 Yunfeng Yu
Yunfeng Yu Xinyu Yang
Xinyu Yang Juan Deng
Juan Deng Jingyi Wu
Jingyi Wu Siyang Bai
Siyang Bai Rong Yu
Rong Yu- 1School of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, China
- 2Department of Endocrinology, The First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, China
- 3The Third School of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
Objective: The role of immune cells in type 1 diabetes (T1D) is unclear. The aim of this study was to assess the causal effect of different immune cells on T1D using Mendelian randomization (MR).
Methods: A dataset of immune cell phenotypes (numbered from GCST0001391 to GCST0002121) was obtained from the European Bioinformatics Institute, while a T1D dataset (numbered finngen_R10_T1D) was obtained from FinnGen. Single nucleotide polymorphisms meeting the conditions were screened stepwise according to the assumptions of association, independence, and exclusivity. Inverse variance weighted was used as the main method for the MR analysis. MR-Egger was used to assess the horizontal pleiotropy of the results. Cochran’s Q and the leave-one-out method were respectively used for the heterogeneity analysis and the sensitivity analysis of the results.
Results: MR analysis showed that effector memory (EM) double-negative (DN) (CD4−CD8−) %T cells [odds ratio (OR) = 1.157, 95% confidence interval (95% CI) = 1.016–1.318, p = 0.028, false discovery rate (FDR) = 0.899], EM CD8br %T cells (OR = 1.049, 95% CI = 1.003–1.098, p = 0.037, FDR = 0.902), CD28 on CD28+CD45RA+CD8br (OR = 1.334, 95% CI = 1.132–1.571, p = 0.001, FDR = 0.044), IgD+CD38dim %lymphocytes (OR = 1.045, 95% CI = 1.002–1.089, p = 0.039, FDR = 0.902), CD80 on monocytes (OR = 1.084, 95% CI = 1.013–1.161, p = 0.020, FDR = 0.834), SSC-A on plasmacytoid dendritic cells (pDCs) (OR = 1.174, 95% CI = 1.004–1.372, p = 0.044, FDR = 0.902), and FSC-A on pDCs (OR = 1.182, 95% CI = 1.011–1.382, p = 0.036, FDR = 0.902) were associated with an increased genetic susceptibility to T1D. Cochran’s Q showed that there was heterogeneity for CD28 on the CD28+CD45RA+CD8br results (p = 0.043), whereas there was no heterogeneity for the other results (p ≥ 0.05). The sensitivity analysis showed that the MR analysis results were robust.
Conclusion: The MR analysis demonstrated that seven immune cell phenotypes were associated with an increased genetic susceptibility to T1D. These findings provide a new direction for the pathogenesis of and the drug development for T1D.
1 Introduction
Diabetes is a chronic metabolic disease that has two main subtypes: type 1 diabetes (T1D) and type 2 diabetes (T2D) (1). T1D, also known as insulin-dependent diabetes, is caused by the destruction of β cells in the pancreas, resulting in an absolute lack of insulin, and is mainly seen in children and adolescents (2). It is generally characterized by the persistent elevation of blood glucose, polydipsia, polyphagia, polyuria, and weight loss (3). Although exogenous insulin supplementation controls the blood glucose levels and relieves the symptoms, it does not reverse or cure T1D (4). Moreover, as the disease progresses, many patients with T1D suffer from diabetic ketoacidosis, severe hypoglycemia, and cardiovascular complications (4). An epidemiological study indicated that T1D has become the third most common chronic disease in children, with more than 1.2 million children and adolescents worldwide suffering from T1D in 2021 (5). As the worldwide prevalence of T1D continues to rise, the direct and indirect healthcare costs it imposes on the country and society also continue to increase, and the resulting global public health problems are becoming increasingly serious (6).
Although the pathogenesis of T1D has not been fully elucidated, previous studies have suggested that genetic variants and environmental factors increase the genetic susceptibility to T1D by compromising immune homeostasis (7, 8). Previous research identified 26 genetic loci associated with the risk of T1D in genome-wide association studies (GWAS), 19 of which were associated with immune regulation (9, 10). Related studies have shown that, in addition to the crosstalk between β cells and the environment, a crosstalk between β cells and immunity also plays a key role in the development of T1D (8). For one, T lymphocytes are associated with the autoimmune damage to pancreatic β cells. A study conducted in the United States revealed that pancreatic β cells activate CD4+ T cells by releasing insulin peptide fragments into the bloodstream, subsequently triggering the specific recognition and attack of β cells by T cells (11). Another study from Argentina identified CD8+ T cells as a crucial contributor to T1D and a novel marker of β-cell autoimmunity, emphasizing the role of immune cells in T1D (12). For another, B lymphocytes also play a significant role in the autoimmune damage to pancreatic β cells. A study from the United Kingdom reported that, among the pancreatic lymphocytes of newly diagnosed T1D patients, B lymphocytes were present at a frequency second only to that of CD8+ T lymphocytes (13). This evidence underscores the intricate relationship between immune cells and T1D. However, the evidence to date is mainly observational and primarily concentrates on a few specific immune cells, which does not allow for a full elucidation of the causal relationship between different immune cells and T1D. Therefore, there is a need for novel and comprehensive approaches to assess the effects of different immune cell phenotypes on T1D.
Mendelian randomization (MR) is a novel approach to epidemiological studies that analyzes the causal effects of two factors through genetic variation. Since genotypes follow a random assignment principle in meiosis, MR is less susceptible to reverse causation and confounding variables than traditional methods (14, 15). This study assessed the impact of 731 immune cell phenotypes on the genetic susceptibility to T1D using GWAS data, aiming to identify key immune cells in the pathogenesis of T1D and to offer insights for its prevention and treatment.
2 Materials and methods
2.1 Study design
MR of immune cells and T1D was based on three fundamental assumptions (16) (Figure 1). The association assumption requires that single nucleotide polymorphisms (SNPs) are strongly correlated with exposure. The independence assumption requires that SNPs are independent of confounding variables. The exclusivity assumption requires that SNPs only act on outcomes through the exposure and not other pathways.
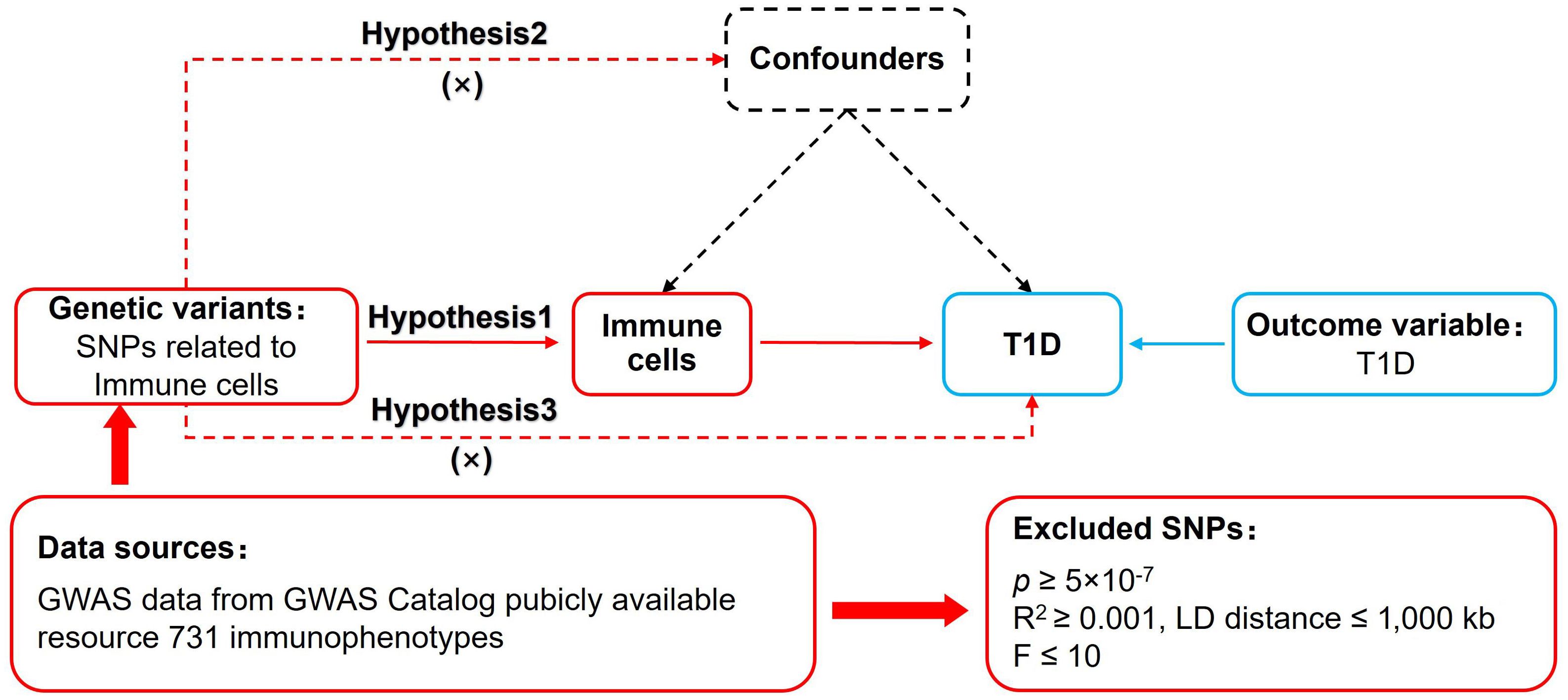
Figure 1. MR design for immune cells on genetic susceptibility to T1D. MR, Mendelian randomization; T1D, type 1 diabetes.
2.2 Data sources
The data sources for exposure and outcome are detailed in Table 1. Immune cell datasets numbered from GCST0001391 to GCST0002121 were acquired from the European Bioinformatics Institute (www.ebi.ac.uk/) (17). These datasets are from a study on 3,757 Sardinian residents that investigated the impact of approximately 22 million variants on 731 immune cell traits, which included 118 absolute cell counts, 389 mean fluorescence intensities of surface antigens, 32 morphological parameters, and 192 relative counts (17). The T1D dataset numbered finngen_R10_T1D was obtained from FinnGen (www.finngen.fi/fi), which contains genetic information on 339,432 Europeans. Due to the exposure data coming from the European Bioinformatics Institute and the outcome data coming from FinnGen, the overlap rate of the samples was extremely small. In addition, as the database is open access, no additional ethical review was required.
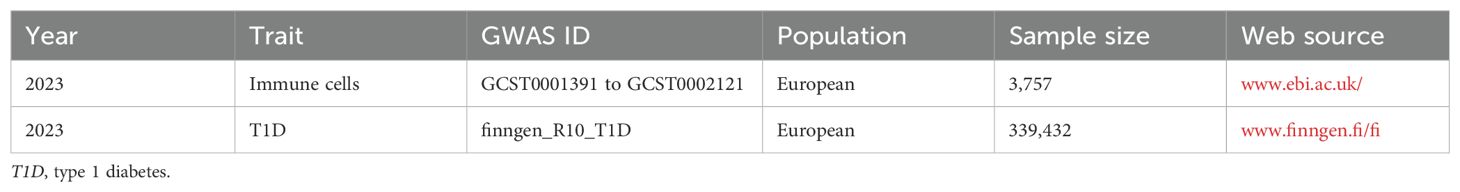
Table 1. Details of the genome-wide association studies (GWAS) included in the Mendelian randomization.
2.3 Selection of genetic instrument variables
Firstly, due to the threshold of p < 5 × 10−8 being insufficient to obtain an adequate number of SNPs and exposures containing SNPs, we established a threshold of p < 5 × 10−7 to search for SNPs closely related to the exposure, thereby enhancing the statistical power and satisfying the association assumption (18, 19). Only 612 immune cells (87.32%) contained at least one SNP at the p < 5 × 10−8 threshold, while 726 immune cells (99.32%) contained at least one SNP at the p < 5 × 10−7 threshold. Secondly, restricted R2 < 0.001 and kb = 1,000 were used to search for independent SNPs to exclude interference from linkage disequilibrium. Thirdly, F > 10 was restricted to search for strongly correlated SNPs to exclude the interference of weakly correlated variables. F was calculated as F = [R2/()]*[)/], where K is the number of paired samples, N is the total number of samples, and R2 is the cumulative explained variance. Fourthly, SNPs containing confounding variables such as age, gender, body mass index, marital status, and education level were excluded using PhenoScanner and Google Scholar to fulfill the independence assumption. Fifthly, mismatched and duplicate SNPs were excluded based on the effect allele frequency when adjusting the allele orientation for exposure and outcome. Lastly, the MR–pleiotropy residual sum and outlier (MR-PRESSO) method was used to exclude SNPs with significant bias (p < 1) to ensure the correctness of causal inference.
2.4 Data analysis
STROBE-MR was used as a guiding methodology (20). R 4.3.1 with the TwoSampleMR (0.5.7) program package installed was used to perform the operations for MR analysis. Firstly, inverse variance weighted (IVW) was set as the main evaluation tool as it allows for an unbiased causal analysis without pleiotropy. Weighted median, which is sensitive to outliers, and MR-Egger, which analyzes data in the presence of pleiotropy, were set as the secondary assessment tools. The statistical significance was set to p < 0.05. In addition, MR-Egger was used to analyze horizontal pleiotropy, which was required to satisfy the exclusivity assumption (p ≥ 0.05). Secondly, false discovery rate (FDR) based on the Benjamini–Hochberg method was used to correct the MR analysis results, with p < 0.05 considered a significant correlation and p ≥ 0.05 considered a potential correlation. Thirdly, Cochran’s Q and the leave-one-out method were used to analyze heterogeneity and sensitivity, respectively. There was no heterogeneity in the results when p ≥ 0.05, and the results were robust when no significant changes in the combined effect sizes were observed.
3 Results
3.1 Genetic instrument variables
We screened SNPs that met the basic assumptions of MR according to the steps enumerated above and excluded SNPs related to the confounding variables. Among them, effector memory (EM) CD8br %T cells excluded rs191753228 (cigarette consumption), rs76668354 (biological sex), and rs4507432 (body mass index); IgD+CD38dim %lymphocytes excluded rs16974449 (body mass index); CD80 on monocytes excluded rs114253672 (smoking initiation) and rs35075155 (body mass index); and SSC-A on plasmacytoid dendritic cells (pDCs) excluded rs17552904 (smoking initiation). The included exposures and SNPs are shown in Supplementary Table S1.
3.2 Two-sample MR analysis
IVW showed that EM double-negative (DN) (CD4−CD8−) %T cells [odds ratio (OR) = 1.157, 95% confidence interval (95% CI) = 1.016–1.318, p = 0.028, FDR = 0.899], EM CD8br %T cells (OR = 1.049, 95% CI = 1.003–1.098, p = 0.037, FDR = 0.902), CD28 on CD28+CD45RA+CD8br (OR = 1.334, 95% CI = 1.132–1.571, p < 0.001, FDR = 0.044), IgD+CD38dim %lymphocytes (OR = 1.045, 95% CI = 1.002–1.089, p = 0.039, FDR = 0.902), CD80 on monocytes (OR = 1.084, 95% CI = 1.013–1.161, p = 0.020, FDR = 0.834), SSC-A on pDCs (OR = 1.174, 95% CI = 1.004–1.372, p = 0.044, FDR = 0.902), and FSC-A on pDCs (OR = 1.182, 95% CI = 1.011–1.382, p = 0.036, FDR = 0.902) were associated with an increased genetic susceptibility to T1D. The forest plot and the scatter plot are shown in Figures 2 and 3, respectively. MR-Egger showed no horizontal pleiotropy for these results (p ≥ 0.05), as shown in Supplementary Table S2.
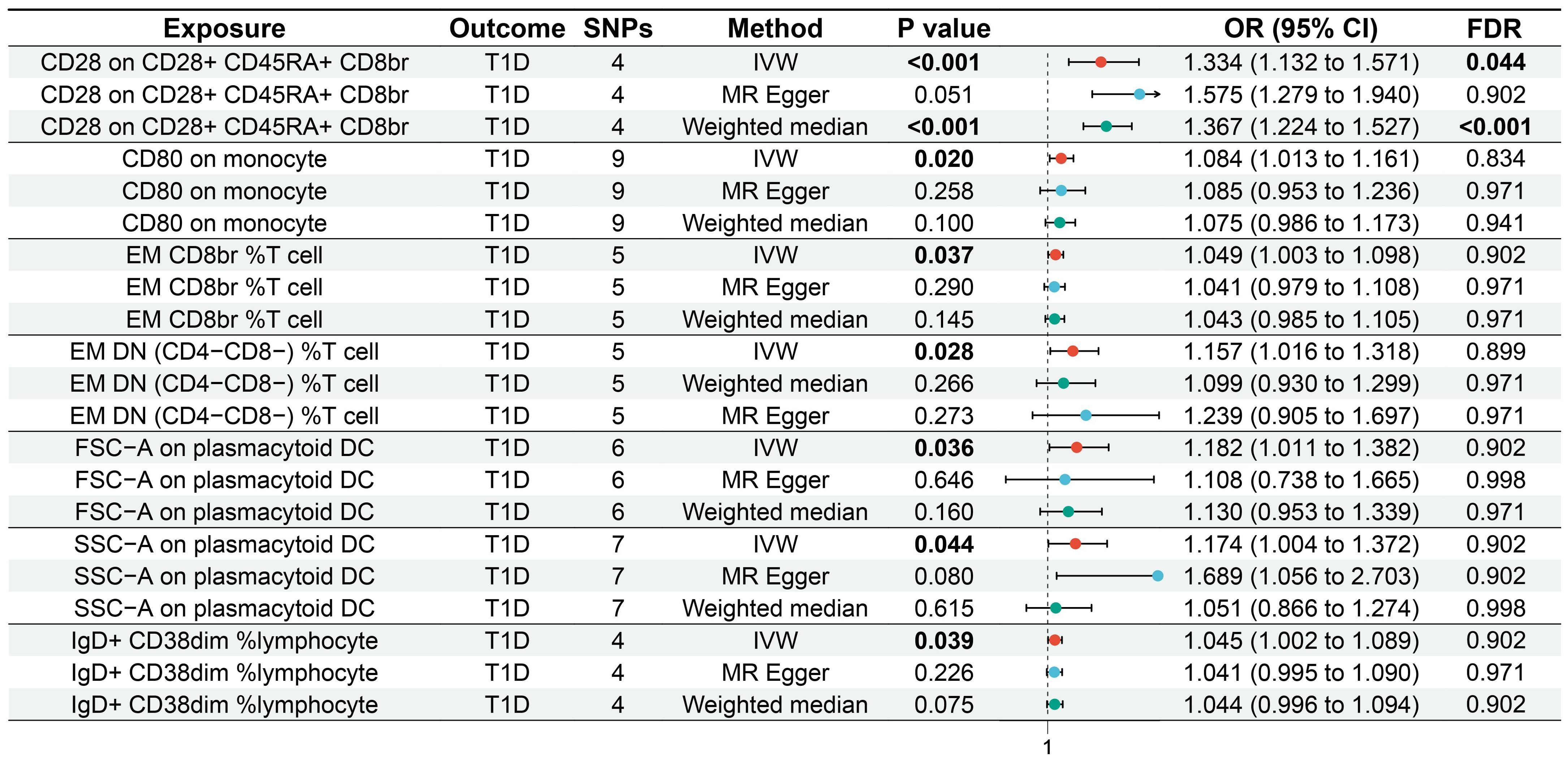
Figure 2. Forest plot of the MR analysis for immune cells on genetic susceptibility to T1D. MR, Mendelian randomization; T1D, type 1 diabetes.
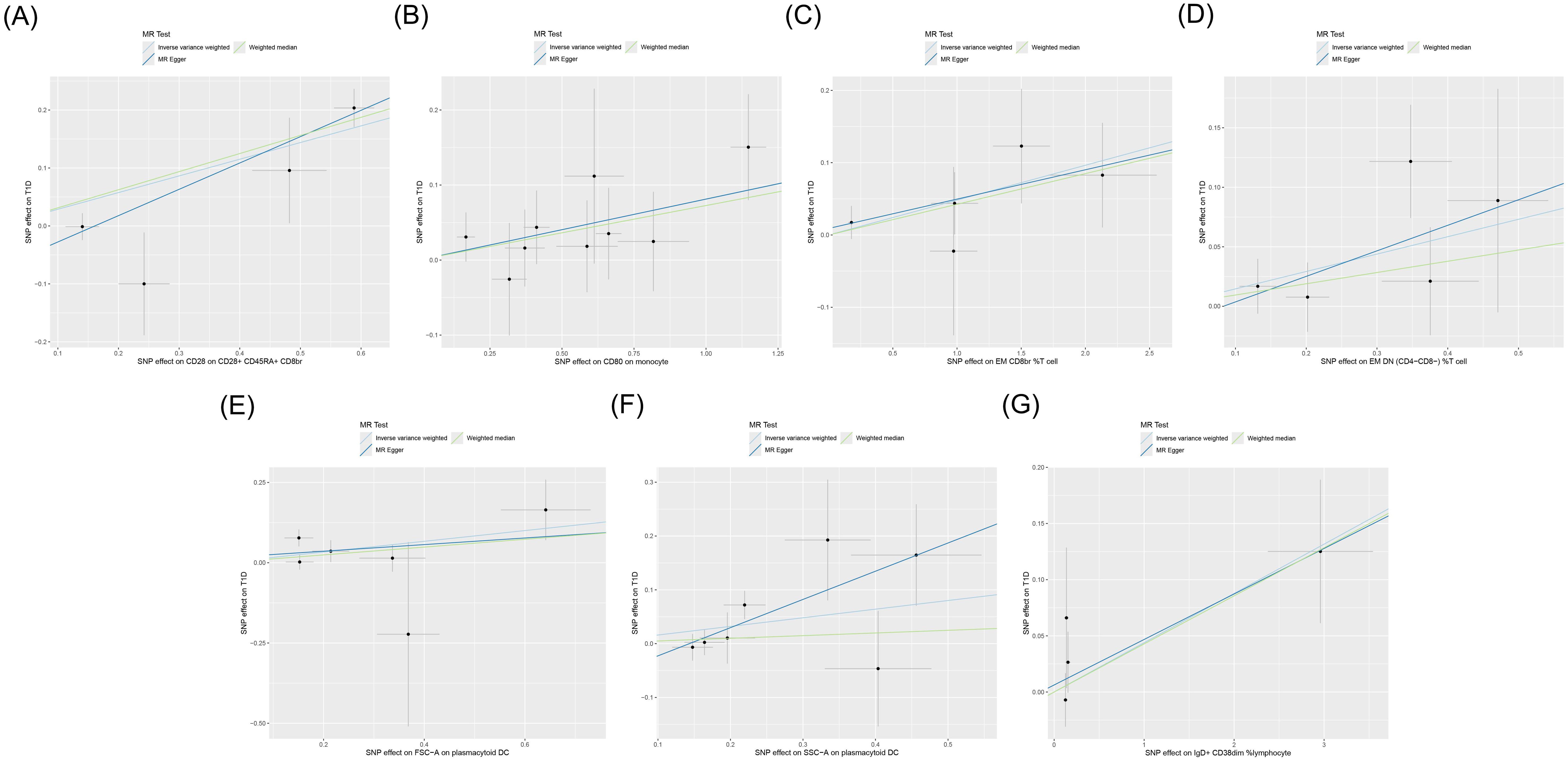
Figure 3. Scatter plot of the MR analysis for immune cells on genetic susceptibility to T1D. MR, Mendelian randomization; T1D, type 1 diabetes. (A) CD28 on CD28+CD45RA+CD8br in T1D. (B) CD80 on monocytes in T1D. (C) Effector memory (EM) CD8br %T cells in T1D. (D) EM double-negative (DN) (CD4−CD8−) %T cells in T1D. (E) FSC-A on plasmacytoid dendritic cells (pDCs) in T1D. (F) SSC-A on pDCs in T1D. (G) IgD+ CD38dim %lymphocytes in T1D. MR, Mendelian randomization; T1D, type 1 diabetes.
3.3 FDR analysis
FDR analysis showed that CD28 on CD28+CD45RA+CD8br was significantly associated with genetic susceptibility to T1D (p = 0.044), while EM DN (CD4−CD8−) %T cells (p = 0.899), EM CD8br %T cells (p = 0.902), IgD+CD38dim %lymphocytes (p = 0.902), CD80 on monocytes (p = 0.834), SSC-A on pDCs (p = 0.902), and FSC-A on pDCs (p = 0.902) were potentially associated with genetic susceptibility to T1D.
3.4 Heterogeneity and sensitivity analyses
Cochran’s Q test showed heterogeneity in the results of CD28 on CD28+CD45RA+CD8br (p = 0.043), whereas no significant heterogeneity was observed in the other results (p ≥ 0.05), as shown in Supplementary Table S3 and Figure 4. Sensitivity analysis demonstrated that the MR analysis results were robust, as illustrated in Figure 5.
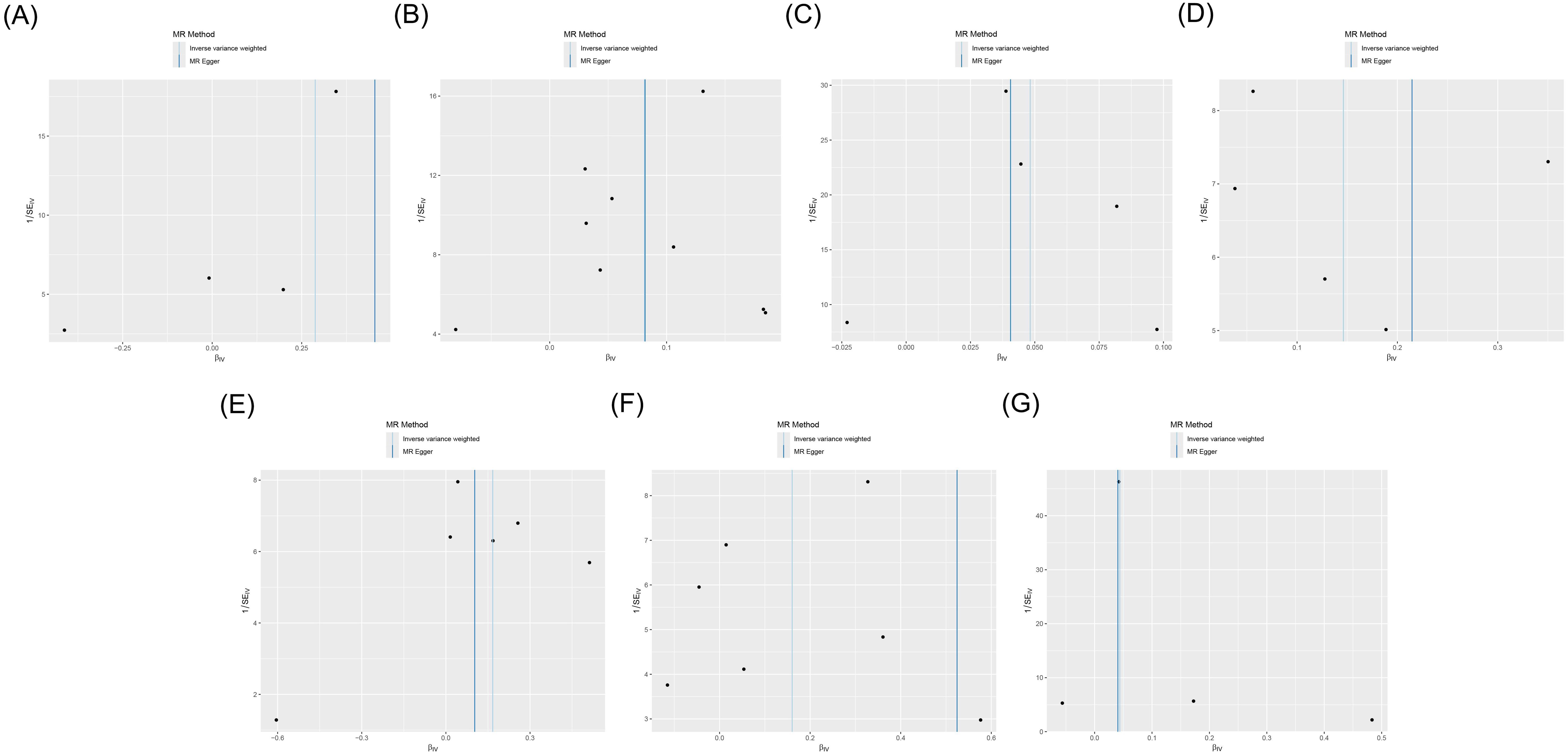
Figure 4. Funnel plot of the MR analysis for immune cells on genetic susceptibility to T1D. (A) CD28 on CD28+CD45RA+CD8br in T1D. (B) CD80 on monocytes in T1D. (C) Effector memory (EM) CD8br %T cells in T1D. (D) EM double-negative (DN) (CD4−CD8−) %T cells in T1D. (E) FSC-A on plasmacytoid dendritic cells (pDCs) in T1D. (F) SSC-A on pDCs in T1D. (G) IgD+ CD38dim %lymphocytes in T1D. MR, Mendelian randomization; T1D, type 1 diabetes.
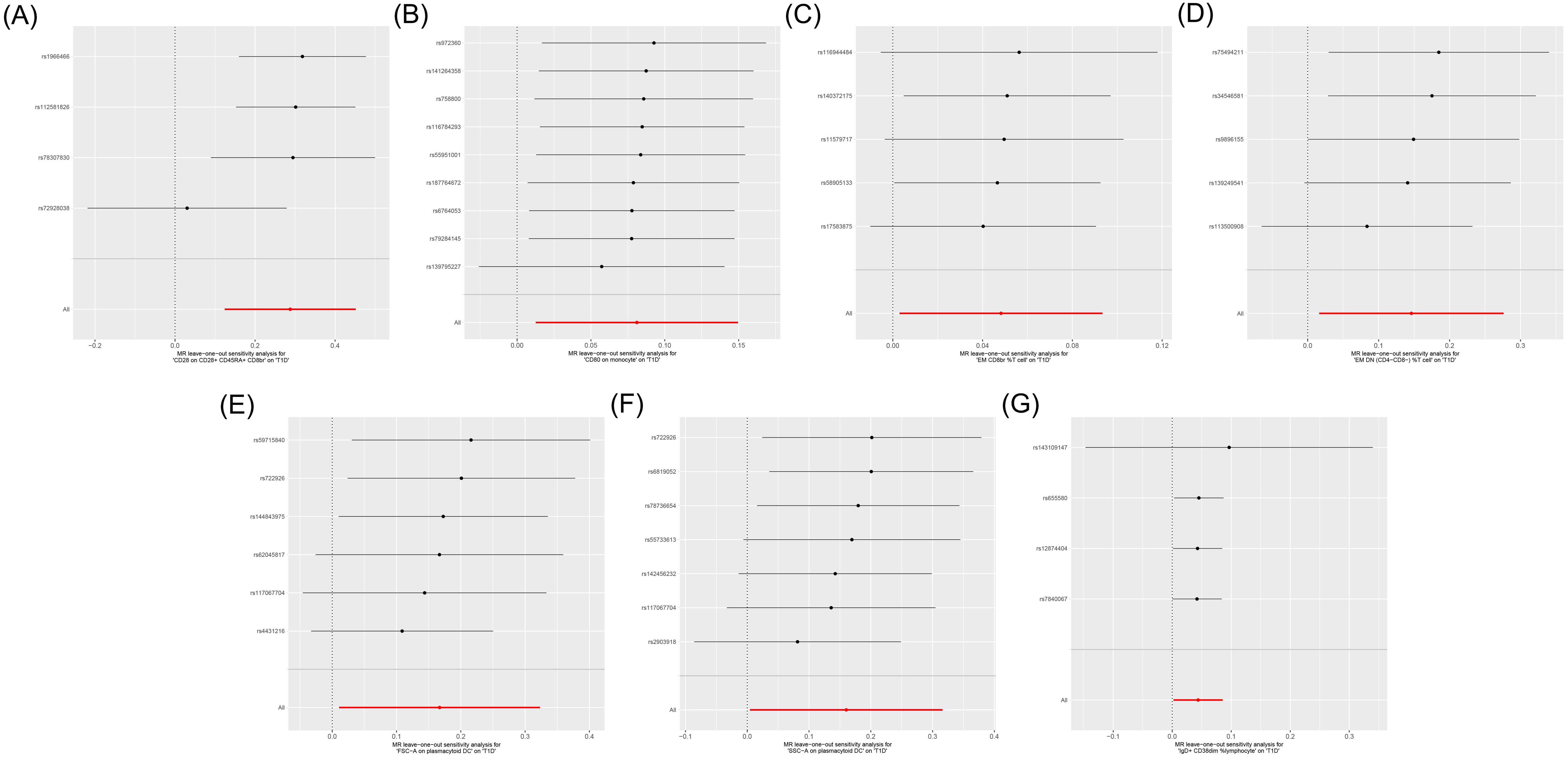
Figure 5. Leave-one-out sensitive analysis for immune cells on genetic susceptibility to T1D. (A) CD28 on CD28+CD45RA+CD8br in T1D. (B) CD80 on monocytes in T1D. (C) Effector memory (EM) CD8br %T cells in T1D. (D) EM double-negative (DN) (CD4−CD8−) %T cells in T1D. (E) FSC-A on plasmacytoid dendritic cells (pDCs) in T1D. (F) SSC-A on pDCs in T1D. (G) IgD+ CD38dim %lymphocytes in T1D. T1D, type 1 diabetes.
4 Discussion
Immune cells reside in tissues and organs within the body, the purpose of which is to perform immune functions and regulate immune homeostasis. Similar to the immune cells in other tissues, those residing in pancreatic islets play a crucial role in the maintenance of immune homeostasis within the islet microenvironment. A previous study has identified differences in the immune cell phenotypes between patients with T1D and the general population (21). However, limited both in quantity and quality of evidence, it is currently challenging to ascertain the roles of various immune cells in the onset process of T1D. To our knowledge, this study represents the first comprehensive assessment of the causal effects of 731 immune cells on T1D. The results revealed that CD28 on CD28+CD45RA+CD8br was significantly associated with genetic susceptibility to T1D, while EM DN (CD4−CD8−) %T cells, EM CD8br %T cells, IgD+CD38dim %lymphocytes, CD80 on monocytes, SSC-A on pDCs, and FSC-A on pDCs were potentially associated with genetic susceptibility to T1D. Cochran’s Q test showed heterogeneity in the results of CD28 on CD28+CD45RA+CD8br, which may be related to the sources of the exposure and outcome. In this MR analysis, the dataset of exposure was from Italians and the dataset of outcome from Finns. Although both Italians and Finns are Europeans, long-term geographical isolation may have resulted in genetic differences between the Finns living in Northern Europe and the Italians living in Southern Europe. However, despite the fact that there was heterogeneity in the results of CD28 on CD28+CD45RA+CD8br, the sensitivity analysis suggested the robustness of these findings.
EM DN (CD4−CD8−) %T cells comprise the proportion of EM T lymphocytes with no expression of CD4 and CD8 out of the total T lymphocytes. DN T cells exhibit distinct effector functions compared with CD4 T cells, amplifying interleukin 2 (IL-2) production (22). Furthermore, overactivated IL-2 might exacerbate the autoimmune response against pancreatic β cells (23). In addition, DN T cells generally refer to CD3+ T lymphocytes with a memory function. CD3 is a cell surface marker molecule that plays a key role in the differentiation, development, and maturation of T cells. During T-cell development, CD3 molecules bind to T-cell receptors (TCRs) to form the TCR/CD3 complex, which, together, participate in signaling and activation. A study has shown that oral administration of anti-CD3 drugs delayed the onset of diabetes and reduced morbidity in non-obese diabetic (NOD) mice, the mechanism of which may be related to the regulation of interferon gamma (IFN-γ) and IL-17 (24). Since IFN-γ and IL-17-mediated pro-inflammatory pathways play a key role in β-cell deletion, blockage of the secretion of IFN-γ and IL-17 helps to delay or reverse the pathogenesis of T1D (25). These pieces of evidence support the association of DN T cells with the increased risk of diabetes, pointing to EM DN (CD4−CD8−) %T cells as a potential risk factor for T1D.
EM CD8br %T cells represent the proportion of EM T lymphocytes with a high CD8 expression in T lymphocytes. CD8+ T cells comprise the main driving factor for the destruction of pancreatic β cells. They specifically recognize and attack pancreatic β cells, thereby leading to insufficient insulin secretion (26). In addition, CD8+ T cells secrete cytokines such as perforin, tumor necrosis factor (TNF), IFN-γ, and IL-2 (27), and the direct action of these cytokines is a key factor leading to β-cell death (28, 29). Relevant studies have shown that pancreatic β-cell death is inhibited by the depletion of CD8 T cells, thereby ameliorating the condition of T1D (30). Previous studies have confirmed the significant infiltration of CD8+ T cells in the pancreas of patients with T1D (31), with the percentage and the absolute count of late peripheral blood EM CD8+ T cells being markedly elevated compared with those in healthy individuals (32). These pieces of evidence suggest EM CD8br %T cells as a potential risk factor for T1D.
CD28 on CD28+CD45RA+CD8br comprise a cell subset characterized by the expression of CD28, CD45RA, and a high level of CD8. CD28 belongs to the immunoglobulin superfamily, and nearly half of them are expressed on CD8+ T cells, serving as co-stimulatory molecules essential for the maintenance of T-cell activation. By binding to B7-1 (CD80) and B7-2 (CD86) on antigen-presenting cells, CD28 provides co-stimulatory signals for optimal T-cell activation (33). Related studies have shown that blockage of CD28 co-stimulation can limit T-cell activation and infiltration in pancreatic islets, thereby preventing and treating T1D (34). CD45RA is an elongated isoform of CD45 that is typically expressed on the surface of naive T cells. After antigen stimulation, CD8+CD45RA+ T cells will transform into TEMRA cells (EM cells that re-express CD45RA) with characteristics of memory T cells and effector T cells (35). TEMRA cells are cytotoxic and can secrete cytokines such as perforin to induce the apoptosis of pancreatic β cells (36). A clinical study conducted in Spain confirmed the connection between TEMRA cells and T1D, reporting that the peripheral blood TEMRA levels in patients with T1D were significantly higher than those in healthy individuals (32). These findings suggest the pivotal roles of CD28, CD45RA, and CD8 in the progression of T1D, supporting CD28 on CD28+CD45RA+CD8br as a potential risk factor for T1D.
IgD+CD38dim %lymphocytes comprise the proportion of lymphocytes with immunoglobulin D (IgD) present on the surface and with a low CD38 expression out of the total lymphocytes. IgD is an antibody in the human immune system that primarily exists in membrane-bound form on the surface of B cells, playing a role in the activation and maturation of B cells. In pancreatic lymphocytes from newly diagnosed T1D patients, B lymphocytes are present at a frequency second only to CD8T lymphocytes (13). After treatment with rituximab, the destruction of pancreatic β cells decreases with the selective depletion of B lymphocytes, thereby preserving the function of pancreatic β cells in newly diagnosed T1D patients as much as possible (37). In addition to B lymphocytes, CD38 also plays a significant role in T1D. A study of Finnish children demonstrated that the anti-CD38 antibody levels increased with disease duration 10 years after the diagnosis of T1D (38). CD38 serves as the primary NAD-consuming enzyme, maintaining cellular NAD+ homeostasis. NAD, a crucial cofactor in the energy metabolism regulation, is involved in processes such as glycolysis, β-oxidation, and oxidative phosphorylation (39). In chronic T1D animal models such as streptozotocin diabetic rats, the NAD levels exhibited a significant decline (40). A related study indicated that high-dose nicotinamide prevents or delays T1D by modulating CD38 and reducing NAD+ depletion to protect pancreatic β cells (41). These pieces of evidence suggest that IgD and CD38 contribute to the progression of T1D, indirectly supporting IgD+CD38dim %lymphocytes as a potential risk factor for T1D.
In addition to lymphocytes, the role of myeloid antigen-presenting cells, such as monocytes, macrophages, and DCs, in T1D has been well documented (42). Monocytes are precursor cells produced by the bone marrow that enter tissues through the circulation and differentiate to form macrophages and DCs (43). In this MR analysis, it was identified that the myeloid cell immunophenotypes CD80 on monocytes, human leukocyte antigen DR isotype (HLA-DR) on CD33brHLA DR+CD14−, SSC-A on pDCs, and FSC-A on pDCs were associated with an increased risk of T1D. CD80 on monocytes refer to monocytes with surface expression of CD80. CD80, also known as B7-1, is widely expressed on the surface of a variety of immune cells and plays a crucial regulatory role in immune response. The expression of CD80 synergizes with pancreas-specific cytotoxic T cells in the rapid destruction of β cells (44). When CD80 was co-expressed with TNF-α or IL-2, a large proportion of mice developed severe pancreatitis and diabetes mellitus (45, 46). In addition, when CD80 was expressed on pancreatic β cells from B6 mice backcrossed once with genetically susceptible NOD, the onset rate of diabetes and the severity of the autoimmune response also increased (47). This suggests that CD80 on monocytes may contribute to the pathogenesis of T1D. HLA-DR on CD33brHLA DR+CD14− represents a subpopulation of immune cells with positive HLA-DR, high expression of CD33, and no expression of CD14. Specific HLA-DR genotypes are associated with an increased genetic susceptibility to T1D, as mentioned above. CD33 is a cell surface molecule expressed widely in bone marrow hematopoietic cells that is involved in the regulation of inflammation, activation of cell proliferation, and immune response (48, 49). The current study demonstrated that CD33 HLA-DR myeloid-derived suppressor cells (MDSCs) were significantly increased in the peripheral blood of patients with T1D and positively correlated with the levels of HbA1C (50). These pieces of evidence support that CD80 on monocytes and HLA-DR on CD33brHLA DR+CD14− could serve as risk factors for T1D.
SSC-A on pDCs and FSC-A on pDCs are two phenotypes of DCs. SSC-A is a parameter used in flow cytometry to characterize the side scatter properties of a cell, which is commonly used to assess the intracellular complexity and particle content. FSC-A is a parameter used in flow cytometry to characterize the forward scatter signal of cells, typically employed to assess the size and morphology of cells. SSC-A on pDCs and FSC-A on pDCs may refer to pDCs with specific scatter properties that act as a bridge between innate and adaptive immunity in T1D development (51). pDCs are predominantly found in the peripheral blood, lymph, and bone marrow, playing roles in the secretion of IFN, the modulation of T-cell immune responses, and in autoimmune diseases (51). A clinical study in the United States showed that the number of pDCs was significantly increased in the peripheral blood of patients with T1D compared with healthy subjects, while that of other DC types remained similar (51). Furthermore, in the NOD mouse model, the injection of IFN-α or its inducers significantly accelerated the onset of T1D (52), while the depletion of pDCs significantly delayed the onset of T1D (53). These pieces of evidence support that pDCs are strongly associated with T1D, indicating that SSC-A on pDCs and FSC-A on pDCs are potential risk factors for T1D.
While this MR analysis provides genetic insights into the causal relationship between immune cell phenotypes and T1D, certain limitations must be acknowledged. Firstly, the socioeconomic status of diverse racial groups significantly contributes to health disparities (54). The GWAS data included in this study were obtained from European populations in high-income countries, restricting the generalization of the findings to low-/middle-income countries and other ethnicities. Secondly, the heterogeneity of the results of naive CD8br %T cells and HLA-DR on CD33brHLA DR+CD14− might amplify the potential risk of bias. Thirdly, although this study identified the causal effects of eight immune cell phenotypes on T1D, the biological mechanisms through which they influence T1D remain unclear. Consequently, we anticipate the following improvements in future studies. Firstly, we should continuously enrich the GWAS data and make efforts to realize MR analysis for diverse ethnic and socioeconomic status groups. Secondly, we look forward to validate the expression levels of the immune cells identified in this study through animal experiments in order to provide more robust biological evidence. Thirdly, the design of high-quality clinical research protocols is expected to enrich the evidence-based support for the causal effect of immune cell phenotypes on T1D.
5 Conclusion
The MR analysis revealed that CD28 on CD28+CD45RA+CD8br was significantly associated with genetic susceptibility to T1D, while EM DN (CD4−CD8−) %T cells, EM CD8br %T cells, IgD+CD38dim %lymphocytes, CD80 on monocytes, SSC-A on pDCs, and FSC-A on pDCs were potentially associated with genetic susceptibility to T1D. These findings provide a new direction for the pathogenesis of and the development of new drugs for T1D. However, further biological studies are necessary in the future to reveal their relationships and mechanisms.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
YY: Conceptualization, Supervision, Writing – original draft. XY: Data curation, Methodology, Writing – original draft. JD: Data curation, Methodology, Writing – original draft. JW: Formal analysis, Writing – original draft. SB: Data curation, Writing – original draft. RY: Conceptualization, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by The Key Support Project of the Regional Innovation and Development Joint Fund of the National Natural Science Foundation of China (U21A20411).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1402956/full#supplementary-material
Abbreviations
CI, confidence interval; GWAS, genome-wide association study; IFN, interferon; IL, interleukin; IVW, inverse variance weighted; MR, Mendelian randomization; NOD, non-obese diabetic; OR, odds ratio; pDCs, plasmacytoid dendritic cells; PRESSO, pleiotropy residual sum and outlier method; SNP, single nucleotide polymorphism; TNF, tumor necrosis factor; T1D, type 1 diabetes; T2D, type 2 diabetes.
References
1. Desai S, Deshmukh A. Mapping of type 1 diabetes mellitus. Curr Diabetes Rev. (2020) 16:438–41. doi: 10.2174/1573399815666191004112647
2. Gillespie KM. Type 1 diabetes: Pathogenesis and prevention. CMAJ. (2006) 175:165–70. doi: 10.1503/cmaj.060244
3. Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Chapter 1: Epidemiology of type 1 diabetes. Endocrinol Metab Clinics North America. (2010) 39:481–97. doi: 10.1016/j.ecl.2010.05.011
5. Magliano DJ, Boyko EJ IDF. Diabetes Atlas 10th edition scientific committee, in: Idf diabetes atlas. (2021). Brussels: International Diabetes Federation. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK581934/ (Accessed February 3, 2024).
6. Sussman M, Benner J, Haller MJ, Rewers M, Griffiths R. Estimated lifetime economic burden of type 1 diabetes. Diabetes Technol Ther. (2020) 22:121–30. doi: 10.1089/dia.2019.0398
7. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
8. Houeiss P, Luce S, Boitard C. Environmental triggering of type 1 diabetes autoimmunity. Front Endocrinol. (2022) 13:933965. doi: 10.3389/fendo.2022.933965
9. Kim S-S, Hudgins AD, Yang J, Zhu Y, Tu Z, Rosenfeld MG, et al. A comprehensive integrated post-GWAS analysis of type 1 diabetes reveals enhancer-based immune dysregulation. PloS One. (2021) 16:e0257265. doi: 10.1371/journal.pone.0257265
10. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. n Engl J Med. (2009) 360:1646-54. doi: 10.1056/NEJMra0808284
11. Wan X, Zinselmeyer BH, Zakharov PN, Vomund AN, Taniguchi R, Santambrogio L, et al. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature. (2018) 560:107. doi: 10.1038/s41586-018-0341-6
12. Mallone R, Martinuzzi E, Blancou P, Novelli G, Afonso G, Dolz M, et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes. (2007) 56:613–21. doi: 10.2337/db06-1419
13. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. (2009) 155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x
14. Birney E. Mendelian randomization. Cold Spring Harbor Perspect Med. (2022) 12:a041302. doi: 10.1101/cshperspect.a041302
15. Smith GD, Ebrahim S. mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. (2003) 32:1–22. doi: 10.1093/ije/dyg070
16. Davies NM, Holmes MV, Davey Smith G. Reading mendelian randomization studies: A guide, glossary, and checklist for clinicians. BMJ (Clinical Res ed). (2018) 362:k601. doi: 10.1136/bmj.k601
17. Orrù V, Steri M, Sidore C, Marongiu M, Serra V, Olla S, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. (2020) 52:1036–45. doi: 10.1038/s41588-020-0684-4
18. Choi KW, Chen C-Y, Stein MB, Klimentidis YC, Wang M-J, Koenen KC, et al. Assessment of bidirectional relationships between physical activity and depression among adults: A 2-sample mendelian randomization study. JAMA Psychiatry. (2019) 76:399–408. doi: 10.1001/jamapsychiatry.2018.4175
19. Choi KW, Stein MB, Nishimi KM, Ge T, Coleman JRI, Chen C-Y, et al. An exposure-wide and mendelian randomization approach to identifying modifiable factors for the prevention of depression. Am J Psychiatry. (2020) 177:944–54. doi: 10.1176/appi.ajp.2020.19111158
20. Skrivankova VW, Richmond RC, Woolf BAR, Davies NM, Swanson SA, VanderWeele TJ, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): Explanation and elaboration. BMJ. (2021) 375:n2233. doi: 10.1136/bmj.n2233
21. Odegard JM, Nepom GT, Wambre E. Biomarkers for antigen immunotherapy in allergy and type 1 diabetes. Clin Immunol (Orlando Fla). (2015) 161:44–50. doi: 10.1016/j.clim.2015.05.023
22. Haug T, Aigner M, Peuser MM, Strobl CD, Hildner K, Mougiakakos D, et al. Human double-negative regulatory T-cells induce a metabolic and functional switch in effector T-cells by suppressing mTOR activity. Front Immunol. (2019) 10:883. doi: 10.3389/fimmu.2019.00883
23. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes. (2012) 61:14–22. doi: 10.2337/db11-1213
24. Kuhn C, Rezende RM, da Cunha AP, Valette F, Quintana FJ, Chatenoud L, et al. Mucosal administration of CD3-specific monoclonal antibody inhibits diabetes in NOD mice and in a preclinical mouse model transgenic for the CD3 epsilon chain. J Autoimmun. (2017) 76:115–22. doi: 10.1016/j.jaut.2016.10.001
25. Marwaha AK, Tan S, Dutz JP. Targeting the IL-17/IFN-γ axis as a potential new clinical therapy for type 1 diabetes. Clin Immunol (Orlando Fla). (2014) 154:84–9. doi: 10.1016/j.clim.2014.06.006
26. Gearty SV, Dündar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, et al. An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature. (2022) 602:156–61. doi: 10.1038/s41586-021-04248-x
27. Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. (2002) 111:837–51. doi: 10.1016/s0092-8674(02)01139-x
28. Barral AM, Thomas HE, Ling EM, Darwiche R, Rodrigo E, Christen U, et al. SOCS-1 protects from virally-induced CD8 T cell mediated type 1 diabetes. J Autoimmun. (2006) 27:166–73. doi: 10.1016/j.jaut.2006.08.002
29. Thomas HE, Trapani JA, Kay TWH. The role of perforin and granzymes in diabetes. Cell Death Differentiation. (2010) 17:577–85. doi: 10.1038/cdd.2009.165
30. Kwong C-TJ, Selck C, Tahija K, McAnaney LJ, Le DV, Kay TW, et al. Harnessing CD8+ T-cell exhaustion to treat type 1 diabetes. Immunol Cell Biol. (2021) 99:486–95. doi: 10.1111/imcb.12444
31. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TWH, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. (2012) 209:51–60. doi: 10.1084/jem.20111187
32. Teniente-Serra A, Pizarro E, Quirant-Sánchez B, Fernández MA, Vives-Pi M, Martinez-Caceres EM. Identifying changes in peripheral lymphocyte subpopulations in adult onset type 1 diabetes. Front Immunol. (2021) 12:784110. doi: 10.3389/fimmu.2021.784110
33. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: From mechanism to therapy. Immunity. (2016) 44:973. doi: 10.1016/j.immuni.2016.04.020
34. Besançon A, Goncalves T, Valette F, Mary C, Vanhove B, Chatenoud L, et al. A selective CD28 antagonist and rapamycin synergise to protect against spontaneous autoimmune diabetes in NOD mice. Diabetologia. (2018) 61:1811–6. doi: 10.1007/s00125-018-4638-7
35. Zhu H, Galdos FX, Lee D, Waliany S, Huang YV, Ryan J, et al. Identification of pathogenic immune cell subsets associated with checkpoint inhibitor-induced myocarditis. Circulation. (2022) 146:316–35. doi: 10.1161/CIRCULATIONAHA.121.056730
36. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
37. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. New Engl J Med. (2009) 361:2143–52. doi: 10.1056/NEJMoa0904452
38. Savola K, Sabbah E, Kulmala P, Vähäsalo P, Ilonen J, Knip M. Autoantibodies associated with type I diabetes mellitus persist after diagnosis in children. Diabetologia. (1998) 41:1293–7. doi: 10.1007/s001250051067
39. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J BioMed Sci. (2019) 26:34. doi: 10.1186/s12929-019-0527-8
40. Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. (2001) 50:537–46. doi: 10.33549/physiolres
41. Kolb H, Burkart V. Nicotinamide in type 1 diabetes. mechanism of action revisited. Diabetes Care. (1999) 22 Suppl 2:B16–20.
42. Unanue ER. Antigen presentation in the autoimmune diabetes of the NOD mouse. Annu Rev Immunol. (2014) 32:579–608. doi: 10.1146/annurev-immunol-032712-095941
43. Coillard A, Segura E. In vivo differentiation of human monocytes. Front Immunol. (2019) 10:1907. doi: 10.3389/fimmu.2019.01907
44. Harlan DM, Hengartner H, Huang ML, Kang YH, Abe R, Moreadith RW, et al. Mice expressing both b7-1 and viral glycoprotein on pancreatic beta cells along with glycoprotein-specific transgenic T cells develop diabetes due to a breakdown of T-lymphocyte unresponsiveness. Proc Natl Acad Sci USA. (1994) 91:3137–41. doi: 10.1073/pnas.91.8.3137
45. Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator b7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci U.S.A. (1994) 91:5138–42. doi: 10.1073/pnas.91.11.5138
46. Allison J, Stephens LA, Kay TW, Kurts C, Heath WR, Miller JF, et al. The threshold for autoimmune T cell killing is influenced by b7-1. Eur J Immunol. (1998) 28:949–60. doi: 10.1002/(SICI)1521-4141(199803)28:03<949::AID-IMMU949>3.0.CO;2-H
47. Wong S, Guerder S, Visintin I, Reich EP, Swenson KE, Flavell RA, et al. Expression of the co-stimulator molecule b7-1 in pancreatic beta-cells accelerates diabetes in the NOD mouse. Diabetes. (1995) 44:326–9. doi: 10.2337/diab.44.3.326
48. Crocker PR, McMillan SJ, Richards HE. CD33-related siglecs as potential modulators of inflammatory responses. Ann New York Acad Sci. (2012) 1253:102–11. doi: 10.1111/j.1749-6632.2011.06449.x
49. Cao H, Crocker PR. Evolution of CD33-related siglecs: Regulating host immune functions and escaping pathogen exploitation? Immunology. (2011) 132:18–26. doi: 10.1111/j.1365-2567.2010.03368.x
50. Hassan M, Raslan HM, Eldin HG, Mahmoud E, Elwajed HAA. CD33+ HLA-DR– myeloid-derived suppressor cells are increased in frequency in the peripheral blood of type1 diabetes patients with predominance of CD14+ subset. Open Access Macedonian J Med Sci. (2018) 6:303–9. doi: 10.3889/oamjms.2018.080
51. Xia C-Q, Peng R, Chernatynskaya AV, Yuan L, Carter C, Valentine J, et al. Increased IFN-α-producing plasmacytoid dendritic cells (pDCs) in human th1-mediated type 1 diabetes: PDCs augment th1 responses through IFN-α production. J Immunol (Baltimore Md: 1950). (2014) 193:1024–34. doi: 10.4049/jimmunol.1303230
52. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. (2008) 105:12439–44. doi: 10.1073/pnas.0806439105
53. Li Q, McDevitt HO. The role of interferon alpha in initiation of type I diabetes in the NOD mouse. Clin Immunol (Orlando Fla). (2011) 140:3–7. doi: 10.1016/j.clim.2011.04.010
Keywords: immune cell, phenotype, type 1 diabetes, GWAS, Mendelian randomization
Citation: Yu Y, Yang X, Deng J, Wu J, Bai S and Yu R (2024) How do immune cells shape type 1 diabetes? Insights from Mendelian randomization. Front. Endocrinol. 15:1402956. doi: 10.3389/fendo.2024.1402956
Received: 18 March 2024; Accepted: 30 November 2024;
Published: 24 December 2024.
Edited by:
Guy A. Rutter, Imperial College London, United KingdomReviewed by:
Guang Chen, Huazhong University of Science and Technology, ChinaGlilciane Morceli, Minas Gerais State University, Brazil
Copyright © 2024 Yu, Yang, Deng, Wu, Bai and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Yu, eXVyb25nMTk2OTA1QDE2My5jb20=
†These authors have contributed equally to this work