 Meihui Song1,2†
Meihui Song1,2† Ying Tang
Ying Tang Ling Qi
Ling Qi Keping Xie
Keping Xie
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 17 May 2024
Sec. Translational and Clinical Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1408312
This article is part of the Research Topic Stem Cell and Translational Medicine Research in Endocrine Diseases View all 6 articles
Pancreatic cancer is difficult to diagnose early and progresses rapidly. Researchers have found that a cytokine called Interleukin-6 (IL-6) is involved in the entire course of pancreatic cancer, promoting its occurrence and development. From the earliest stages of pancreatic intraepithelial neoplasia to the invasion and metastasis of pancreatic cancer cells and the appearance of tumor cachexia, IL-6 drives oncogenic signal transduction pathways and immune escape that accelerate disease progression. IL-6 is considered a biomarker for pancreatic cancer diagnosis and prognosis, as well as a potential target for treatment. IL-6 antibodies are currently being explored as a hot topic in oncology. This article aims to systematically explain how IL-6 induces the deterioration of normal pancreatic cells, with the goal of finding a breakthrough in pancreatic cancer diagnosis and treatment.
IL-6 is a glycoprotein, initially known as B cell stimulating factor 2 due to its capability to induce B cells to produce immunoglobulin upon its early discovery (1, 2). Upon further study of this factor, it was revealed that its functions extend beyond immune regulation to encompass areas such as inflammation, hematopoiesis, and tumors, prompting its renaming as IL-6. Within healthy individuals, IL-6 is produced by a range of normal cells including hematopoietic cells, monocytes, macrophages, epithelial cells, and muscle cells, contributing to neural development, immune response, and angiogenesis (3). IL-6 is promptly and transiently generated in reaction to infections and tissue damage, aiding host defense by triggering acute phase responses, promoting hematopoiesis, and enhancing immune reactions. While its production is tightly regulated by, persistent dysregulated synthesis of IL-6 can lead to pathological effects such as chronic inflammation and autoimmunity (4).
Pancreatic cancer stands out as the most malignant tumor within the digestive system and is a leading cause of cancer-related deaths globally. This disease often progresses without early symptoms, leading to late-stage detection and a poor prognosis (5, 6). The tumor microenvironment (TME) significantly influences tumor progression, with inflammatory cells releasing tumor-promoting cytokines. Prolonged exposure to these cytokines can lead to chronic pancreatitis, contributing to pancreatic cancer development and progression. With the increasing incidence and mortality rates of pancreatic cancer, there is a growing emphasis on identifying effective diagnostic and treatment approaches. Recent studies suggest that biotherapy holds promise as a crucial breakthrough in managing pancreatic tumors, garnering considerable attention from clinicians and researchers (7).
Biotherapy involves identifying targets at the immune system and molecular level, which relies on cytokines to facilitate cellular communication. Research has demonstrated that IL-6, a multifunctional cytokine, is overexpressed in pancreatic cancer (8). The signal transduction facilitated by IL-6 plays a vital role in mediating interactions between tumor cells and stromal cells. This is particularly significant in shaping the microenvironment of pancreatic cancer, providing conducive conditions for cancer progression and metastasis (9). In this paper, we aim to comprehensively expound upon the mechanisms through which IL-6 contributes to the onset and progression of pancreatic cancer, while also highlighting potential directions for clinical treatment of the disease.
Pancreatic intraepithelial neoplasia (PanIN) is the most common precursor lesion of pancreatic cancer (10), often resulting from Kras protein mutation. In cases where the Kras protein remains unmutated, it can facilitate tissue repair when pancreatic lesions arise, promoting the swift restoration of normal cell morphology and pancreatic function. Conversely, the mutated Kras protein transforms into an oncogene, functioning as a molecular switch that transmits signals downstream through two pathways. One involves the RAF/MEK/ERK signal pathway within the MAPK family, while the other entails the PI3K/AKT signal pathway. Ultimately, the phosphorylated substrate triggers the onset of pancreatic cancer (11, 12). However, simply relying on the Kras gene is insufficient to drive pancreatic carcinogenesis. Recent studies suggesting that the role of IL-6 in PanIN acts synergistically with Kras protein to promote pancreatic carcinogenesis have generated considerable debate and discussion.
In a genetically engineered mouse model of pancreatic cancer known as the iKras* mouse (p48-Cre; R26-rtTa IRES-EGFP; TetO-KrasG12D), characterized by pancreas-specific, inducible, and reversible expression of oncogenic KrasG12D (Kras*), the pancreas exhibited significantly elevated levels of IL-6 mRNA expression compared to the wild control group. Interestingly, a decrease in PanIN formation was observed in IL-6 deficient iKras* mice, indicating a clear link between IL-6 and PanIN. The absence of IL-6 led to reduced levels of MAPK and PI3K/Akt signaling, which correlated with a decrease in PanIN formation. This suggests that IL-6 is essential for the downstream signaling of the Kras gene (13–15).
PanIN relies on IL-6 to trigger the reactive oxygen species (ROS) detoxification program. Accumulation of ROS poses a major threat to tumor cells, therefore, scavenging ROS is a critical step in tumorigenesis (16). Despite mutant Kras playing a pivotal role in regulating redox homeostasis (17), IL-6 remains essential for bolstering the antioxidant stress capacity of tumor cells. IL-6 increases the expression of Nrf2, a crucial transcription factor that facilitates the ROS detoxification program (13). IL-6 and Kras genes collaborate to advance the progression of PanIN to pancreatic cancer, with the signals they generate playing a crucial role in tumor growth.
The metabolic rate of cancer cells surpasses that of normal cells, resulting in a frequently hypoxic environment. To ensure survival, the upregulation of the IL-6/STAT3 pathway triggers the synthesis of vascular endothelial growth factor (VEGF) and promotes the proliferation of tumor blood vessels, creating favorable conditions to meet the heightened nutritional demands of tumor cell (18). Furthermore, with the overactivation of the STAT3 protein through the IL-6 signaling pathway, there is an increase in the expression of cyclin D1 and Bcl-2 genes (19).
The IL-6 signaling pathway is a key factor in the development of pancreatic cancer, with the IL-6/JAK/STAT pathway playing a particularly crucial role. This pathway facilitates signal transduction through two distinct mechanisms (Figure 1) (20, 21). When IL-6 binds to the membrane-bound IL-6 receptor (mIL-6R) to activate STAT protein, it’s known as classical signal transduction (22, 23). In contrast, when IL-6 binds to soluble IL-6R (sIL-6R), resulting from the cleavage and shedding of mIL-6R, and transmits signals downstream, it’s termed trans signal transduction (24, 25). IL-6 can bind mIL-6R or sIL-6R, forming a dimer and initiating activation of transmembrane gp130, leading to the association of the box region in gp130 with JAK2 to trigger the activation of the STAT3 protein (15, 26).
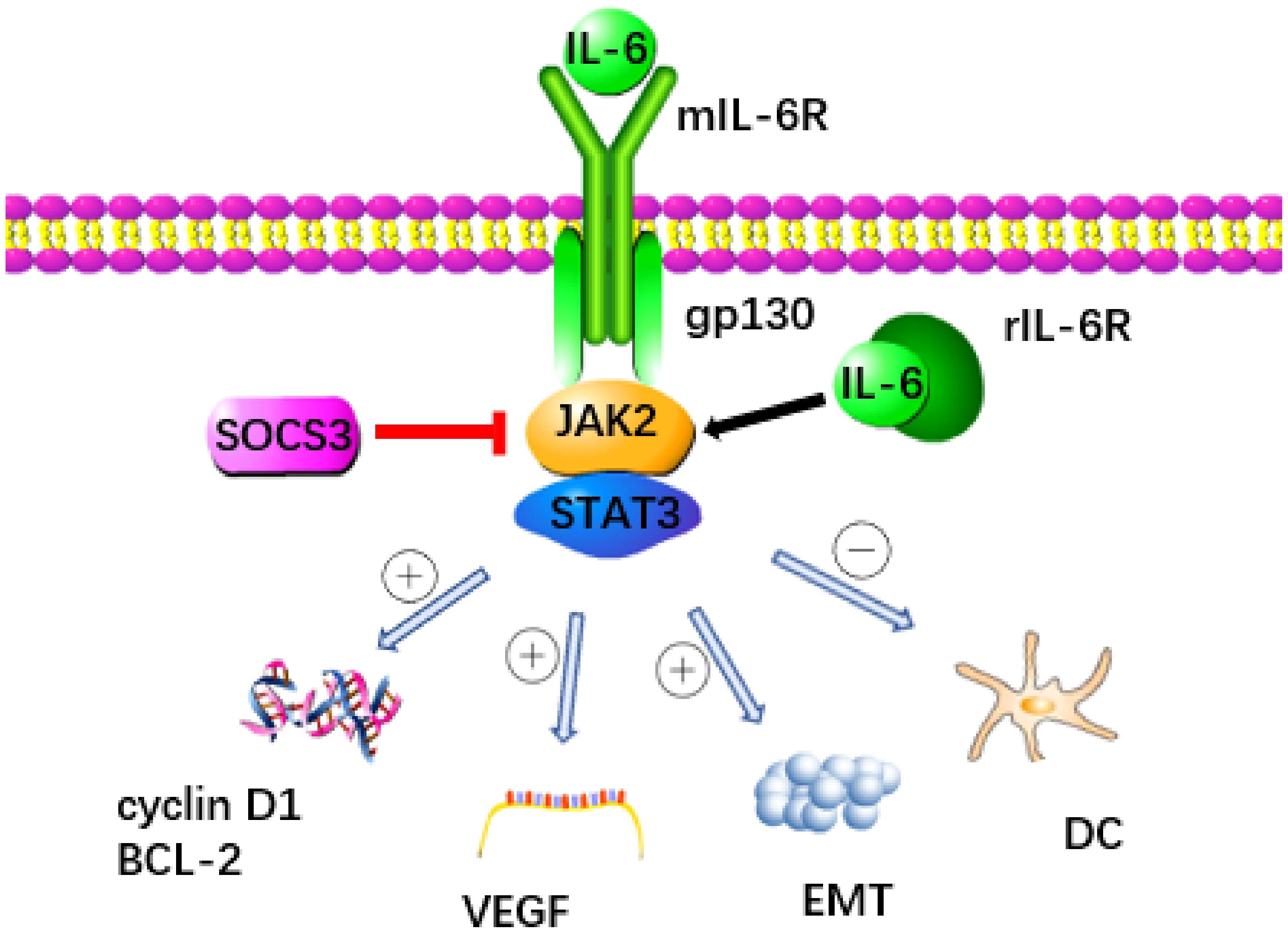
Figure 1 The signaling pathway triggered by IL-6 is recognized as a key contributor to the development of pancreatic cancer. IL-6 binds to mIL-6R or rIL-6R to activate JAK2 and transmit signal to downstream STAT3 protein. This process was inhibited by SOCS3. STAT3 promotes the progression of pancreatic cancer through a variety of ways: (1) Promoting the expression of cyclin D1 and Bcl-2 genes, (2) Up regulating VEGF, (3) Accelerating the occurrence of EMT, (4) Inhibiting the immune function of DC.
Cytokine signal transduction inhibitor 3 (SOCS3) functions as a crucial negative regulator within the IL-6/JAK2/STAT3 pathway, effectively curbing STAT3 overactivation and maintaining equilibrium (19). SOCS3 not only directly decreases the catalytic activity of JAKs but also enhances the ubiquitination of gp130 and JAK2 by increasing components of an E3 ubiquitin ligase complex, thereby impeding downstream signal transmission from IL-6 (27, 28). Moreover, the E3 ubiquitin ligase NEDD4L interacts with GP130, promoting its ubiquitination in tumors and thwarting the activation of the IL-6/GP130/STAT3 signaling axis, thereby exhibiting anti-cancer properties (29). Various members of the membrane-bound E3 ubiquitin ligase family, including MARCH2, MARCH3, MARCH4, and MARCH9, target cell surface receptors for degradation, leading to a reduction in IL-6 responsiveness, primarily through a decrease in cell surface expression levels of IL-6R (30). Recent research has pinpointed C1q/TNF-related protein 4 (CTRP4) as a natural regulator of the IL-6 receptor signaling pathway. CTRP4 competes with IL-6 for binding to IL-6R, thereby dampening IL-6-induced activation of the STAT3 pathway (31). These negative regulatory pathways governing IL6 activity are pivotal in preventing excessive responses to infections and maintaining endogenous IL6 levels. Manipulating these pathways holds promise for therapeutic interventions in tumors, as well as a broad spectrum of human autoimmune and inflammatory conditions (32).
Cancer cells frequently evade attacks from the immune system and encroach upon the territory of normal cells, particularly in the case of pancreatic cancer. This immune evasion is attributed to issues within various immune regulatory pathways, including an imbalance in the Th1/Th2 ratio of helper T cells (33). Both Th1 and Th2 cells originate from pre-CD4+T cells, and the differentiation into each type of cell is largely influenced by the cytokine environment (34). Th1 cells serve as the primary force of the immune response, rallying other cells (macrophages, NK cells, and T cells) to ward off foreign cell invasion. On the other hand, Th2 cells are generated from CD4+T cells under the influence of Th2 cytokines (such as IL-4, IL-5, IL-10, and IL-13). In contrast to the function of Th1 cells, Th2 cells can suppress the combat effectiveness of immune cells and protect their own cells from destruction (35). In normal conditions, Th1 and Th2 cells exert inhibitory effects on each other’s proliferation. Maintaining a balance between them is crucial for preserving overall bodily health. Any disruption in this balance, leading to a biased shift toward one side (referred to as “Th1/Th2 drift”), can contribute to the onset of disease (36).
Elena discovered that CD4+T cells in the serum of pancreatic cancer patients exhibited increased production of the Th2 cytokine IL-5, resulting in a phenotype of tumor-infiltrating lymphocytes that favored Th2 cells (37). An excessive presence of Th2 cells may result in the suppression of Th1 cells, indicating that an imbalance in Th1/Th2 cell levels could potentially play a role in the onset and progression of pancreatic cancer (38, 39). Hence, understanding the cause of the upregulation of Th2 cells is a potential avenue to improve outcomes for pancreatic cancer patients. Louis W and his colleagues investigated the cytokine environment surrounding Th2 cells and observed a significant increase in IL-13 and IL-10 secretion induced by IL-6. Their findings suggest that IL-6 can stimulate pancreatic cancer cells to release Th2 cytokines, consequently fostering the generation of Th2 cells and indirectly facilitating immune evasion by tumor cells (40). Furthermore, there is evidence indicating that IL-6 can enhance the expression of the Th2 cytokine IL-4 gene by activating NFAT transcription factors, thereby facilitating the differentiation of T cells into Th2 cells (41). It is important to counteract the influence of IL-6 on helper T cells in order to preserve the balance of Th1/Th2 and bolster anti-tumor immunity.
Altered energy metabolism is a characteristic of pancreatic cancer. In order to fulfill the growth requirements of tumor cells, there is an observed increase in glycolytic flux. When blood glucose levels are low or cellular carbohydrate reserves (e.g., glycogen) are depleted by malignant cells, liver cells metabolize fatty acids into ketones to generate additional energy and maintain the normal function of tissue cells. This self-protective response is known as the action of ketone bodies. It has been suggested that the metabolic reprogramming induced by ketone bodies may mitigate the cachexia associated with pancreatic cancer (42). However, patients with pancreatic cancer experience a reduced capacity for ketogenesis.
Flint et al. discovered that in cachexia mice, IL-6 diminished ketogenesis in the liver by inhibiting PPAR α (43). PPAR α plays a crucial role in activating ketogenic genes and serves as an essential transcription factor to initiate the process of ketogenic transcription (44). When IL-6 downregulates PPAR α, it restricts the ketogenic effect, leading to greater energy deficiency in cancer patients. Interestingly, this energy deficit can result in systemic metabolic stress, characterized by a notable increase in glucocorticoid levels. While synthetic glucocorticoids (such as dexamethasone) can be employed as apoptosis-inducing agents in clinical cancer treatment, endogenous glucocorticoids typically promote cancer progression (45). In a mouse model of pancreatic cancer, the injection of recombinant IL-6 not only elevated corticosterone levels but also led to the depletion of immune cells such as CD4+T cells, CD8+T cells, and NK cells (43). This evidence demonstrates that the hormone dysfunction induced by IL-6-driven metabolic reprogramming further influences anti-tumor immunity. It is conceivable that pancreatic cancer cells alter the host’s metabolism by secreting IL-6, and subsequently leverage the influence of hormones to bolster their defense. This positive feedback cycle is likely to result in the failure of immunotherapy (46).
Cancer-related cachexia is present in over 85% of individuals with pancreatic cancer, significantly contributing to the low survival rates among these patients (47). In 2011, an international consensus defined cachexia as a wasting syndrome marked by progressive depletion of skeletal muscle mass (33), his may be accompanied by severe anorexia, significant weight loss, muscle weakness, cognitive decline, and other detrimental manifestations (48). Nevertheless, the impact of cachexia extends beyond muscle loss, affecting nearly all tissues and organs in the body (49). Tian and colleagues noted myocardial fibrosis and myocardial atrophy in a mouse model of cancer cachexia, indicating that cachexia may diminish cardiac contractility (50). Rosa discovered that cachexia triggered collagen accumulation in the liver, resulting in disrupted liver metabolism (51). Additionally, dysfunctions of the reproductive system, imbalances in intestinal flora, and endocrine changes in the islets are also associated with cachexia (49, 52). Consequently, researchers have started redirecting their focus toward the mediators of cancer cachexia in order to prevent these outcomes.
In recent years, cachexia has been found to be intricately linked to the host itself and numerous tumor-derived cytokines (53), with IL-6 being identified as one of the factors related to cachexia in pancreatic cancer. Clinical data statistics indicate that progressive weight loss, fatigue, and other cachexia symptoms in patients with advanced pancreatic cancer are often positively associated with serum IL-6 levels (54). Martinoni and colleagues conducted immunohistochemical staining on tissue sections from pancreatic cancer patients, they found that IL-6 exhibited greater immunoreactivity in tissue regions of patients with cachexia compared to those without cachexia (55). Furthermore, the administration of anti-IL-6 monoclonal antibody in mice was found to partially inhibit the progression of cancer cachexia (56). This indicates that IL-6 contributes to the development of cachexia in pancreatic cancer.
Cachexia primarily arises from the imbalance between catabolic and anabolic signals. Notably, IL-6 is implicated in both of these signaling pathways. During the early stages of cachexia, prior to skeletal muscle atrophy, fat depletion occurs as white adipose tissue (WAT) transforms into brown adipose tissue (BAT). Unlike WAT, BAT efficiently breaks down triglycerides stored in adipocytes into free fatty acids and glycerol, leading to impaired energy storage over time (57, 58). The process of white adipose tissue browning is viewed as the foundation of cachexia, and it is actually triggered by IL-6 (59, 60). Elevated levels of IL-6 can enhance the expression of uncoupling protein 1 (UCP1) (57), by disrupting the link between electron transport and phosphorylation in a segment of the typical respiratory chain, resulting in halted ATP synthesis. An anti-IL-6 receptor antibody was employed in treating cachexia in mice, this led to the inhibition of white adipose tissue browning and lipolysis (60).
Apart from catabolism, the depletion of skeletal muscle also stems from disruptions in anabolism. The IGF/Akt/mTOR signal transduction pathway is crucial for protein and lipid synthesis. Ribosomal S6K and eukaryotic initiation factor 4EBP-1, activated by mTOR, significantly contribute to sustaining muscle growth, making them potentially potent defenses against cachexia (61, 62). Nevertheless, IL-6 induced AMP activated protein kinase (AMPK) showed a negative correlation with mTOR (63), which was detrimental to muscle preservation. In pancreatic cancer, pancreatic stellate cells release significant quantities of IL-6, which signals downstream activation of AMPK. AMPK evidently suppresses mTOR activity (64), ultimately resulting in muscle atrophy. Notably, exercise training could serve as an effective method to suppress AMPK, thereby delaying the advancement of cachexia (65). Additionally, it has been proposed that the IL-6/JAK/STAT3 pathway is a potential contributor to muscle loss, particularly through the phosphorylation of STAT3 (Figure 2). Introducing STAT3 into the tibialis anterior of tumor-bearing mice using a plasmid as a vector resulted in noticeable muscle atrophy. This process was accompanied by an increase in the protein degradation gene atrogin-1 (66), although further confirmation is required to establish whether atrogin-1 is associated with the activation of STAT3.
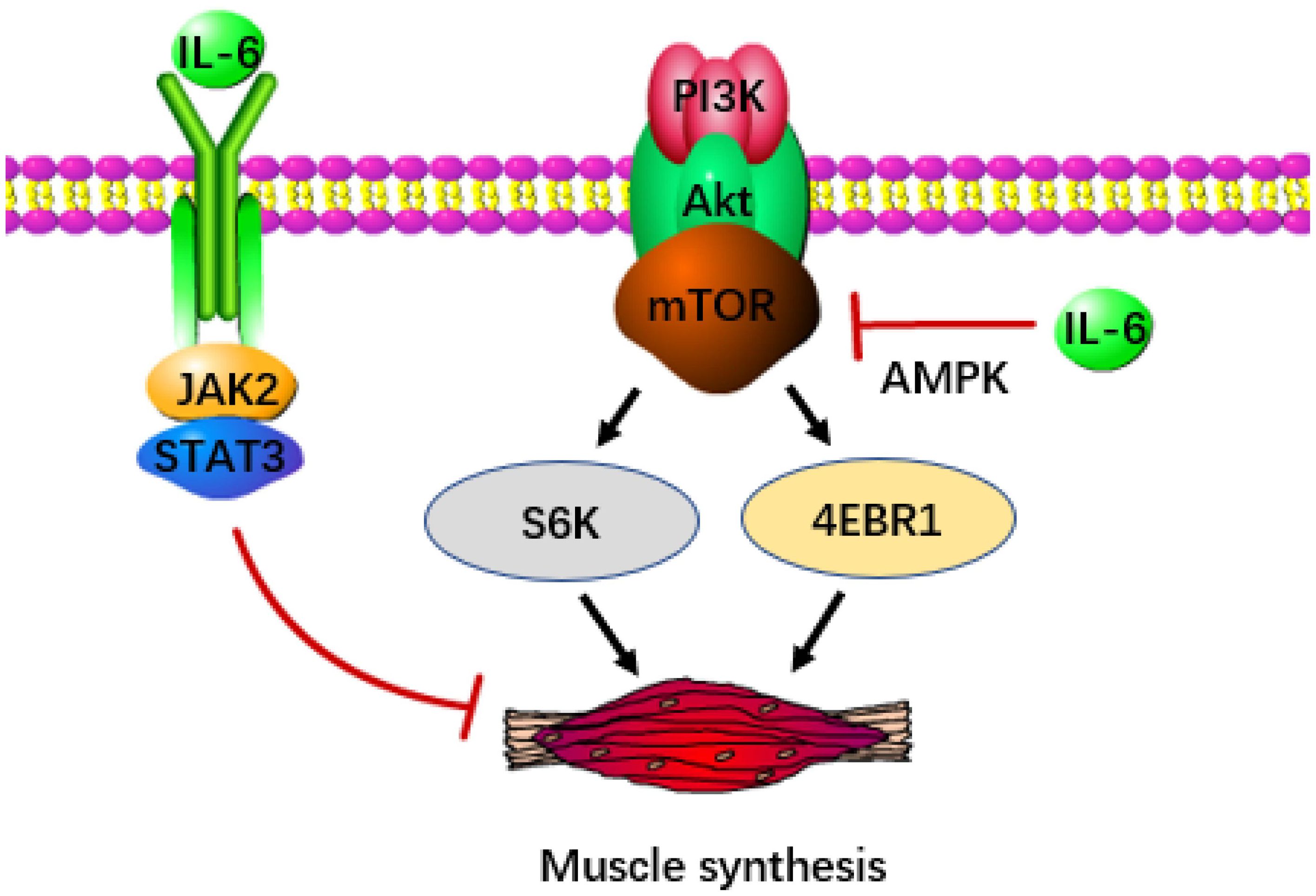
Figure 2 IL-6 accelerates the occurrence of cachexia in pancreatic cancer. IL-6 can inhibit the release of signals from the IGF/Akt/mTOR pathway, thereby reducing the production of S6K and 4EBP-1 (the key transcription factor of muscle synthesis). IL-6 can also directly inhibit muscle synthesis through JAK/STAT3.
In individuals with lung cancer, Pettersen discovered that the combination of rIL-6R and IL-6 expedited the autophagy of muscle cells through trans-signaling, potentially contributing to muscle loss in patients with cachexia (67). While elevated levels of branched-chain amino acids and protein metabolites are evident in the plasma of pancreatic cancer patients, pointing to autophagy in the advanced stages of pancreatic cancer (68), there is no evidence yet to confirm that this is also triggered by the anti-signal of IL-6. In any event, it can be determined that IL-6 acts as an inducer of cachexia in pancreatic cancer and suppressing IL-6 release represents a target for enhancing prognosis.
Using ELISA, it was found that the plasma IL-6 levels in untreated pancreatic cancer patients were significantly higher than those in healthy individuals, and IL-6 level were positively correlated with the tumor stage (8, 69). This finding indicates that IL-6 serves as a valuable diagnostic biomarker for pancreatic cancer. In a study aimed at finding immunobiomarkers, plasma from 73 patients with untreated metastatic pancreatic cancer was tested. Those with high IL-6 expression tended to experience faster metastasis and shorter survival times, highlighting the potential of plasma IL-6 levels in predicting the survival rate of patients with metastatic pancreatic cancer (70).
Considering IL-6’s pivotal role in driving tumorigenesis and metastasis, one might wonder whether inhibiting IL-6 could be a viable strategy in managing the progression of pancreatic cancer. While IL-6 targeted drugs are currently utilized in pancreatic cancer treatment, the effectiveness of individual drugs seems to be limited, likely due to variations among individuals and small sample sizes. Recognizing that tumor development is influenced by multiple factors, it becomes imperative to explore combinations of IL-6 targeted drugs with other medications. Preclinical studies indicate that targeted IL-6 inhibition can enhance the efficacy of anti-PD-L1 therapy in pancreatic cancer (71). Additionally, substances like raloxifene have shown promise in potentiating the effectiveness of chemotherapy agents such as paclitaxel by targeting IL-6 signaling pathways (72). Notably, IL-6 receptor blockade has been shown to improve chemotherapy outcomes in pancreatic cancer. Considering these promising findings, there is strong rationale to believe that IL-6 antibodies hold significant potential in combination therapies for pancreatic cancer (73).
After the discovery of IL-6, its role in promoting the growth, invasion, and spread of cancer has been widely recognized. The use of IL-6 monoclonal antibody has shown promising results in treating renal cell carcinoma, prostate cancer, lymphoma, multiple myeloma, and other diseases (74–76). However, clinical trials for pancreatic cancer treatment are still pending. It’s important to note that due to the short half-life of the IL-6 antibody, repeated administration is necessary to maintain effective blood concentration (77). Research into the development of IL-6 receptor antibodies could potentially improve therapeutic effectiveness and enhance patient convenience (78).
MS: Conceptualization, Funding acquisition, Project administration, Writing – original draft. YT: Validation, Investigation, Writing – original draft. KC: Writing – original draft, Data curation. KX: Supervision, Visualization, Writing – review & editing. LQ: Writing – review & editing, Conceptualization, Project administration, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82203351), Guangdong Basic and Applied Basic Research Foundation (2021A1515111095), and Scientific Research Project of Guangdong Bureau of Traditional Chinese Medicine (20231412).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. (2010) 22:347–52. doi: 10.1093/intimm/dxq030
2. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. (1986) 324:73–6. doi: 10.1038/324073a0
3. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
4. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. (2014) 26:54–74. doi: 10.1016/j.smim.2014.01.001
5. McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. (2018) 24:4846–61. doi: 10.3748/wjg.v24.i43.4846
6. Tempero MA. NCCN guidelines updates: pancreatic cancer. J Natl Compr Canc Netw. (2019) 17:603–5. doi: 10.6004/jnccn.2019.5007
7. Heinrich S, Lang H. Neoadjuvant therapy of pancreatic cancer: definitions and benefits. Int J Mol Sci. (2017) 18:1622. doi: 10.3390/ijms18081622
8. Miura T, Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Takahashi H, et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas. (2015) 44:756–63. doi: 10.1097/MPA.0000000000000335
9. Holmer R, Goumas FA, Waetzig GH, Rose-John S, Kalthoff H. Interleukin-6: a villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat Dis Int. (2014) 13:371–80. doi: 10.1016/s1499-3872(14)60259-9
10. Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. (2006) 97:1–7. doi: 10.1111/j.1349-7006.2005.00134.x
11. Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. (2016) 168:29–42. doi: 10.1016/j.pharmthera.2016.09.003
12. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2020) 17:153–68. doi: 10.1038/s41575-019-0245-4
13. Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. (2013) 73:6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T
14. Pop V-V, Seicean A, Lupan I, Samasca G, Burz C-C. IL-6 roles - Molecular pathway and clinical implication in pancreatic cancer - A systemic review. Immunol Lett. (2017) 181:45–50. doi: 10.1016/j.imlet.2016.11.010
15. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374:1–20. doi: 10.1042/BJ20030407
16. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. (2011) 475:106–9. doi: 10.1038/nature10189
17. Ferino A, Rapozzi V, Xodo LE. The ROS-KRAS-Nrf2 axis in the control of the redox homeostasis and the intersection with survival-apoptosis pathways: Implications for photodynamic therapy. J Photochem Photobiol B. (2020) 202:111672. doi: 10.1016/j.jphotobiol.2019.111672
18. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. (2002) 21:2000–8. doi: 10.1038/sj.onc.1205260
19. Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. (2005) 392:335–44. doi: 10.1042/BJ20050465
20. Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. (2017) 108:1947–52. doi: 10.1111/cas.13332
21. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
22. Knüpfer H, Preiss R. sIL-6R: more than an agonist? Immunol Cell Biol. (2008) 86:87–91. doi: 10.1038/sj.icb.7100113
23. Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. (2015) 34:75–82. doi: 10.1016/j.coi.2015.02.008
24. Bharadwaj U, Marin-Muller C, Li M, Chen C, Yao Q. Mesothelin overexpression promotes autocrine IL-6/sIL-6R trans-signaling to stimulate pancreatic cancer cell proliferation. Carcinogenesis. (2011) 32:1013–24. doi: 10.1093/carcin/bgr075
25. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). (2012) 122:143–59. doi: 10.1042/CS20110340
26. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. (2012) 8:1237–47. doi: 10.7150/ijbs.4989
27. Kershaw NJ, Laktyushin A, Nicola NA, Babon JJ. Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: identification of the active components and JAK2 and gp130 as substrates. Growth Factors. (2014) 32:1–10. doi: 10.3109/08977194.2013.877005
28. Zhang M-Y, Wang J, Guo J. Role of regenerating islet-derived protein 3A in gastrointestinal cancer. Front Oncol. (2019) 9:1449. doi: 10.3389/fonc.2019.01449
29. Liu H, Wang N, Yang R, Luan J, Cao M, Zhai C, et al. E3 ubiquitin ligase NEDD4L negatively regulates skin tumorigenesis by inhibiting IL-6/GP130 signaling pathway. J Invest Dermatol. (2024) 4:S0022–202X(24)00272–0. doi: 10.1016/j.jid.2024.03.030
30. Babon JJ, Stockwell D, DiRago L, Zhang J-G, Laktyushin A, Villadangos J, et al. (MARCH) proteins down-regulate cell surface expression of the interleukin-6 receptor alpha chain (IL6Rα). Biochem J. (2019) 476:2869–82. doi: 10.1042/BCJ20190577
31. Cao L, Deng J, Chen W, He M, Zhao N, Huang H, et al. CTRP4/interleukin-6 receptor signaling ameliorates autoimmune encephalomyelitis by suppressing Th17 cell differentiation. J Clin Invest. (2023) 134:e168384. doi: 10.1172/JCI168384
32. Orange ST, Leslie J, Ross M, Mann DA, Wackerhage H. The exercise IL-6 enigma in cancer. Trends Endocrinol Metab. (2023) 34:749–63. doi: 10.1016/j.tem.2023.08.001
33. Zhang Y, Zhang Y, Gu W, He L, Sun B. Th1/Th2 cell’s function in immune system. Adv Exp Med Biol. (2014) 841:45–65. doi: 10.1007/978-94-017-9487-9_3
34. Zhang Y, Zhang Y, Gu W, Sun B. TH1/TH2 cell differentiation and molecular signals. Adv Exp Med Biol. (2014) 841:15–44. doi: 10.1007/978-94-017-9487-9_2
35. Chraa D, Naim A, Olive D, Badou A. T lymphocyte subsets in cancer immunity: Friends or foes. J Leukoc Biol. (2019) 105:243–55. doi: 10.1002/JLB.MR0318-097R
36. Hirahara K, Nakayama T. CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol. (2016) 28:163–71. doi: 10.1093/intimm/dxw006
37. Tassi E, Gavazzi F, Albarello L, Senyukov V, Longhi R, Dellabona P, et al. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. (2008) 181:6595–603. doi: 10.4049/jimmunol.181.9.6595
38. Protti MP, De Monte L. Cross-talk within the tumor microenvironment mediates Th2-type inflammation in pancreatic cancer. Oncoimmunology. (2012) 1:89–91. doi: 10.4161/onci.1.1.17939
39. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. (2011) 208:469–78. doi: 10.1084/jem.20101876
40. Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, et al. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther. (2007) 6:1096–100. doi: 10.4161/cbt.6.7.4328
41. Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. (2002) 39:531–6. doi: 10.1016/s0161-5890(02)00210-9
42. Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. (2014) 2:18. doi: 10.1186/2049-3002-2-18
43. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. (2016) 24:672–84. doi: 10.1016/j.cmet.2016.10.010
44. Grabacka M, Pierzchalska M, Dean M, Reiss K. Regulation of ketone body metabolism and the role of PPARα. Int J Mol Sci. (2016) 17:2093. doi: 10.3390/ijms17122093
45. Yao Y, Yao Q-Y, Xue J-S, Tian X-Y, An Q-M, Cui L-X, et al. Dexamethasone inhibits pancreatic tumor growth in preclinical models: Involvement of activating glucocorticoid receptor. Toxicol Appl Pharmacol. (2020) 401:115118. doi: 10.1016/j.taap.2020.115118
46. Flint TR, Fearon DT, Janowitz T. Connecting the metabolic and immune responses to cancer. Trends Mol Med. (2017) 23:451–64. doi: 10.1016/j.molmed.2017.03.001
47. Henderson SE, Makhijani N, Mace TA. Pancreatic cancer-induced cachexia and relevant mouse models. Pancreas. (2018) 47:937–45. doi: 10.1097/MPA.0000000000001124
48. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. (2011) 12:489–95. doi: 10.1016/S1470-2045(10)70218-7
49. Schmidt SF, Rohm M, Herzig S, Berriel Diaz M. Cancer cachexia: more than skeletal muscle wasting. Trends Cancer. (2018) 4:849–60. doi: 10.1016/j.trecan.2018.10.001
50. Xu H, Crawford D, Hutchinson KR, Youtz DJ, Lucchesi PA, Velten M, et al. Myocardial dysfunction in an animal model of cancer cachexia. Life Sci. (2011) 88:406–10. doi: 10.1016/j.lfs.2010.12.010
51. Rosa-Caldwell ME, Brown JL, Lee DE, Wiggs MP, Perry RA, Haynie WS, et al. Hepatic alterations during the development and progression of cancer cachexia. Appl Physiol Nutr Metab. (2020) 45:500–12. doi: 10.1139/apnm-2019-0407
52. Costa RGF, Caro PL, de Matos-Neto EM, Lima JDCC, Radloff K, Alves MJ, et al. Cancer cachexia induces morphological and inflammatory changes in the intestinal mucosa. J Cachexia Sarcopenia Muscle. (2019) 10:1116–27. doi: 10.1002/jcsm.12449
53. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers. (2018) 4:17105. doi: 10.1038/nrdp.2017.105
54. Suh S-Y, Choi YS, Yeom CH, Kwak SM, Yoon HM, Kim DG, et al. Interleukin-6 but not tumour necrosis factor-alpha predicts survival in patients with advanced cancer. Support Care Cancer. (2013) 21:3071–7. doi: 10.1007/s00520-013-1878-4
55. Martignoni ME, Kunze P, Hildebrandt W, Künzli B, Berberat P, Giese T, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin Cancer Res Off J Am Assoc Cancer Res (2005) 11:5802–8. doi: 10.1158/1078-0432.CCR-05-0185
56. Barber MD, Ross JA, Fearon KC. Disordered metabolic response with cancer and its management. World J Surg. (2000) 24:681–9. doi: 10.1007/s002689910110
57. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. (2014) 20:433–47. doi: 10.1016/j.cmet.2014.06.011
58. Park A, Kim WK, Bae K-H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J Stem Cells. (2014) 6:33–42. doi: 10.4252/wjsc.v6.i1.33
59. Daas SI, Rizeq BR, Nasrallah GK. Adipose tissue dysfunction in cancer cachexia. J Cell Physiol. (2018) 234:13–22. doi: 10.1002/jcp.26811
60. Han J, Meng Q, Shen L, Wu G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. (2018) 17:14. doi: 10.1186/s12944-018-0657-0
61. Duval AP, Jeanneret C, Santoro T, Dormond O. mTOR and tumor cachexia. Int J Mol Sci. (2018) 19:2225. doi: 10.3390/ijms19082225
62. Miyamoto Y, Hanna DL, Zhang W, Baba H, Lenz H-J. Molecular pathways: cachexia signaling-A targeted approach to cancer treatment. Clin Cancer Res. (2016) 22:3999–4004. doi: 10.1158/1078-0432.CCR-16-0495
63. White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab. (2013) 304:E1042–1052. doi: 10.1152/ajpendo.00410.2012
64. Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. (2002) 277:23977–80. doi: 10.1074/jbc.C200171200
65. Puppa MJ, White JP, Velázquez KT, Baltgalvis KA, Sato S, Baynes JW, et al. The effect of exercise on IL-6-induced cachexia in the Apc ( Min/+) mouse. J Cachexia Sarcopenia Muscle. (2012) 3:117–37. doi: 10.1007/s13539-011-0047-1
66. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab. (2012) 303:E410–421. doi: 10.1152/ajpendo.00039.2012
67. Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci Rep. (2017) 7:2046. doi: 10.1038/s41598-017-02088-2
68. Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. (2014) 20:1193–8. doi: 10.1038/nm.3686
69. Sarris EG, Syrigos KN, Saif MW. Pancreatic cancer: updates on translational research and future applications. JOP. (2013) 14:145–8. doi: 10.6092/1590-8577/1466
70. Farren MR, Mace TA, Geyer S, Mikhail S, Wu C, Ciombor K, et al. Systemic immune activity predicts overall survival in treatment-naïve patients with metastatic pancreatic cancer. Clin Cancer Res. (2016) 22:2565–74. doi: 10.1158/1078-0432.CCR-15-1732
71. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. (2018) 67:320–32. doi: 10.1136/gutjnl-2016-311585
72. Hering NA, Günzler E, Arndt M, Zibell M, Lauscher JC, Kreis ME, et al. Targeting interleukin-6/glycoprotein-130 signaling by raloxifene or SC144 enhances paclitaxel efficacy in pancreatic cancer. Cancers (Basel). (2023) 15:456. doi: 10.3390/cancers15020456
73. Long KB, Tooker G, Tooker E, Luque SL, Lee JW, Pan X, et al. IL6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol Cancer Ther. (2017) 16:1898–908. doi: 10.1158/1535-7163.MCT-16-0899
74. Rossi J-F, Négrier S, James ND, Kocak I, Hawkins R, Davis H, et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br J Cancer. (2010) 103:1154–62. doi: 10.1038/sj.bjc.6605872
75. Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN, Van Veldhuizen PJ, et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. (2010) 16:3028–34. doi: 10.1158/1078-0432.CCR-09-3122
76. Trikha M, Corringham R, Klein B, Rossi J-F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. (2003) 9:4653–65.
77. Puchalski T, Prabhakar U, Jiao Q, Berns B, Davis HM. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin Cancer Res. (2010) 16:1652–61. doi: 10.1158/1078-0432.CCR-09-2581
Keywords: IL-6, pancreatic cancer, PanIN, tumor immunity, cachexia, diagnostic biomarker
Citation: Song M, Tang Y, Cao K, Qi L and Xie K (2024) Unveiling the role of interleukin-6 in pancreatic cancer occurrence and progression. Front. Endocrinol. 15:1408312. doi: 10.3389/fendo.2024.1408312
Received: 28 March 2024; Accepted: 06 May 2024;
Published: 17 May 2024.
Edited by:
Guifang Zhao, Mayo Clinic Florida, United StatesReviewed by:
Xuan Yang, Shandong Maternal and Child Health Hospital, ChinaCopyright © 2024 Song, Tang, Cao, Qi and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keping Xie, bWN4aWVrZXBpbmdAc2N1dC5lZHUuY24=; Ling Qi, cWlsaW5nMTcxOEBnemhtdS5lZHUuY24=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.