 J. S. Saavedra T.1
J. S. Saavedra T.1 Humberto Alejandro Nati-Castillo2
Humberto Alejandro Nati-Castillo2 L. A. Valderrama Cometa3
L. A. Valderrama Cometa3 Wilfredo A. Rivera-Martínez4
Wilfredo A. Rivera-Martínez4 Josué Asprilla5C. M. Castaño-Giraldo6Leonardo Sánchez S.7Mishell Heredia-Espín7
Josué Asprilla5C. M. Castaño-Giraldo6Leonardo Sánchez S.7Mishell Heredia-Espín7 Marlon Arias-Intriago7
Marlon Arias-Intriago7 Juan S. Izquierdo-Condoy7*
Juan S. Izquierdo-Condoy7*- 1Family Medicine Department, Universidad Javeriana, Cali, Colombia
- 2Interinstitutional Group on Internal Medicine (GIMI 1), Department of Internal Medicine, Universidad Libre, Cali, Colombia
- 3Organ and Tissue Transplant Unit, Clínica Imbanaco, Cali, Colombia
- 4Facultad de Medicina, Universidad de Antioquia, Medellin, Colombia
- 5Division of Pathology, Clínica Imbanaco, Grupo Quirónsalud, Cali, Colombia
- 6Facultad de ciencias de la Salud, Universidad del Quindío, Armenia, Colombia
- 7One Health Research Group, Universidad de las Americas, Quito, Ecuador
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors derived from chromaffin cells, with 80–85% originating in the adrenal medulla and 15–20% from extra-adrenal chromaffin tissues (paragangliomas). Approximately 30–40% of PPGLs have a hereditary component, making them one of the most genetically predisposed tumor types. Recent advances in genetic research have classified PPGLs into three molecular clusters: pseudohypoxia-related, kinase-signaling, and WNT-signaling pathway variants. Specifically, the detection of SDHB-related tumors indicates an increased risk of metastatic disease, which may impact decisions regarding functional imaging in patients with high suspicion of metastasis and influence targeted treatment strategies. Diagnosis of PPGLs primarily relies on biochemical testing, measuring catecholamines or their metabolites in plasma or urine. However, molecular testing, functional imaging, and targeted therapies have greatly enhanced diagnostic precision and management. Personalized treatment approaches based on genetic profiling are becoming integral to the clinical management of these tumors. In South American countries like Colombia, functional imaging techniques such as positron emission tomography/computed tomography (PET/CT) with tracers like 18F-DOPA, 18F-fluorodeoxyglucose (18F-FDG), and 68Ga-DOTA-conjugated somatostatin receptor-targeting peptides (68Ga-DOTA-SST) are used to guide follow-up and treatment strategies. Radionuclide therapy with lutetium-177 DOTATATE is employed for patients showing uptake in 68Ga-DOTA-SST PET/CT scans, while access to 131-MIBG therapy remains limited due to high costs and availability. Recent clinical trials have shown promise for systemic therapies such as sunitinib and cabozantinib, offering potential new options for patients with slow or moderate progression of PPGLs. These advancements underscore the potential of personalized and targeted therapies to improve outcomes in this challenging patient population.
1 Introduction
Pheochromocytomas and paragangliomas (PPGLs) are closely related tumors that originate from neuroendocrine cells, arising from chromaffin cells in the adrenal medulla and neural crest progenitors located outside of adrenal gland, respectively (1). These tumors are characterized by a proliferation of chromaffin cells arranged in clustered or trabecular patterns (2, 3), Although rare, occurring in fewer than 0.1% of individuals per million (4, 5), pheochromocytomas and sympathetic paragangliomas in particular require prompt treatment to reduce associated morbidity and mortality (3, 6).
Paragangliomas (PGLs) arise in sympathetic and parasympathetic paraganglia (7–9). Those in the head and neck are predominantly parasympathetic, typically non-metastatic, and often present as palpable masses, while abdominal PGLs arise from the sympathetic neuroendocrine system and share origins with pheochromocytomas (2). Notably, carotid body tumors, a type of PGL, are highly vascularized glomus tumors located at the carotid bifurcation, where the external and internal carotid arteries diverge (10–13).
PPGL, whether located in the adrenal medulla or at extramedullary sites, secrete excessive amounts of catecholamines, adrenaline, noradrenaline, and/or dopamine. There is a subdivision based on the genotype of PPGLs in cluster 1 for variants in pseudohypoxia genes, cluster 2 for alterations in the kinase pathway and cluster 3 in WNT signaling. This classification in cases such as SDHB-related tumors defines the prognosis of developing metastatic disease and can modify the conduct of treatment and surveillance (14–17). Cluster 1 tumors such as VHL typically produce norepinephrine, whereas cluster 2, MEN2, or NF1 tumors are more likely to produce epinephrine, and SDHB, SDHC, and SDHD-related tumors may secrete dopamine and norepinephrine (18, 19).
2 Epidemiology
Pheochromocytomas occur with an estimated incidence of 0.05%, primarily in adults aged 30-50 (20). They constitute around 4% of incidental adrenal masses and are implicated in approximately 0.1% of hypertension cases. Demographically, pheochromocytoma affects adults of both sexes, typically between 30 and 50 years of age, presenting, and they appear with similar frequency in both adrenal glands (2, 4, 5).
Around 20% of PPGL present metastases. 70% of pheochromocytomas are sporadic, while the remaining 30% are associated with hereditary syndromes such as multiple endocrine neoplasia type 2 (MEN II), von Hippel-Lindau disease (VHL), neurofibromatosis type 1 (NF1), and familial paraganglioma syndrome (21, 22).
Traditionally, pheochromocytomas were managed under the “10% rule,” (23), which suggested that 10% occur in children, 10% are extra-adrenal, 10% are familial, 10% are bilateral in adrenal glands, and 10% are metastatic (24). However, it is currently not recommended to refer to it as “the 10 percent tumor,” since approximately 25% of patients with apparently sporadic pheochromocytoma may carry pathogenic variants (18).
3 Clinical predictor of metastasis
All PPGLs have metastatic potential, however histological characteristics do not allow differentiating “benign” from “malignant” tumors, so the latest WHO update recommends changing these terms to metastatic, when there is evidence of distant tumor (25). The ESMO guidelines, for their part, suggest using the definition of “high risk of metastasis” when one or more of the following criteria are present: (a) tumor size greater than or equal to 5 cm; (b) any extra-adrenal PPGL; (c) known germline SDHB pathogenic variant; or (d) plasma 3MT > 3 times above the upper limit of normal (26). Certain tumors, particularly SDHB-related PGLs of the head and neck, can produce dopamine. In such cases, detecting its metabolite, methoxytyramine, in blood has shown only a modest improvement in detecting head and neck PPGL (27, 28). Although its use as a metastatic risk marker has been considered, its performance remains limited (29), and it is unavailable in most countries around the world. Management guidelines do not recommend its measurement (30, 31).
Histologically, pheochromocytomas are characterized by pleomorphic cellular nests, with large ball-shaped chromaffins that show strong positivity for chromogranin A, synaptophysin, CD56, and focal S100 (Figure 1). There are no definitive histological criteria for malignancy; thus, the term “metastatic tumor” is preferred when chromaffin tissue invasion is confirmed beyond the site of origin into distant organs (32, 33). It is also important to assess histological criteria for aggressive biological behavior, including an insular pattern of growth, mitotic activity, and invasion of capsular blood and lymphatic vessels (34). Scores like PASS (Pheochromocytoma of the Adrenal Gland Scaled Score), Grading of Adrenal Pheochromocytoma and Paraganglioma, COOPS (Composite Pheochromocytoma/Paraganglioma Prognostic Score) and multivariate predictive models (SGAP-Score and ASES/ASS-Score) have been developed to identify PPGL patients with increased metastatic potential. However, immunohistochemistry may yield inconsistent results, lacks molecular testing which is more reliable, and scores such as PASS lack clinical validation studies supporting their application (35). These methods are not currently utilized in Colombia or Ecuador (30, 36).
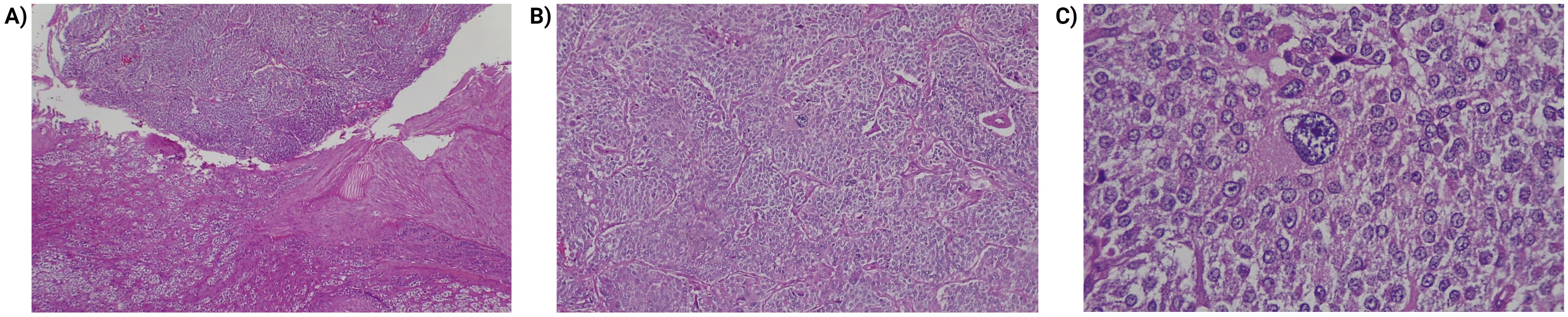
Figure 1. Histopathologic findings of pheochromocytoma. (A) Transition between the usual histology adrenal cortex (lower part of the image) and the tumor (upper part). Hematoxylin and eosin 40x. (B) Pheochromocytoma, cells arranged in a pattern in nests (zellballen) and trabeculae. Hematoxylin and eosin 40x. (C) Pheochromocytoma, lesion cells are large, polygonal, show fine, granular cytoplasm. Note the presence of pleomorphism. Hematoxylin and eosin 400x.
On the other hand, there are more promising clinical predictors, TNM staging may be correlated with overall survival in PPGL. Jimenez et al. (37), found that a large primary tumor, an extra-adrenal location, infiltration of surrounding tissues by the primary tumor, and regional lymph node metastasis are associated with a higher risk of distant metastasis and consequently decreased overall survival, and in turn, patients with distant metastasis (stage IV) have the worst prognosis.
4 Hereditary and phenotypic pattern
4.1 Susceptibility genes
PPGL are associated with germline pathogenic variants at higher rates than any other solid tumor. Rates of germline pathogenic variants vary by tumor type: 25% in pheochromocytoma, 40% in PGL, and up to 50% in patients presenting with metastatic disease. Patients with pathogenic variants generally present with PPGL at a younger age and are more likely to have multifocal disease (18). 30% to 40% of cases occur in the context of a genetic syndrome, however, in almost half of apparently sporadic PPGLs somatic pathogenic variants are found in one of the susceptibility genes, which means that at least three quarters can be classified into a defined cluster.
The susceptibility to pheochromocytoma can be linked to germline pathogenic variants in the RET proto-oncogene and tumor suppressor genes such as von Hippel-Lindau (VHL), and Neurofibromatosis type 1 (NF1) (2, 38, 39). Hereditary paraganglioma syndromes are caused by pathogenic variants in the succinate dehydrogenase subunit (SDHx) genes: SDHD, SDHAF2, SDHC, SDHB, SDHA (Paraganglioma syndromes 1-5, respectively) (40). The VHL gene, located on the short arm of chromosome 3 (3p25.3), has over 1,500 identified pathogenic variants in patients with VHL disease, with 20% of these being de novo pathogenic variants, as observed in both PGLs and pheochromocytomas (8, 23, 41). Other pathogenic variants in susceptibility genes such as TMEM127, MAX, FH and MDH2 are associated with PPGL syndromes with a lower pathogenic variant frequency (42, 43).
The discovery of syndromes linked to pathogenic variants in genes has shown that the probability of metastasis may vary, being no higher than 12% in SDHC, THEM127, NF1, VHL, RET and MAX, but for SDHD, SDHA and SDHB it reaches 29, 66 and 75%, respectively (30, 42, 44–46).
4.2 Inheritance pattern
Pheochromocytoma is currently known to result from disorders with an autosomal dominant inheritance pattern in most cases, such as multiple endocrine neoplasia type 2 (MEN-2) (presenting manifestation is medullary thyroid cancer in 60%, medullary thyroid carcinoma and synchronous pheochromocytoma in 34%, and pheochromocytoma in 6%. 72% have bilateral pheochromocytoma, 82% of tumors are synchronous and are unlikely to be metastatic) and von Hippel-Lindau disease (with retinal angioma, central nervous system hemangioblastoma, renal cell carcinoma, pancreatic cysts, and epididymal cystadenoma) (24, 43, 47). The precise frequency of these syndromes in patients with pheochromocytoma is not fully known (2, 38, 39, 43). The exception are pathogenic variants in the SDHAF2 and SDHD genes, in which maternal imprinting occurs with silencing of the maternal allele and therefore only pathogenic variants inherited from the father will cause the disease, they probably have the highest penetrance, greater than 50% and are usually associated with PPGL in the head, neck and chest (30, 44–46). On the other hand, the inheritance pattern for variants in the MAX gene is not clear; it is believed that its penetrance is high and it usually presents as bilateral pheochromocytomas and abdominal PGL (48).
4.3 Molecular phenotype
The genome of PPGL has been characterized, providing valuable insights into the genetic factors driving tumorigenesis and the degree of genetic instability (49, 50) (Figure 2). These tumors are generally characterized by a relatively low degree of genetic instability at both the nucleotide and chromosome levels. Although there are rare tumors that behave differently from others, most of the genes associated with the development of PPGL are categorized into three clusters based on the mechanism of tumorigenesis (49–51):
● Pseudohypoxia pathway (cluster 1), tumors that infiltrate stromal cells, which have been associated with pathogenic variants in the genes EGLN1, EGLN2, DLST, FH, IDH3B, MDH2, SDHA, SDHAF2, SDHB, SDHC, SDHD, VHL, EPAS1, IDH1, and IDH2.
● Signaling kinase pathway (cluster 2) has been associated with pathogenic variants in the NF1, MAX, MERTK, MET, MYCN, RET, or TMEM127 genes.
● In cluster 3, MAML3 fusion gene and CSDE1 somatic pathogenic variants affect and overactivate the Wnt/β-catenin pathway, which is responsible for the regulation of metabolism, angiogenesis, proliferation, and invasion.
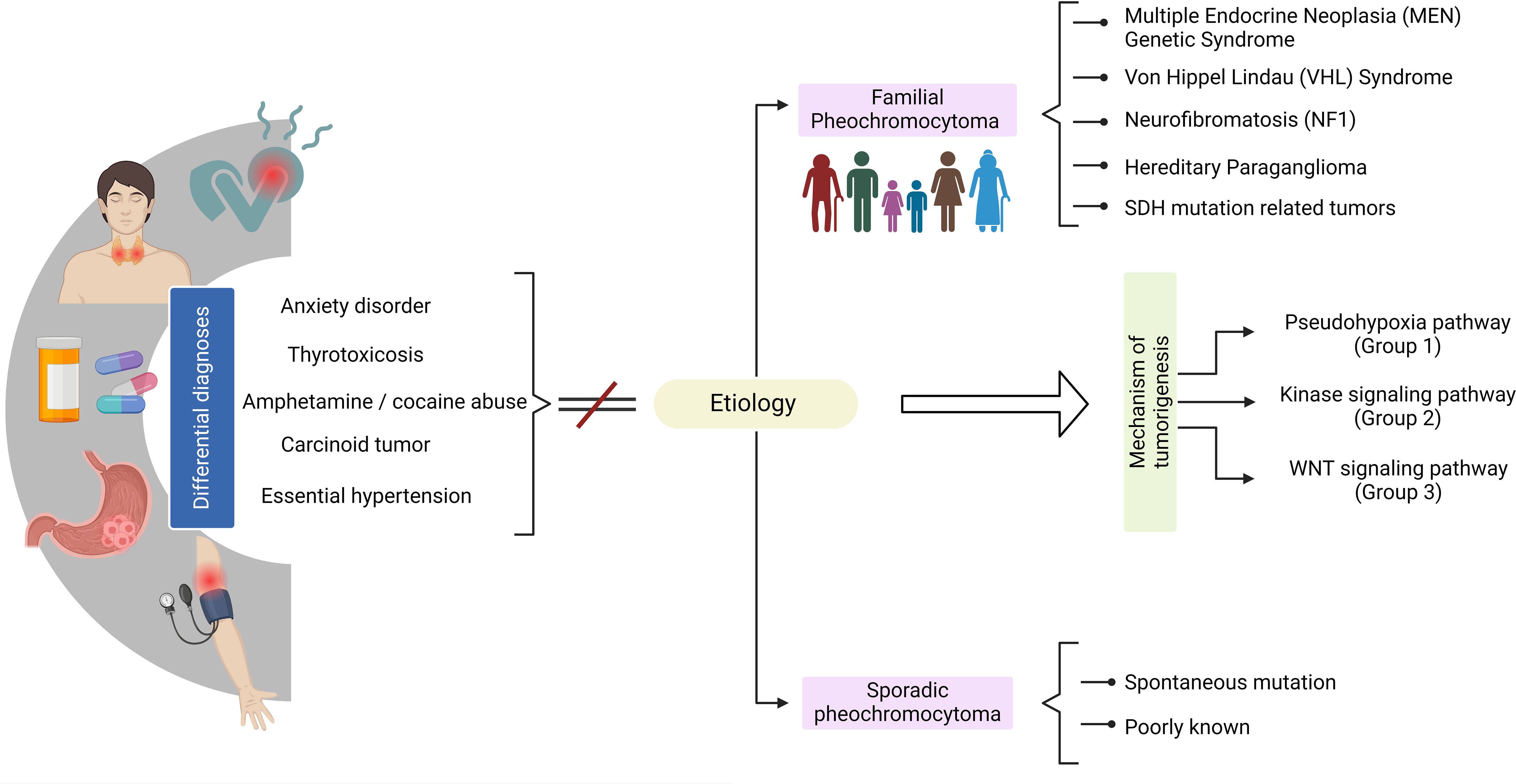
Figure 2. Clinical overview of pheochromocytoma diagnosis.
Given the above pathogenic variants, one of them stands out for its potential usefulness in the prognosis of the disease. SDH (succinate dehydrogenase) is an important enzyme in energy formation pathways, taking action in the Krebs cycle and in the Electron Transfer chain within the mitochondria, where is conforming the complex ll with four functional subunits: A, B, C, and D. Inactivating germline pathogenic variants results in loss of function of SDH and, therefore, an elevation in succinate levels which diffuses back to the cytoplasm and inhibits prolyl hydroxylases, resulting in further stabilization of the Hypoxia-Inducible Factor (HIF) pathway of tumorigenesis. SDHB related tumors, are commonly found in the abdomen, have a high potential for recurrence, local and distant metastasis compared to SDHD and SDHC tumors, which are commonly found in the head and neck areas, each of them requiring follow-up for the possibility of relapse or extension of the disease (52, 53).
5 Clinical presentation
Patients with pheochromocytoma are often described as presenting with the “classic triad” of diaphoresis, headache, and palpitations, typically accompanied by hypertension (22). In a review of 200 cases, Ando et al. found that PPGL attacks are associated with multisystem involvement, 99% cardiac damage, 44% pulmonary damage, and 21.5% renal damage (54). Sustained hypertension is observed in approximately 50-55% of cases, while paroxysmal hypertension occurs in 30-45% (24, 48, 55). Additionally, hyperhidrosis occurs in 60% of cases, often accompanied by hypertensive crises (56, 57). Headache is one of the most common symptoms, and may occur in up to 40% of cases (4, 54, 58). Headache is related to the transient elevation of blood pressure. Instead of sustained hypertension, PPGL headache can occur suddenly as cluster headache, however they are usually bilateral, associated with hypertension and hyperhidrosis (Figure 3) (54, 59, 60).
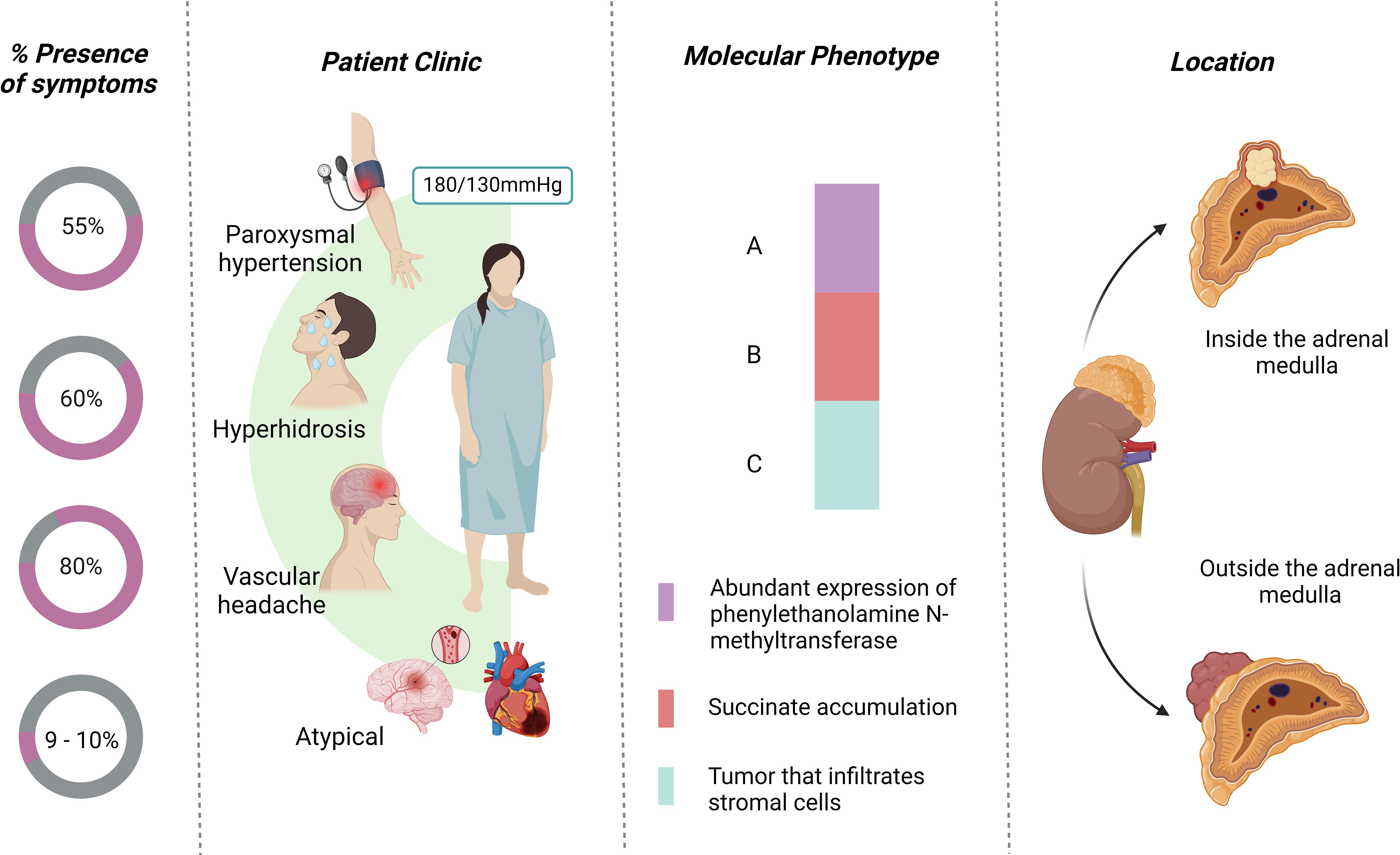
Figure 3. Characteristics of the presentation of pheochromocytoma.
PPGL is presented with sustained or episodic hypertension, sweating, palpitations, hyperglycemia, and glycosuria (5). Although some tumors produce dopamine, the majority secrete noradrenaline and adrenaline (5, 61). PPGLs may experience episodes of severe hypertensive crisis, with an increased likelihood of developing acute kidney injury. Over time, the vasoconstrictive effect of catecholamines released by the tumor leads to chronic kidney disease being a potential complication in PPGLs (62).
Giant pheochromocytomas, defined as those larger than 7 cm, are rare (63–65). These tumors often do not present with classic symptoms; most patients report vague discomfort, and a few may present with a palpable abdominal mass (34, 65, 66).
Table 1 summarizes the clinical translation of the conditions in patients with pheochromocytoma and sympathetic paragangliomas, and their possible complications.
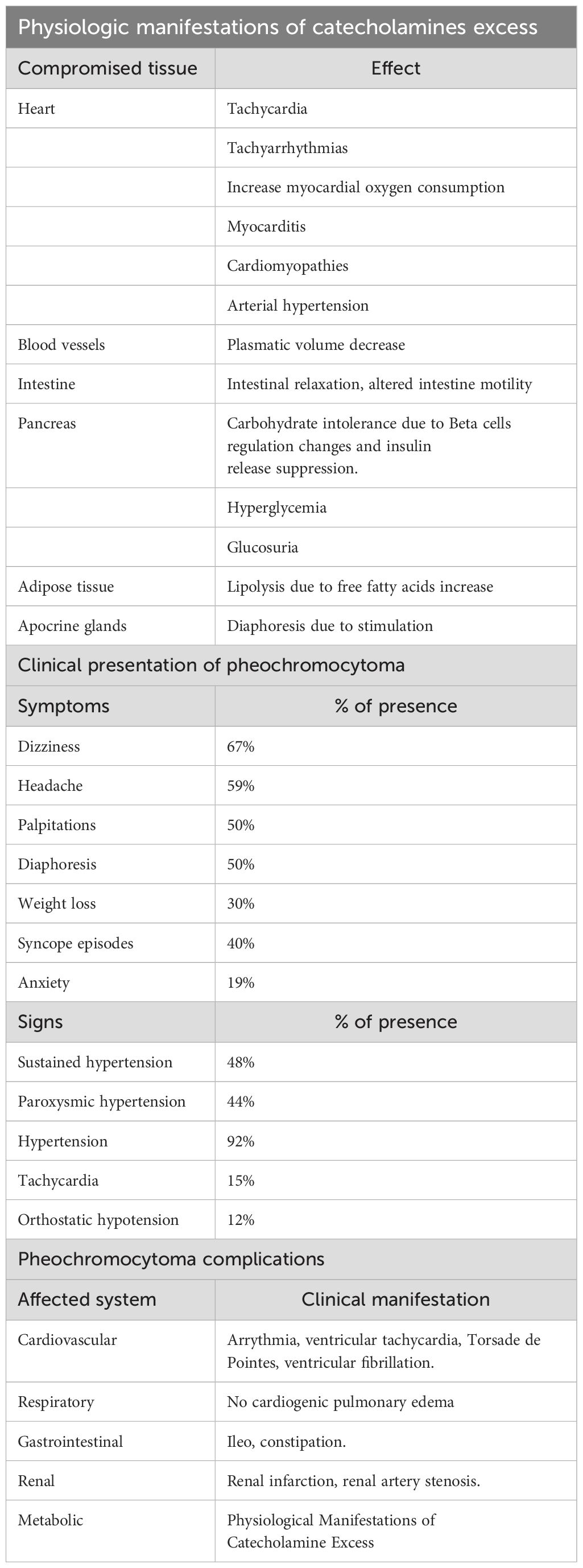
Table 1. Synthesis of clinical expression from diagnosis to complications of pheochromocytoma and sympathetic paragangliomas.
Signs and symptoms alone are often nonspecific, and relying solely on them can lead to diagnostic errors. To improve diagnostic accuracy, efforts have been made to develop diagnostic scores that emphasize the most specific clinical features, such as pallor, hyperhidrosis, and palpitations (Table 2) (67).
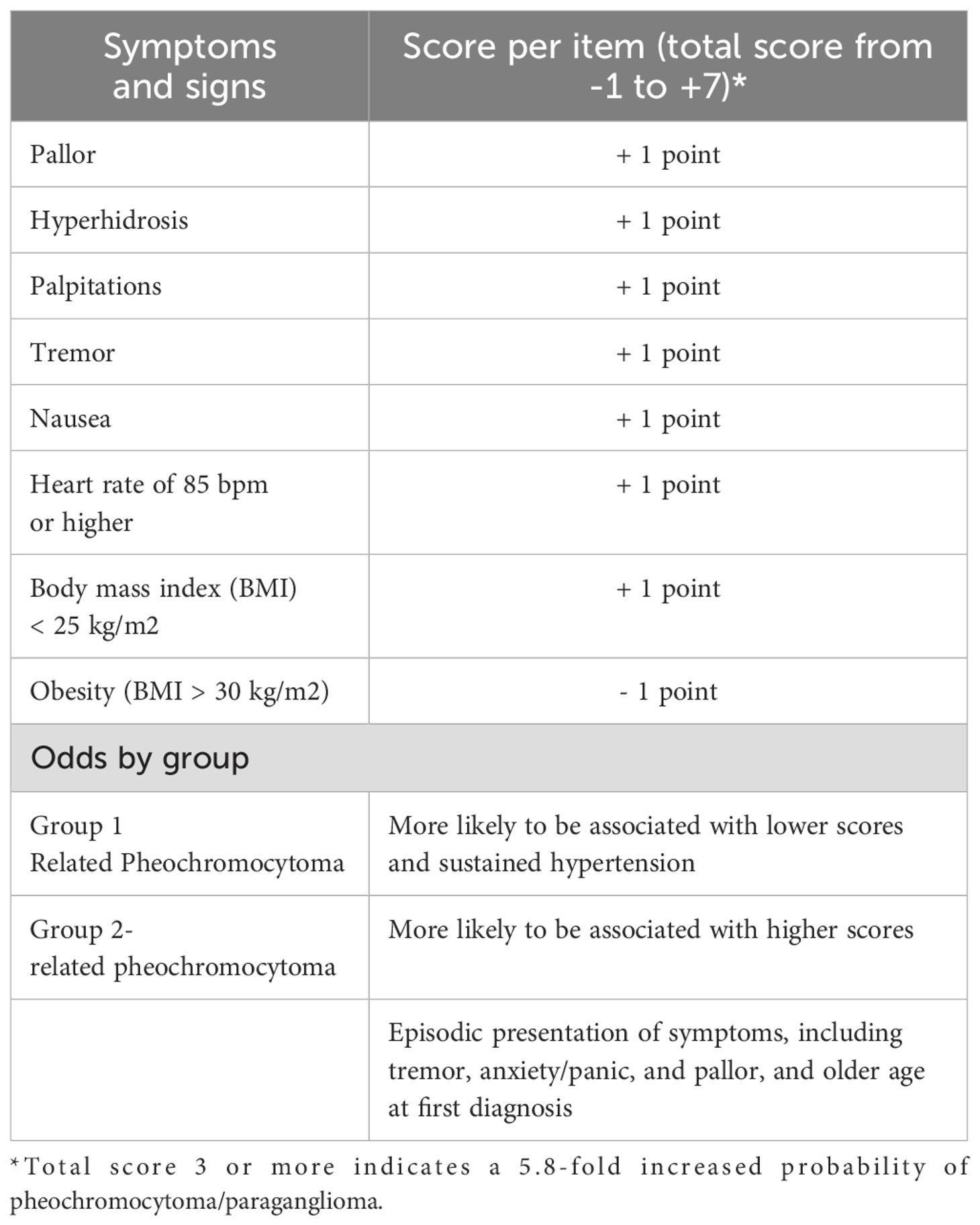
Table 2. Scoring system to classify the probability of pheochromocytoma/paraganglioma according to symptoms and clinical signs.
6 Diagnosis
6.1 Laboratory tests
Diagnostic testing is crucial for confirming PPGL, although it is typically performed under specific conditions, such as the presence of known germline pathogenic variants, a history of PPGL, detection of an incidental adrenal or extra-adrenal mass suggestive of these tumors, or presentation with relevant clinical signs and symptoms (17). The 24-hour plasma metanephrine test offers the highest sensitivity (96%) and a specificity of 85% (68, 69). Studies have shown that plasma normetanephrine levels above 2.5 pmol/mL or metanephrine levels above 1.4 pmol/mL are highly indicative of pheochromocytoma, with 100% specificity. To reduce false-positive results, it is recommended that blood samples be collected with the patient in a supine position after being recumbent for at least 30 minutes (2).
The 24-hour urinary collection for catecholamines and metanephrines is another accessible test, providing a sensitivity of 87.5% and a specificity of 99.7%. Linking urinary metanephrine levels to urinary creatinine further enhances accuracy (70).
It is important to note, however, that catecholamine measurements are only informative when levels are elevated, as in a catecholaminergic crisis, which is typically not present at diagnosis. Physical activity and psychological stress can increase plasma and urinary metanephrine levels; thus, minimizing these factors before sampling is recommended. Additionally, various commonly used medications—including tricyclic antidepressants, monoamine oxidase inhibitors, atypical antipsychotics, selective adrenergic receptor blockers, stimulants, sympathomimetics, paracetamol, sulfasalazine, and amoxicillin—can interfere with test results, so these should be avoided prior to testing, if possible (71).
The clonidine suppression test helps differentiate elevated plasma norepinephrine levels due to sympathetic nerve release from those due to pheochromocytoma (70, 72). Clonidine, a centrally acting alpha-2 agonist, suppresses neuronal norepinephrine release (73, 74). However, chromaffin cells in pheochromocytomas are not regulated by clonidine and continue to release catecholamines inappropriately (73, 75). This test is thus useful in evaluating false-positive results for pheochromocytoma (76), and to reliably differentiate pheochromocytomas from essential hypertension. This test has a sensitivity of 97% and a specificity of 100%. A decrease in plasma norepinephrine levels below 50% following clonidine administration is considered normal, while persistent elevations suggest pheochromocytoma (73, 74).
6.2 Imaging study
Once biochemical analyses suggest the presence of pheochromocytoma, imaging studies are recommended to locate the tumor. A computed tomography (CT) scan of the abdomen and pelvis is typically the initial imaging test of choice (2). CT has a sensitivity of 88%, being useful in the localization of pheochromocytomas larger than 1.3 cm in diameter with an accuracy of 90 to 95%, and represents the most common imaging method used in the diagnosis of pheochromocytomas (77). In comparison, magnetic resonance imaging, with differences in access and costs, offers better spatial resolution (65). Another imaging modality, 123I-metaiodobenzylguanidine (MIBG) scintigraphy, is especially effective for detecting adrenal and extra-adrenal pheochromocytomas (78). Magnetic resonance imaging in the diagnosis shows a differentiated appearance with a sensitivity of 100%, as well as Scintigraphy131 -MBG (sensitivity of 100%), this analogue is located in the adrenergic tissue, it is especially useful to locate extra-adrenal pheochromocytomas (78, 79).
In functional imaging, 68Ga-DOTA-conjugated somatostatin receptor-targeting peptide (68Ga-DOTA-SST) positron emission tomography (PET/CT) has a detection rate of 93% (95% IC 91-95%) (80), making it the first line of functional imaging for patients without known germline pathogenic variants. However, due to the limited availability of 68Ga-DOTA-SST in Latin America, 18F-fluorodeoxyglucose (18F-FDG) PET/CT is often used as a second imaging option in these populations, with a detection rate of 74% (95% CI, 46-91%). 18F-L3,4-dihydroxiphenylalanine (18F-DOPA) PET/CT is available in Colombia and is the first choice in Hereditary pheochromocytoma cluster 2, with an 80% detection rate (95% CI, 69-88%) (17). Alternatively, a vena cava sample can be used to determine plasma catecholamines and metanephrines (81).
6.3 Genetic testing
Since 35-45% of PPGL patients may harbor pathogenic germline variants, genetic testing is recommended for all diagnosed cases, regardless of patient or family history (42, 44, 68). Bilateral tumors and early-onset cases are often associated with inherited syndromes such as VHL, MEN2, and NF1. At a minimum, testing should include FH, NF1, RET, SDHB, SDHD, and VHL genes. Testing for MEN1, SDHA, SDHAF2, SDHC, TMEM127, and MAX is also advised. Carrier testing should be offered to asymptomatic first-degree relatives (and to second-degree relatives in the case of SDHD and SDHAF2, which exhibit maternal imprinting) (42, 44). Following identification of a pathogenic variant, first-degree relatives should be screened (82).
6.4 Immunohistochemistry
Pathogenic variants in SDHB in PPGL patients are associated with a higher risk of tumor progression, and several studies have shown that SDHB pathogenic variation can be detected by the loss of SDHB staining in immunohistochemistry (IHC). This staining loss can serve as an independent IHC biomarker for prognosis. However, this approach is not universally applicable; under normal conditions, SDHB functions as part of the succinate dehydrogenase complex. Pathogenic variants in any gene encoding other complex subunits or auxiliary factors (such as SDHD, SDHC, SDHA, or SDHAF2) disrupt the assembly and functionality of the entire complex, resulting in an absence of SDHB staining on IHC (83).
7 Management and treatment
7.1 Surgical resection of the tumor
In most cases, resection of pheochromocytomas smaller than 5 cm can be effectively performed using minimally invasive laparoscopic surgery. This approach offers significant advantages, including reduced blood loss, less pain, precise dissection, shorter hospital stays, and fewer postoperative complications. The transabdominal approach provides a broader field of view and more space for maneuvering, making it suitable for bilateral tumor removal. Conversely, the retroperitoneal approach allows for unilateral resection with shorter distance to the tumor, minimizing the risk of injury to abdominal organs (2, 30, 84, 85).
Evidence suggests that patients with PPGL associated with malignancy predictors—such as primary tumor size over 5 cm, extra-adrenal location, or SDHB germline pathogenic variants—and those undergoing resection of a primary tumor with synchronous metastases may benefit more from open laparotomy with lymph node dissection (86). In cases with larger tumors or evidence of local invasion, open adrenalectomy may be preferable to ensure complete resection and minimize the risk of capsular rupture, which can lead to tumor seeding, fragmentation, peritoneal dissemination, and local recurrence due to periadrenal invasion (18, 30).
7.2 Preoperative stabilization
Preoperative stabilization is crucial to reducing the risk of uncontrolled hypertension, tachycardia, and volume expansion during surgery (11, 24, 59). According to the Endocrine Society, the preferred preoperative preparation involves alpha-adrenergic receptor blockers. A typical regimen includes phenoxybenzamine, starting at 10 mg orally twice daily and carefully increasing to a maximum of 1 mg/kg/day; however, its availability is limited in many countries (87). Alternatively, selective alpha-1 antagonists like doxazosin are commonly used in regions such as Latin America. They reduce the risk of postoperative hypotension but require close monitoring due to the potential for orthostatic hypotension upon initiation. Beta-blockers (e.g., propranolol, atenolol) are added 3–4 days after starting alpha-blockers to control tachycardia (87). Calcium channel blockers, such as amlodipine or nifedipine, can also be used as additional agents to manage hypertension. Increased water and salt intake is recommended 10–14 days before surgery to prevent postoperative hypotension (2, 20, 81).
During perioperative management, surgeons should minimize tumor manipulation to prevent catecholamine surges and avoid tumor spillage, especially in cystic lesions. Early control of the adrenal vein is also recommended to manage the sudden decrease in peripheral vascular resistance following tumor removal (88). Preoperative biopsies are generally not recommended (89).
7.3 Surveillance and restaging of patients with metastatic PPGL
For patients with secretory metastatic PPGL, biochemical monitoring of 24-hour urinary fractionated metanephrines or free plasma is recommended at least every six months, as large increases may indicate disease progression. In nonsecretory metastatic PPGL, further measurements of plasma or 24-hour urinary metanephrines are generally unnecessary unless there is a germline pathogenic variant indicating persistent risk or if signs and symptoms of secretory disease appear (30).
Expert guidelines, such as those from NANETS, recommend surveillance imaging for metastatic PPGL with cross-sectional anatomic imaging (CT or MRI) every 3–6 months during the first year, and if disease remains stable, every 6–12 months thereafter. For liver metastases, triple-phase CT or MRI with contrast is recommended. In metastatic PPGL cases on systemic therapies, surveillance imaging with CT or MRI is suggested every 3–6 months. Functional imaging is not typically recommended for patients with primary PPGL before or after surgery; however, it can more accurately detect metastatic disease if strongly suspected (30). In such cases, PET/CT has been studied with various radiotracers, including 18F-DOPA, 18F-FDG, and 68Ga-DOTATATE. These tracers are superior to MIBG scintigraphy, which is only used when 131I-MIBG treatment is being considered. Tracer efficacy in cluster 1 PPGL depends on somatostatin receptor uptake, making 68Ga-DOTATATE highly specific and effective for detecting small tumors, and tumor glucose metabolism, which enhances the effectiveness of 18F-FDG in aggressive, undifferentiated tumors (90). For bone-only metastatic disease, both SSTR PET/CT and FDG may be useful for routine imaging surveillance (30). 18F-DOPA’s effectiveness is based on tissue uptake via amino acid transporters, making it particularly suitable for non-metastatic cluster 2 tumors (90).
7.4 Non-surgical and novel therapies
Various local and regional therapies, including debulking surgery, cementoplasty, radiotherapy (including stereotactic and CyberKnife), radiofrequency ablation, cryotherapy, and tumor embolization, can manage symptoms associated with catecholamine production, tumor burden, or bone involvement (91–93). For patients with stable disease, low tumor burden, and oligometastatic disease, active surveillance is indicated (30, 92, 93). For resectable lesions, options include primary tumor surgery, oligometastatic disease surgery, and debulking surgery. When surgery is not feasible, and disease progression is rapid, with a high visceral tumor burden or severe symptoms, systemic chemotherapy is recommended to control disease progression and alleviate symptoms (14, 15, 92–94). Current chemotherapy regimens include cyclophosphamide, doxorubicin, dacarbazine, and vincristine, though no first-line drug has been defined. Approximately 37% of patients respond to chemotherapy, although complete responses are uncommon (95, 96). Temozolamide, an oral alternative to dacarbazine, may be an option for patients with pathogenic variants SDHB with methylation of the O(6)-methylguanine-DNA methyltransferase (MGMT) promoter. However, chemotherapy generally reduces tumor size and helps control blood pressure in only one-third of patients with metastatic pheochromocytoma-sympathetic paraganglioma (97, 98).
The only FDA-approved treatment for metastatic PPGL, approved in 2018, is high-specific activity iodine-131 metaiodobenzylguanidine (HSA-I-131-MIBG), targeting neuroendocrine cells with a response rate of 30–40% (35). In a multicenter phase 2 trial, 68 patients with advanced PPGL received at least one dose, with 25% (95% CI, 16%-37%) showing durable reductions in antihypertensive medication. Among evaluable patients, 92% achieved either partial response or stable disease within 12 months. Elevated serum chromogranin levels (≥1.5 times the baseline upper limit) decreased in 68% of patients (19 of 28), and median overall survival was 36.7 months (95% CI, 29.9-49.1 months). Common side effects included nausea, myelosuppression, and fatigue, with no hypertensive events (99). A real-world study by Al-Ward et al. reported a 38% objective response rate and an 83% disease control rate in 24 patients with metastatic PPGL, with complete response in two cases, 30% metanephrine normalization, and >50% improvement in 46% of cases. Blood pressure normalized in 56%, though seven patients had reversible grade 3–4 myelosuppression, and one experienced fatal pneumonitis (100). Ultratrace iobenguane 131I, a highly specific 131I-MIBG, is no longer available. In Colombia, 131I-MIBG can be imported, though its high cost—greater than tyrosine kinase inhibitors and chemotherapy—limits availability. Additionally, 50% of patients do not show 131I-MIBG uptake, further restricting its use in Latin America (101). Lutetium-177 DOTATATE/TOC has emerged as an alternative management option in patients with advanced PPGL, however its evidence so far comes from retrospective studies (102, 103). In Colombia, Lutetium-177 DOTATATE/TOC is approved for use in neuroendocrine tumors. In a phase II clinical trial conducted by Reyes et al. Lutetium was shown to be safe and effective in a population of 13 patients with inoperable and progressing advanced neuroendocrine tumors, but no patients with PPGL were included (104).
In addition, for patients with slow/moderate progression, not candidates for radionuclide therapy angiogenesis and proliferative signaling inhibitors have been tested as novel treatments, focusing on the interaction between several growth factors including vascular endothelial growth factor [VEGF], platelet-derived growth factors [PDGF] and others, with tyrosine kinase receptors (105). Sunitinib, which inhibits VEGF1, VEGF2, VEGF3, PDGF-alpha, PDGF-beta, c-kit, fms-related tyrosine kinase 3, and RET proto-oncogene receptors, has demonstrated potential in reducing angiogenesis and tumor cell growth. Small studies have shown a disease control rate of 57–83%, with median progression-free survival ranging from 4 to 13 months (106). The FIRSTMAPP study, a phase II randomized placebo-controlled trial, recently reported that Sunitinib achieved the primary endpoint of 12-month progression-free survival in 36% of patients with progressive metastatic PPGL (90% CI, 23–50%), compared to 19% in the placebo group (90% CI, 11–31%). Grade 3 or 4 adverse effects included asthenia, hypertension, and bone or back pain (107). Another drug, Cabozantinib was evaluated in the Natalie Trial, a single-arm phase II trial with 17 patients and a median follow-up of 25 months. The overall response rate was 25.0% (95% CI, 7.3–52.4), with responses observed in 4 out of 16 patients. Grade 3 adverse events included hand-foot syndrome, hypertension, rectal fistula, QT prolongation, and asymptomatic hypomagnesemia. Additionally, two cases of asymptomatic elevations in amylase and lipase were reported (108). On the other hand, Axitinib, a VEGFR2 inhibitor, has also shown promise, particularly in metastatic pheochromocytomas where the pseudohypoxic tumor environment stimulates VEGF synthesis, promoting angiogenesis. Phase II trials of Axitinib reported a partial response in 36% of patients (105).
New therapies for metastatic PPGL under investigation include Belzutifan, a HIF-2 inhibitor used in VHL disease, currently in a phase II trial (NCT04924075) for PPGL (108). Another HIF-2 inhibitor, DFF332, is currently in a phase I/Ib trial (NCT04895748) as monotherapy and in combination with agents like everolimus, spartalizumab, and taminadenant.
Additional investigational drugs include Olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor involved in DNA repair, being tested in combination with temozolomide (NCT04394858), and Tipifarnib, a farnesyl transferase inhibitor that supports tumor cell survival (NCT04284774).
Finally, Imipridone, a promising agent targeting caseinolytic protease P (ClpP) and acting as a dopamine-like receptor antagonist, has also garnered interest. In a phase II trial, 10 patients received Imipridone at 625 mg weekly; of these, five showed partial responses, and two had stable disease. In a second cohort, where patients received two doses on consecutive days weekly, one achieved a partial response, and seven maintained stable disease (109).
8 Conclusions
PPLG are rare neuroendocrine tumors with relevant clinical implications, characterized predominantly by the production of catecholamines, which can manifest in a spectrum of clinical symptoms.
Pheochromocytomas have the potential to be part of inherited syndromes such as MEN-2, VHL, and NF1, which implicate a variety of other pathologies and necessitate genetic screening of affected individuals and their family members. This genetic association requires a robust approach to diagnosis and treatment, integrating advanced imaging techniques, accurate laboratory testing, and detailed genetic analysis.
Treatment strategies for pheochromocytomas involve a multidisciplinary approach, including surgical intervention as the primary therapeutic option. Preoperative preparation with alpha-blockers and beta-blockers is crucial to mitigate the risks associated with catecholamine secretion during tumor manipulation. Non-surgical approaches, including chemotherapy and novel therapies such as tyrosine kinase inhibitors, play a role in the treatment of metastatic or inoperable cases, offering symptomatic relief and possible disease control.
Although PPGL are rare, their complex clinical presentations and potential genetic basis make them a significant challenge in endocrine and oncologic practice. Early diagnosis, a thorough understanding of the genetic landscape, and a comprehensive treatment strategy are critical to improving outcomes for patients with these potentially life-threatening conditions.
Author contributions
JT: Writing – original draft, Validation, Software, Resources, Project administration, Methodology, Investigation, Data curation, Conceptualization. HN-C: Writing – original draft, Validation, Resources, Methodology, Investigation, Data curation, Conceptualization. LC: Writing – original draft, Validation, Resources, Methodology, Investigation, Data curation, Conceptualization. WR-M: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Methodology, Investigation, Conceptualization. JA: Writing – original draft, Validation, Software, Resources, Methodology, Investigation. CC-G: Writing – original draft, Validation, Resources, Investigation, Data curation. LS: Writing – review & editing, Writing – original draft, Validation, Resources, Methodology, Investigation. MH-E: Writing – original draft, Visualization, Software, Project administration, Methodology, Investigation. MA-I: Writing – review & editing, Writing – original draft, Visualization, Validation, Resources, Methodology, Investigation. JI-C: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Data curation.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Klöppel G. Tumoren des nebennierenmarks und der paraganglien. Pathologe. (2003) 24:280–6. doi: 10.1007/s00292-003-0635-8
2. Lenders JWM, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SKG, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
3. Eisenhofer G, Pamporaki C, Lenders JWM. Biochemical assessment of pheochromocytoma and paraganglioma. Endocr Rev. (2023) 44:862–909. doi: 10.1210/endrev/bnad011
4. Aygun N, Uludag M. Pheochromocytoma and paraganglioma: from epidemiology to clinical findings. Sisli Etfal Hastan Tip Bul. (2020) 54:159–68. doi: 10.14744/SEMB.2020.18794
5. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol. (2022) 33:90–114. doi: 10.1007/s12022-022-09704-6
6. Tischler AS, Pacak K, Eisenhofer G. The adrenal medulla and extra-adrenal paraganglia: then and now. Endocr Pathol. (2014) 25:49–58. doi: 10.1007/s12022-013-9286-3
7. Glenn F, Gray GF. Functional tumors of the organ of Zuckerkandl. Ann Surg. (1976) 183:578–85. doi: 10.1097/00000658-197605000-00015
8. Ilias I, Pacak K. Diagnosis and management of tumors of the adrenal medulla. Horm Metab Res. (2005) 37:717–21. doi: 10.1055/s-2005-921091
9. Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. (2004) 292:943–51. doi: 10.1001/jama.292.8.943
10. Kihara C, Patel S, Moss R. A rapidly progressing carotid body tumor: A case report. Cureus. (2023) 15:e43654. doi: 10.7759/cureus.43654
11. Santarpia L, Habra MA, Jiménez C. Malignant pheochromocytomas and paragangliomas: molecular signaling pathways and emerging therapies. Horm Metab Res. (2009) 41:680–6. doi: 10.1055/s-0029-1214381
12. Jadhav SS, Dhok AP, Mitra KR. Carotid body paraganglioma: a case report. Pan Afr Med J. (2023) 44:182. doi: 10.11604/pamj.2023.44.182.38636
13. Karatas E, Sirikci A, Baglam T, Mumbuc S, Durucu C, Tutar E, et al. Synchronous bilateral carotid body tumor and vagal paraganglioma: A case report and review of literature. Auris Nasus Larynx. (2008) 35:171–5. doi: 10.1016/j.anl.2007.05.007
14. Bracigliano A, Marretta AL, Guerrera LP, Simioli R, Clemente O, Granata V, et al. The management of phaeochromocytomas and paragangliomas in the era of precision medicine: where are we now? Evidence-based systemic treatment options and future cluster oriented perspectives. Pharmaceuticals. (2024) 17:354. doi: 10.3390/ph17030354
15. Sharma S, Fishbein L. Diagnosis and management of pheochromocytomas and paragangliomas: A guide for the clinician. Endocr Pract. (2023) 29:999–1006. doi: 10.1016/j.eprac.2023.07.027
16. Lima JV, Kater CE. The Pheochromocytoma/Paraganglioma syndrome: an overview on mechanisms, diagnosis and management. Int Braz J Urol. (2023) 49:307–19. doi: 10.1590/S1677-5538.IBJU.2023.0038
17. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. (2022) 43:199–239. doi: 10.1210/endrev/bnab019
18. Passman JE, Wachtel H. Management of pheochromocytomas and paragangliomas. Surg Clin North Am. (2024) 104:863–81. doi: 10.1016/j.suc.2024.02.014
19. Neumann HPH, Young WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
20. Strosberg JR. Update on the management of unusual neuroendocrine tumors: pheochromocytoma and paraganglioma, medullary thyroid cancer and adrenocortical carcinoma. Semin Oncol. (2013) 40:120–33. doi: 10.1053/j.seminoncol.2012.11.009
21. Almeida MQ, Bezerra-Neto JE, Mendonça BB, Latronico AC, Fragoso MCBV. Primary Malignant tumors of the adrenal glands. Clinics (Sao Paulo). (2018) 73:e756s. doi: 10.6061/clinics/2018/e756s
22. Lorenzo S, Luis J. Diagnóstico y tratamiento de los feocromocitomas y paragangliomas. Rev Finlay. (2021) 11:307–15.
23. Gimenez-Roqueplo A-P, Burnichon N, Amar L, Favier J, Jeunemaitre X, Plouin P-F. Recent advances in the genetics of phaeochromocytoma and functional paraganglioma. Clin Exp Pharmacol Physiol. (2008) 35:376–9. doi: 10.1111/j.1440-1681.2008.04881.x
24. Korpershoek E, Petri B-J, Van Nederveen FH, Dinjens WNM, Verhofstad AA, De Herder WW, et al. Candidate gene mutation analysis in bilateral adrenal pheochromocytoma and sympathetic paraganglioma. Endocr Relat Cancer. (2007) 14:453–62. doi: 10.1677/ERC-06-0044
25. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. (2017) 134:521–35. doi: 10.1007/s00401-017-1769-8
26. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HR, et al. Adrenocortical carcinomas and Malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2020) 31:1476–90. doi: 10.1016/j.annonc.2020.08.2099
27. Mustafa K, Zadeh S, Culver SA. Dopamine-secreting carotid body paraganglioma in a patient with SDHB mutation. AACE Clin Case Rep. (2024) 10:109–12. doi: 10.1016/j.aace.2024.03.003
28. Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein F, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol. (2017) 177:103–13. doi: 10.1530/EJE-17-0077
29. Pamporaki C, Berends AMA, Filippatos A, Prodanov T, Meuter L, Prejbisz A, et al. Prediction of metastatic pheochromocytoma and paraganglioma: a machine learning modelling study using data from a cross-sectional cohort. Lancet Digit Health. (2023) 5:e551–9. doi: 10.1016/S2589-7500(23)00094-8
30. Fishbein L, Del Rivero J, Else T, Howe JR, Asa SL, Cohen DL, et al. The north american neuroendocrine tumor society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. (2021) 50:469–93. doi: 10.1097/MPA.0000000000001792
31. Mihai R, De Crea C, Guerin C, Torresan F, Agcaoglu O, Simescu R, et al. Surgery for advanced adrenal Malignant disease: recommendations based on European Society of Endocrine Surgeons consensus meeting. Br J Surg. (2024) 111:znad266. doi: 10.1093/bjs/znad266
32. Roman-Gonzalez A, Jimenez C. Malignant pheochromocytoma-paraganglioma: pathogenesis, TNM staging, and current clinical trials. Curr Opin Endocrinol Diabetes Obes. (2017) 24:174–83. doi: 10.1097/MED.0000000000000330
33. Jandou I, Moataz A, Dakir M, Debbagh A, Aboutaieb R. Malignant pheochromocytoma: A diagnostic and therapeutic dilemma. Int J Surg Case Rep. (2021) 83:106009. doi: 10.1016/j.ijscr.2021.106009
34. Sangoi AR, McKenney JK. A tissue microarray-based comparative analysis of novel and traditional immunohistochemical markers in the distinction between adrenal cortical lesions and pheochromocytoma. Am J Surg Pathol. (2010) 34:423–32. doi: 10.1097/PAS.0b013e3181cfb506
35. Jimenez C, Erwin W, Chasen B. Targeted radionuclide therapy for patients with metastatic pheochromocytoma and paraganglioma: from low-specific-activity to high-specific-activity iodine-131 metaiodobenzylguanidine. Cancers (Basel). (2019) 11:1018. doi: 10.3390/cancers11071018
36. Lam AK. Update on adrenal tumours in 2017 world health organization (WHO) of endocrine tumours. Endocr Pathol. (2017) 28:213–27. doi: 10.1007/s12022-017-9484-5
37. Jimenez C, Ma J, Roman Gonzalez A, Varghese J, Zhang M, Perrier N, et al. TNM staging and overall survival in patients with pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab. (2023) 108:1132–42. doi: 10.1210/clinem/dgac677
38. Shine B. Gene targeting through O-methylated catecholamine metabolite patterns. Clin Chem. (2011) 57:361–2. doi: 10.1373/clinchem.2010.159178
39. Astuti D, Latif F, Dallol A, Dahia PLM, Douglas F, George E, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. (2001) 69:49–54. doi: 10.1086/321282
40. Mercado-Asis LB, Wolf KI, Jochmanova I, Taïeb D. Pheochromocytoma: A genetic and diagnostic update. Endocrine Pract. (2018) 24:78–90. doi: 10.4158/EP-2017-0057
41. Reda SA, Japp EA, Galati S-J, Krakoff LR, Levine AC. Pheochromocytoma. In: Davies TF, editor. A Case-Based Guide to Clinical Endocrinology. Springer International Publishing, Cham (2022). p. 167–82. doi: 10.1007/978-3-030-84367-0_19
42. Muth A, Crona J, Gimm O, Elmgren A, Filipsson K, Stenmark Askmalm M, et al. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med. (2019) 285:187–204. doi: 10.1111/joim.12869
43. Bruel A-L, Vitobello A, Thiffault I, Manwaring L, Willing M, Agrawal PB, et al. ITSN1: a novel candidate gene involved in autosomal dominant neurodevelopmental disorder spectrum. Eur J Hum Genet. (2022) 30:111–6. doi: 10.1038/s41431-021-00985-9
44. Giacché M, Tacchetti MC, Castellano M. Genetics and Molecular Biology of Pheochromocytoma and Paraganglioma. In: Tiberio GAM, editor. Primary Adrenal Malignancies. Springer Nature Switzerland, Cham (2024). p. 23–30. doi: 10.1007/978-3-031-62301-1_4
45. Lian B, Lu J, Fang X, Zhang Y, Wang W, He Y, et al. Genotype and clinical phenotype characteristics of MAX germline mutation–associated pheochromocytoma/paraganglioma syndrome. Front Endocrinol (Lausanne). (2024) 15:1442691. doi: 10.3389/fendo.2024.1442691
46. Szabo Yamashita T, Tame-Elorduy A, Skefos CM, Varghese JM, Habra MA, Fisher SB, et al. SDHB-associated pheochromocytomas: what is their clinical behavior? Ann Surg Oncol. (2024) 31:9007–13. doi: 10.1245/s10434-024-16120-z
47. Thosani S, Ayala-Ramirez M, Palmer L, Hu MI, Rich T, Gagel RF, et al. The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. (2013) 98:E1813–9. doi: 10.1210/jc.2013-1653
48. Meyer-Rochow GY, Smith JM, Richardson A-L, Marsh DJ, Sidhu SB, Robinson BG, et al. Denaturing high performance liquid chromatography detection of SDHB, SDHD, and VHL germline mutations in pheochromocytoma. J Surg Res. (2009) 157:55–62. doi: 10.1016/j.jss.2008.07.043
49. Björklund P, Pacak K, Crona J. Precision medicine in pheochromocytoma and paraganglioma: current and future concepts. J Intern Med. (2016) 280:559–73. doi: 10.1111/joim.12507
50. Sarkadi B, Saskoi E, Butz H, Patocs A. Genetics of pheochromocytomas and paragangliomas determine the therapeutical approach. Int J Mol Sci. (2022) 23:1450. doi: 10.3390/ijms23031450
51. Eid M, Foukal J, Sochorová D, Tuček Š, Starý K, Kala Z, et al. Management of pheochromocytomas and paragangliomas: Review of current diagnosis and treatment options. Cancer Med. (2023) 12:13942–57. doi: 10.1002/cam4.6010
52. Assadipour Y, Sadowski SM, Alimchandani M, Quezado M, Steinberg SM, Nilubol N, et al. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery. (2017) 161:230–9. doi: 10.1016/j.surg.2016.05.050
53. Kantorovich V, King KS, Pacak K. SDH-related pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. (2010) 24:415–24. doi: 10.1016/j.beem.2010.04.001
54. Ando Y, Ono Y, Sano A, Fujita N, Ono S, Tanaka Y. Clinical characteristics and outcomes of pheochromocytoma crisis: a literature review of 200 cases. J Endocrinol Invest. (2022) 45:2313–28. doi: 10.1007/s40618-022-01868-6
55. Klein RD, Jin L, Rumilla K, Young WF, Lloyd RV. Germline SDHB mutations are common in patients with apparently sporadic sympathetic paragangliomas. Diagn Mol Pathol. (2008) 17:94–100. doi: 10.1097/PDM.0b013e318150d67c
56. Stein PP. A simplified diagnostic approach to pheochromocytoma. A Rev literature Rep One institution’s experience. (1991) 70:46–66. doi: 10.1097/00005792-199101000-00004
57. Greenleaf CE, Griffin LA, Shake JG, Orr WS. Hypertensive crisis secondary to pheochromocytoma. Proc (Bayl Univ Med Cent). (2017) 30:314–5. doi: 10.1080/08998280.2017.11929629
58. Mannelli M. Diagnostic problems in pheochromocytoma. J Endocrinol Invest. (1989) 12:739–57. doi: 10.1007/BF03350050
59. Lance JW, Hinterberger H. Symptoms of pheochromocytoma, with particular reference to headache, correlated with catecholamine production. Arch Neurol. (1976) 33:281–8. doi: 10.1001/archneur.1976.00500040065011
60. Cortelli P, Grimaldi D, Guaraldi P, Pierangeli G. Headache and hypertension. Neurol Sci. (2004) 25 Suppl 3:S132–134. doi: 10.1007/s10072-004-0271-y
61. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
62. Roth MA, Leyba K, Garg I, Madrid WH, Quazi MA, Sohail AH, et al. Mortality and in-patient outcomes in pheochromocytoma patients with hypertensive emergency in the United States: A propensity score matched analysis. Curr Probl Cardiol. (2024) 49:102578. doi: 10.1016/j.cpcardiol.2024.102578
63. Rupala K, Mittal V, Gupta R, Yadav R. Atypical presentation of pheochromocytoma: Central nervous system pseudovasculitis. Indian J Urol. (2017) 33:82–4. doi: 10.4103/0970-1591.195760
64. Li C, Chen Y, Wang W, Teng L. A case of clinically silent giant right pheochromocytoma and review of literature. Can Urol Assoc J. (2012) 6:E267–9. doi: 10.5489/cuaj.11195
65. Sarveswaran V, Kumar S, Kumar A, Vamseedharan M. A giant cystic pheochromocytoma mimicking liver abscess an unusual presentation – a case report. Clin Case Rep. (2015) 3:64–8. doi: 10.1002/ccr3.149
66. Staren ED, Prinz RA. Selection of patients with adrenal incidentalomas for operation. Surg Clinics North America. (1995) 75:499–509. doi: 10.1016/S0039-6109(16)46636-3
67. Geroula A, Deutschbein T, Langton K, Masjkur J, Pamporaki C, Peitzsch M, et al. Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol. (2019) 181:409–20. doi: 10.1530/EJE-19-0159
68. Neumann HP, Young WF, Krauss T, Bayley J-P, Schiavi F, Opocher G, et al. 65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocr Relat Cancer. (2018) 25:T201–19. doi: 10.1530/ERC-18-0085
69. de Jong WHA, Eisenhofer G, Post WJ, Muskiet FAJ, de Vries EGE, Kema IP. Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors. J Clin Endocrinol Metab. (2009) 94:2841–9. doi: 10.1210/jc.2009-0303
70. Kim HJ, Lee JI, Cho YY, Lee SY, Kim JH, Jung BC, et al. Diagnostic accuracy of plasma free metanephrines in a seated position compared with 24-hour urinary metanephrines in the investigation of pheochromocytoma. Endocr J. (2015) 62:243–50. doi: 10.1507/endocrj.EJ14-0384
71. Boot CS. A laboratory medicine perspective on the investigation of phaeochromocytoma and paraganglioma. Diagnostics (Basel). (2023) 13:2940. doi: 10.3390/diagnostics13182940
72. Wan W, Nguyen B, Graybill S, Kim J. Clonidine suppression testing for pheochromocytoma in neurofibromatosis type 1. BMJ Case Rep. (2019) 12:e228263. doi: 10.1136/bcr-2018-228263
73. Tsiomidou S, Pamporaki C, Geroula A, Van Baal L, Weber F, Dralle H, et al. Clonidine suppression test for a reliable diagnosis of pheochromocytoma: When to use. Clin Endocrinol (Oxf). (2022) 97:541–50. doi: 10.1111/cen.14724
74. Anderson GH, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. (1994) 12:609–15. doi: 10.1097/00004872-199405000-00015
75. Wakabayashi T. Mechanism of the calcium-regulation of muscle contraction — In pursuit of its structural basis. Proc Jpn Acad Ser B Phys Biol Sci. (2015) 91:321–50. doi: 10.2183/pjab.91.321
76. Shen S-J, Cheng H-M, Chiu AW, Chou C-W, Chen J-Y. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. (2005) 28:44–50.
77. Čtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. (2018) 15:3151–60. doi: 10.3892/etm.2018.5871
78. Berglund AS, Hulthén UL, Manhem P, Thorsson O, Wollmer P, Törnquist C. Metaiodobenzylguanidine (MIBG) scintigraphy and computed tomography (CT) in clinical practice. Primary and secondary evaluation for localization of phaeochromocytomas. J Intern Med. (2001) 249:247–51. doi: 10.1046/j.1365-2796.2001.00792.x
79. Bhatia KSS, Ismail MM, Sahdev A, Rockall AG, Hogarth K, Canizales A, et al. 123I-metaiodobenzylguanidine (MIBG) scintigraphy for the detection of adrenal and extra-adrenal phaeochromocytomas: CT and MRI correlation. Clin Endocrinol (Oxf). (2008) 69:181–8. doi: 10.1111/j.1365-2265.2008.03256.x
80. Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of 68Ga-DOTA-conjugated somatostatin receptor-targeting peptide PET in detection of pheochromocytoma and paraganglioma: A systematic review and metaanalysis. J Nucl Med. (2019) 60:369–76. doi: 10.2967/jnumed.118.211706
81. Pacak K, Eisenhofer G, Carrasquillo JA, Chen CC, Li ST, Goldstein DS. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension. (2001) 38:6–8. doi: 10.1161/01.hyp.38.1.6
82. Neumann HPH, Hoegerle S, Manz T, Brenner K, Iliopoulos O. How many pathways to pheochromocytoma? Semin Nephrol. (2002) 22:89–99. doi: 10.1053/snep.2002.30207
83. Su T, Yang Y, Jiang L, Xie J, Zhong X, Wu L, et al. SDHB immunohistochemistry for prognosis of pheochromocytoma and paraganglioma: A retrospective and prospective analysis. Front Endocrinol (Lausanne). (2023) 14:1121397. doi: 10.3389/fendo.2023.1121397
84. Dogrul AB, Cennet O, Dincer AH. Minimally invasive techniques in benign and Malignant adrenal tumors. World J Clin cases. (2022) 10:12812–21. doi: 10.12998/wjcc.v10.i35.12812
85. Wang X, Zhao Y, Liao Z, Zhang Y. Surgical strategies of complicated pheochromocytomas/paragangliomas and literature review. Front Endocrinol (Lausanne). (2023) 14:1129622. doi: 10.3389/fendo.2023.1129622
86. Roman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, et al. Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann Surg. (2018) 268:172–8. doi: 10.1097/SLA.0000000000002195
87. Román-González A, Padilla-Zambrano H, Jimenez LFV. Perioperative management of pheocromocytoma/paraganglioma: a comprehensive review. Colombian J Anesthesiology. (2021) 49:1–12. doi: 10.5554/22562087.e958
88. Munakomi S, Rajbanshi S, Adhikary PS. Case Report: A giant but silent adrenal pheochromocytoma – a rare entity. F1000Res. (2016) 5:290. doi: 10.12688/f1000research.8168.1
89. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JWM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. (2016) 174:G1–G10. doi: 10.1530/EJE-16-0033
90. Marcus C, Subramaniam RM. Paragangliomas and pheochromocytomas: positron emission tomography/computed tomography diagnosis and therapy. PET Clin. (2023) 18:233–42. doi: 10.1016/j.cpet.2022.11.006
91. Breen W, Bancos I, Young WF, Bible KC, Laack NN, Foote RL, et al. External beam radiation therapy for advanced/unresectable Malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol. (2018) 3:25–9. doi: 10.1016/j.adro.2017.11.002
92. Garcia-Carbonero R, Matute Teresa F, Mercader-Cidoncha E, Mitjavila-Casanovas M, Robledo M, Tena I, et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin Transl Oncol. (2021) 23:1995–2019. doi: 10.1007/s12094-021-02622-9
93. Wang K, Crona J, Beuschlein F, Grossman AB, Pacak K, Nölting S. Targeted therapies in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. (2022) 107:2963–72. doi: 10.1210/clinem/dgac471
94. Jimenez C, Xu G, Varghese J, Graham PH, Campbell MT, Lu Y. New directions in treatment of metastatic or advanced pheochromocytomas and sympathetic paragangliomas: an american, contemporary, pragmatic approach. Curr Oncol Rep. (2022) 24:89–98. doi: 10.1007/s11912-022-01197-0
95. Huang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, et al. Treatment of Malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. (2008) 113:2020–8. doi: 10.1002/cncr.23812
96. Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EPM. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for Malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). (2014) 81:642–51. doi: 10.1111/cen.12542
97. Hadoux J, Favier J, Scoazec J-Y, Leboulleux S, Al Ghuzlan A, Caramella C, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. (2014) 135:2711–20. doi: 10.1002/ijc.28913
98. Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N, et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. (2012) 118:2804–12. doi: 10.1002/cncr.26577
99. Pryma DA, Chin BB, Noto RB, Dillon JS, Perkins S, Solnes L, et al. Efficacy and safety of high-specific-activity 131I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. J Nucl medicine: Off publication Soc Nucl Med. (2019) 60:623–30. doi: 10.2967/jnumed.118.217463
100. Al-Ward R, Brondani VB, Sawani S, Potter CL, Xu G, Waguespack SG, et al. High-specific-activity 131 I-MIBG for the treatment of advanced pheochromocytoma and paraganglioma. Clin Nucl Med. (2024) 49:610–20. doi: 10.1097/RLU.0000000000005184
101. Román-González A. Nuevos conceptos en feocromocitoma y paraganglioma en el 2017. Rev Colombiana Endocrinología Diabetes Metabolismo. (2017) 4:19–23. doi: 10.53853/encr.4.4.148
102. Tang CYL, Chua WM, Huang HL, Lam WW-C, Loh LM, Tai D, et al. Safety and efficacy of peptide receptor radionuclide therapy in patients with advanced pheochromocytoma and paraganglioma: A single-institution experience and review of the literature. J Neuroendocrinol. (2023) 35:e13349. doi: 10.1111/jne.13349
103. Kornerup LS, Andreassen M, Knigge U, Arveschoug AK, Poulsen PL, Kjær A, et al. Effects of peptide receptor radiotherapy in patients with advanced paraganglioma and pheochromocytoma: A nation-wide cohort study. Cancers (Basel). (2024) 16:1349. doi: 10.3390/cancers16071349
104. De los Reyes A, Llamas-Olier A, Martí A, Fierro F, Rojas L, Martínez MC, et al. Eficacia de lutecio-177 DOTATATE/TOC en pacientes con tumores neuroendocrinos bien diferenciados en estado avanzado. Ensayo clínico fase II. Rev Colombiana Cancerología. (2021) 25:13–24. doi: 10.35509/01239015.132
105. Jimenez C. Treatment for patients with Malignant pheochromocytomas and paragangliomas: A perspective from the hallmarks of cancer. Front Endocrinol (Lausanne). (2018) 9:277. doi: 10.3389/fendo.2018.00277
106. O’Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer. (2019) 120:1113–9. doi: 10.1038/s41416-019-0474-x
107. Baudin E, Goichot B, Berruti A, Hadoux J, Moalla S, Laboureau S, et al. Sunitinib for metastatic progressive phaeochromocytomas and paragangliomas: results from FIRSTMAPPP, an academic, multicentre, international, randomised, placebo-controlled, double-blind, phase 2 trial. Lancet (London England). (2024) 403:1061–70. doi: 10.1016/S0140-6736(23)02554-0
108. Jimenez C, Habra MA, Campbell MT, Tamsen G, Cruz-Goldberg D, Long J, et al. Cabozantinib in patients with unresectable and progressive metastatic phaeochromocytoma or paraganglioma (the Natalie Trial): a single-arm, phase 2 trial. Lancet Oncol. (2024) 25:658–67. doi: 10.1016/S1470-2045(24)00133-5
Keywords: pheochromocytoma, paraganglioma, hormonal imbalance, diagnosis, management, treatment
Citation: Saavedra T. JS, Nati-Castillo HA, Valderrama Cometa LA, Rivera-Martínez WA, Asprilla J, Castaño-Giraldo CM, Sánchez S. L, Heredia-Espín M, Arias-Intriago M and Izquierdo-Condoy JS (2024) Pheochromocytoma: an updated scoping review from clinical presentation to management and treatment. Front. Endocrinol. 15:1433582. doi: 10.3389/fendo.2024.1433582
Received: 16 May 2024; Accepted: 15 November 2024;
Published: 13 December 2024.
Edited by:
Farhadul Islam, University of Rajshahi, BangladeshReviewed by:
Sergei Tevosian, University of Florida, United StatesCamilo Jimenez, University of Texas MD Anderson Cancer Center, United States
Copyright © 2024 Saavedra T., Nati-Castillo, Valderrama Cometa, Rivera-Martínez, Asprilla, Castaño-Giraldo, Sánchez S., Heredia-Espín, Arias-Intriago and Izquierdo-Condoy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan S. Izquierdo-Condoy, anVhbjFpenF1aWVyZG8xMUBnbWFpbC5jb20=