, 44) as demonstrated by Caria et al. (45) in one BRAFmutpTERTmut PTC cell line, B-CPAP. Recent studies suggest that the complex interplay between cancer stem cells and the immune microenvironment results in immunosuppression, and additional work is necessary to demonstrate if our findings can be explained through such mechanisms (12, 46). Moreover, double mutant tumors maintain relatively high levels of two potential immune-therapeutic targets, B7-H3 and PD-L1, which may inform novel therapeutic approaches for management of these more aggressive tumors. This is consistent with existing literature which reports that PDL1 expression is elevated in CD44+ breast and head and neck squamous cancer stem cells (37, 47, 48) and CD133+ colon cancer stem cells (49).
On the other hand, these data yield little insight into why double mutant PTC tumors are more aggressive than BRAFwtpTERTwt tumors. Very few immune markers were repressed in BRAFmutpTERTmut samples compared to BRAFwtpTERTwt samples, so there does not seem to be a compelling immune explanation for clinical differences in aggressiveness between these two genotypes. This observation lends itself to speculation that we may be asking the wrong question. The key question may be: why is BRAFmut not more oncogenic (ie, why are BRAFmut PTC tumors not more aggressive than BRAFwt tumors)? BRAFmut constitutively activates growth factor/MAPK signaling, and from a signal transduction perspective, it is not obvious why BRAFmut is not a powerful oncogene, in line with many well-known MAPK-activating oncogenic mutations to RAS, RAF, ERBB2, EGFR, etc. The answer to this enigma may lie in the observation that BRAFmut engenders an environment that is immune-activated, thereby suppressing the transforming potential of the BRAFV600E oncogene. If this hypothesis is correct, one may understand how the pTERT mutation makes BRAFmut tumors more aggressive, by suppressing the immune-activating properties of BRAFmut while essentially unfettering the oncogenic driver stimulus that would normally be expected from a mutation that potentiates growth factor/MAPK signaling.
Although our data suggests that TERT is an important immune modulator in the tumor microenvironment, our methodology has two notable limitations. First, because both the NanoString Immune Profiling Panel and LM22 were comprised of a selected set of genes which did not overlap completely, the results of the deconvolution analysis of the Mayo samples may be under representative of certain immune cell types. However, the TCGA dataset deconvolution analysis was carried out with a 95% gene overlap. Second, our FFPE samples were macrodissected with the purpose of reducing the amount of normal tissue but did not remove it at a microscopic level. This prevented us from pursuing comparisons between immune gene expression in tumor versus background to further understand whether our findings are due to the result of gene alterations at the level of tumor cells, the surrounding immune cells or both. The answer to this question, as well as whether similar changes are present in other BRAFmutpTERTmut thyroid cancer histologies will be the subject of future studies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Mayo Clinic Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
A-MS-C: Visualization, Validation, Supervision, Project administration, Methodology, Investigation, Funding acquisition, Data curation, Conceptualization, Writing – review & editing, Writing – original draft. MR: Writing – review & editing, Methodology, Investigation, Formal analysis. YM: Software, Formal analysis, Writing – review & editing. AN: Software, Formal analysis, Writing – review & editing. YL: Software, Formal analysis, Writing – review & editing. XW: Software, Formal analysis, Writing – review & editing. BN: Methodology, Writing – review & editing, Investigation, Formal analysis. JK: Writing – review & editing, Methodology, Investigation. JC: Funding acquisition, Writing – review & editing. CB: Data curation, Writing – review & editing. AR: Data curation, Writing – review & editing. VB: Supervision, Methodology, Writing – review & editing. JC: Methodology, Writing – review & editing. KK: Software, Methodology, Visualization, Formal analysis, Writing – review & editing. ET: Project administration, Supervision, Funding acquisition, Formal analysis, Conceptualization, Writing – review & editing, Writing – original draft. RS: Supervision, Investigation, Funding acquisition, Formal analysis, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Mayo Clinic’s Center for Clinical and Translational Science and Team Science Pilot award, the Georg Haub Family Career Development Award in Cancer Research Honoring Richard F. Emslander and Mayo Clinic Comprehensive Cancer Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1440722/full#supplementary-material
References
1. Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. (2017) 317:1338–48. doi: 10.1001/jama.2017.2719
2. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
3. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. (2014) 32:2718–26. doi: 10.1200/JCO.2014.55.5094
4. Song YS, Yoo SK, Kim HH, Jung G, Oh AR, Cha JY, et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr Relat Cancer. (2019) 26:629–41. doi: 10.1530/ERC-17-0562
5. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. (2013) 309:1493–501. doi: 10.1001/jama.2013.3190
6. Gan X, Shen F, Deng X, Feng J, Lu J, Cai W, et al. Prognostic implications of the BRAF-V600(E) mutation in papillary thyroid carcinoma based on a new cut-off age stratification. Oncol Lett. (2020) 19:631–40. doi: 10.3892/ol.2019.11132
7. Xing M, Alzahrani AS, Carson KA, Shong YK, Kim TY, Viola D, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J Clin Oncol. (2015) 33:42–50. doi: 10.1200/JCO.2014.56.8253
8. Huang Y, Qu S, Zhu G, Wang F, Liu R, Shen X, et al. BRAF V600E mutation-assisted risk stratification of solitary intrathyroidal papillary thyroid cancer for precision treatment. J Natl Cancer Inst. (2018) 110:362–70. doi: 10.1093/jnci/djx227
9. Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell. (2014) 159:676–90. doi: 10.1016/j.cell.2014.09.050
10. Liu X, Qu S, Liu R, Sheng C, Shi X, Zhu G, et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J Clin Endocrinol Metab. (2014) 99:E1130–6. doi: 10.1210/jc.2013-4048
11. Qasem E, Murugan AK, Al-Hindi H, Xing M, Almohanna M, Alswailem M, et al. TERT promoter mutations in thyroid cancer: a report from a Middle Eastern population. Endocr Relat Cancer. (2015) 22:901–8. doi: 10.1530/ERC-15-0396
12. Babli S, Payne RJ, Mitmaker E, Rivera J. Effects of chronic lymphocytic thyroiditis on the clinicopathological features of papillary thyroid cancer. Eur Thyroid J. (2018) 7:95–101. doi: 10.1159/000486367
13. Sulaieva O, Selezniov O, Shapochka D, Belemets N, Nechay O, Chereshneva Y, et al. Hashimoto’s thyroiditis attenuates progression of papillary thyroid carcinoma: deciphering immunological links. Heliyon. (2020) 6:e03077. doi: 10.1016/j.heliyon.2019.e03077
14. Dvorkin S, Robenshtok E, Hirsch D, Strenov Y, Shimon I, Benbassat CA. Differentiated thyroid cancer is associated with less aggressive disease and better outcome in patients with coexisting Hashimotos thyroiditis. J Clin Endocrinol Metab. (2013) 98:2409–14. doi: 10.1210/jc.2013-1309
15. Kuo C-Y, Liu T-P, Yang P-S, Cheng S-P. Characteristics of lymphocyte-infiltrating papillary thyroid cancer. J Cancer Res Practice. (2017) 4:95–9. doi: 10.1016/j.jcrpr.2017.03.003
16. Cunha LL, Marcello MA, Ward LS. The role of the inflammatory microenvironment in thyroid carcinogenesis. Endocr Relat Cancer. (2014) 21:R85–R103. doi: 10.1530/ERC-13-0431
17. Huang BY, Hseuh C, Chao TC, Lin KJ, Lin JD. Well-differentiated thyroid carcinoma with concomitant Hashimoto’s thyroiditis present with less aggressive clinical stage and low recurrence. Endocr Pathol. (2011) 22:144–9. doi: 10.1007/s12022-011-9164-9
18. French JD, Kotnis GR, Said S, Raeburn CD, McIntyre RC Jr., Klopper JP, et al. Programmed death-1+ T cells and regulatory T cells are enriched in tumor-involved lymph nodes and associated with aggressive features in papillary thyroid cancer. J Clin Endocrinol Metab. (2012) 97:E934–43. doi: 10.1210/jc.2011-3428
19. French JD, Weber ZJ, Fretwell DL, Said S, Klopper JP, Haugen BR. Tumor-associated lymphocytes and increased FoxP3+ regulatory T cell frequency correlate with more aggressive papillary thyroid cancer. J Clin Endocrinol Metab. (2010) 95:2325–33. doi: 10.1210/jc.2009-2564
20. Means C, Clayburgh DR, Maloney L, Sauer D, Taylor MH, Shindo ML, et al. Tumor immune microenvironment characteristics of papillary thyroid carcinoma are associated with histopathological aggressiveness and BRAF mutation status. Head Neck. (2019) 41:2636–46. doi: 10.1002/hed.25740
21. Kebebew E, Treseler PA, Ituarte PH, Clark OH. Coexisting chronic lymphocytic thyroiditis and papillary thyroid cancer revisited. World J Surg. (2001) 25:632–7. doi: 10.1007/s002680020165
22. Wang K, Zhang Y, Xing Y, Wang H, He M, Guo R. Current and future of immunotherapy for thyroid cancer based on bibliometrics and clinical trials. Discovery Oncol. (2024) 15:50. doi: 10.1007/s12672-024-00904-6
23. Smallridge RC, Chindris AM, Asmann YW, Casler JD, Serie DJ, Reddi HV, et al. RNA sequencing identifies multiple fusion transcripts, differentially expressed genes, and reduced expression of immune function genes in BRAF (V600E) mutant vs BRAF wild-type papillary thyroid carcinoma. J Clin Endocrinol Metab. (2014) 99:E338–47. doi: 10.1210/jc.2013-2792
24. Kim K, Jeon S, Kim TM, Jung CK. Immune gene signature delineates a subclass of papillary thyroid cancer with unfavorable clinical outcomes. Cancers (Basel). (2018) 10. doi: 10.3390/cancers10120494
25. Zhi J, Zhang P, Zhang W, Ruan X, Tian M, Guo S, et al. Inhibition of BRAF sensitizes thyroid carcinoma to immunotherapy by enhancing tsMHCII-mediated immune recognition. J Clin Endocrinol Metab. (2021) 106:91–107. doi: 10.1210/clinem/dgaa656
26. Ghossein RA, Katabi N, Fagin JA. Immunohistochemical detection of mutated BRAF V600E supports the clonal origin of BRAF-induced thyroid cancers along the spectrum of disease progression. J Clin Endocrinol Metab. (2013) 98:E1414–21. doi: 10.1210/jc.2013-1408
27. Hu S, Liu X, Li T, Li Z, Hu F. LAG3 (CD223) and autoimmunity: Emerging evidence. J Autoimmun. (2020) 112:102504. doi: 10.1016/j.jaut.2020.102504
28. Andrews LP, Butler SC, Cui J, Cillo AR, Cardello C, Liu C, et al. LAG-3 and PD-1 synergize on CD8(+) T cells to drive T cell exhaustion and hinder autocrine IFN-gamma-dependent anti-tumor immunity. Cell. (2024) 187:4355–72 e22. doi: 10.1016/j.cell.2024.07.016
29. Farinola MA, Weir EG, Ali SZ. CD56 expression of neuroendocrine neoplasms on immunophenotyping by flow cytometry: a novel diagnostic approach to fine-needle aspiration biopsy. Cancer. (2003) 99:240–6. doi: 10.1002/cncr.11458
30. Salem A, Alotaibi M, Mroueh R, Basheer HA, Afarinkia K. CCR7 as a therapeutic target in Cancer. Bba-Rev Cancer. (2021) 1875. doi: 10.1016/j.bbcan.2020.188499
31. Hickman E, Smyth T, Cobos-Uribe C, Immormino R, Rebuli ME, Moran T, et al. Expanded characterization of in vitro polarized M0, M1, and M2 human monocyte-derived macrophages: Bioenergetic and secreted mediator profiles. PloS One. (2023) 18:e0279037. doi: 10.1371/journal.pone.0279037
32. Liu Q, Sun W, Zhang H. Roles and new insights of macrophages in the tumor microenvironment of thyroid cancer. Front Pharmacol. (2022) 13:875384. doi: 10.3389/fphar.2022.875384
33. Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. (2010) 185:642–52. doi: 10.4049/jimmunol.1000413
34. Steen CB, Liu CL, Alizadeh AA, Newman AM. Profiling cell type abundance and expression in bulk tissues with CIBERSORTx. Methods Mol Biol. (2020) 2117:135–57. doi: 10.1007/978-1-0716-0301-7_7
35. Song YS, Park YJ. Mechanisms of TERT reactivation and its interaction with BRAFV600E. Endocrinol Metab (Seoul). (2020) 35:515–25. doi: 10.3803/EnM.2020.304
36. Han J, Chen M, Wang Y, Gong B, Zhuang T, Liang L, et al. Identification of biomarkers based on differentially expressed genes in papillary thyroid carcinoma. Sci Rep. (2018) 8:9912. doi: 10.1038/s41598-018-28299-9
37. Chien MN, Yang PS, Hsu YC, Liu TP, Lee JJ, Cheng SP. Transcriptome analysis of papillary thyroid cancer harboring telomerase reverse transcriptase promoter mutation. Head Neck. (2018) 40:2528–37. doi: 10.1002/hed.v40.11
38. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
39. Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes. (2012) 61:2718–27. doi: 10.2337/db11-1595
40. Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. (2018) 9:888. doi: 10.3389/fimmu.2018.00888
41. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
42. Mikami N, Matsushita H, Kato T, Kawasaki R, Sawazaki T, Kishimoto T, et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J Immunol. (2011) 186:6886–93. doi: 10.4049/jimmunol.1100028
43. Rodríguez-Rodríguez N RF, Crispín F. CD8 is down(regulated) for tolerance. Trends Immunol. (2024) 45:12. doi: 10.1016/j.it.2024.04.012
44. Hannen R, Bartsch JW. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. (2018) 592:2023–31. doi: 10.1002/feb2.2018.592.issue-12
45. Caria P, Pillai R, Dettori T, Frau DV, Zavattari P, Riva G, et al. Thyrospheres from B-CPAP Cell Line with BRAF and TERT Promoter Mutations have Different Functional and Molecular Features than Parental Cells. J Cancer. (2017) 8:1629–39. doi: 10.7150/jca.18855
46. Bayik D, Lathia JD. Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer. (2021) 21:526–36. doi: 10.1038/s41568-021-00366-w
47. Wu Y, Chen M, Wu P, Chen C, Xu ZP, Gu W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol. (2017) 44:602–4. doi: 10.1111/cep.2017.44.issue-5
48. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ Cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. (2016) 22:3571–81. doi: 10.1158/1078-0432.CCR-15-2665
Keywords: papillary thyroid cancer (PTC), BRAF mutation V600 E, TERT promoter mutation (pTERT), immune genes, lymphocytic infiltration
Citation: Sigarteu Chindris A-M, Rivera M, Ma Y, Nair A, Liu Y, Wang X, Necela BM, Kachergus JM, Casler JD, Brett C, Rivas Mejia AM, Bernet VJ, Copland JA III, Knutson KL, Thompson EA and Smallridge RC (2024) BRAFV600E/pTERT double mutated papillary thyroid cancers exhibit immune gene suppression. Front. Endocrinol. 15:1440722. doi: 10.3389/fendo.2024.1440722
Received: 29 May 2024; Accepted: 18 November 2024;
Published: 09 December 2024.
Edited by:
Cesidio Giuliani, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Jeffrey A. Knauf, Cleveland Clinic, United StatesClara Ugolini, University of Pisa, Italy
Copyright © 2024 Sigarteu Chindris, Rivera, Ma, Nair, Liu, Wang, Necela, Kachergus, Casler, Brett, Rivas Mejia, Bernet, Copland, Knutson, Thompson and Smallridge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana-Maria Sigarteu Chindris, Y2hpbmRyaXMuYW5hbWFyaWFAbWF5by5lZHU=