 Kyung Eun Lee1†Miyuki Nishi2†Jongsoo Kim1†
Kyung Eun Lee1†Miyuki Nishi2†Jongsoo Kim1† Takashi Murayama3
Takashi Murayama3 Zachary Dawson1Xiaoliang Wang1
Zachary Dawson1Xiaoliang Wang1 Xinyu Zhou1
Xinyu Zhou1 Tao Tan1
Tao Tan1 Chuanxi Cai1Hiroshi Takeshima2*
Chuanxi Cai1Hiroshi Takeshima2* Ki Ho Park1*
Ki Ho Park1*- 1Division of Surgical Sciences, Department of Surgery, University of Virginia, Charlottesville, VA, United States
- 2Department of Biological Chemistry, Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto, Japan
- 3Department of Cellular and Molecular Pharmacology, Graduate School of Medicine, Juntendo University, Tokyo, Japan
Rationale: MG53’s known function in facilitating tissue repair and anti-inflammation has broad applications to regenerative medicine. There is controversy regarding MG53’s role in the development of type 2 diabetes mellitus.
Objective: This study aims to address this controversy – whether MG53’s myokine function contributes to inhibition of insulin signaling in muscle, heart, and liver tissues.
Study design: We determined the binding affinity of the recombinant human MG53 (rhMG53) to the insulin receptor extracellular domain (IR-ECD) and found low affinity of interaction with Kd (>480 nM). Using cultured C2C12 myotubes and HepG2 cells, we found no effect of rhMG53 on insulin-stimulated Akt phosphorylation (p-Akt). We performed in vivo assay with C57BL/6J mice subjected to insulin stimulation (1 U/kg, intraperitoneal injection) and observed no effect of rhMG53 on insulin-stimulated p-Akt in muscle, heart and liver tissues.
Conclusion: Overall, our data suggest that rhMG53 can bind to the IR-ECD, however has a low likelihood of a physiologic role, as the Kd for binding is ~10,000 higher than the physiologic level of MG53 present in the serum of rodents and humans (~10 pM). Our findings question the notion proposed by Xiao and colleagues – whether targeting circulating MG53 opens a new therapeutic avenue for type 2 diabetes mellitus and its complications.
1 Introduction
Since its discovery as a cell membrane repair protein in 2009 (1), MG53/TRIM72 has been the subject of significant research, shedding light on its mechanistic actions in regenerative medicine and its role in regulating metabolic syndromes (2, 3). Beyond its role in tissue repair, MG53 also exhibits anti-inflammatory properties, offering potential applications in aging biology, organ failure, viral infection, and wound healing (4–6). However, the debate surrounding MG53’s involvement in the development of type 2 diabetes mellitus persists (3, 7–12).
Xiao and colleagues proposed that MG53-mediated downregulation of IRS-1 could serve as a causative factor for development of type II diabetes (10, 12). However, other studies demonstrated that even total ablation of IRS-1 is not sufficient to induce type II diabetes in mice (13, 14). Moreover, many other investigators have presented data that challenge their conclusion (15–18). We previously generated db/db mice with knockout or overexpression of MG53 and found no changes in insulin signaling or glucose metabolism, suggesting that “MG53 does not manifest the development of diabetes in db/db mice” (11). Recently, Philouze et al. (9) conducted an extensive series of biochemical and physiological assays and also concluded that “MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle”. In our recent publication in JCI Insight (19), we demonstrated that MG53 suppresses NF-κB activation to mitigate age-related heart failure in mice, and repetitive administration of recombinant human MG53 protein (rhMG53) did not produce adverse effects on major vital organs.
Wu et al. (12) proposed that MG53 has dual actions as a myokine and an E3 ligase to inhibit insulin signaling in muscle, heart, and liver tissues. Here we present new data that found no evidence of a physiologic interaction of MG53 with insulin receptor, and negative result for MG53 in controlling insulin-stimulated Akt phosphorylation in muscle, heart and liver tissues. These findings further challenge the notion proposed by Wu et al. that intervention to reduce circulating MG53 can be beneficial to diabetes and its complications.
2 Research design and methods
Biolayer Interferometry (BLI) Assays were utilized to analyze protein binding affinity. The binding affinity of rhMG53 with human insulin receptor extracellular domain (IR-ECD)-His (INR-H52Ha, Acro Biosystems) was determined using Anti-Penta-His (HIS1K) biosensors in an Octet Red 96 instrument (ForteBio Inc., Menlo Park, CA). IR-ECD-His was immobilized onto the surface of HIS1K biosensors. Increasing concentrations of rhMG53 were allowed to interact with the immobilized IR-ECD at room temperature in running buffer (10 mM Hepes, 150 mM NaCl, 0.005% Tween-20 pH 7.4). The final volume of all solutions was 200 µL. Assays were performed in black solid 96-well flat bottom plates on a shaker set at 1,000 rpm/min. The association and dissociation of IR-ECD was measured for 250 sec intervals. All data were analyzed using the Octet Data Analysis 9.0 software (ForteBio).
Cell culture experiments were conducted using C2C12 myotubes and HepG2 cells to investigate the effects of recombinant human MG53 (rhMG53) on insulin-stimulated phosphorylation of Akt (p-Akt). Cell lines were maintained at 37°C, 95% air, and 5% CO2 in a humidified incubator. C2C12 cells (ATCC, CRL-1772) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal bovine serum (FBS, GibcoTM, 16140071) and 1% penicillin-streptomycin (GibcoTM, 15140122). For differentiation, C2C12 cells were cultured in the DMEM with 2.5% horse serum (GibcoTM, 26050088) for 5 days. HepG2 cells (ATCC, HB-8065) were cultured in RPMI1640 medium with 10% FBS and 1% penicillin-streptomycin. HepG2 and differentiated C2C12 cells were pre-treated with 10, 100, or 500 nM of rhMG53 (or 500 nM of BSA as control) for 60 minutes, and then stimulated with 10 nM of insulin (GibcoTM, 12585014) for 10 minutes. Cells were washed using ice-cold PBS and detached mechanically using a scraper, and then collected.
In vivo studies were performed on C57BL/6J male mice to assess the impact of intravenous administration of rhMG53 on insulin-stimulated p-Akt levels in muscle, heart, and liver tissues. Animal handling and experimental procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of University of Virginia and were compliant with guidelines of the American Association for the Accreditation of Laboratory Animal Care. All experimental mice purchased from The Jackson Laboratory were 8-10 weeks of age (C57BL/6J, The Jackson Lab stock No: 000664). For in vivo treatment, after fasting overnight (13-15 hours), mice were administered rhMG53 and BSA intravenously at doses of 1 mg/kg and 6 mg/kg, respectively. 10 minutes later, they were subjected to stimulation with 1 U/kg of insulin via intraperitoneal injection. The heart, liver, and skeletal muscles were collected 10 minutes after insulin stimulation, and snap-frozen in liquid nitrogen, and stored in -80°C for later processing and analysis.
Whole lysates were isolated from cells (C2C12 and HepG2) and mouse tissues (heart, liver, and skeletal muscles) with RIPA lysis buffer containing protease inhibitor (Sigma, P8340) and phosphatase inhibitors (Sigma, P0044). The denatured proteins (20 µg/well) were separated by 4-12% protein gel (Thermo Scientific) and transferred onto PVDF membranes (MiliporeSigma, IPVH00010). Membranes were incubated in 5% (w/v) nonfat dry milk for 1 hour at room temperature, further probed with primary antibody and incubated at 4°C with gentle shaking overnight. Then, they were washed with Tris-buffered saline with 0.1% Tween 20 detergent (TBST) and probed with a HRP (horseradish peroxidase)-conjugated secondary antibody. Immunodetection was performed by iBrightTM CL 1500 Imaging System (Invitrogen) using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, 34580). Primary antibodies used were as follows: Akt (Cell Signaling Technology, 9272S, 1:1000 dilute), phospho-Akt (Ser 473) (Cell Signaling Technology, 4060S, 1:2000 dilute), IRS-1 (Cell Signaling Technology, 2382S, 1:1000 dilute), phospho-IRS-1 (Ser 636/639) (Cell Signaling Technology, 2388S, 1:1000 dilute), anti-MG53 (custom-made, 914, 1:2000), GAPDH (Cell Signaling Technology, 2118S, 1:5000 dilute) was diluted in 2.5% (w/v) nonfat dry milk.
ANOVA test (single factor) as statistical analysis was used to determine statistical difference between groups. All data were analyzed using Excel and GraphPad Prism 10 software.
3 Results and discussion
Wu et al. (12) reported elevated serum levels of MG53 (~2 fold) in diabetic mouse models and human diabetic patients, concluding that MG53 has dual actions as a myokine and an E3 ligase to inhibit insulin signaling in muscle, heart and liver tissues. Using BiACore assay, they showed recombinant human MG53 (rhMG53) protein binds to the extracellular domain of insulin receptor (IR-ECD) to block insulin signaling, with an estimated Kd of 8 nM [Figure 6C Wu et al (12)]. Compared to the rapid koff/kon of insulin binding, the kinetics for rhMG53 binding to IR-ECD is atypical; rhMG53 has slow reaction and fails to reach equilibration or saturation [Figure 6B Wu et al. (12)].
We have followed the exact protocol as described and found that rhMG53 can interact with IR-ECD but at a much higher Kd (>480 nM) (Figures 1A, B). Moreover, we confirmed pre-loading of the Octet sensorgrams with rhMG53 does not impact insulin’s affinity with IR-ECD (Figure 1C). Our results indicate that rhMG53 does not directly interfere with insulin binding to IR-ECD, and rhMG53 has a much lower competitive/allosteric threshold than previously described.
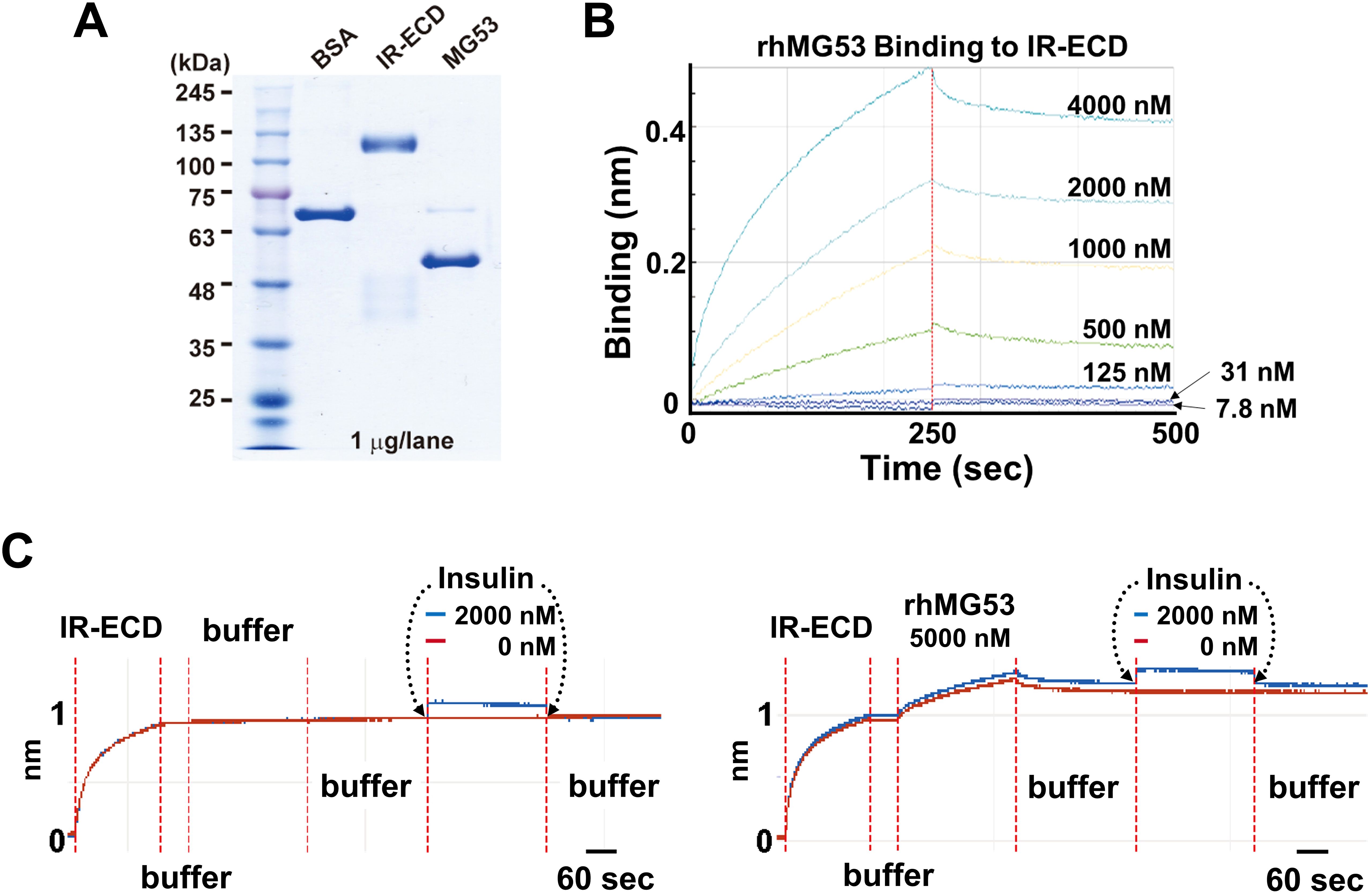
Figure 1. Biolayer interferometry (BLI) assays. (A) SDS-polyacrylamide gel electrophoresis (SDS-PAGE) of bovine serum albumin (BSA, Sigma A1470), IR-ECD (Acro Biosystem), and rhMG53 (see ref (2) for preparation). (B) In the protein-protein interaction assay using Octet RED96 instrument (ForteBio Inc., Menlo Park, CA), Octet sensorgrams of rhMG53 binding to IR-ECD revealed a Kd > 480 nM (n=3). (C) BLI assay of insulin binding to IR-ECD without rhMG53 (left panel) or with 5,000 nM rhMG53 preloaded onto the biosensor chip (right panel). Insulin binding to IR-ECD was not affected by rhMG53.
Wu et al. (12) further suggested that “… extracellular MG53 acts as an allosteric, rather than a competitive, blocker of the IR”, based on their findings in liver cell culture where incubation with rhMG53 (not BSA) inhibited insulin-stimulated phosphorylation of Akt (p-Akt) [Figure 7A Wu et al. (12)]; and intravenous administration of rhMG53 blocked insulin-stimulated p-Akt in mouse tissues [Figure 5A Wu et al. (12)]. We were puzzled by these results, as a recent report by Philouze et al. (9) showed negative findings with rhMG53 on insulin-stimulated p-Akt [Figures 3, 4 Philouze et al. (9)]. Thus, we conducted a study to determine whether rhMG53 alters insulin-stimulated p-Akt in a dose-dependent manner. We treated C2C12 myotubes and HepG2 cells with 10 nM of insulin, then quantified the impact of rhMG53 in a wide range of concentrations (10, 100 and 500 nM). As shown in Figures 2A, B, we saw no effect of rhMG53 on insulin-stimulated p-Akt in all concentrations tested.
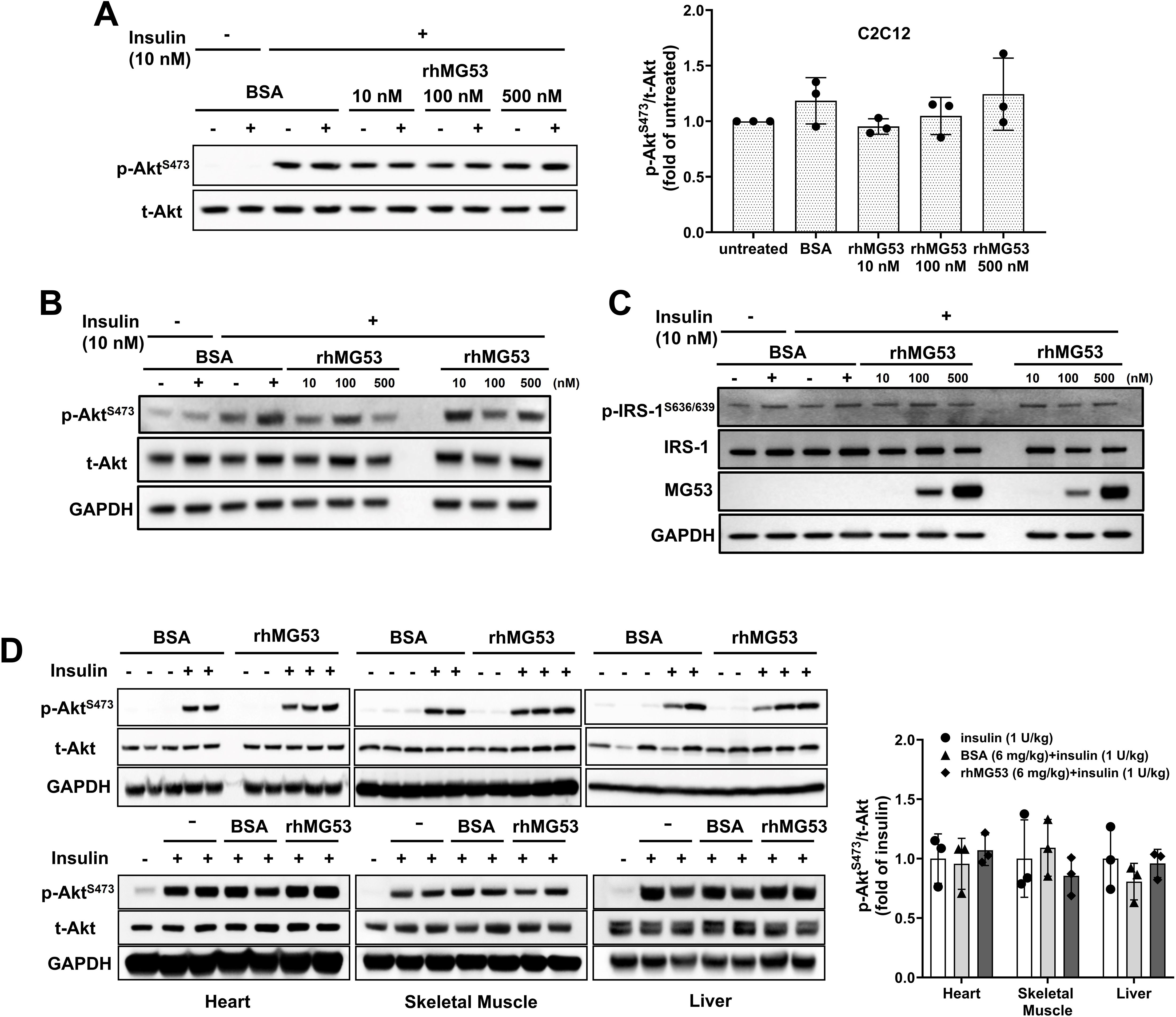
Figure 2. Role of MG53 in insulin signaling pathways in C2C12 myotubes, HepG2 cells, and mouse tissues. (A) C2C12 myotubes (5 days post differentiation) (n=3) and (B) HepG2 cells were treated with varying concentrations of rhMG53 for 1 hour (or BSA as control), followed by 10 nM insulin for 10 min. Western blot were conducted with antibody against p-Akt [Cat #4060, Cell Signaling Technology (CST)] and total Akt (t-Akt) (Cat #9272, CST) (n=2). (C) HepG2 cells were treated with varying concentrations of rhMG53 for 1 hour (or BSA as control), followed by 10 nM insulin for 10 min. Western blot were conducted with antibody against total IRS-1 (Cat #2382S, CST), p-IRS-1 (Cat #2388S, CST), anti-MG53 (custom-made, 914), GAPDH (Cat #2118S, CST) (n=2). (D) 1 mg/kg rhMG53 (upper panel) and 6 mg/kg rhMG53 (lower panel) treatment did not alter the level of p-Akt in skeletal muscle, heart and liver derived from mice subjected to 1 U/kg insulin treatment. Sample sizes were as follows: 1 mg/kg BSA + 1 U/kg insulin (n=2), 1 mg/kg rhMG53 + 1 U/kg insulin (n=3); 1 U/kg insulin (n=3), 6 mg/kg BSA + 1 U/kg insulin (n=3), 6 mg/kg rhMG53 + 1 U/kg insulin (n=3).
While our previous study (17) and study from other investigators (10) have found physical interaction between MG53 and IRS-1, and MG53 can facilitate ubiquitination of IRS-1, there is discrepancy on the in vivo role of MG53-mediated degradation of IRS-1 in muscle and heart tissues (8, 11). Our tests showed rhMG53 does not appear to impact the protein level for IRS-1 or insulin-stimulated IRS-1 phosphorylation (Figure 2C).
These findings prompted us to conduct in vivo assays to determine whether intravenous administration of rhMG53 has any impact on insulin-stimulated p-Akt in muscle, heart and liver tissues. Following the protocol of Wu et al. (12), C57BL6J mice (male, 8-10 weeks old, The Jackson Laboratory) were treated with 1 U/kg of insulin (intraperitoneal injection); 10 min prior to insulin injection, 1 mg/kg or 6 mg/kg of rhMG53 was administered into the tail vein of the mice. Administration of 1 mg/kg rhMG53 would result in a peak level of MG53 in the blood (~10 µg/ml) which should be 1000-fold and 500-fold higher than the resting level of MG53 observed in healthy and diabetic animals. We would also expect this to further saturate the IR-ECD if any allosteric interaction was occurring. However, we were surprised to find that rhMG53 has no impact on p-Akt level in skeletal muscle, heart or liver tissues (Figure 2D), which contrasts the data presented by Wu et al. (12), who notably used 6 mg/kg of rhMG53 in their study.
Overall, our data suggest that rhMG53 can bind to IR-ECD, however has low likelihood of a physiologic role, as the Kd for binding is ~10,000 higher than the physiologic level of MG53 present in the serum of rodents and humans (~10 pM). Consistent with the data shown by Wu et al. (12), rhMG53 clearly does not interfere with insulin binding at the IR. Based on our current findings and those of Philouze et al. (9), it is unlikely that MG53 has any allosteric impact on insulin-signaling in muscle, heart and liver tissues. All together, these findings question the notion proposed by Wu et al. (12) – whether targeting circulating MG53 opens a new therapeutic avenue for type 2 diabetes mellitus and its complications.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by The Institutional Animal Care and Use Committee at University of Virginia. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
KL: Writing – original draft, Writing – review & editing. MN: Writing – original draft, Writing – review & editing. JK: Writing – original draft, Writing – review & editing. TM: Writing – original draft, Writing – review & editing. ZD: Writing – review & editing. XW: Writing – review & editing. XZ: Writing – review & editing. TT: Writing – review & editing. CC: Writing – review & editing. HT: Writing – original draft, Writing – review & editing. KP: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project is supported by the Department of Surgery at University of Virginia, Kyoto University, and Juntendo University. KP was supported by a NIH grant (R01AG071676).
Conflict of interest
TT has equity interest in TRIM-edicine, Inc., which develops MG53 for regenerative medicine application.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BLI, biolayer interferometry; IR-ECD, insulin receptor extracellular domain; BSA, bovine serum albumin; Kd, dissociation constant; koff/kon, dissociation rate constant/association rate constant; MG53, mitsugumin 53; p-Akt, Akt phosphorylation; rhMG53, recombinant human MG53; t-Akt, total Akt; IRS-1, insulin receptor substrate 1; p-IRS-1, IRS-1 phosphorylation.
References
1. Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. (2009) 11:56–64. doi: 10.1038/ncb1812
2. Li Z, Wang L, Yue H, Whitson BA, Haggard E, Xu X, et al. MG53, A tissue repair protein with broad applications in regenerative medicine. Cells. (2021) 10:122. doi: 10.3390/cells10010122
3. Ma H, Liu J, Bian Z, Cui Y, Zhou X, Zhou X, et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PloS One. (2015) 10:e0124128. doi: 10.1371/journal.pone.0124128
4. Kenney AD, Li Z, Bian Z, Zhou X, Li H, Whitson BA, et al. Recombinant MG53 protein protects mice from lethal influenza virus infection. Am J Respir Crit Care Med. (2021) 203:254–7. doi: 10.1164/rccm.202007-2908LE
5. Park KH, He X, Jiang L, Zhu H, Liang J, Wang Y, et al. Activation of MG53 enhances cell survival and engraftment of human induced pluripotent stem cell-derived cardiomyocytes in injured hearts. Stem Cell Rev Rep. (2023) 19:2420–8. doi: 10.1007/s12015-023-10596-0
6. Whitson BA, Tan T, Gong N, Zhu H, Ma J. Muscle multiorgan crosstalk with MG53 as a myokine for tissue repair and regeneration. Curr Opin Pharmacol. (2021) 59:26–32. doi: 10.1016/j.coph.2021.04.005
7. Andac B, Ozgun E, Bulbul BY, Colak SY, Okur M, Yekdes AC, et al. Association of MG53 with presence of type 2 diabetes mellitus, glycemic control, and diabetic complications. PloS One. (2023) 18:e0291333. doi: 10.1371/journal.pone.0291333
8. Bian Z, Wang Q, Zhou X, Tan T, Park KH, Kramer HF, et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat Commun. (2019) 10:4659. doi: 10.1038/s41467-019-12483-0
9. Philouze C, Turban S, Cremers B, Caliez A, Lamarche G, Bernard C, et al. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PloS One. (2021) 16:e0245179. doi: 10.1371/journal.pone.0245179
10. Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature. (2013) 494:375–9. doi: 10.1038/nature11834
11. Wang Q, Bian Z, Jiang Q, Wang X, Zhou X, Park KH, et al. MG53 does not manifest the development of diabetes in db/db mice. Diabetes. (2020) 69:1052–64. doi: 10.2337/db19-0807
12. Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, et al. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation. (2019) 139:901–14. doi: 10.1161/CIRCULATIONAHA.118.037216
13. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. (1994) 372:182–6. doi: 10.1038/372182a0
14. Terauchi Y, Iwamoto K, Tamemoto H, Komeda K, Ishii C, Kanazawa Y, et al. Development of non-insulin-dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate-1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J Clin Invest. (1997) 99:861–6. doi: 10.1172/JCI119250
15. Ma LL, Zhang FJ, Kong FJ, Qian LB, Ma H, Wang JA, et al. Hypertrophied myocardium is refractory to sevoflurane-induced protection with alteration of reperfusion injury salvage kinase/glycogen synthase kinase 3beta signals. Shock. (2013) 40:217–21. doi: 10.1097/SHK.0b013e3182a0674b
16. Xu Y, Ma LL, Zhou C, Zhang FJ, Kong FJ, Wang WN, et al. Hypercholesterolemic myocardium is vulnerable to ischemia-reperfusion injury and refractory to sevoflurane-induced protection. PloS One. (2013) 8:e76652. doi: 10.1371/journal.pone.0076652
17. Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun. (2013) 4:2354. doi: 10.1038/ncomms3354
18. Yuan H, Niu Y, Liu X, Yang F, Niu W, Fu L. Proteomic analysis of skeletal muscle in insulin-resistant mice: response to 6-week aerobic exercise. PloS One. (2013) 8:e53887. doi: 10.1371/journal.pone.0053887
Keywords: Akt phosphorylation, C2C12 cells, type 2 diabetes mellitus, HepG2 cells, MG53/TRIM72, mice, myokines, insulin receptor
Citation: Lee KE, Nishi M, Kim J, Murayama T, Dawson Z, Wang X, Zhou X, Tan T, Cai C, Takeshima H and Park KH (2024) MG53’s non-physiologic interaction with insulin receptor: lack of effect on insulin-stimulated Akt phosphorylation in muscle, heart and liver tissues. Front. Endocrinol. 15:1425426. doi: 10.3389/fendo.2024.1425426
Received: 29 April 2024; Accepted: 15 August 2024;
Published: 17 September 2024.
Edited by:
Francoise Koumanov, University of Bath, United KingdomReviewed by:
Kakali Ghoshal, Vanderbilt University Medical Center, United StatesCopyright © 2024 Lee, Nishi, Kim, Murayama, Dawson, Wang, Zhou, Tan, Cai, Takeshima and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ki Ho Park, S2lIby5QYXJrQHZpcmdpbmlhLmVkdQ==; Hiroshi Takeshima, dGFrZXNoaW1hLmhpcm9zaGkuOG1Aa3lvdG8tdS5hYy5qcA==
†These authors share first authorship