 Takashi Shoji1
Takashi Shoji1 Ichiro Yamauchi1*
Ichiro Yamauchi1* Hidenori Kawasaki2
Hidenori Kawasaki2 Kogoro Iwanaga3Takuro Hakata1Daisuke Tanaka1
Kogoro Iwanaga3Takuro Hakata1Daisuke Tanaka1 Junji Fujikura1Toshihiko Masui4
Junji Fujikura1Toshihiko Masui4 Hisato Suzuki5Mamiko Yamada5Kenjiro Kosaki5Yosuke Kasai6Etsuro Hatano6Akira Inaba7
Hisato Suzuki5Mamiko Yamada5Kenjiro Kosaki5Yosuke Kasai6Etsuro Hatano6Akira Inaba7 Takahito Wada7Shinji Kosugi7Yohei Ueda1Toshihito Fujii1
Takahito Wada7Shinji Kosugi7Yohei Ueda1Toshihito Fujii1 Daisuke Taura1Nobuya Inagaki8
Daisuke Taura1Nobuya Inagaki8- 1Department of Diabetes, Endocrinology and Nutrition, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 2Department of Genomic Medicine, Kyoto University School of Public Health, Kyoto, Japan
- 3Department of Pediatrics, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 4Department of Surgery, Kurashiki Central Hospital, Kurashiki, Japan
- 5Center for Medical Genetics, Keio University School of Medicine, Tokyo, Japan
- 6Department of Surgery, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 7Clinical Genetics Unit, Kyoto University Hospital, Kyoto, Japan
- 8Medical Research Institute KITANO HOSPITAL, PIIF Tazuke-kofukai, Osaka, Japan
Silver-Russell syndrome (SRS) is a syndrome characterized by prenatal and postnatal growth retardation, facial features, and body asymmetry. SRS is often complicated with hypoglycemia, whose etiology is unclear. We describe the clinical course of 25-year-old man with hypoglycemia. We diagnosed him with hyperinsulinemic hypoglycemia (HH) and treated him with laparoscopic distal pancreatectomy. Histological examination led to a diagnosis of nesidioblastosis. The juvenile onset of his nesidioblastosis and its slowly progressive course suggested a genetic etiology. Whole-exome sequencing (WES) identified the heterozygous NR0B2 Ala195Ser variant, which alone was unlikely to cause nesidioblastosis because this variant is sometimes detected in the Japanese population. Copy number analysis using WES data suggested duplication in chromosome 7, and subsequent G-banding chromosome analysis confirmed mos dup(7)(p11.2p14). We determined that the patient had SRS-like phenotype based on his clinical features and this duplication. Furthermore, we found that the duplicated region contained the GCK gene, whose gain-of function variants could cause HH. Taken together, the patient’s HH may have been caused by duplication of the GCK gene, which could be a novel cause of nesidioblastosis.
Introduction
Silver-Russell syndrome (SRS) is an imprinting disorder that causes growth retardation, characteristic facial features, and body asymmetry (1). The Netchine-Harbison clinical scoring system (NH-CSS) is used to diagnose SRS based on six features: small for gestational age, postnatal growth failure, relative macrocephaly at birth, protruding forehead, body asymmetry, and feeding difficulties and/or low body mass index (2). While the clinical features of SRS are well known, the genetic background is complicated. Loss of methylation of chromosome 11p15 (11p15 LOM) is observed in 30–60% of patients with SRS (1), while maternal uniparental disomy of chromosome 7 (upd(7)mat) is seen in 10%. Patients with SRS due to upd(7)mat often present with less typical features than those due to 11p15 LOM (3, 4). In addition to its usual characteristics, SRS is often accompanied by hypoglycemia. Previous reports demonstrated that 15–26% of patients with SRS experienced hypoglycemia (4, 5). The pathogenesis of hypoglycemia in this context is unclear, but it is thought to involve feeding difficulties and growth hormone insufficiency (6).
Hypoglycemia has a variety of causes in adults. Drugs (e.g., insulin and insulin secretagogue), clinical illnesses, hormone deficiencies (e.g., glucocorticoids, growth hormone, and thyroid hormone), and non-islet cell tumors should be considered. Endogenous hyperinsulinemic hypoglycemia (HH) is also an important differential diagnosis (7). A key pathophysiological feature of HH is inappropriate secretion of insulin when fasting plasma glucose levels are low (7). The diagnosis is based on the results of the classical 72-hour fasting test: plasma insulin level ≥ 6 µU/mL, plasma C-peptide level ≥ 0.2 nmol/L, and plasma proinsulin level ≥ 5 pmol/L at end-of-fast glucose < 45 mg/dL (8). The majority of adult-onset HH is caused by insulinoma, whose estimated prevalence is 0.16 per 100,000 population (9).
Other causes of HH in adults include functional β-cell disorders, gastric surgery, and insulin autoimmune hypoglycemia. Functional β-cell disorders belong to a group of ill-defined entities (10). Among them, nesidioblastosis is histologically characterized by β cells with enlarged hyperchromatic nuclei and abundant clear cytoplasm (11). Congenital hyperinsulinism (CHI) is a common cause of HH during infancy and childhood. Several genes causing CHI have been identified: ABCC8, KCNJ11, GCK, GLUD-1, HADH1, SLC16A1, HNF1A, HNF4A UCP2, HK1, PGM1, and PMM2 (12).
Here, we report an adult male patient with hypoglycemia who was diagnosed clinically and pathologically with HH due to nesidioblastosis. Genetic analyses showed that he had somatic mosaicism for segmental duplication of 7p11.2–p14. We reviewed his clinical history and phenotype, and then determined that he had SRS-like phenotype. Although previously reported patients with SRS-like phenotype caused by duplication of similar sites also had hypoglycemia (13) and hyperinsulinemia (14), the causes were unclear. The patient in the present case was confirmed to have HH, which suggests that duplication of the GCK gene located in 7p11.2–p14 is a novel cause of HH.
Case description
Clinical course
A 20-year-old male patient was incidentally found to have asymptomatic hypoglycemia. His plasma glucose level was 59 mg/dL and his immunoreactive insulin (IRI) level was 20.6 µU/mL. His plasma glucose level after a 72-hour fast was 64 mg/dL. He was referred to Kyoto University Hospital for further examination. As no pancreatic tumors were detected by contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), or endoscopic ultrasonography (EUS), we began careful follow-up. During the follow-up period of 5 years, his plasma glucose levels gradually decreased and his body weight increased by 5 kg. We decided to re-evaluate his hypoglycemia at 25 years of age.
His past medical history was notable for mental retardation and surgery for squinting, but he had no significant family history including his two younger brothers. His body mass index was calculated as 25.4 kg/m2 based on a height of 159.1 cm and a body weight of 64.3 kg. The final height of his father was 170 cm and that of his mother was 160 cm. Blood test results at 25 years of age are listed in Supplementary Table 1. We diagnosed him with HH on the basis of the following results after 66-hour fasting: low plasma glucose (44 mg/dL) and unsuppressed IRI (6.2 µU/mL) (Supplementary Table 2) (8). We performed contrast-enhanced CT, MRI, and EUS again, which did not detect any pancreatic tumors. Selective arterial calcium injection identified an increase in IRI when calcium gluconate was injected into the proximal portion of the splenic artery: the IRI before the injection was 18.1 µU/mL and the peak IRI was 60.9 µU/mL at 20 s after the injection (Figure 1A). We performed a 75-g oral glucose tolerance test (OGTT) and found a striking elevation of IRI: the IRI before intake was 22.1 µU/mL, with a plasma glucose level of 73 mg/dL, and the peak IRI was 780.3 µU/mL after 90 min (Figures 1B, C).
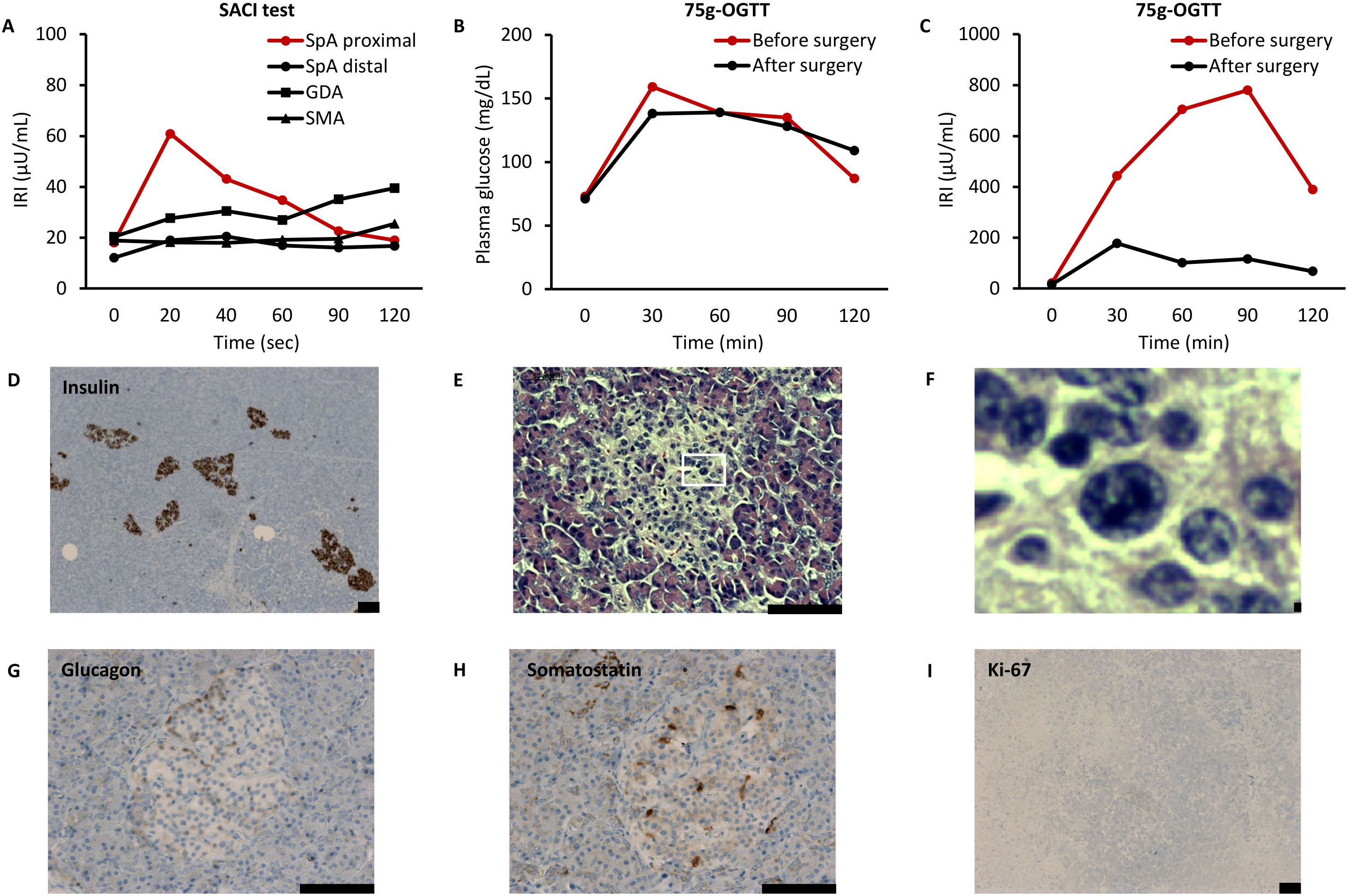
Figure 1. Results of clinical examinations in the present case. (A) Selective arterial calcium injection (SACI) test. IRI, immunoreactive insulin; SpA, splenic artery; GDA, gastroduodenal artery; and SMA, superior mesenteric artery. (B, C) 75-g oral glucose tolerance test (OGTT). Vertical axes show plasma glucose (B) and immunoreactive insulin (IRI) (C). (D–I) Histological examinations of resected pancreas. Immunohistochemistry of insulin (D: magnification, ×50), hematoxylin & eosin staining (E: magnification, ×200, F: ×10 expansion of E), immunohistochemistry of glucagon (G: magnification, ×200), immunohistochemistry of somatostatin (H: magnification, ×200), and immunohistochemistry of Ki-67 (I: magnification, ×50). Scale bars indicate 100 µm.
The fact that his fasting test results became abnormal indicated the progressive nature of his HH. Furthermore, he had become symptomatic, as seen by his increased body weight and elevated transaminases: aspartate aminotransferase (AST) was 101 U/L and alanine aminotransferase (ALT) was 234 U/L (Supplementary Table 1). CT imaging revealed the presence of fatty liver, with a CT value of 36.8 Hounsfield Unit (HU). We prescribed 75 mg of diazoxide in three divided doses. His increased appetite subsided, and his body weight decreased to 60.0 kg 6 weeks after initiating diazoxide. In addition, AST decreased to 47 IU/L and ALT decreased to 78 IU/L. Thus, improvement of his HH appeared to be significant. We decided to perform laparoscopic distal pancreatectomy because we considered this could cure his HH regardless of whether it was caused by nesidioblastosis or occult insulinoma.
As expected, HH improved postoperatively, with a 72-h fasting test resulting in a plasma glucose level of 58 mg/dL and an IRI of 5.2 µU/mL (Supplementary Table 2). The IRI was markedly decreased during a repeat 75 g-OGTT: before intake the IRI was 16.0 µU/mL, with a plasma glucose level of 71 mg/dL, and the peak IRI was 177.8 µU/mL after 30 min (Figures 1B, C). In addition, the area under the curve of the IRI during the 75-g OGTT was 65.7% lower than that before surgery, which almost corresponded to the extent of the resected pancreas. Improvement of HH was maintained even 2 years after surgery: in a 75 g-OGTT, before intake the IRI was 7.4 µU/mL, with a plasma glucose level of 64 mg/dL, and the peak IRI was 121.5 µU/mL after 60 min. His body weight decreased to 51.5kg, AST decreased to 25 IU/L, and ALT decreased to 38 IU/L. Furthermore, CT imaging revealed improvement in fatty liver, with an elevated CT value of 50.4 HU.
Histological examinations of the resected pancreas showed no neoplastic lesions or any invasion of pancreatic islets into the pancreatic ducts (Figure 1D). There were no histological findings of nesidioblastosis that are characteristically seen in newborns, such as diffuse or disseminated proliferation of islet cells (15). Meanwhile, some β cells exhibited enlarged hyperchromatic nuclei and clear cytoplasm (Figures 1E, F). The islets contained glucagon-positive cells and somatostatin-positive cells with normal spatial distribution (Figures 1G, H). There seemed to be an absence of proliferative activity, as almost no islet cells were stained with Ki-67 (Figure 1I). These findings were compatible with the pathological criteria of adult nesidioblastosis (11). Thus, we concluded that the patient’s HH was caused by nesidioblastosis.
Genetic analysis
The juvenile onset of his nesidioblastosis and its slowly progressive nature suggested a congenital etiology. Therefore, we initiated genetic examinations at Kyoto University. After obtaining informed consent from the patient, his genomic DNA was extracted from peripheral blood leukocytes using a NucleoSpin Blood kit (Macherey-Nagel, Düren, Germany). We outsourced whole-exome sequencing (WES) to Macrogen Japan (Tokyo, Japan). The sequencing library was generated using a SureSelect Human All ExonV6 kit (Agilent Technologies, Santa Clara, CA) and was sequenced using the NovaSeq 6000 platform (Illumina, Inc., San Diego, CA). No pathogenic variants were called in the following genes known to be associated with HH secondary to CHI: ABCC8, KCNJ11, GCK, GLUD-1, HADH1, SLC16A1, HNF1A, HNF4A, UCP2, HK1, PGM1, and PMM2. Interestingly, we found a heterozygous variant in the NR0B2 gene: c.583G>T p.Ala195Ser. Specific amplifications of this gene were performed using a polymerase chain reaction (PCR) technique with the following primers: NR0B2 forward, 5′-CAGATCTTGGGCCAGTCTTG-3′; NR0B2 reverse, 5′-CTCCAGGAGCATTGGGTCAC-3′. Sanger sequencing of PCR products, conducted by Macrogen Japan, confirmed this NR0B2 variant (Figure 2A). His mother did not carry this variant. We could not examine his father’s genotype because he was uncontactable. Likewise, we outsourced WES of the resected pancreas specimen to Macrogen Japan, which revealed that no pathogenic variants were called in the HH-associated genes: ABCC8, KCNJ11, GCK, GLUD-1, HADH1, SLC16A1, HNF1A, HNF4A, UCP2, HK1, PGM1, and PMM2.
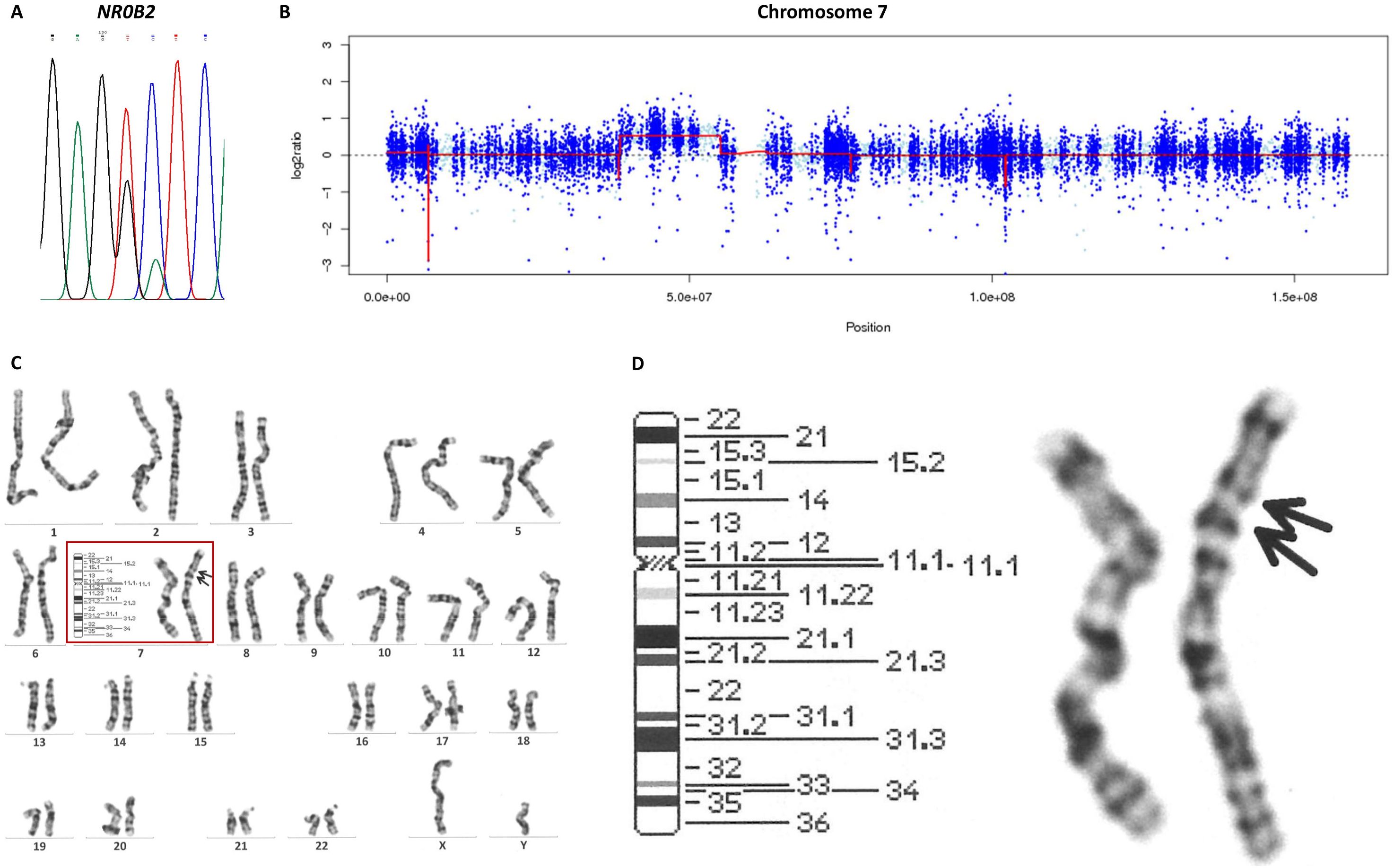
Figure 2. Genetic analyses in the present case. (A) Sanger sequencing of the NR0B2 gene revealed the heterogenous c.583G>T variant. (B) A copy number analysis with EXCAVATOR2 suggested duplication of chromosome 7p11.2–p14.1. (C) Images of all chromosomes in G-banding karyotyping. (D) Chromosome 7 in G-banding karyotyping revealed dup(7)(p11.2p14).
The Initiative on Rare and Undiagnosed Diseases (IRUD), a unified all-Japan diagnostic and research program for rare and undiagnosed diseases, also conducted WES (16). After obtaining informed consent from the patient and the patient’s mother, their genomic DNA was extracted from peripheral blood leukocytes using the standard phenol extraction protocol. All the exons were captured using a SureSelect XT Human All Exon V6 kit (Agilent Technologies); then, exome analyses were performed using the NovaSeq 6000 platform (Illumina, Inc.). EXCAVATOR2 software, which screens for copy number variants using aligned exome data in bam format (17, 18), revealed that the patient had a heterozygous duplication of approximately 18 Mbp in chromosome 7p11.2–p14.1 (location: 38,530,605–55,137,273) (Figure 2B) and his mother did not. G-banding karyotyping of peripheral lymphocytes by LSI Medience Corporation (Tokyo, Japan) confirmed this abnormal mosaic karyotype: mos 46,XY,dup(7)(p11.2p14) (12)/46,XY (18) (Figures 2C, D). To examine whether the patient’s chromosome with the duplication was inherited from his mother, we analyzed single nucleotide polymorphisms (SNPs) located near a GRB10 gene on chromosome 7p11.2–p14.1, which were heterozygous in the patient and homozygous in the mother. If there were no duplication in these regions, the read ratio would converge around 50%. However, the read ratios of the maternal SNPs in the patient ranged from 63.3–71.9%, suggesting that the chromosome with the duplication is maternally derived (Supplementary Table 3).
We suspected with SRS-like phenotype from this mosaic karyotype because duplication of regions including 7p11.2–p14 was reported to cause SRS-like phenotype (13, 14, 19, 20). No point mutations were called from WES data in the following genes associated with SRS: H19, IGF2, CDKN1C, GRB10, and MEST (1). We reviewed the patient’s clinical histories and physical findings. His birth weight was 2410 g (−2.06 SD), height was 47.0 cm (−1.28 SD), and head circumference was 29.0 cm (−3.25 SD), indicating that he was small for his gestational age (39 weeks and 6 days). His growth curves showed that he experienced postnatal growth failure affecting both his height and body weight with feeding difficulty not requiring up to tube feeding (Supplementary Figure 1A). There was no macrocephaly, and in fact his head circumference was rather small (Supplementary Figure 1B). There was also no body asymmetry. As a result, he met two NH-CSS criteria. Additional relevant features included bilateral fifth finger clinodactyly (Supplementary Figures 2A, B) and intellectual disability. Regarding etiology of his intellectual disability, neither hypoglycemia nor convulsions were noted in infancy. In addition, global developmental delay was reported in 34% cases of SRS, and upd(7)mat was more likely to present with global developmental delay than 11p15 LOM (4). Since pathophysiology of dup(7) is speculated to be similar to that of upd(7)mat, we believe that his intellectual disability originated from SRS-like phenotype. However, occult continuous hypoglycemia remains a potential contributing factor. Thus, we determined that he had SRS-like phenotype on the basis of clinical and genetic features.
Discussion
Here we report an adult male patient with SRS-like phenotype and concurrent HH caused by nesidioblastosis. Genetic analyses identified the pathogenic background as the heterozygous NR0B2 Ala195Ser variant and mosaic dup(7)(p11.2p14). A relationship between these findings must be considered because both nesidioblastosis and SRS-like phenotype are rare.
The NR0B2 gene encodes the so-called small heterodimer partner, an orphan member of the mammalian nuclear receptor superfamily. Its endogenous ligands have not been identified, though its regulatory function is mediated by protein-protein interactions with other nuclear receptors and transcription factors (21). NR0B2 acts as a negative regulator of insulin secretion by islets and regulates the synthesis of bile acid and the metabolism of cholesterol and lipid (21, 22). Nishigori et al. reported that patients with early-onset diabetes who harbored NR0B2 variants presented with hyperinsulinemia and obesity, and that mutant proteins, including Ala195Ser, exhibited weaker suppression of the transactivation of HNF-4α, a positive modulator of insulin secretion (23). Thus, the pathogenicity of NR0B2 Ala195Ser variant is classified to “likely pathogenic (PS3 and PM6)” according to the variant classification guidelines from the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) (24).
The frequency of the NR0B2 Ala195Ser variant in the Genome Aggregation Database (gnomAD, v4.0.0) is extremely low, at only 7.4×10-5. However, its frequency in gnomAD is not as low in East Asian individuals (1.4×10-3, 58/41274), and it is not seen in non-East Asian people (0, 0/739670). Furthermore, in 54KJPN, a Japanese whole-genome reference panel, the frequency of this variant in the Japanese population is 4 × 10-3 (429/108604). Considering that HH due to adult nesidioblastosis is extremely rare, the NR0B2 Ala195Ser variant is unlikely to cause HH alone. It should be noted that this variant was unlikely to be related to the etiology of his HH, though the true association could not be determined.
In this study, we detected dup(7)(p11.2p14) and observed that the patient had SRS-like phenotype. His mother did not have dup(7)(p11.2p14), but this did not conflict with his genotype. In this case, the duplication site was presumed to be maternally oriented and not silenced, as the SRS-like phenotype was present and the analysis of SNPs near the GRB10 gene, one of the candidate gene for SRS, also suggested maternal origin. It is speculated that duplication of maternal origin occurred due to somatic mosaicism. The molecular mechanism of SRS-like phenotype due to dup(7) is considered to be similar to that associated with upd(7)mat. We performed a literature review of SRS-like phenotype due to dup(7), and list the identified studies in Table 1 (13, 14, 19, 20). The NH-CSS score in our case was only two, which was not high, and in six previous cases of SRS-like phenotype caused by dup(7), it was also low, ranging from 0 to 2. SRS-like phenotype due to dup(7) seems to present with mild phenotypes as SRS due to upd(7)mat presents with fewer typical clinical features than 11p15 LOM (3, 4). All reported cases of SRS-like phenotype due to dup(7) included GCK gene in the duplication region (13, 14, 19, 20). Interestingly, the patients in one previous case had hypoglycemia (13), and one also had hyperinsulinemia (14). Other reports did not mention hypoglycemia or nesidioblastosis.
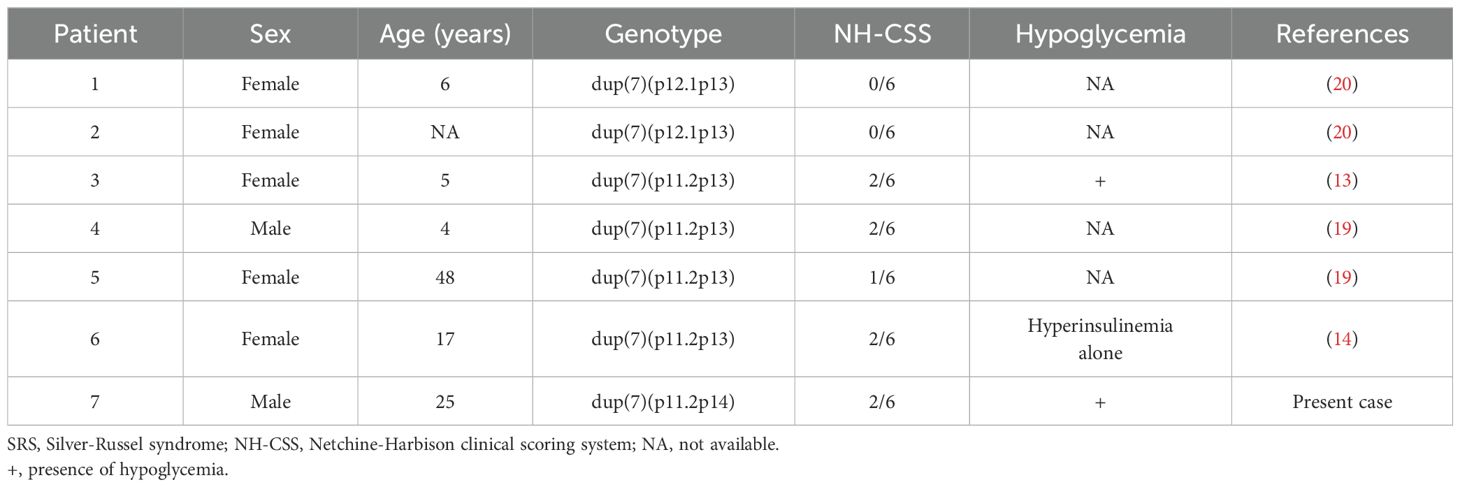
Table 1. A literature review of cases with SRS-like phenotype due to duplication of chromosome 7 (dup7).
Wakeling et al. reported the prevalence of hypoglycemia in SRS according to karyotype (4): 24% for 11p15 LOM and 29% for upd(7)mat. Thus, hypoglycemia is a frequent complication of SRS but its etiology is not well clarified. Azcona et al. examined 24 patients with both SRS and hypoglycemia, and concluded that the most likely causes of hypoglycemia were accelerated starvation and/or growth hormone insufficiency (6). They found no hormonal abnormalities except for growth hormone insufficiency, but their study lacked information on karyotypes (6). Thus it remains unclear if the etiology of hypoglycemia in SRS differs between patients with 11p15LOM, upd(7)mat, or dup(7).
It is informative that in the present case, the cause of hypoglycemia was confirmed to be nesidioblastosis. Furthermore, his hypoglycemia was not noted during childhood, which is atypical for SRS and does not suggest involvement of starvation and/or growth hormone insufficiency. To identify a potential association between nesidioblastosis and dup(7), we searched for genes located in the duplicated region and found the GCK gene at 7p13 (Figure 3). The GCK gene encodes glucokinase, which controls glycogen synthesis in the liver and acts as a primary glucose sensor that regulates insulin secretion in pancreatic islets (25). In pancreatic β cells, glucokinase catalyzes the phosphorylation of glucose to glucose-6-phosphate in the glycolytic pathway; this results in an increase in the ATP: ADP ratio, which in turn leads to the closing of KATP channels and the promotion of insulin secretion (26, 27). Importantly, heterozygous activating mutations in the GCK gene cause mild HH (28). Furthermore, previous reports have shown hypoglycemia and increased GCK activity in transgenic mice that have one additional copy of the GCK gene though the difference of plasma insulin level was not detected (29, 30); this genetic background is similar to that our case, in which the GCK gene itself was duplicated but not mutated.
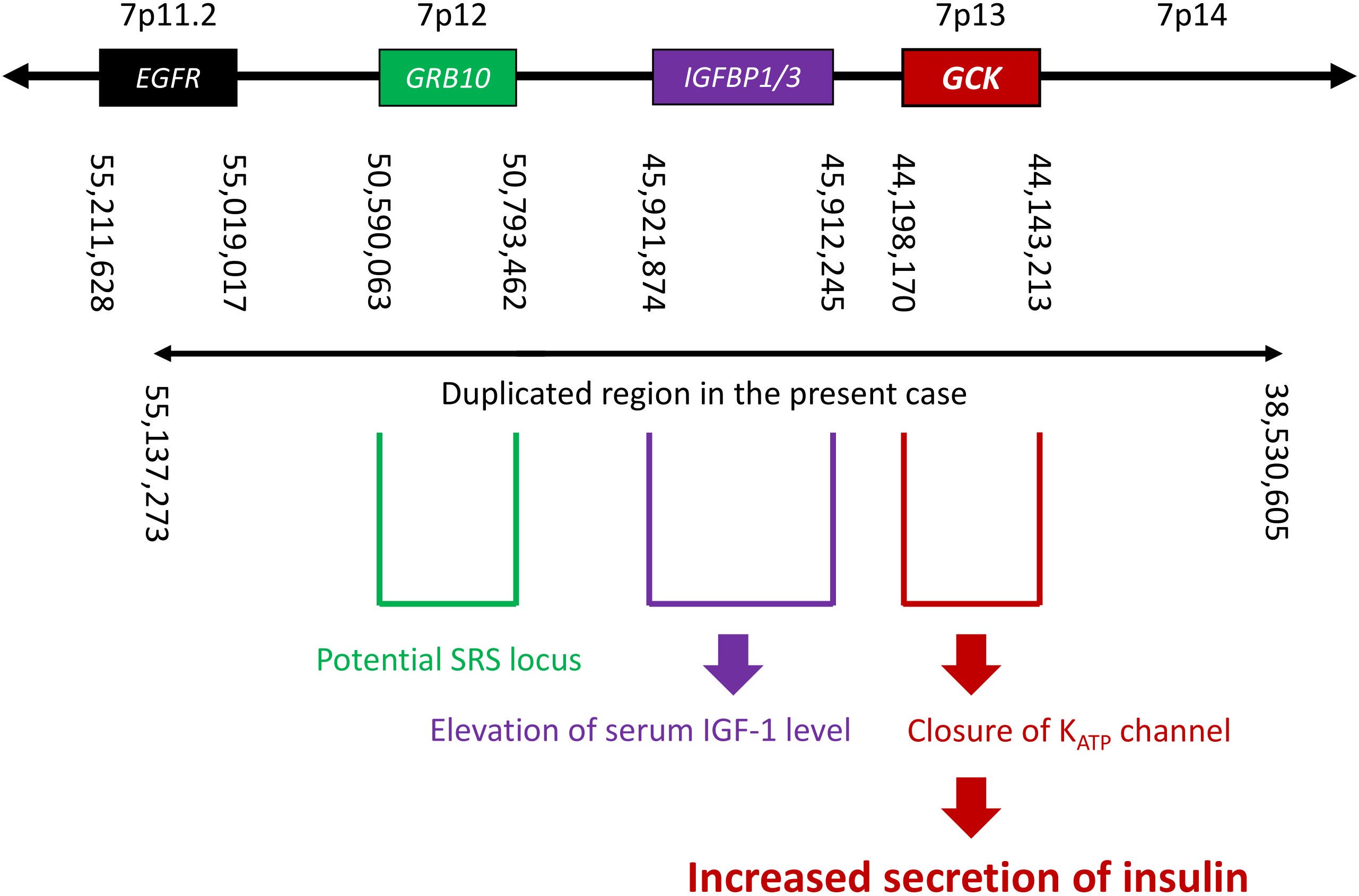
Figure 3. A schema of the genome around 7p11.2–p14, showing the locations of nearby genes, breakpoints suggested by EXCAVATOR2 analysis, and estimated correlations between genotype and phenotype. SRS, Silver-Russell syndrome.
In our patient, dup(7) was a mosaic, detected in only 12 of 30 (40%) peripheral blood mononuclear cells. Islet β cells are heterogenous, and Hub cells with high GCK expression preferentially respond to insulin secretory stimuli, which results in the propagation of signals throughout the islet via inter-β-cell gap junctions (31). Furthermore, the activation of GCK in a minority of β cells is sufficient to change the glucose threshold for insulin secretion in the entire islet and thereby cause HH (32). Taken together, we consider that in our patient, duplication of the GCK gene caused HH due to nesidioblastosis, even though he had mosaic duplications. However, a limitation remains in that we could not obtain direct evidence confirming that the patient’s pancreas has dup(7). Although we analyzed using EXCAVATOR2 software and quantitative PCR, these analyses were unsuccessful due to the low quality of the genomic DNA extracted from the pancreatic specimen, which had deteriorated over time.
Given the pathophysiology in our patient, it is reasonable to assume that the GCK gene could also mediate hypoglycemia in SRS due to upd(7)mat. Hypoglycemia in SRS due to 11p15 LOM seems unrelated to the GCK gene, as demonstrated by a previous study (6). The relationship between elevated transaminases and duplication of the GCK gene remains unclear. The duplication of the IGFBP1/3 gene can increase their expression (Figure 3). When IGFBP3 binds to IGF1, bound IGF1 is inactivated, leading to a decrease in free IGF1, the active form (33). The binding complex extends the half-life of IGF1 in circulation. Total IGF1 increases due to this prolonged half-life, while free IGF1 may be reduced, potentially contributing to his short stature.
Conclusion
Our patient presented with SRS-like phenotype complicated with HH due to nesidioblastosis. Genetic analyses revealed that his SRS-like phenotype was caused by mosaic dup(7)(p11.2p14). While the concomitant heterozygous NR0B2 Ala195Ser variant might have contributed to some extent, duplication of the GCK gene along with dup(7)(p11.2p14) was the most likely cause of his nesidioblastosis. Although further studies are required to confirm the etiology of hypoglycemia in SRS-like phenotype due to dup(7) and SRS due to upd(7)mat, duplication of the GCK gene is suggested as a novel cause of nesidioblastosis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board and Ethics Committee of the Kyoto University Graduate School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
TS: Data curation, Writing – original draft. IY: Data curation, Formal analysis, Writing – original draft. HK: Data curation, Writing – review & editing. KI: Data curation, Writing – review & editing. TH: Writing – review & editing, Data curation. DTn: Formal analysis, Writing – review & editing. JF: Data curation, Writing – review & editing. TM: Data curation, Writing – review & editing. HS: Formal analysis, Writing – review & editing. MY: Formal analysis, Writing – review & editing. KK: Formal analysis, Writing – review & editing. YK: Data curation, Writing – review & editing. EH: Data curation, Writing – review & editing. AI: Writing – review & editing. TW: Writing – review & editing. SK: Writing – review & editing. YU: Writing – review & editing. TF: Writing – review & editing. DTu: Writing – review & editing. NI: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Japan Agency for Medical Research and Development (grant number JP24ek0109760 to KK)
Acknowledgments
We thank the medical staff of Kyoto University Hospital for their clinical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1431547/full#supplementary-material
References
1. Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. (2017) 13:105–24. doi: 10.1038/nrendo.2016.138
2. Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, Netchine I, et al. Original article: A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. (2015) 52:446–53. doi: 10.1136/jmedgenet-2014-102979
3. Bartholdi D, Krajewska-Walasek M, Õunap K, Gaspar H, Chrzanowska KH, Ilyana H, et al. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet. (2009) 46:192–7. doi: 10.1136/jmg.2008.061820
4. Wakeling EL, Abu Amero S, Alders M, Bliek J, Forsythe E, Kumar S, et al. Epigenotype–phenotype correlations in Silver–Russell syndrome. J Med Genet. (2010) 47:760–8. doi: 10.1136/jmg.2010.079111
5. Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, Perin L, et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical russell-silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Cli Endocrinol Metab. (2007) 92:3148–54. doi: 10.1210/jc.2007-0354
6. Azcona C, Stanhope R. Hypoglycaemia and russell-silver syndrome. J Pediatr Endocrinol Metab. (2005) 18:663–70. doi: 10.1515/JPEM.2005.18.7.663
7. Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2009) 94:709–28. doi: 10.1210/jc.2008-1410
8. Service FJ. Hypoglycemic disorders. N Engl J Med. (1995) 332:1144–52. doi: 10.1056/NEJM199504273321707
9. Yamada Y, Kitayama K, Oyachi M, Higuchi S, Kawakita R, Kanamori Y, et al. Nationwide survey of endogenous hyperinsulinemic hypoglycemia in Japan (2017-2018): Congenital hyperinsulinism, insulinoma, non-insulinoma pancreatogenous hypoglycemia syndrome and insulin autoimmune syndrome (Hirata’s disease). J Diabetes Investig. (2020) 11:554–63. doi: 10.1111/jdi.13180
10. Dieterle MP, Husari A, Prozmann SN, Wiethoff H, Stenzinger A, Röhrich M, et al. Diffuse, adult-onset nesidioblastosis/non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS): review of the literature of a rare cause of hyperinsulinemic hypoglycemia. Biomedicines. (2023) 11:1732. doi: 10.3390/biomedicines11061732
11. Anlauf M, Wieben D, Perren A, Sipos B, Komminoth P, Raffel A, et al. Persistent hyperinsulinemic hypoglycemia in 15 adults with diffuse nesidioblastosis: Diagnostic criteria, incidence, and characterization of β-cell changes. Am J Surg Pathol. (2005) 29:524–33. doi: 10.1097/01.pas.0000151617.14598.ae
12. Demirbilek H, Hussain K. Congenital hyperinsulinism: diagnosis and treatment update. J Clin Res Pediatr Endocrinol. (2017) 9:69–87. doi: 10.4274/jcrpe.2017.S007
13. Monk D, Wakeling EL, Proud V, Hitchins M, Abu-Amero SN, Stanier P, et al. Duplication of 7p11.2-p13, including GRB10, in silver-russell syndrome. Am J Hum Genet. (2000) 66:36–46. doi: 10.1086/302717
14. Catusi I, Bonati MT, Mainini E, Russo S, Orlandini E, Larizza L, et al. Recombinant chromosome 7 driven by maternal chromosome 7 pericentric inversion in a girl with features of silver-russell syndrome. Int J Mol Sci. (2020) 21:1–11. doi: 10.3390/ijms21228487
15. Yakovac WC, Baker L, Hummeler K. Beta cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr. (1971) 79:226–31. doi: 10.1016/S0022-3476(71)80105-1
16. Takahashi Y, Date H, Oi H, Adachi T, Imanishi N, Kimura E, et al. Six years’ accomplishment of the Initiative on Rare and Undiagnosed Diseases: nationwide project in Japan to discover causes, mechanisms, and cures. J Hum Genet. (2022) 67:505–13. doi: 10.1038/s10038-022-01025-0
17. D’Aurizio R, Pippucci T, Tattini L, Giusti B, Pellegrini M, Magi A, et al. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. (2016) 44:e154. doi: 10.1093/nar/gkw695
18. Suzuki H, Yamada M, Uehara T, Takenouchi T, Kosaki K. Parallel detection of single nucleotide variants and copy number variants with exome analysis: Validation in a cohort of 700 undiagnosed patients. Am J Med Genet A. (2020) 182:2529–32. doi: 10.1002/ajmg.a.v182.11
19. Monk D, Bentley L, Hitchins M, Myler RA, Clayton-Smith J, Ismail S, et al. Chromosome 7p disruptions in Silver Russell syndrome: delineating an imprinted candidate gene region. Hum Genet. (2002) 111:376–87. doi: 10.1007/s00439-002-0777-4
20. Joyce CA, Sharp A, Walker JM, Bullman H, Temple IK. Duplication of 7p12.1-p13, including GRB10 and IGFBP1, in a mother and daughter with features of Silver-Russell syndrome. Hum Genet. (1999) 105:273–80. doi: 10.1007/s004390051101
21. Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta. (2011) 1812:893–908. doi: 10.1016/j.bbadis.2010.10.006
22. Chanda D, Park JH, Choi HS. Molecular basis of endocrine regulation by orphan nuclear receptor Small Heterodimer Partner. Endocr J. (2008) 55:253–68. doi: 10.1507/endocrj.K07E-103
23. Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, Sho K, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci U.S.A. (2001) 98:575–80. doi: 10.1073/pnas.98.2.575
24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
25. Sternisha SM, Miller BG. Molecular and cellular regulation of human glucokinase. ArchBiochem Biophys. (2019) 663:199–213. doi: 10.1016/j.abb.2019.01.011
26. Senniappan S, Arya VB, Hussain K. The molecular mechanisms, diagnosis and management of congenital hyperinsulinism. Indian J Endocrinol Metab. (2013) 17:19–30. doi: 10.4103/2230-8210.107822
27. Rahman SA, Nessa A, Hussain K. Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol. (2015) 54:R119–129. doi: 10.1530/JME-15-0016
28. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. (2009) 30:1512–26. doi: 10.1002/humu.v30:11
29. Niswender KD, Postic C, Jetton TL, Bennett BD, Piston DW, Efrat S, et al. Cell-specific expression and regulation of a glucokinase gene locus transgene. J Biol Chem. (1997) 272:22564–9. doi: 10.1074/jbc.272.36.22564
30. Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem. (1997) 272:22570–5. doi: 10.1074/jbc.272.36.22570
31. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. (2016) 24:389–401. doi: 10.1016/j.cmet.2016.06.020
32. Chen KH, Doliba N, May CL, Roman J, Ustione A, Tembo T, et al. Genetic activation of glucokinase in a minority of pancreatic beta cells causes hypoglycemia in mice. Life Sci. (2022) 309:120952. doi: 10.1016/j.lfs.2022.120952
Keywords: nesidioblastosis, Silver-Russell Syndrome, hyperinsulinemic hypoglycemia, chromosome 7, glucokinase
Citation: Shoji T, Yamauchi I, Kawasaki H, Iwanaga K, Hakata T, Tanaka D, Fujikura J, Masui T, Suzuki H, Yamada M, Kosaki K, Kasai Y, Hatano E, Inaba A, Wada T, Kosugi S, Ueda Y, Fujii T, Taura D and Inagaki N (2024) Case report: Duplication of the GCK gene is a novel cause of nesidioblastosis: evidence from a case with Silver-Russell syndrome-like phenotype related to chromosome 7. Front. Endocrinol. 15:1431547. doi: 10.3389/fendo.2024.1431547
Received: 12 May 2024; Accepted: 13 November 2024;
Published: 10 December 2024.
Edited by:
Yukihiro Hasegawa, Tokyo Metropolitan Children’s Medical Center, JapanReviewed by:
Masayo Kagami, National Center for Child Health and Development (NCCHD), JapanShigeru Suzuki, Asahikawa Medical University, Japan
Copyright © 2024 Shoji, Yamauchi, Kawasaki, Iwanaga, Hakata, Tanaka, Fujikura, Masui, Suzuki, Yamada, Kosaki, Kasai, Hatano, Inaba, Wada, Kosugi, Ueda, Fujii, Taura and Inagaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ichiro Yamauchi, aWNoaXJveUBrdWhwLmt5b3RvLXUuYWMuanA=