 Yongxiang Yang
Yongxiang Yang Wanlin Liang1,2†
Wanlin Liang1,2† Tao Yang
Tao Yang- 1Department of Neurosurgery, The General Hospital of Western Theater Command, Chengdu, China
- 2School of Clinical Medicine &The First Affiliated Hospital of Chengdu Medical College, Chengdu, China
Introduction: Pituitary carcinoma (PC) is an extremely rare tumor of the adenohypophysis, which manifests as craniospinal dissemination and/or systemic metastasis. The diagnosis of PC is particularly difficult, as the clinical diagnosis only can be made after the metastasis is found. Owing to the complex diagnostic process and less effective treatments, the clinical prognosis of PC is usually very poor. Hence, it is of great significance to illustrate the diagnosis and treatment course of PC.
Methods: In this case report, we described a 48-year-old male patient who was diagnosed with pituitary adenoma (PA) initially and then was diagnosed with PC eventually after spinal cord metastasis was found, and we illustrated the treatment course as well. Furthermore, we summarized all the published case reports until now and provided a comprehensive review of the diagnosis, treatment, prediction, and clinical outcome of PC.
Results and Conclusions: We found that most PC patients had adrenocorticotropic hormone/prolactin (ACTH/PRL)-secreting tumors, Ki-67 ≥ 10%, and P53 positivity, which may have the potential to predict the transformation from PA to PC; surgery excision combined with temozolomide (TMZ) and radiotherapy is helpful to prolong the survival of PC patients.
Introduction
Pituitary adenomas (PAs) originate from the endocrine cells of the anterior pituitary, which are the second most common intracranial tumors and account for approximately 15% of intracranial neoplasms (1, 2). Most PAs are regarded as benign, growing slowly and rarely invading into the surrounding tissues. Nevertheless, 20% to 25% of PAs grow invasively and even aggressively infiltrate the dura mater basilar bone, cavernous sinuses, or sphenoid sinus (3, 4). In extremely rare cases, some PAs show malignant biological behaviors such as craniospinal dissemination and/or systemic metastasis, which can be called pituitary carcinomas (PCs) (5). PCs had been defined by the 2017 World Health Organization (WHO) classification of PAs and the 2017 European Society of Endocrinology (ESE) guidelines on aggressive PAs and PCs based on the presence of metastasis (5–7).
PCs account for 0.1% to 0.2% of PAs more or less; the clinical diagnosis of this rare disease is particularly difficult because it only can be made when primary PAs present and delayed metastasis in the brain, spinal cord, or other distant organs is found (8, 9). Moreover, the clinical manifestations of PCs are related to the functional state of the primary lesion and the location and size of metastatic lesions, which are highly variable and difficult to identify from other non-specific presentations (6, 10). Herein, the early diagnosis of PCs is extremely challenging. Currently, it is almost impossible to predict the clinical features and outcomes according to existing histological results including invasiveness, cellular pleomorphism, nuclear type, mitosis, and necrosis (11). Owing to the difficulty of early diagnosis of PCs and the absence of reliable prognostic criteria or pathological markers of PCs, the treatment and management of PCs are facing many challenges. At present, most PC patients survive less than 1 year after diagnosis, as the existing treatments including surgery, chemotherapy, radiotherapy, and immunotherapy are usually partially effective or not effective (12, 13). Hence, the clinical prognosis of PCs is very poor, and it is of great significance to illustrate the diagnosis and treatment course of PCs.
Up to now, the knowledge about PCs almost entirely comes from case reports or case series, which is far from enough. Although the ESE published clinical practice guidelines for the management of aggressive PAs and PCs in 2017 (5), its guiding value for clinical practice is limited. Accordingly, it is very meaningful to illustrate the diagnosis and treatment process of PCs and review the literature. This study described a 48-year-old male patient who was diagnosed with an atypical pituitary tumor initially and then was diagnosed with PCs eventually after spinal cord metastasis was found. Furthermore, all the published works of literature about PCs including case reports and reviews until now were summarized to review the epidemiology and diagnosis, clinical characteristics, available predictive markers and potential factors implicated in their aggressiveness, current and emerging therapeutic approaches, and outcomes of PCs. The aims of this study were to explore the key clinical aspects of PCs, further enrich the knowledge about PCs, and improve the clinical outcomes of this rare disease.
Methods
First, we retrospectively described the diagnosis, treatment, and outcome of a 48-year-old male patient with PC with spinal cord metastasis. Then, we comprehensively searched all the published English literature with the keyword “pituitary carcinomas” through PubMed and summarized the clinical data of the patients in the literature, including gender, age of onset, pathological results, site of metastasis, interval time of metastasis, treatment method, and final clinical outcome. In the clinical data of patients reported in the literature, we focused on analyzing the correlation between the index of P53 and Ki-67, the interval time of pituitary metastasis, and the clinical outcome of PC patients. In addition, we analyzed the correlation between the treatment methods including surgery, radiotherapy, chemotherapy, and the clinical outcome of PC patients. Finally, we comprehensively summarized the diagnosis, prognosis, treatment, and final outcome of PC by reviewing the literature. In the analysis process, we analyzed the enumeration data (P53, Ki-67, and 2-year survival state) by chi-squared test using SPSS version 18.0 software (SPSS Inc., USA), and two-tailed p < 0.05 was considered statistically significant.
Case description
A 48-year-old male patient came to our hospital and sought diagnosis and treatment on September 7, 2023, mainly due to “headache, bilateral blurry vision and narrowed visual field for more than 6 months”. The patient began to have headaches and blurry vision in both eyes in March 2023 without obvious cause, and his situation did not improve after treatment in the ophthalmology department of other hospitals. Later on, the patient manifested a gradual aggravation of bilateral blurry vision and began to have narrowed visual field little by little, and his condition did not improve after searching for a cure in the ophthalmology departments of other hospitals many times. Then, the patient received a head MRI examination at another hospital on September 1, 2023; an occupying lesion was found in the sellar area and suprasellar cisterna, and the diagnosis was considered to be a pituitary tumor, craniopharyngioma, epidermoid cyst, or other neoplastic lesions. In order to seek further diagnosis and treatment, the patient came to our hospital and was admitted to our neurosurgery department on September 7, 2023. The patient was in good health previously and had no family history of tumors or other hereditary diseases. Physical examination revealed that the patient had binocular vision and temporal hemianopsia. The MRI examination of the head on September 8, 2023, indicated there was an occupying lesion in the sellar area and suprasellar cisterna, the size was 4.4 × 3.9 × 2.6 cm, and the diagnosis was considered to be a pituitary tumor, craniopharyngioma, epidermoid cyst, or other neoplastic lesions (Figure 1). As the lesion invaded bilateral cavernous sinuses and crossed the internal carotid artery, the grade of this PA was Knosp3. Laboratory test results showed that the serum levels of prolactin (PRL), adrenocorticotropic hormone (ACTH), growth hormone (GH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), IGF-1, and thyroid-stimulating hormone (TSH) were all normal. Considering the medical history, clinical symptoms, physical signs, and examination results of the patient, we made a possible diagnosis of a pituitary tumor, craniopharyngioma, or other tumor lesions. After communicating with the patient and his family members about the condition and treatment plan, they asked for a “craniotomy to remove intracranial tumor” and signed the informed consent. Then, a craniotomy sellar tumor resection was successfully performed on September 12. 2023. The postoperative pathological diagnosis was a non-functional PA; the immune markers were as follows: ACTH(−), CD56(+), CK(+), CK20(−), CK8/18(+), CgA(−), FSH(−), glial fibrillary acidic protein (GFAP) (−), GH(−), Ki-67(+,3-5%), LH(−), neuron specific endase (NSE) (+), P53(+), P63(−), PIT-1(−), progesterone receptor (PR) (−), PRL(−), S-100(+), SF-1(−), Syn(−), TSH(−), TTF-1(−), Tg(−), Villin(−), Vim(+), and WT-1(+) (Figure 2). The patient’s headache symptoms were relieved, bilateral vision and visual field improved gradually after the operation, and he was discharged on September 23, 2023. On November 18, 2023, the patient was readmitted to our hospital due to “headache combined with blurred vision and vomiting for 2 weeks”. Physical examination revealed that the patient had blurred vision. A head MRI examination on November 21, 2023, indicated there was no tumor recurrence in the sellar area, but the third ventricle and bilateral lateral ventricles were dilated (Figure 3). Cerebrospinal fluid examination showed that the pressure was 350 mmH2O, and the biochemical/routine index was normal. The diagnosis was considered to be hydrocephalus, and a ventriculoperitoneal shunt surgery was performed on December 7, 2023. The patient’s headache symptoms were relieved, and bilateral vision improved gradually after the operation. A head MRI examination on December 9, 2023, indicated that the volume of the third ventricle and bilateral lateral ventricles was smaller than before (Figure 4). On December 12, 2023, the patient began to manifest severe pain in the left hip and left lower limb. An MRI examination of the lumbar spine on December 15, 2023, indicated that there was an occupying lesion in the L4–S1 intraspinal area, the size was 1.5 × 1.4 × 9.2 cm, and the diagnosis was considered to be a metastatic tumor (Figure 5). Considering the medical history, clinical symptoms, physical signs, and examination results of the patient, we made a diagnosis of PC with spinal cord metastasis. After communicating with the patient and his family members about the therapeutic strategy for PC with spinal cord metastasis, they refused to accept further treatments including surgery, radiotherapy, and chemotherapy, and he was discharged on January 11, 2024. In the follow-up survey, we learned that the patient underwent intraspinal metastatic resection at another hospital on January 29, 2024, and the postoperative pathological diagnosis was considered to be a tumor metastasized from the PA; his immune markers were as follows: GFAP(partial+), Oligo-(2−), EMA(dot +), P53(in +), Syn(+), CgA(−), SOX-2(+) and SOX-2(−), ATRX IDH-1(+), H3 K27M(−), INI1 (without missing), and Ki-67 (MIB) (+5% to 10%). Then, the patient received temozolomide (TMZ) therapy for 3 months, but his result was not satisfactory; his eyesight was becoming worse, and the muscle strength of both lower limbs gradually decreased.
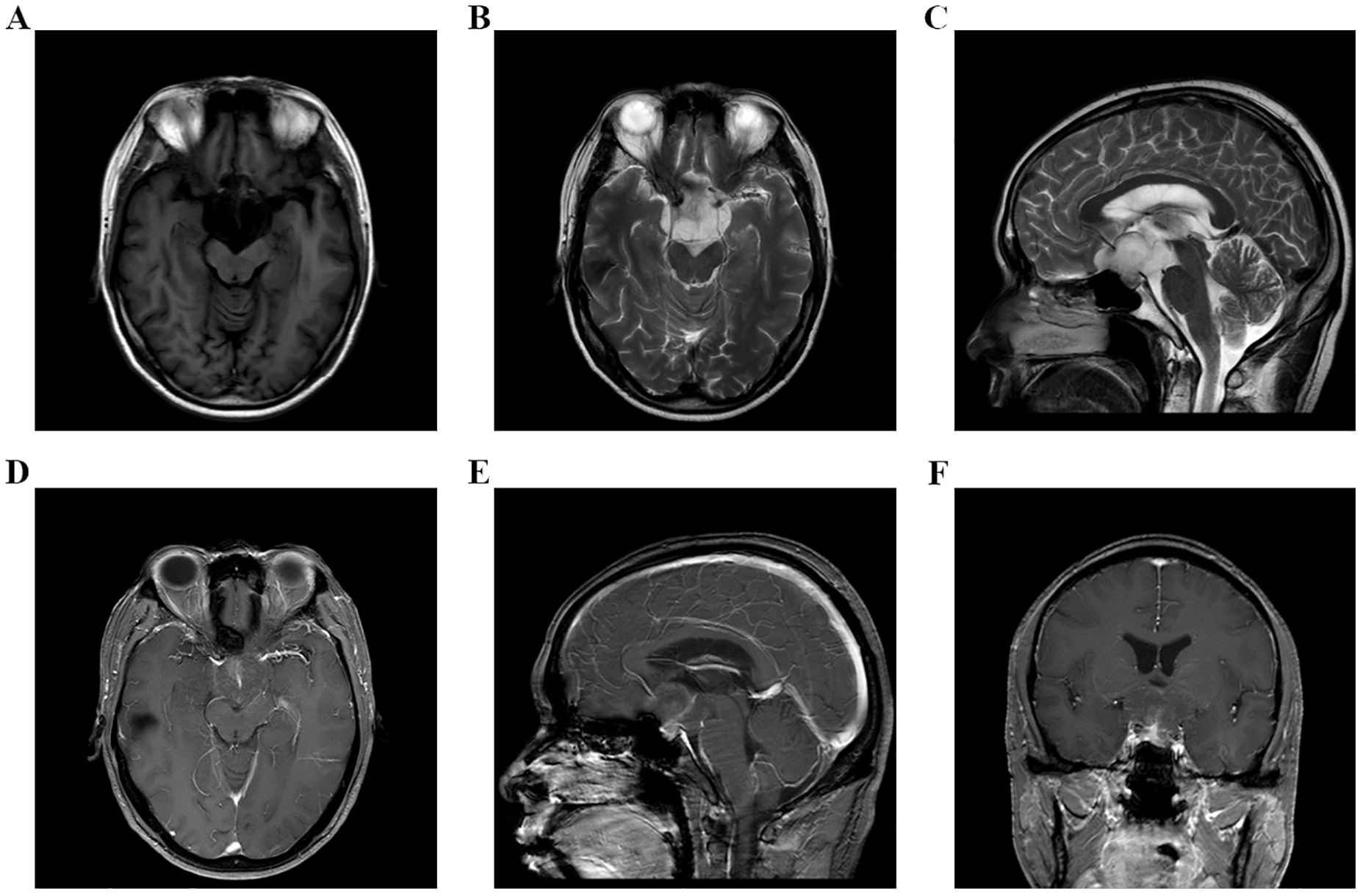
Figure 1. (A–F) MRI examination of head on September 8, 2023, indicated that there was an occupying lesion in the sellar area and suprasellar cisterna, the size was 4.4 × 3.9 × 2.6 cm, and the grade was Knosp3.
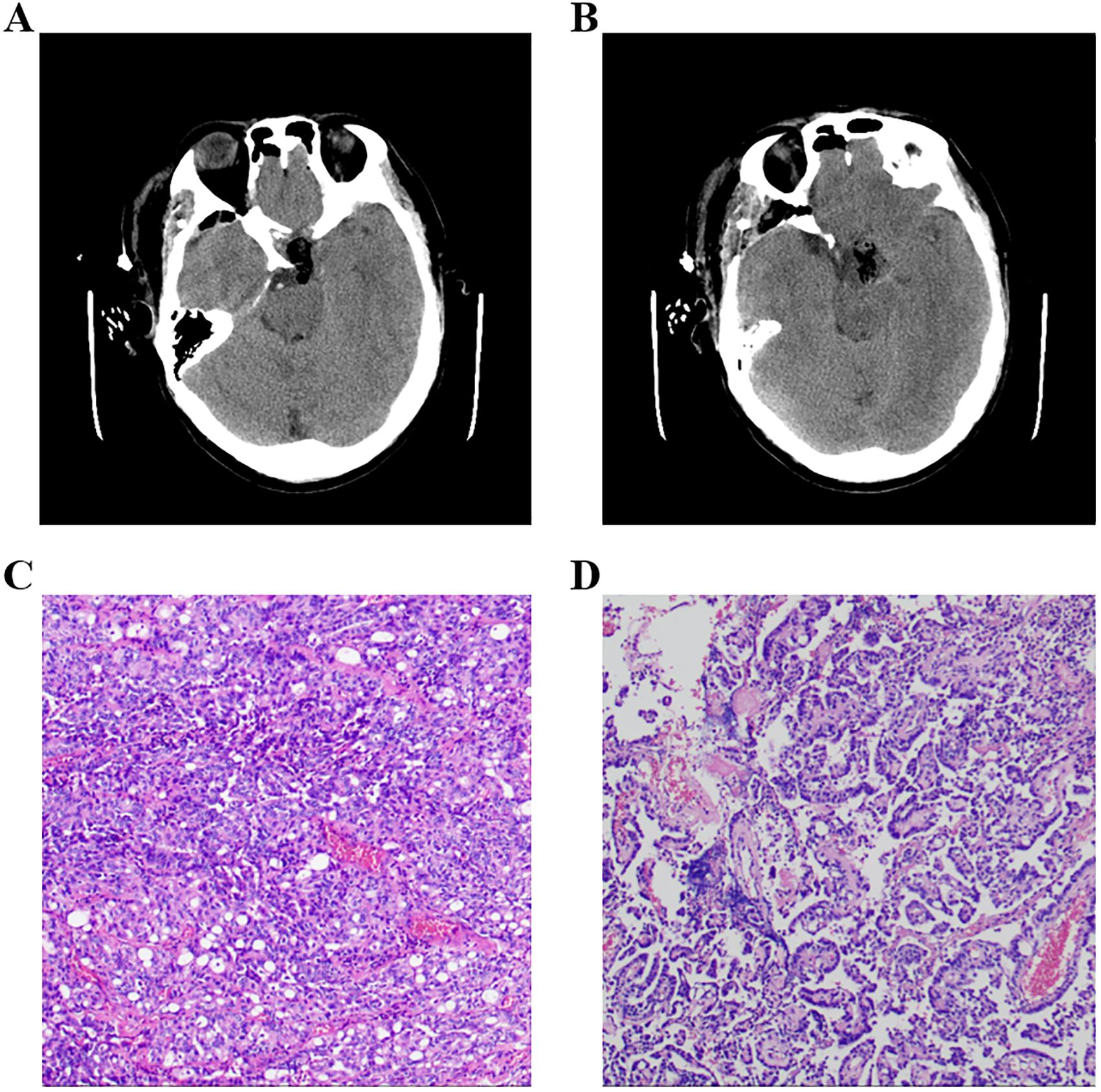
Figure 2. (A, B) Postoperational CT examination of the head. (C, D) Pathology results of the tumor, and the immune markers were as follows: ACTH(−), CD56(+), CK(+), CK20(−), CK8/18(+), CgA(−), FSH(−), GFAP(−), GH(−), Ki-67(+,3-5%), LH(−), NSE(+), P53(+), P63(−), PIT-1(−), PR(−), PRL(−), S-100(+), SF-1(−), Syn(−), TSH(−), TTF-1(−), Tg(−), Villin(−), Vim(+), and WT-1(+). ACTH, adrenocorticotropic hormone; PRL, prolactin; GH, growth hormone; TSH, thyroid-stimulating hormone; FSH, follicle-stimulating hormone; LH, luteinizing hormone.
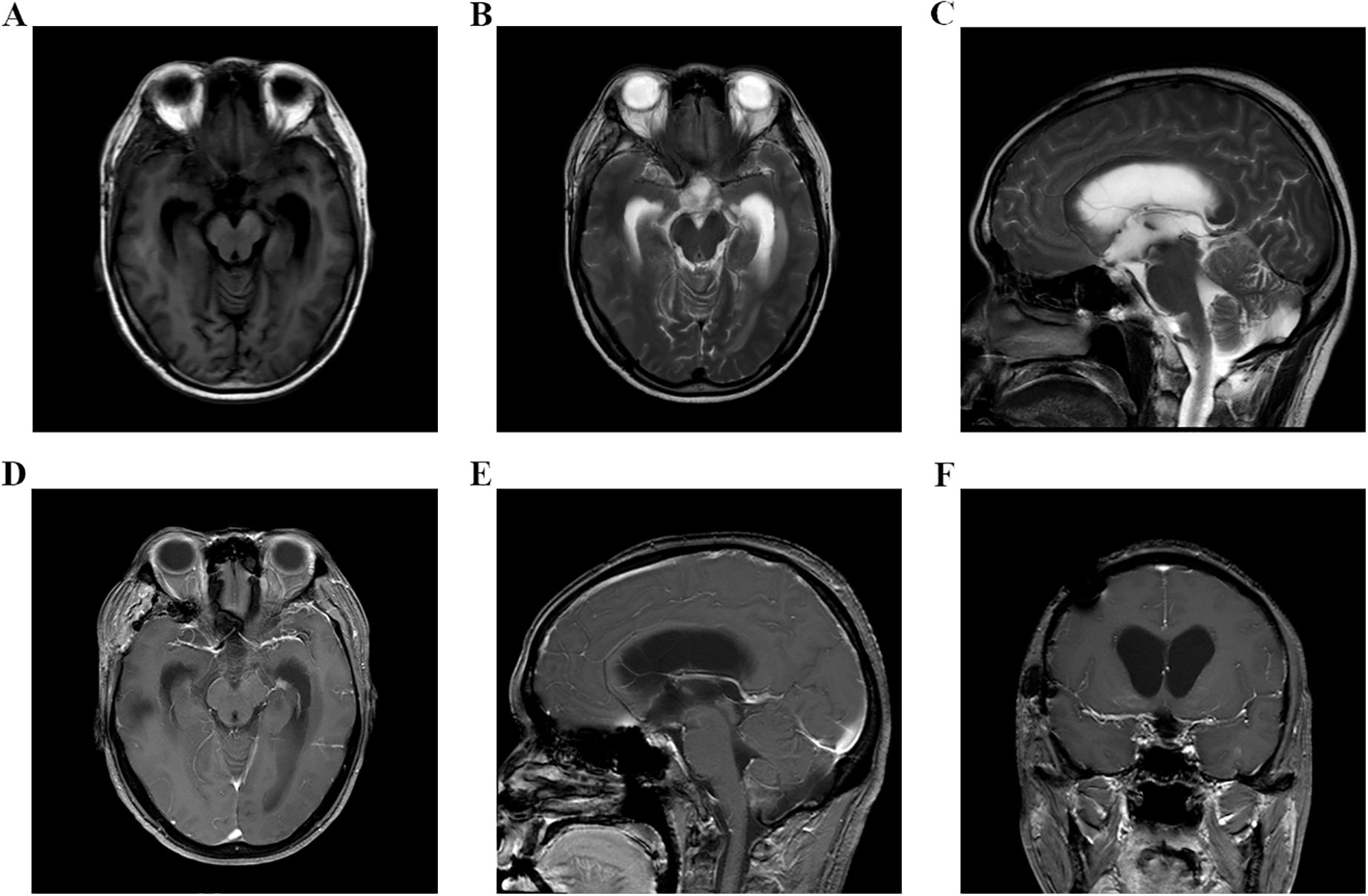
Figure 3. (A–F) Head MRI examination on November 21, 2023, indicated that there was no tumor recurrence in the sellar area, but the third ventricle and bilateral lateral ventricles were dilated.
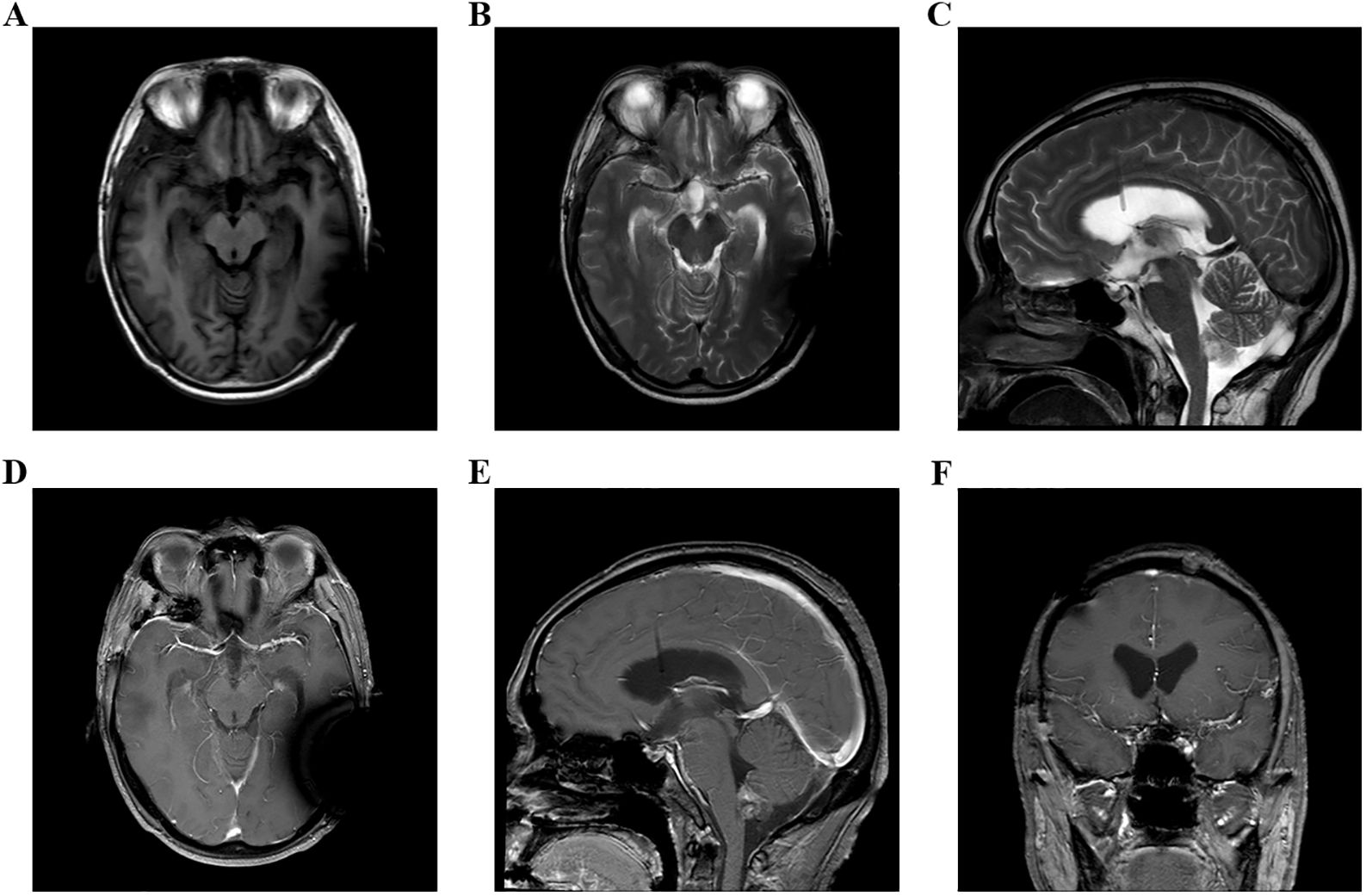
Figure 4. (A–F) Head MRI examination on December 9, 2023, indicated that the volume of third ventricle and bilateral lateral ventricles were smaller than before.
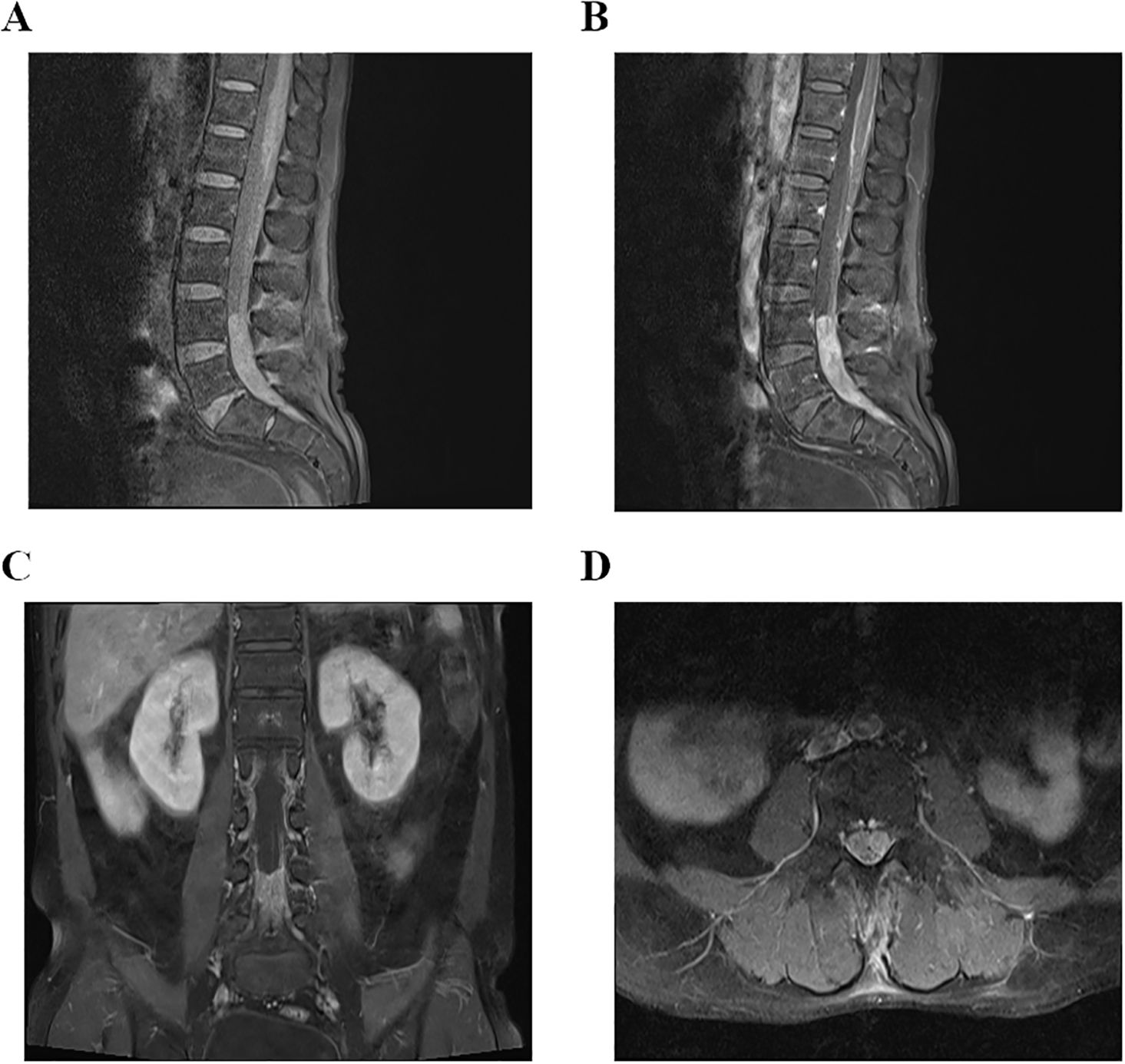
Figure 5. (A–D) MRI examination of lumbar spine on December 15, 2023, indicated that there was an occupying lesion in the L4–S1 intraspinal area, and the size was 1.5 × 1.4 × 9.2 cm.
Literature summary and analysis results
“PubMed” was searched using the keyword “pituitary carcinoma” until the time of May 25, 2024. A total of 116 articles were reviewed, and 128 PC patients were included in the literature eventually (8, 13–124). An additional 41 articles were excluded from this summary due to the inaccessibility of the full article. Most of the articles were published at the time interval of 2010–2019 and in the USA, China, and the United Kingdom (Figure 6). The clinical data of the included patients are illustrated in Table 1. Among the 128 PC patients, 64 were men and 64 were women, and their average age at diagnosis was 48.2 years and ranged from 9 to 75 years. The average time interval from the diagnosis of PAs to PCs was 9.7 years within 0–31 years. The most common pathological type of PC was ACTH with 50 cases (39.1%), followed by PRL with 24 cases (18.8%) and non-functional PC with 25 cases (19.5%). Moreover, there were seven patients who had tumors that secreted two hormones. The most common metastasis site was intracranial with 48 patients (37.5%), followed by spinal metastasis with 29 patients (22.7%), liver metastasis with 18 patients (14.1%), cervical lymph node with 10 patients, and bone metastasis (7.8%) with nine patients (7%). In addition, there were 38 PC patients who had multiple metastatic sites. The treatment of PCs is usually multimodal including surgery, radiotherapy, and chemotherapy. The average surgery time of reviewed patients was 2.7, and 17 patients received five to eight surgical procedures throughout the course of treatment. The transsphenoidal surgical approach was applied in 50 patients (41.3%), of which the endoscopic approach was used in 40 patients (80%). The transcranial approach was applied in 27 patients (22.3%), and the transcranial combined with transsphenoidal approach was applied in 44 patients (36.4%). Radiation therapy was employed in the treatment of 112 patients (87.5%), and chemotherapy was given to 61 patients (47.7%). Among the patients who received chemotherapy, 37 patients (60.7%) were treated with TMZ. Of the 128 patients, 63 patients (49.2%) died as reported, 42 patients (32.8%) were alive at the time of publication, and 23 patients (18.0%) were ambiguous in their survival status. As for the deaths reported, 37 patients died within 1 year of diagnosis, and 18 patients died within 4 years of diagnosis. The average survival time since the diagnosis of PC was 10.5 months, ranging from 6 months to 18 years. The 2-year survival rate of PC patients who received TMZ treatment was increased by a minor degree compared to that of patients who did not receive TMZ therapy, but without statistical significance (Table 2). The Ki-67 and P53 were detected in 43 and 26 patients, respectively; the rate of Ki-67 ≥ 10% and P53 positive both increased significantly “after metastasis” than “before metastasis” (Table 3).
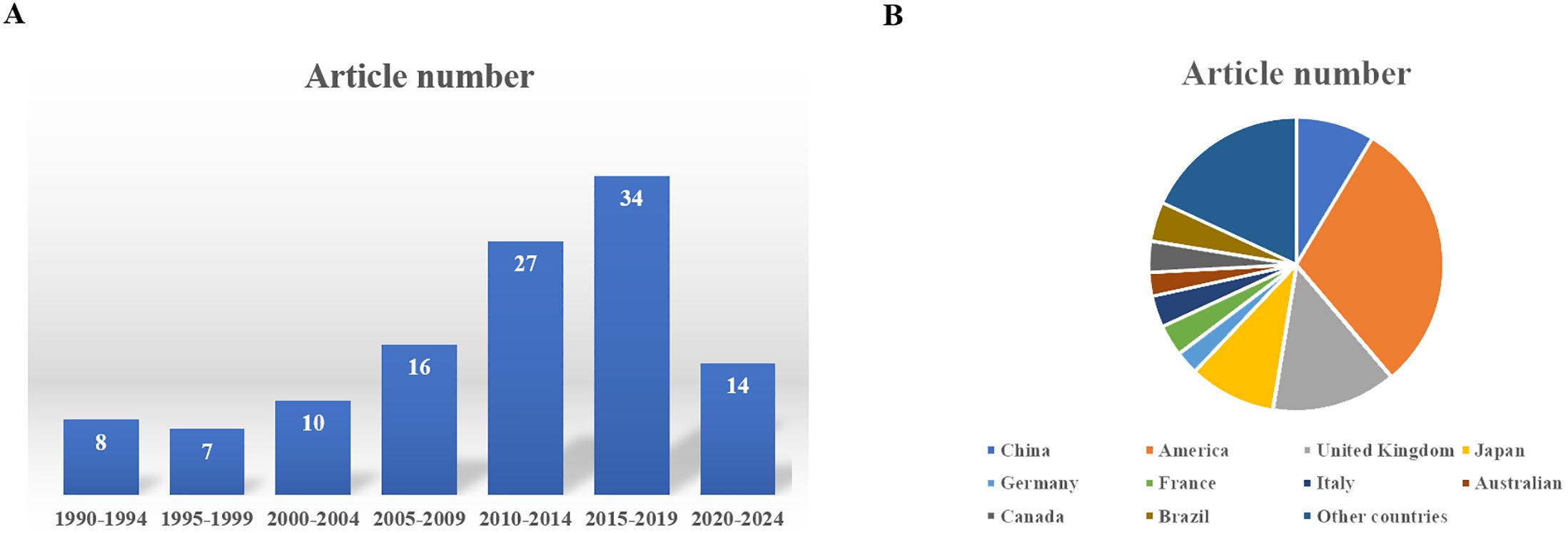
Figure 6. (A) The distribution of the publishing time of these articles. (B) The distribution of the country of authors of these articles.
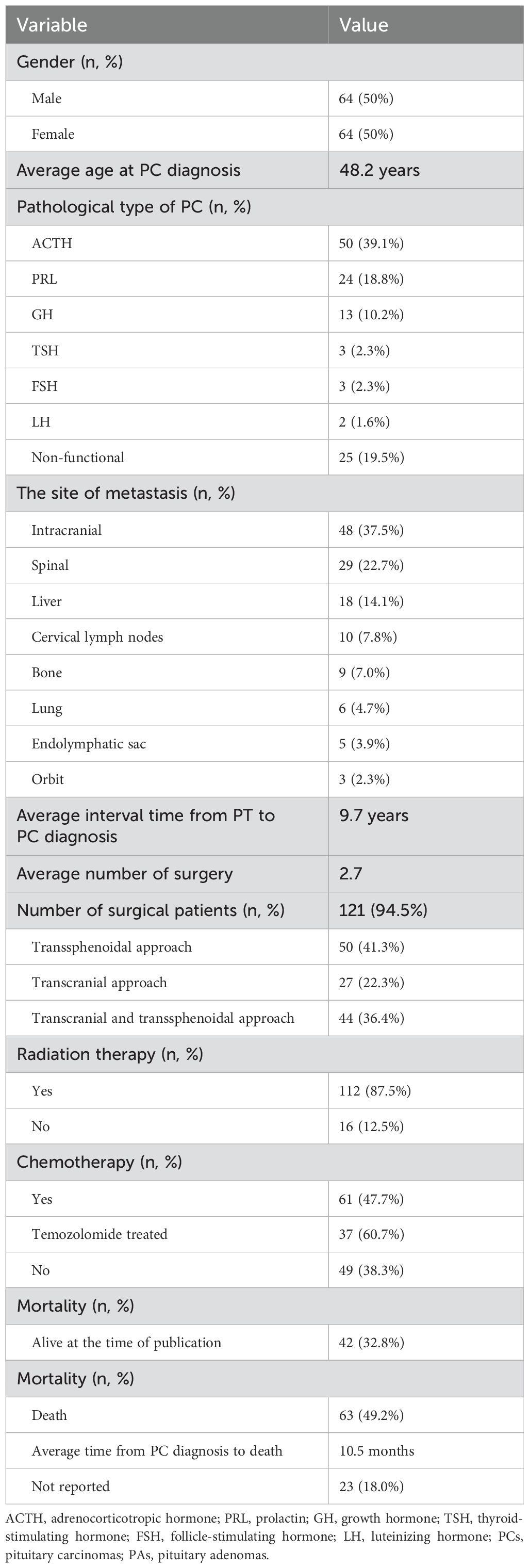
Table 1. Clinical characteristics of pituitary carcinoma patient cases.
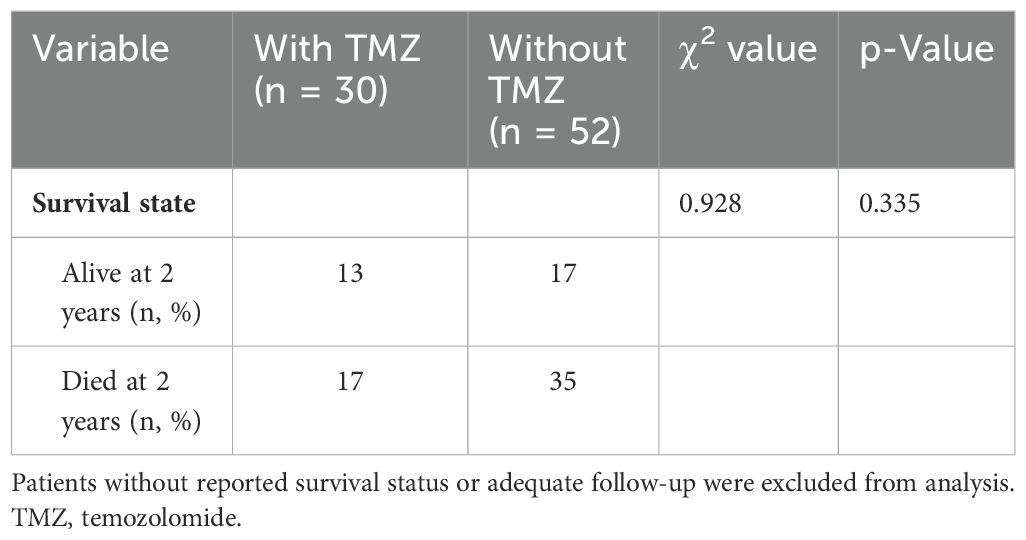
Table 2. Comparison of survival state between those treated with TMZ and those without TMZ.
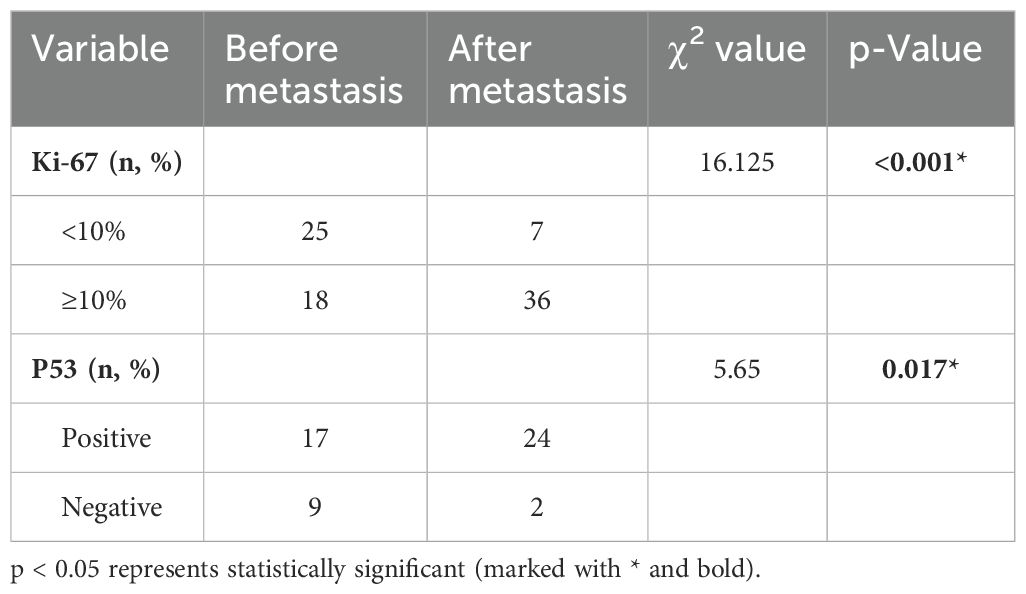
Table 3. The Ki-67 and P53 index before and after the metastasis.
Literature review and discussions
PAs are relatively common sellar tumors accounting for approximately 15% of intracranial tumors, whereas PCs are exceedingly rare diseases with an incidence of approximately 0.1% to 0.2% of PAs (125, 126). In this case report, we described the diagnosis and treatment course of a PC patient with spinal cord metastasis. We also summarized the case reports/series in the literature and reviewed the clinical features, epidemiology and diagnosis, available predictive markers and potential factors implicated in their aggressiveness, current and emerging therapeutic approaches, and outcomes of PCs.
PCs usually progress from PAs after a very long or short latency period following the diagnosis of original PAs (57, 127). Most PC patients manifest a similar clinical course as PA but with repeated recurrence and delayed metastasis; a few PC patients present rapid malignant behavior, multiple recurrences, and early metastasis (128). The clinical course of our reported patient falls in the latter group, with a manifestation of PC within 6 months after the diagnosis of PA. According to the reviewed literature, the mean latency period between the diagnosis of PA and the identification of PC in the 128 cases is 9.7 years, which is longer than the reported “6 years” in a case series by the University of Texas, MD Anderson Cancer Center. Only 28 patients were diagnosed with PC within 1–3 years of the diagnosis of PA. Moreover, the time of latency period is likely to be related to the hormone-secreting type of PC. Most PCs have endocrine activity and are transformed from ACTH/PRL-secreting PAs, whose average latency period is 9.5 and 4.7 years, respectively (129). The fifth edition of the WHO classifications (2021 World Health Organization Classification of Central Nervous System Tumors and 2022 World Health Organization Classification of Endocrine and Neuroendocrine Tumors) has introduced significant changes to the classification of PAs (130). It is worth noting that the anterior pituitary hormone-secreting cells are categorized into three major groups based on their corresponding transcription factors: PIT1, TPIT, and SF1. In the new WHO classification, the PIT1 group comprises somatotroph tumors, lactotroph tumors, mammosomatotroph tumors, thyrotroph tumors, other mature plurihormonal PIT1-lineage tumors, immature PIT1-lineage tumors, acidophil stem cell tumors, mixed somatotroph, and lactotroph tumors. Corticotroph tumors are categorized in the TPIT group, while somatotroph tumors belong to the SF1 group. Moreover, mammosomatotroph tumors and acidophil stem cell tumors are classified into the PIT1 group, and plurihormonal PIT-1-positive PA was divided into immature PIT1-lineage and mature plurihormonal PIT1-lineage tumors.
The diagnosis of PCs is exceedingly difficult because it only can be made when original malignant PAs are present and there is combined metastasis in the brain, spinal cord, or other distant organs. The absence of histological characteristics of malignancy and the delayed presentation of metastasis make the early diagnosis of PC problematic (131). The majority of PCs originate from functional PAs and are mostly represented by ACTH/PRL-secreting tumors, and a small part of PCs evolve from functional PAs switched from nonfunctional tumors (8, 57, 132, 133). The transformation from PAs to PCs is usually combined with some clinical features such as abnormal hormone secretion, cranial nerve palsy, neck and back pain, obstructive hydrocephalus, and discordance between biochemical and radiological findings (133). This phenomenon was also observed in our report, as the patient first showed the symptom of hydrocephalus and then manifested severe pain in the left hip and left lower limb. The most common metastasis localizations are intracranial (43.1%) and spinal (37.5%), followed by the liver (13.9%), cervical lymph node (11.1%), and bone (9.1%), and rarely in the lung (4.2%), endolymphatic sac (2.8%), or orbit (1.4%) (8), which is similar with our research results. To search for the metastasis localizations, ESE guidelines recommend performing the examination of CT/MRI and/or FDG/SSTPET/CT when PC patients present with some specific symptoms such as neurological complaints or neck and/or back pain or discordances between biochemical and radiological findings (5). The patient in our case report first presented with hydrocephalus and severe pain in the left hip and left lower limb; he then received a lumbar spine MRI examination and was found to have an occupying lesion in the L4–S1 intraspinal area, and the diagnosis was considered to be PC with spinal cord metastasis ultimately.
Owing to the majority of PCs being transformed from aggressive PAs, it is significant to distinguish aggressive PAs and identify PAs with potential for metastasis using some specific molecular markers. Now, the most commonly reported and intensively studied molecular markers are Ki-67 and P53. Ki-67 labeling index uses the MIB-1 antibody, and its mean level was reported to be 11.9% ± 3.4% in PCs compared to 1.4% ± 0.15% in non-invasive PAs (12, 134, 135). As 35%–61% of PAs and PCs had a Ki-67 index of ≥10% (6, 136–140), this criterion was considered to be indicative of PCs. PC patients with a Ki-67 index of ≥10% were proposed to be malignant potential including clinically aggressive, invasive, and highly proliferative (6). Though many authors considered that PAs with a Ki-67 index of more than 10% should be diagnosed as primary PCs (96, 128), the Ki-67 still could not be established as a prognostic marker due to the lack validation of a large number of clinical data (141). Moreover, Ki-67 is an imperfect marker of malignant potential, as the Ki-67 level variably ranged from 0% to 21.9%, and not all studies showed an association between Ki-67 and invasiveness (142, 143). P53 is encoded by the tumor suppressor TP53 gene, which is another protein implicated in PCs and has the prognostic value for the malignant potential (3, 144) but almost never mutates in PAs. Nevertheless, a part of reported PC cases were without P53 immunopositivity (12, 135, 142), and some authors did not consider P53 to be a regular basis for the prediction of the malignant potential (145). Moreover, some case series reports demonstrate that the use of P53 immunodetection as a prognostic tool is controversial (136, 145–147). A recent ESE survey showed that Ki-67 ≥ 10% was frequently seen in 85% and P53 was positive in 78% of 34 PC patients, indicating these two markers had a strong prognostic value for PCs (5). The 2017 WHO classification of PAs does not recommend using the Ki-67 index and P53 to predict tumor invasion, but these two markers are recommended in the ESE clinical practice guidelines to predict the tumor behavior of PAs (5, 145). In addition, the 2017 and 2022 WHO classifications of PAs both encourage the use of transcription factors to predict the tumor behavior of PAs (129, 145, 148). Herein, it is not advisable to predict the malignant potential of PAs solely based on existing pathological markers Ki-67 and P53, and the combination of clinical investigations, radiological manifestations, and further pathological indexes is very essential. Our research based on the reviewed literature showed that the rate of Ki-67 ≥ 10% and P53 positivity after metastasis were significantly higher than those before metastasis. These results indicated the potential value of Ki-67 and P53 in the prediction of PAs transforming into PCs.
The treatment of PCs is usually multimodal including surgery, radiotherapy, and chemotherapy. Surgical therapy is the mainstay treatment of PCs, which can alleviate acute mass effects with debulking through gross total or subtotal resecting of the sellar tumor (134, 135). The transsphenoidal surgical approach, especially the endoscopic approach, is considered to be the first-line treatment of most PC patients, which allows more extensive resections of tumors invading the cavernous sinus and parasellar structures (149). The transcranial approach may exhibit an advantage of obtaining a near-total tumor resection when the tumor presents with intracranial extension. In some cases, multiple repeated surgeries may be performed to resect recurrence and extension lesions, and locoregional therapeutic surgery should be considered to address metastatic sites if amenable to resection (5, 150). Our reported patient received transcranial surgery, and his bilateral vision and visual field improved gradually after the operation, indicating that the decompression effect on the optic nerve is remarkable. According to the reviewed literature, PC patients underwent 2.7 surgical interventions on average, and the transsphenoidal surgical approach accounted for approximately 41.3% of all initial sellar operations. Radiation therapy is recommended by the ESE guidelines and commonly employed for the primary treatment of PCs, which can be delivered as stereotactic radiosurgery, adjuvant radiation therapy, or fractionated radiation therapy over a 5–6-week course (133). In clinical practice, both stereotactic radiosurgery (SRS) and fractionated stereotactic radiotherapy (FSRT) are being used to obtain good disease control, while the success rate is varied and difficult to assess due to the variation in technique and doses administered in reported cases (151). The rate of long-term tumor control and hormone level normalization of the radiation therapy was reported to be 80%–97% and 40%–70%, respectively (152). The most frequent complication of radiation therapy is hypopituitarism, whose incidence is approximately 30%–60% 5–10 years after the treatment (153, 154). The incidence of other rare complications such as radiation-induced optic neuropathy, cerebrovascular accidents, and secondary tumors is approximately 0%–3% (152, 155, 156). According to the reviewed literature, 87.5% of PC patients had undergone SRS/FSRT. Chemotherapies are frequently used in the treatment of PC, of which the most common one is TMZ. TMZ is an oral alkylating agent that can lead to the irreversible impairment of DNA through methylation. TMZ monotherapy was first reported as a successful treatment for PC in 2006, which was then recommended by the ESE guideline as a first-line chemotherapy for PCs after the failure of standard therapy in 2018 (5). The recommended protocol of ESE guideline was using TMZ monotherapy (150–200 mg/m2 daily in consecutively repeated cycles (treatment given for 5 days every 28 days) in the case of documented tumor growth and suggested using the Stupp protocol [that is, concomitant administration of TMZ 75 mg/m2 daily and radiotherapy, followed by TMZ alone 150–200 mg/m2 daily (treatment given for 5 days every 28 days)] in the case of rapid tumor growth in patients who did not previously receive maximal doses of radiotherapy (5, 157, 158). In case reports and cohort studies, TMZ was reported to reduce tumor volume and hormonal levels in 47% of PC patients and induce partial or complete response in 71 of 149 patients in aggregate with populations ranging from 3 to 43 patients (5, 43, 51, 55, 126, 139, 140, 159–167). Our research indicated that the 2-year survival rate of PCs in patients who received TMZ treatment was increased by a minor degree compared to that of patients who did not receive TMZ but without statistical significance. It is suggested that the identification of responder and non-responder should be evaluated after three cycles of TMZ therapy, and the duration of continuing treatment for PC patients responding to first-line TMZ should be at least 6 months in total. MGMT methylation and DNA mismatch repair (MMR) proteins are known predictors of response to TMZ; therefore, it is strongly recommended that the precise selection of patients to receive TMZ treatment should be accomplished via the evaluation of MGMT (5, 131, 167–169). TMZ is often combined with radiotherapy, as it is a known radiosensitizer, and an ESE survey demonstrated that PC patients treated with concomitant chemoradiotherapy had a better tumor response (132). TMZ combined with radiotherapy may be a promising treatment for PC, as TMZ combined with whole-brain and spinal cord radiotherapy could induce the shrinkage of metastatic lesions. A new study indicated that TMZ was an effective medical treatment of PC, but was sometimes followed by tumor progression, and the co-administration with radiotherapy following the Stupp protocol may increase the progression-free survival rate (170). In addition to the above-mentioned treatments, other potential treatments include targeted therapies such as tyrosine kinase inhibitors against EGFR/HER2 such as lapatinib (171), the anti-VEGF antibody such as bevacizumab as a rescue treatment in combination with TMZ (5), the immune-checkpoint inhibitors (ICIs) such as inhibition of programmed death 1 (PD-1) and/or cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) (172). The pharmacological mechanism of ICIs is based on the fact that PCs contain tumorinfiltrating lymphocytes and express PDL1, which is a potential predictor of response, as well as on emerging preclinical data on the efficacy of ICIs in murine models of PCs (158). Up to now, eight patients with PC were reported to be treated with ICIs, partial radiological response was observed in five patients (112, 113, 173), stable condition was observed in two patients (113, 174), and progressive condition was observed in one patient (113, 173), indicating that ICIs may be a new and promising treatment of PCs.
The mortality rate of reported PC patients was approximately 66% in 1 year and 80% in 8 years (142). Our research based on the reviewed literature demonstrated that 63 patients (49.2%) died as reported, 42 patients (32.8%) were alive at the time of publication, and 23 patients (18.0%) were ambiguous in their survival status. Among the patients who died, 37 patients died within 1 year of diagnosis, and 18 patients died within 4 years of diagnosis. The average survival time since the diagnosis of PC was 10.5 months, ranging from 6 months to 18 years. Though most PC patients died within 1 year after the metastasis was found, it was rare that some patients still showed survival greater than 5 years (35, 45, 48, 62). Due to the lack of large-scale clinical studies, there is still no universally recognized uniform standard for the mortality of PC patients.
Conclusions
We described a 48-year-old male patient who was diagnosed with an atypical pituitary tumor initially and then was diagnosed with PC eventually after spinal cord metastasis was found, and the treatment course was illustrated as well. Furthermore, we summarized all the published case reports until now and provided a comprehensive review of the diagnosis, treatment, prediction, and clinical outcome of PC. We found that most PCs had ACTH/PRL-secreting tumors, Ki-67 ≥ 10%, and P53 positive, which may have the potential to predict the transformation from PAs to PCs; surgery excision combined with TMZ and radiotherapy is helpful to prolong the survival of PC patients. These results may enrich the knowledge about PCs and improve the clinical outcomes of this rare disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by The General Hospital of Western Theater Command. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YY: Writing – original draft, Writing – review & editing. WL: Investigation, Methodology, Writing – review & editing. KF: Investigation, Writing – original draft. TY: Conceptualization, Methodology, Writing – review & editing. JC: Conceptualization, Data curation, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (82101465 to YY) and the Natural Science Foundation of Sichuan Province (2022NSFSC1444 to YY).
Acknowledgments
We would like to thank the patient for providing written informed consent for publication, as well as the research staff involved in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Melmed S. Pituitary-tumor endocrinopathies. N Engl J Med. (2020) 382:937–50. doi: 10.1056/NEJMra1810772
2. Saeger W, Ludecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S. Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur J Endocrinol. (2007) 156:203–16. doi: 10.1530/eje.1.02326
3. Raverot G, Dantony E, Beauvy J, Vasiljevic A, Mikolasek S, Borson-Chazot F, et al. Risk of recurrence in pituitary neuroendocrine tumors: A prospective study using a five-tiered classification. J Clin Endocrinol Metab. (2017) 102:3368–74. doi: 10.1210/jc.2017-00773
4. Meij BP, Lopes MB, Ellegala DB, Alden TD, Laws EJ. The long-term significance of microscopic dural invasion in 354 patients with pituitary adenomas treated with transsphenoidal surgery. J Neurosurg. (2002) 96:195–208. doi: 10.3171/jns.2002.96.2.0195
5. Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. (2018) 178:G1–24. doi: 10.1530/EJE-17-0796
6. Trouillas J, Jaffrain-Rea ML, Vasiljevic A, Dekkers O, Popovic V, Wierinckx A, et al. Are aggressive pituitary tumors and carcinomas two sides of the same coin? Pathologists reply to clinician's questions. Rev Endocr Metab Disord. (2020) 21:243–51. doi: 10.1007/s11154-020-09562-9
7. Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer. (2017) 24:C5–08. doi: 10.1530/ERC-17-0004
8. Yoo F, Kuan EC, Heaney AP, Bergsneider M, Wang MB. Corticotrophic pituitary carcinoma with cervical metastases: case series and literature review. Pituitary. (2018) 21:290–301. doi: 10.1007/s11102-018-0872-8
9. Xu L, Khaddour K, Chen J, Rich KM, Perrin RJ, Campian JL. Pituitary carcinoma: Two case reports and review of literature. World J Clin Oncol. (2020) 11:91–102. doi: 10.5306/wjco.v11.i2.91
10. Trouillas J, Burman P, McCormack A, Petersenn S, Popovic V, Dekkers O, et al. Aggressive pituitary tumours and carcinomas: two sides of the same coin? Eur J Endocrinol. (2018) 178:C7–09. doi: 10.1530/EJE-18-0250
11. Wurth R, Thellung S, Corsaro A, Barbieri F, Florio T. Experimental evidence and clinical implications of pituitary adenoma stem cells. Front Endocrinol (Lausanne). (2020) 11:54. doi: 10.3389/fendo.2020.00054
12. Heaney A. Management of aggressive pituitary adenomas and pituitary carcinomas. J Neurooncol. (2014) 117:459–68. doi: 10.1007/s11060-014-1413-6
13. Pinchot SN, Sippel R, Chen H. ACTH-producing carcinoma of the pituitary with refractory Cushing's Disease and hepatic metastases: a case report and review of the literature. World J Surg Oncol. (2009) 7:39. doi: 10.1186/1477-7819-7-39
14. Sakamoto T, Itoh Y, Fushimi S, Kowada M, Saito M. Primary pituitary carcinoma with spinal cord metastasis–case report. Neurol Med Chir (Tokyo). (1990) 30:763–67. doi: 10.2176/nmc.30.763
15. Atienza DM, Vigersky RJ, Lack EE, Carriaga M, Rusnock EJ, Tsou E, et al. Prolactin-producing pituitary carcinoma with pulmonary metastases. Cancer. (1991) 68:1605–10. doi: 10.1002/1097-0142(19911001)68:7<1605::aid-cncr2820680723>3.0.co;2-d
16. Tonner D, Belding P, Moore SA, Schlechte JA. Intracranial dissemination of an ACTH secreting pituitary neoplasm–a case report and review of the literature. J Endocrinol Invest. (1992) 15:387–91. doi: 10.1007/BF03348759
17. Cannavo S, Curto L, Fazio R, Paterniti S, Blandino A, Marafioti T, et al. Coexistence of growth hormone-secreting pituitary adenoma and intracranial meningioma: a case report and review of the literature. J Endocrinol Invest. (1993) 16:703–08. doi: 10.1007/BF03348915
18. Petterson T, MacFarlane IA, MacKenzie JM, Shaw MD. Prolactin secreting pituitary carcinoma. J Neurol Neurosurg Psychiatry. (1992) 55:1205–06. doi: 10.1136/jnnp.55.12.1205
19. Yamashita S, Izumi M, Nagataki S. Acromegaly and pituitary carcinoma. Ann Intern Med. (1992) 117:1057–58. doi: 10.7326/0003-4819-117-12-1057
20. Mixson AJ, Friedman TC, Katz DA, Feuerstein IM, Taubenberger JK, Colandrea JM, et al. Thyrotropin-secreting pituitary carcinoma. J Clin Endocrinol Metab. (1993) 76:529–33. doi: 10.1210/jcem.76.2.8432799
21. Cusimano MD, Ohori P, Martinez AJ, Jungreis C, Wright DC. Pituitary carcinoma. Skull Base Surg. (1994) 4:46–51. doi: 10.1055/s-2008-1058989
22. Beauchesne P, Trouillas J, Barral F, Brunon J. Gonadotropic pituitary carcinoma: case report. Neurosurgery. (1995) 37:810–15, 815-16. doi: 10.1227/00006123-199510000-00027
23. Dayan C, Guilding T, Hearing S, Thomas P, Nelson R, Moss T, et al. Biochemical cure of recurrent acromegaly by resection of cervical spinal canal metastases. Clin Endocrinol (Oxf). (1996) 44:597–602. doi: 10.1046/j.1365-2265.1996.685507.x
24. Greenman Y, Woolf P, Coniglio J, O'Mara R, Pei L, Said JW, et al. Remission of acromegaly caused by pituitary carcinoma after surgical excision of growth hormone-secreting metastasis detected by 111-indium pentetreotide scan. J Clin Endocrinol Metab. (1996) 81:1628–33. doi: 10.1210/jcem.81.4.8636379
25. Lormeau B, Miossec P, Sibony M, Valensi P, Attali JR. Adrenocorticotropin-producing pituitary carcinoma with liver metastasis. J Endocrinol Invest. (1997) 20:230–36. doi: 10.1007/BF03346909
26. Garrao AF, Sobrinho LG, Pedro-Oliveira, Bugalho MJ, Boavida JM, Raposo JF, et al. ACTH-producing carcinoma of the pituitary with haematogenic metastases. Eur J Endocrinol. (1997) 137:176–80. doi: 10.1530/eje.0.1370176
27. Kemink SA, Wesseling P, Pieters GF, Verhofstad AA, Hermus AR, Smals AG. Progression of a Nelson's adenoma to pituitary carcinoma; a case report and review of the literature. J Endocrinol Invest. (1999) 22:70–5. doi: 10.1007/BF03345482
28. Masuda T, Akasaka Y, Ishikawa Y, Ishii T, Isshiki I, Imafuku T, et al. An ACTH-producing pituitary carcinoma developing Cushing's disease. Pathol Res Pract. (1999) 195:183–87. doi: 10.1016/S0344-0338(99)80032-6
29. McCutcheon IE, Pieper DR, Fuller GN, Benjamin RS, Friend KE, Gagel RF. Pituitary carcinoma containing gonadotropins: treatment by radical excision and cytotoxic chemotherapy: case report. Neurosurgery. (2000) 46:1233–39, 1239-40. doi: 10.1097/00006123-200005000-00042
30. Zahedi A, Booth GL, Smyth HS, Farrell WE, Clayton RN, Asa SL, et al. Distinct clonal composition of primary and metastatic adrencorticotrophic hormone-producing pituitary carcinoma. Clin Endocrinol (Oxf). (2001) 55:549–56. doi: 10.1046/j.1365-2265.2001.01322.x
31. le Roux CW, Mulla A, Meeran K. Pituitary carcinoma as a cause of acromegaly. N Engl J Med. (2001) 345:1645–46. doi: 10.1056/NEJM200111293452216
32. Sironi M, Cenacchi G, Cozzi L, Tonnarelli G, Iacobellis M, Trere D, et al. Progression on metastatic neuroendocrine carcinoma from a recurrent prolactinoma: a case report. J Clin Pathol. (2002) 55:148–51. doi: 10.1136/jcp.55.2.148
33. Pichard C, Gerber S, Laloi M, Kujas M, Clemenceau S, Ponvert D, et al. Pituitary carcinoma: report of an exceptional case and review of the literature. J Endocrinol Invest. (2002) 25:65–72. doi: 10.1007/BF03343963
34. Suzuki K, Morii K, Nakamura J, Kaneko S, Ukisu J, Hanyu O, et al. Adrenocorticotropin-producing pituitary carcinoma with metastasis to the liver in a patient with Cushing's disease. Endocr J. (2002) 49:153–58. doi: 10.1507/endocrj.49.153
35. Landman RE, Horwith M, Peterson RE, Khandji AG, Wardlaw SL. Long-term survival with ACTH-secreting carcinoma of the pituitary: a case report and review of the literature. J Clin Endocrinol Metab. (2002) 87:3084–89. doi: 10.1210/jcem.87.7.8667
36. Roncaroli F, Nose V, Scheithauer BW, Kovacs K, Horvath E, Young WJ, et al. Gonadotropic pituitary carcinoma: HER-2/neu expression and gene amplification. Rep two cases J Neurosurg. (2003) 99:402–08. doi: 10.3171/jns.2003.99.2.0402
37. Tysome J, Gnanalingham KK, Chopra I, Mendoza N. Intradural metastatic spinal cord compression from ACTH-secreting pituitary carcinoma. Acta Neurochir (Wien). (2004) 146:1251–54. doi: 10.1007/s00701-004-0350-0
38. Ayuk J, Natarajan G, Geh JI, Mitchell RD, Gittoes NJ. Pituitary carcinoma with a single metastasis causing cervical spinal cord compression. Case Rep J Neurosurg Spine. (2005) 2:349–53. doi: 10.3171/spi.2005.2.3.0349
39. Yamashita H, Nakagawa K, Tago M, Nakamura N, Shiraishi K, Yamauchi N, et al. Pathological changes after radiotherapy for primary pituitary carcinoma: a case report. J Neurooncol. (2005) 75:209–14. doi: 10.1007/s11060-005-2887-z
40. Brown RL, Muzzafar T, Wollman R, Weiss RE. A pituitary carcinoma secreting TSH and prolactin: a non-secreting adenoma gone awry. Eur J Endocrinol. (2006) 154:639–43. doi: 10.1530/eje.1.02141
41. Lim S, Shahinian H, Maya MM, Yong W, Heaney AP. Temozolomide: a novel treatment for pituitary carcinoma. Lancet Oncol. (2006) 7:518–20. doi: 10.1016/S1470-2045(06)70728-8
42. Sivan M, Nandi D, Cudlip S. Intramedullary spinal metastasis (ISCM) from pituitary carcinoma. J Neurooncol. (2006) 80:19–20. doi: 10.1007/s11060-006-9156-7
43. Fadul CE, Kominsky AL, Meyer LP, Kingman LS, Kinlaw WB, Rhodes CH, et al. Long-term response of pituitary carcinoma to temozolomide. Report of two cases. J Neurosurg. (2006) 105:621–26. doi: 10.3171/jns.2006.105.4.621
44. Tena-Suck ML, Salinas-Lara C, Sanchez-Garcia A, Rembao-Bojorquez D, Ortiz-Plata A. Late development of intraventricular papillary pituitary carcinoma after irradiation of prolactinoma. Surg Neurol. (2006) 66:527–33, 533. doi: 10.1016/j.surneu.2006.02.039
45. Koyama J, Ikeda K, Shose Y, Kimura M, Obora Y, Kohmura E. Long-term survival with non-functioning pituitary carcinoma - case report -. Neurol Med Chir (Tokyo). (2007) 47:475–78. doi: 10.2176/nmc.47.475
46. Choi G, Choi HJ, Kim YM, Choi SH, Cho YC, Kim Y, et al. Pituitary carcinoma with mandibular metastasis: a case report. J Korean Med Sci. (2007) 22 Suppl:S145–48. doi: 10.3346/jkms.2007.22.S.S145
47. Goh KP, Lee HY, Rajasoorya RC. Triple jeopardy in the pituitary. Pituitary. (2008) 11:331–36. doi: 10.1007/s11102-007-0075-1
48. Scheithauer BW, Kovacs K, Nose V, Lombardero M, Osamura YR, Lloyd RV, et al. Multiple endocrine neoplasia type 1-associated thyrotropin-producing pituitary carcinoma: report of a probable de novo example. Hum Pathol. (2009) 40:270–78. doi: 10.1016/j.humpath.2008.06.013
49. Figueiredo EG, Paiva WS, Teixeira MJ. Extremely late development of pituitary carcinoma after surgery and radiotherapy. J Neurooncol. (2009) 92:219–22. doi: 10.1007/s11060-008-9748-5
50. Guzel A, Tatli M, Senturk S, Guzel E, Cayli SR, Sav A. Pituitary carcinoma presenting with multiple metastases: case report. J Child Neurol. (2008) 23:1467–71. doi: 10.1177/0883073808319078
51. Hagen C, Schroeder HD, Hansen S, Hagen C, Andersen M. Temozolomide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur J Endocrinol. (2009) 161:631–37. doi: 10.1530/EJE-09-0389
52. Ilkhchoui Y, Appelbaum DE, Pu Y. FDG-PET/CT findings of a metastatic pituitary tumor. Cancer Imaging. (2010) 10:114–16. doi: 10.1102/1470-7330.2010.0015
53. Bode H, Seiz M, Lammert A, Brockmann MA, Back W, Hammes HP, et al. SOM230 (pasireotide) and temozolomide achieve sustained control of tumour progression and ACTH secretion in pituitary carcinoma with widespread metastases. Exp Clin Endocrinol Diabetes. (2010) 118:760–63. doi: 10.1055/s-0030-1253419
54. Yakoushina TV, Lavi E, Hoda RS. Pituitary carcinoma diagnosed on fine needle aspiration: Report of a case and review of pathogenesis. Cytojournal. (2010) 7:14. doi: 10.4103/1742-6413.67108
55. Curto L, Torre ML, Ferrau F, Pitini V, Altavilla G, Granata F, et al. Temozolomide-induced shrinkage of a pituitary carcinoma causing Cushing's disease–report of a case and literature review. ScientificWorldJournal. (2010) 10:2132–38. doi: 10.1100/tsw.2010.210
56. Murakami M, Mizutani A, Asano S, Katakami H, Ozawa Y, Yamazaki K, et al. A mechanism of acquiring temozolomide resistance during transformation of atypical prolactinoma into prolactin-producing pituitary carcinoma: case report. Neurosurgery. (2011) 68:E1761–67, discussion E1767. doi: 10.1227/NEU.0b013e318217161a
57. Dudziak K, Honegger J, Bornemann A, Horger M, Mussig K. Pituitary carcinoma with Malignant growth from first presentation and fulminant clinical course–case report and review of the literature. J Clin Endocrinol Metab. (2011) 96:2665–69. doi: 10.1210/jc.2011-1166
58. Moshkin O, Syro LV, Scheithauer BW, Ortiz LD, Fadul CE, Uribe H, et al. Aggressive silent corticotroph adenoma progressing to pituitary carcinoma: the role of temozolomide therapy. Hormones (Athens). (2011) 10:162–67. doi: 10.14310/horm.2002.1307
59. Annamalai AK, Dean AF, Kandasamy N, Kovacs K, Burton H, Halsall DJ, et al. Temozolomide responsiveness in aggressive corticotroph tumours: a case report and review of the literature. Pituitary. (2012) 15:276–87. doi: 10.1007/s11102-011-0363-7
60. Lee W, Cheung AS, Freilich R. TSH-secreting pituitary carcinoma with intrathecal drop metastases. Clin Endocrinol (Oxf). (2012) 76:604–06. doi: 10.1111/j.1365-2265.2011.04288.x
61. Shastri BR, Nanda A, Fowler M, Levine SN. Adrenocorticotropic hormone-producing pituitary carcinoma with intracranial metastases. World Neurosurg. (2013) 79:404–11. doi: 10.1016/j.wneu.2011.04.018
62. Arnold PM, Ratnasingam D, O'Neil MF, Johnson PL. Pituitary carcinoma recurrent to the lumbar intradural extramedullary space: case report. J Spinal Cord Med. (2012) 35:118–21. doi: 10.1179/2045772311Y.0000000055
63. Zhou Q, Chang H, Gao Y, Cui L. Tumor-to-tumor metastasis from pituitary carcinoma to radiation-induced meningioma. Neuropathology. (2013) 33:209–12. doi: 10.1111/j.1440-1789.2012.01343.x
64. Sreenan S, Sengupta E, Tormey W, Landau R. Metastatic pituitary carcinoma in a patient with acromegaly: a case report. J Med Case Rep. (2012) 6:322. doi: 10.1186/1752-1947-6-322
65. Morokuma H, Ando T, Hayashida T, Horie I, Inoshita N, Murata F, et al. A case of nonfunctioning pituitary carcinoma that responded to temozolomide treatment. Case Rep Endocrinol. (2012) 2012:645914. doi: 10.1155/2012/645914
66. Kovacs GL, Goth M, Rotondo F, Scheithauer BW, Carlsen E, Saadia A, et al. ACTH-secreting Crooke cell carcinoma of the pituitary. Eur J Clin Invest. (2013) 43:20–6. doi: 10.1111/eci.12010
67. Zemmoura I, Wierinckx A, Vasiljevic A, Jan M, Trouillas J, Francois P. Aggressive and Malignant prolactin pituitary tumors: pathological diagnosis and patient management. Pituitary. (2013) 16:515–22. doi: 10.1007/s11102-012-0448-y
68. Phillips J, East HE, French SE, Melcescu E, Hamilton RD, Nicholas WC, et al. What causes a prolactinoma to be aggressive or to become a pituitary carcinoma? Hormones (Athens). (2012) 11:477–82. doi: 10.14310/horm.2002.1380
69. Miller BA, Tanaka T, Ioachimescu AG, Vincentelli C, Appin CL, Oyesiku NM. Transformation of a silent adrencorticotrophic pituitary tumor into central nervous system melanoma. J Investig Med High Impact Case Rep. (2013) 1:1565008136. doi: 10.1177/2324709613494008
70. Balili I, Sullivan S, Mckeever P, Barkan A. Pituitary carcinoma with endolymphatic sac metastasis. Pituitary. (2014) 17:210–13. doi: 10.1007/s11102-013-0489-x
71. Lall RR, Shafizadeh SF, Lee KH, Mao Q, Mehta M, Raizer J, et al. Orbital metastasis of pituitary growth hormone secreting carcinoma causing lateral gaze palsy. Surg Neurol Int. (2013) 4:59. doi: 10.4103/2152-7806.110658
72. Park KS, Hwang JH, Hwang SK, Kim S, Park SH. Pituitary carcinoma with fourth ventricle metastasis: treatment by excision and Gamma-knife radiosurgery. Pituitary. (2014) 17:514–18. doi: 10.1007/s11102-013-0537-6
73. Maclean J, Aldridge M, Bomanji J, Short S, Fersht N. Peptide receptor radionuclide therapy for aggressive atypical pituitary adenoma/carcinoma: variable clinical response in preliminary evaluation. Pituitary. (2014) 17:530–38. doi: 10.1007/s11102-013-0540-y
74. Cornell RF, Kelly DF, Bordo G, Carroll TB, Duong HT, Kim J, et al. Chemotherapy-induced regression of an adrenocorticotropin-secreting pituitary carcinoma accompanied by secondary adrenal insufficiency. Case Rep Endocrinol. (2013) 2013:675298. doi: 10.1155/2013/675298
75. Lee HH, Hung SH, Tseng TM, Lin YH, Cheng JC. Undifferentiated carcinoma of the pituitary gland: A case report and review of the literature. Oncol Lett. (2014) 7:778–80. doi: 10.3892/ol.2014.1796
76. Lin GC, Adams ME, Arts HA. Lesions of the petrous ridge: metastatic pituitary carcinoma with discussion of differential diagnosis. Otol Neurotol. (2014) 35:645–48. doi: 10.1097/MAO.0000000000000326
77. Mendola M, Passeri E, Ambrosi B, Corbetta S. Multiple cerebral hemorrhagic foci from metastases during temozolomide treatment in a patient with corticotroph pituitary carcinoma. J Clin Endocrinol Metab. (2014) 99:2623–24. doi: 10.1210/jc.2014-1183
78. Takeuchi K, Hagiwara Y, Kanaya K, Wada K, Shiba M, Kato Y. Drop metastasis of adrenocorticotropic hormone-producing pituitary carcinoma to the cauda equina. Asian Spine J. (2014) 8:680–83. doi: 10.4184/asj.2014.8.5.680
79. Borba CG, Batista RL, Musolino NR, MaChado VC, Alcantara AE, Da SG, et al. Progression of an invasive ACTH pituitary macroadenoma with cushing's disease to pituitary carcinoma. Case Rep Oncol Med. (2015) 2015:810367. doi: 10.1155/2015/810367
80. Wang YQ, Fan T, Zhao XG, Liang C, Qi XL, Li JY. Pituitary carcinoma with intraspinal metastasis: report of two cases and review of the literature. Int J Clin Exp Pathol. (2015) 8:9712–17.
81. Wei Z, Zhou C, Liu M, Yao Y, Sun J, Xiao J, et al. MicroRNA involvement in a metastatic non-functioning pituitary carcinoma. Pituitary. (2015) 18:710–21. doi: 10.1007/s11102-015-0648-3
82. Novruzov F, Aliyev JA, Jaunmuktane Z, Bomanji JB, Kayani I. The use of (68)Ga DOTATATE PET/CT for diagnostic assessment and monitoring of (177)Lu DOTATATE therapy in pituitary carcinoma. Clin Nucl Med. (2015) 40:47–9. doi: 10.1097/RLU.0000000000000589
83. Halevy C, Whitelaw BC. How effective is temozolomide for treating pituitary tumours and when should it be used? Pituitary. (2017) 20:261–66. doi: 10.1007/s11102-016-0745-y
84. Kamiya-Matsuoka C, Cachia D, Waguespack SG, Crane CH, Mahajan A, Brown PD, et al. Radiotherapy with concurrent temozolomide for the management of extraneural metastases in pituitary carcinoma. Pituitary. (2016) 19:415–21. doi: 10.1007/s11102-016-0721-6
85. Grandidge C, Hall A, Kitchen N. Secreting follicle-stimulating hormone pituitary carcinoma with cervical metastasis. World Neurosurg. (2016) 93:413–90. doi: 10.1016/j.wneu.2016.05.095
86. Seltzer J, Carmichael JD, Commins D, Liu CS, Omura E, Chang E, et al. Prolactin-secreting pituitary carcinoma with dural metastasis: diagnosis, treatment, and future directions. World Neurosurg. (2016) 91:623–76. doi: 10.1016/j.wneu.2016.04.112
87. Wang H, Liang J, Yong WH, Sullivan P. Metastatic pituitary carcinoma to cervical lymph node: diagnosis by fine needle aspiration and review of the literature. Acta Cytol. (2017) 61:242–46. doi: 10.1159/000467384
88. Tufton N, Roncaroli F, Hadjidemetriou I, Dang MN, Denes J, Guasti L, et al. Pituitary carcinoma in a patient with an SDHB mutation. Endocr Pathol. (2017) 28:320–25. doi: 10.1007/s12022-017-9474-7
89. AbdelBaki MS, Waguespack SG, Salceda V, Jones J, Stapleton SL, Baskin DS, et al. Significant response of pituitary carcinoma to carboplatin, leucovorin and fluorouracil chemotherapy: a pediatric case report and review of the literature. J Neurooncol. (2017) 135:213–15. doi: 10.1007/s11060-017-2554-1
90. Chang EA, Shah R, Smith SV, Sadaka A, Ortiz GJ, Chevez-Barrios P, et al. Neuro-ophthalmic manifestations of pituitary carcinoma. J Neuroophthalmol. (2018) 38:339–41. doi: 10.1097/WNO.0000000000000620
91. Chandler CM, Lin X. Cytomorphology of metastatic pituitary carcinoma to the bone. Diagn Cytopathol. (2017) 45:645–50. doi: 10.1002/dc.23702
92. Bengtsson D, Joost P, Aravidis C, Askmalm SM, Backman AS, Melin B, et al. Corticotroph pituitary carcinoma in a patient with lynch syndrome (LS) and pituitary tumors in a nationwide LS cohort. J Clin Endocrinol Metab. (2017) 102:3928–32. doi: 10.1210/jc.2017-01401
93. Yang J, Liu S, Yang Z, Shi YB. Ectopic thyrotropin secreting pituitary adenoma concomitant with papillary thyroid carcinoma: Case report. Med (Baltimore). (2017) 96:e8912. doi: 10.1097/MD.0000000000008912
94. Touma W, Hoostal S, Peterson RA, Wiernik A, SantaCruz KS, Lou E. Successful treatment of pituitary carcinoma with concurrent radiation, temozolomide, and bevacizumab after resection. J Clin Neurosci. (2017) 41:75–7. doi: 10.1016/j.jocn.2017.02.052
95. Rutkowski MJ, Alward RM, Chen R, Wagner J, Jahangiri A, Southwell DG, et al. Atypical pituitary adenoma: a clinicopathologic case series. J Neurosurg. (2018) 128:1058–65. doi: 10.3171/2016.12.JNS162126
96. Endo T, Ogawa Y, Watanabe M, Tominaga T. A case of pituitary carcinoma initially diagnosed as an ectopic growth hormone producing pituitary adenoma with a high ki-67 labeling index. J Neurol Surg Cent Eur Neurosurg. (2018) 79:90–5. doi: 10.1055/s-0037-1600515
97. Kiatpanabhikul P, Shuangshoti S, Chantra K, Navicharern P, Kingpetch K, Houngngam N, et al. A case of coexistence of TSH/GH-secreting pituitary tumor and papillary thyroid carcinoma: Challenges in pathogenesis and management. J Clin Neurosci. (2017) 41:78–80. doi: 10.1016/j.jocn.2017.02.050
98. Garmes HM, Carvalheira J, Reis F, Queiroz LS, Fabbro MD, Souza V, et al. Pituitary carcinoma: A case report and discussion of potential value of combined use of Ga-68 DOTATATE and F-18 FDG PET/CT scan to better choose therapy. Surg Neurol Int. (2017) 8:162. doi: 10.4103/sni.sni_498_16
99. Joehlin-Price AS, Hardesty DA, Arnold CA, Kirschner LS, Prevedello DM, Lehman NL. Case report: ACTH-secreting pituitary carcinoma metastatic to the liver in a patient with a history of atypical pituitary adenoma and Cushing's disease. Diagn Pathol. (2017) 12:34. doi: 10.1186/s13000-017-0624-5
100. Bilbao I, Egana N, Garcia C, Olaizola I. Failure of a second temozolomide cycle in a patient with a prolactin-secreting pituitary carcinoma. Endocrinol Diabetes Nutr. (2017) 64:564–66. doi: 10.1016/j.endinu.2017.08.007
101. Krueger EM, Seibly J. Seeding of a pituitary adenoma or atypical pituitary carcinoma? Cureus. (2017) 9:e1211. doi: 10.7759/cureus.1211
102. Bettencourt-Silva R, Pereira J, Belo S, Magalhaes D, Queiros J, Carvalho D. Prolactin-producing pituitary carcinoma, hypopituitarism, and graves' Disease-report of a challenging case and literature review. Front Endocrinol (Lausanne). (2018) 9:312. doi: 10.3389/fendo.2018.00312
103. Souza MJ, de Sa CA, de Araujo CNA, Dos SFM, Pereira SC. Pituitary metastasis of thyroid carcinoma: A case report. Am J Case Rep. (2018) 19:896–902. doi: 10.12659/AJCR.909523
104. Moscote-Salazar LR, Satyarthee GD, Calderon-Miranda WG, Matus JA, Pacheco-Hernandez A, Puac-Polanco PC, et al. Prolactin secreting pituitary carcinoma with extracranial spread presenting with pathological fracture of femur. J Neurosci Rural Pract. (2018) 9:170–73. doi: 10.4103/jnrp.jnrp_325_17
105. Guo F, Wang G, Wang F, Xu D, Liu X. Identification of novel genes involved in the pathogenesis of an ACTH-secreting pituitary carcinoma: A case report and literature review. Front Oncol. (2018) 8:510. doi: 10.3389/fonc.2018.00510
106. Rotman LE, Vaughan TB, Hackney JR, Riley KO. Long-term survival after transformation of an adrenocorticotropic hormone-secreting pituitary macroadenoma to a silent corticotroph pituitary carcinoma. World Neurosurg. (2019) 122:417–23. doi: 10.1016/j.wneu.2018.11.011
107. Alshaikh OM, Asa SL, Mete O, Ezzat S. An institutional experience of tumor progression to pituitary carcinoma in a 15-year cohort of 1055 consecutive pituitary neuroendocrine tumors. Endocr Pathol. (2019) 30:118–27. doi: 10.1007/s12022-019-9568-5
108. Donofrio CA, Pizzimenti C, Djoukhadar I, Kearney T, Gnanalingham K, Roncaroli F. Colorectal carcinoma to pituitary tumour: tumour to tumour metastasis. Br J Neurosurg. (2023) 37:1367–70. doi: 10.1080/02688697.2020.1823937
109. Soon WC, Czyz M, Dhir J. Metastatic pituitary carcinoma causing cord compression. World Neurosurg. (2020) 139:266–67. doi: 10.1016/j.wneu.2020.03.154
110. Venable ER, Kerr SE, Lopes M, Jones KA, Bellizzi AM, Mounajjed T, et al. Liver metastases from pituitary carcinomas mimicking visceral well-differentiated neuroendocrine tumors: a series of four cases. Diagn Pathol. (2020) 15:81. doi: 10.1186/s13000-020-00997-x
111. Buchfelder M, Schlaffer SM. Surgical treatment of aggressive pituitary adenomas and pituitary carcinomas. Rev Endocr Metab Disord. (2020) 21:253–61. doi: 10.1007/s11154-020-09563-8
112. Lamb LS, Sim HW, McCormack AI. Case report: A case of pituitary carcinoma treated with sequential dual immunotherapy and vascular endothelial growth factor inhibition therapy. Front Endocrinol (Lausanne). (2020) 11:576027. doi: 10.3389/fendo.2020.576027
113. Majd N, Waguespack SG, Janku F, Fu S, Penas-Prado M, Xu M, et al. Efficacy of pembrolizumab in patients with pituitary carcinoma: report of four cases from a phase II study. J Immunother Cancer. (2020) 8(2):e001532. doi: 10.1136/jitc-2020-001532
114. Dai C, Sun B, Guan S, Wang W, Liu H, Li Y, et al. Evolution of a refractory prolactin-secreting pituitary adenoma into a pituitary carcinoma: report of a challenging case and literature review. BMC Endocr Disord. (2021) 21:217. doi: 10.1186/s12902-021-00874-8
115. Sakata K, Ono T, Koga M, Kikuchi J, Komaki S, Akiba J, et al. Primary pituitary adenoid cystic carcinoma: A rare salivary gland-like tumor in the sella. Head Neck Pathol. (2021) 15:1289–98. doi: 10.1007/s12105-020-01256-7
116. Remon-Ruiz P, Venegas-Moreno E, Dios-Fuentes E, Moreno J, Fernandez PI, Garcia MA, et al. A silent corticotroph pituitary carcinoma: lessons from an exceptional case report. Front Endocrinol (Lausanne). (2021) 12:784889. doi: 10.3389/fendo.2021.784889
117. Du Four S, van der Veken J, Duerinck J, Vermeulen E, Andreescu CE, Bruneau M, et al. Pituitary carcinoma - case series and review of the literature. Front Endocrinol (Lausanne). (2022) 13:968692. doi: 10.3389/fendo.2022.968692
118. Demir MK, Yapicier O, Oral A, Yilmaz B, Kilic T. Non-functional recurrent pituitary adenoma with intracranial metastasis-Pituitary carcinoma: A case report and review of the literature. Neurochirurgie. (2022) 68:106–12. doi: 10.1016/j.neuchi.2021.02.006
119. Sumislawski P, Rotermund R, Klose S, Lautenbach A, Wefers AK, Soltwedel C, et al. ACTH-secreting pituitary carcinoma with TP53, NF1, ATRX and PTEN mutations Case report and review of the literature. Endocrine. (2022) 76:228–36. doi: 10.1007/s12020-021-02954-0
120. Stelmachowska-Banas M, Maksymowicz M, Kolasinska-Cwikla A, Zielinski G, Korbonits M, Zgliczynski W. Pituitary carcinoma as a rare cause of liver metastases successfully treated with temozolomide. Pol Arch Intern Med. (2022) 132(3):16178. doi: 10.20452/pamw.16178
121. Zhang H, Li J, Lee M, Ho CL. Pituitary carcinoma in a patient with cowden syndrome. Am J Case Rep. (2022) 23:e934846. doi: 10.12659/AJCR.934846
122. Kwok MM, Virk JS, Michael M, McKinley M, Magarey M. Cervical nodal metastatic pituitary carcinoma: A case report. Ear Nose Throat J. (2022) 101:110–13. doi: 10.1177/0145561320944649
123. Yearley AG, Chalif EJ, Gupta S, Chalif JI, Bernstock JD, Nawabi N, et al. Metastatic pituitary tumors: an institutional case series. Pituitary. (2023) 26:561–72. doi: 10.1007/s11102-023-01341-4
124. Stewart PM, Carey MP, Graham CT, Wright AD, London DR. Growth hormone secreting pituitary carcinoma: a case report and literature review. Clin Endocrinol (Oxf). (1992) 37:189–94. doi: 10.1111/j.1365-2265.1992.tb02306.x
125. Dekkers OM, Karavitaki N, Pereira AM. The epidemiology of aggressive pituitary tumors (and its challenges). Rev Endocr Metab Disord. (2020) 21:209–12. doi: 10.1007/s11154-020-09556-7
126. Liu JK, Patel J, Eloy JA. The role of temozolomide in the treatment of aggressive pituitary tumors. J Clin Neurosci. (2015) 22:923–29. doi: 10.1016/j.jocn.2014.12.007
127. Philippon M, Morange I, Barrie M, Barlier A, Taieb D, Dufour H, et al. Long-term control of a MEN1 prolactin secreting pituitary carcinoma after temozolomide treatment. Ann Endocrinol (Paris). (2012) 73:225–29. doi: 10.1016/j.ando.2012.03.001
128. Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB. Clinical review: Diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. (2005) 90:3089–99. doi: 10.1210/jc.2004-2231
129. Yang Z, Zhang T, Gao H. Genetic aspects of pituitary carcinoma: A systematic review. Med (Baltimore). (2016) 95:e5268. doi: 10.1097/MD.0000000000005268
130. Tsukamoto T, Miki Y. Imaging of pituitary tumors: an update with the 5th WHO Classifications-part 1. Pituitary neuroendocrine tumor (PitNET)/pituitary adenoma. Jpn J Radiol. (2023) 41:789–806. doi: 10.1007/s11604-023-01400-7
131. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol. (2022) 33:6–26. doi: 10.1007/s12022-022-09703-7
132. McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. (2018) 178:265–76. doi: 10.1530/EJE-17-0933
133. Kasuki L, Raverot G. Definition and diagnosis of aggressive pituitary tumors. Rev Endocr Metab Disord. (2020) 21:203–08. doi: 10.1007/s11154-019-09531-x
134. Heaney AP. Clinical review: Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab. (2011) 96:3649–60. doi: 10.1210/jc.2011-2031
135. Scheithauer BW, Kurtkaya-Yapicier O, Kovacs KT, Young WJ, Lloyd RV. Pituitary carcinoma: a clinicopathological review. Neurosurgery. (2005) 56:1066–74.
136. Thapar K, Kovacs K, Scheithauer BW, Stefaneanu L, Horvath E, Pernicone PJ, et al. Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an analysis using the MIB-1 antibody. Neurosurgery. (1996) 38:99–106, 106-07. doi: 10.1097/00006123-199601000-00024
137. Scheithauer BW, Gaffey TA, Lloyd RV, Sebo TJ, Kovacs KT, Horvath E, et al. Pathobiology of pituitary adenomas and carcinomas. Neurosurgery. (2006) 59:341–53. doi: 10.1227/01.NEU.0000223437.51435.6E
138. McCormack AI, Wass JA, Grossman AB. Aggressive pituitary tumours: the role of temozolomide and the assessment of MGMT status. Eur J Clin Invest. (2011) 41:1133–48. doi: 10.1111/j.1365-2362.2011.02520.x
139. Hirohata T, Asano K, Ogawa Y, Takano S, Amano K, Isozaki O, et al. DNA mismatch repair protein (MSH6) correlated with the responses of atypical pituitary adenomas and pituitary carcinomas to temozolomide: the national cooperative study by the Japan Society for Hypothalamic and Pituitary Tumors. J Clin Endocrinol Metab. (2013) 98:1130–36. doi: 10.1210/jc.2012-2924
140. Bengtsson D, Schroder HD, Andersen M, Maiter D, Berinder K, Feldt RU, et al. Long-term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab. (2015) 100:1689–98. doi: 10.1210/jc.2014-4350
141. Kontogeorgos G. Update on pituitary adenomas in the 2017 World Health Organization classification: innovations and perspectives. Hormones (Athens). (2021) 20:287–91. doi: 10.1007/s42000-020-00269-9
142. Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, Young WJ, et al. Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer. (1997) 79:804–12. doi: 10.1002/(sici)1097-0142(19970215)79:4<804::aid-cncr18>3.0.co;2-3
143. Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M. Ki-67 in pituitary neoplasms: a review–part I. Neurosurgery. (2009) 65:429–37. doi: 10.1227/01.NEU.0000349930.66434.82
144. Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol. (2013) 126:123–35. doi: 10.1007/s00401-013-1084-y
145. Lopes M. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. (2017) 134:521–35. doi: 10.1007/s00401-017-1769-8
146. Burger PC, Shibata T, Kleihues P. The use of the monoclonal antibody Ki-67 in the identification of proliferating cells: application to surgical neuropathology. Am J Surg Pathol. (1986) 10:611–17. doi: 10.1097/00000478-198609000-00003
147. Paek KI, Kim SH, Song SH, Choi SW, Koh HS, Youm JY, et al. Clinical significance of Ki-67 labeling index in pituitary macroadenoma. J Korean Med Sci. (2005) 20:489–94. doi: 10.3346/jkms.2005.20.3.489
148. Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol. (2017) 28:228–43. doi: 10.1007/s12022-017-9498-z
149. Ng S, Messerer M, Engelhardt J, Bruneau M, Cornelius JF, Cavallo LM, et al. Aggressive pituitary neuroendocrine tumors: current practices, controversies, and perspectives, on behalf of the EANS skull base section. Acta Neurochir (Wien). (2021) 163:3131–42. doi: 10.1007/s00701-021-04953-6
150. Veit JA, Boehm B, Luster M, Scheuerle A, Rotter N, Rettinger G, et al. Detection of paranasal ectopic adrenocorticotropic hormone-secreting pituitary adenoma by Ga-68-DOTANOC positron-emission tomography-computed tomography. Laryngoscope. (2013) 123:1132–35. doi: 10.1002/lary.23867
151. Jahangiri A, Wagner JR, Pekmezci M, Hiniker A, Chang EF, Kunwar S, et al. A comprehensive long-term retrospective analysis of silent corticotrophic adenomas vs hormone-negative adenomas. Neurosurgery. (2013) 73:8–17, 17-18. doi: 10.1227/01.neu.0000429858.96652.1e
152. Minniti G, Clarke E, Scaringi C, Enrici RM. Stereotactic radiotherapy and radiosurgery for non-functioning and secreting pituitary adenomas. Rep Pract Oncol Radiother. (2016) 21:370–78. doi: 10.1016/j.rpor.2014.09.004
153. Sheehan JP, Starke RM, Mathieu D, Young B, Sneed PK, Chiang VL, et al. Gamma Knife radiosurgery for the management of nonfunctioning pituitary adenomas: a multicenter study. J Neurosurg. (2013) 119:446–56. doi: 10.3171/2013.3.JNS12766
154. Deng WC, Yan JL, Chuang CC, Wei KC, Chang CN, Wu CT, et al. Adjuvant radiation therapy compared with observation alone for postoperative residual nonfunctional pituitary adenomas. World Neurosurg. (2019) 128:e1024–33. doi: 10.1016/j.wneu.2019.05.066
155. Ding D, Starke RM, Sheehan JP. Treatment paradigms for pituitary adenomas: defining the roles of radiosurgery and radiation therapy. J Neurooncol. (2014) 117:445–57. doi: 10.1007/s11060-013-1262-8
156. Minniti G, Flickinger J, Tolu B, Paolini S. Management of nonfunctioning pituitary tumors: radiotherapy. Pituitary. (2018) 21:154–61. doi: 10.1007/s11102-018-0868-4
157. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
158. Raverot G, Ilie MD, Lasolle H, Amodru V, Trouillas J, Castinetti F, et al. Aggressive pituitary tumours and pituitary carcinomas. Nat Rev Endocrinol. (2021) 17:671–84. doi: 10.1038/s41574-021-00550-w
159. Campdera M, Palacios N, Aller J, Magallon R, Martin P, Saucedo G, et al. Temozolomide for aggressive ACTH pituitary tumors: failure of a second course of treatment. Pituitary. (2016) 19:158–66. doi: 10.1007/s11102-015-0694-x
160. Losa M, Bogazzi F, Cannavo S, Ceccato F, Curto L, De Marinis L, et al. Temozolomide therapy in patients with aggressive pituitary adenomas or carcinomas. J Neurooncol. (2016) 126:519–25. doi: 10.1007/s11060-015-1991-y
161. Bruno OD, Juarez-Allen L, Christiansen SB, Manavela M, Danilowicz K, Vigovich C, et al. Temozolomide therapy for aggressive pituitary tumors: results in a small series of patients from Argentina. Int J Endocrinol. (2015) 2015:587893. doi: 10.1155/2015/587893
162. Whitelaw BC, Dworakowska D, Thomas NW, Barazi S, Riordan-Eva P, King AP, et al. Temozolomide in the management of dopamine agonist-resistant prolactinomas. Clin Endocrinol (Oxf). (2012) 76:877–86. doi: 10.1111/j.1365-2265.2012.04373.x
163. Bush ZM, Longtine JA, Cunningham T, Schiff D, Jane JJ, Vance ML, et al. Temozolomide treatment for aggressive pituitary tumors: correlation of clinical outcome with O(6)-methylguanine methyltransferase (MGMT) promoter methylation and expression. J Clin Endocrinol Metab. (2010) 95:E280–90. doi: 10.1210/jc.2010-0441
164. Raverot G, Sturm N, de Fraipont F, Muller M, Salenave S, Caron P, et al. Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: a French multicenter experience. J Clin Endocrinol Metab. (2010) 95:4592–99. doi: 10.1210/jc.2010-0644
165. Losa M, Mazza E, Terreni MR, McCormack A, Gill AJ, Motta M, et al. Salvage therapy with temozolomide in patients with aggressive or metastatic pituitary adenomas: experience in six cases. Eur J Endocrinol. (2010) 163:843–51. doi: 10.1530/EJE-10-0629
166. Ceccato F, Lombardi G, Manara R, Emanuelli E, Denaro L, Milanese L, et al. Temozolomide and pasireotide treatment for aggressive pituitary adenoma: expertise at a tertiary care center. J Neurooncol. (2015) 122:189–96. doi: 10.1007/s11060-014-1702-0
167. Lasolle H, Cortet C, Castinetti F, Cloix L, Caron P, Delemer B, et al. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. Eur J Endocrinol. (2017) 176:769–77. doi: 10.1530/EJE-16-0979
168. Syro LV, Rotondo F, Ortiz LD, Kovacs K. 65 YEARS OF THE DOUBLE HELIX: Treatment of pituitary tumors with temozolomide: an update. Endocr Relat Cancer. (2018) 25:T159–69. doi: 10.1530/ERC-18-0015
169. Kontogeorgos G, Thodou E, Koutourousiou M, Kaltsas G, Seretis A. MGMT immunohistochemistry in pituitary tumors: controversies with clinical implications. Pituitary. (2019) 22:614–19. doi: 10.1007/s11102-019-00993-5
170. Lamas C, Camara R, Fajardo C, Remon-Ruiz P, Biagetti B, Guerrero-Perez F, et al. Efficacy and safety of temozolomide in the treatment of aggressive pituitary neuroendocrine tumours in Spain. Front Endocrinol (Lausanne). (2023) 14:1204206. doi: 10.3389/fendo.2023.1204206
171. Varlamov EV, McCartney S, Fleseriu M. Functioning pituitary adenomas - current treatment options and emerging medical therapies. Eur Endocrinol. (2019) 15:30–40. doi: 10.17925/EE.2019.15.1.30
172. Wang PF, Wang TJ, Yang YK, Yao K, Li Z, Li YM, et al. The expression profile of PD-L1 and CD8(+) lymphocyte in pituitary adenomas indicating for immunotherapy. J Neurooncol. (2018) 139:89–95. doi: 10.1007/s11060-018-2844-2
173. Duhamel C, Ilie MD, Salle H, Nassouri AS, Gaillard S, Deluche E, et al. Immunotherapy in corticotroph and lactotroph aggressive tumors and carcinomas: two case reports and a review of the literature. J Pers Med. (2020) 10(3):88. doi: 10.3390/jpm10030088
Keywords: pituitary carcinoma, metastasis, temozolomide, radiotherapy, Ki-67, p53
Citation: Yang Y, Liang W, Fan K, Yang T and Cheng J (2024) Clinical features of pituitary carcinoma: analysis based on a case report and literature review. Front. Endocrinol. 15:1440247. doi: 10.3389/fendo.2024.1440247
Received: 29 May 2024; Accepted: 09 October 2024;
Published: 31 October 2024.
Edited by:
Chandrasekaran Kaliaperumal, University of Edinburgh, United KingdomReviewed by:
Stephanie Du Four, University Hospital Brussels, BelgiumYuan Shan, Baylor Scott and White Health, United States
Lyu Louie Hao, Shenzhen Second People’s Hospital, China
Copyright © 2024 Yang, Liang, Fan, Yang and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingmin Cheng, ZG9jdG9yX2NqbUAxNjMuY29t; Tao Yang, eWFuZy10YW9AMTM5LmNvbQ==
†These authors have contributed equally to this work