 Marianna Rita Stancampiano
Marianna Rita Stancampiano Silvia Laura Carla Meroni
Silvia Laura Carla Meroni- Department of Pediatrics, IRCCS San Raffaele Scientific Institute, Milan, Italy
The term ‘differences of sex development’ (DSD) refers to a group of congenital conditions that are associated with atypical development of chromosomal, gonadal, and/or anatomical sex. DSD in individuals with a 46,XX karyotype can occur due to fetal or postnatal exposure to elevated amount of androgens or maldevelopment of internal genitalia. Clinical phenotype could be quite variable and for this reason these conditions could be diagnosed at birth, in newborns with atypical genitalia, but also even later in life, due to progressive virilization during adolescence, or pubertal delay. Understand the physiological development and the molecular bases of gonadal and adrenal structures is crucial to determine the diagnosis and best management and treatment for these patients. The most common cause of DSD in 46,XX newborns is congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, determining primary adrenal insufficiency and androgen excess. In this review we will focus on the other rare causes of 46,XX DSD, outside CAH, summarizing the most relevant data on genetic, clinical aspects, puberty and fertility outcomes of these rare diseases.
1 Introduction
Phenotypic sex is the result of a complex and multistep developmental process, controlled by different gene pathways and transcriptional factors, determining specific hormone secretion during critical stages of fetal life (1, 2). Sexual development involves two different developmental processes: sex determination (divided into chromosomal sex determination and gonadal sex determination) and sex differentiation (3, 4). Chromosomal sex determination already occurs during fertilization, when a sperm contributes either an X or Y chromosome to the X chromosome in the oocyte, determining an XX or XY zygote (5). Gonadal sex determination is a complex and dynamic multistep event, regulated by different genetic pathways, leading the bipotential indifferent gonad either into an ovary or a testis (4, 6). Sex differentiation occurs once the gonad has developed and is induced by the hormones produced by gonadal tissues, establishing the internal and external genitalia and a male or female phenotype (6–10).
Differences of sex development (DSD) are defined as congenital conditions with atypical development of chromosomal, gonadal, and/or anatomic sex (11). According to the Chicago Consensus (11), recently revised in a European Consensus Statement (12), DSD could be classified into chromosomal DSD, 46, XY DSD and 46,XX DSD. The 46,XX DSD group includes a wide spectrum of conditions due to atypical gonadal development and altered hormonal secretion; in the same group are also classified patients with atypical differentiation of Mullerian structures, affected by the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH).
The impact of DSD diagnosis in the affected individuals and their families is quite huge: patients and parents are facing a complex set of circumstances when they first hear about the term DSD. For this reason, these conditions require a multidisciplinary team, at diagnosis and during each stage of life (11–13). Diagnosis and then follow-up are quite challenging for clinicians, including crucial aspects like sex assignment (and re-assignment in some cases), gonadal management (including the decision of gonadectomy or gonadal surveillance) and pubertal induction (if needed). A correct and preferable early diagnosis could represent a key factor for improving quality of life of patients and their families, followed by a constant psychological support (12, 14, 15).
The most common condition in 46,XX DSD is represented by Congenital Adrenal Hyperplasia (CAH) due to 21-hydroxylase deficiency, with an overall incidence in the Caucasian population of 1:30.000 female newborns (16). Other forms of CAH include 11ß-hydroxylase deficiency, 3ß-hydroxysteroid dehydrogenase type 2 deficiency, and the newest P450 oxidoreductase deficiency, characterized by DSD also often associated with skeletal defects (16).
However, although rarer, other defects involving specific genetic and/or steroidogenesis pathways could determine a DSD condition in 46,XX subjects.
The aim of this review is to summarize the most relevant data on the pathogenesis, diagnosis, clinical and therapeutic management of 46,XX DSD, outside CAH. The first part will focus on sexual differentiation, with a special emphasis on genetic pathways involved in gonadal development, highlighting their role on 46,XX DSD. In the second part the author will discuss clinical management of specific diseases.
2 Sexual development in 46,XX
Sexual development is a complex multistep process through which the undifferentiated embryonic structures develop toward a male or female phenotype (7, 8). Until approximately the fifth-sixth week post-fertilization, no sexual differences are observable in a XX or XY fetus. Gonadal determination into either ovaries or testes (or ‘atypical’ gonads in DSD) plays a key role in sexual differentiation, influencing individual’s internal and external genitalia and secondary sexual characteristics (7–10, 17–19).
2.1 Gonadal determination
In the last decades there has been a considerable increase in our knowledge of the genes and mechanism involved in the development and function of human gonads.
In 1940s, after Jost’s discovery that the precocious removal of embryonic gonads in rabbits leads to a female phenotype (regardless chromosomal assessment) (20), the ovarian development was long thought to be just a passive or default process. In 1990, the discovery of SRY gene (21), also contribute to this “classic theory” for which ovarian development has been considered just as a consequence of the absence of the SRY. So, the crucial event in sex determination was whether or not a testis developed. This prompted the researchers to focus on the discovery of genes eventually leading to testis differentiation (22–25).
However, the occurrence of male phenotype in XX individuals in the absence of SRY gene, or the cases of XY sex reversal in the presence of SRY, led to hypothesize that SRY could act as a repressor of the female pathway, also highlighting the hypothesis of the existence of a gene ‘Z’ repressing male development and/or activating female development (26, 27). This model emphasized the theory of the antagonistic nature of male and female specific genetic pathways in the ‘battle of sexes’ during embryonic development (8).
Although to date the precise genetic mechanism of sexual development is still not completely understood, especially in females, it is widely believed that the genetic programs of male and female differentiation are closely intertwined and determined by antagonistic pathways (8, 9, 17, 19).
In humans, the gonadal primordium (also known as the genital ridge) first appears at the 4th week of gestational age, arising from the urogenital ridge comprising of the pronephros (from which the adrenal primordium derives), the mesonephros (from which the gonadal primordium derives) and the metanephros (from which the reno-urinary system derives) (28).
Since the 90s, there has been a significant increase in our knowledge of the genes required for this early step of sexual development, that is the formation of the bipotential undifferentiated gonad. The genes identified as vital for the initial process of gonadal ridge formation include nuclear receptor subfamily 5, group A, member 1 (NR5A1); Wilms’ tumor suppressor 1 (WT1); GATA-binding protein 4 (GATA4); chromobox homolog 2 (CBX2); LIM homeobox gene 9 (LHX9); empty-spiracles homeobox gene 2 (EMX2) (2, 6, 8–10, 28–30) (Figure 1).
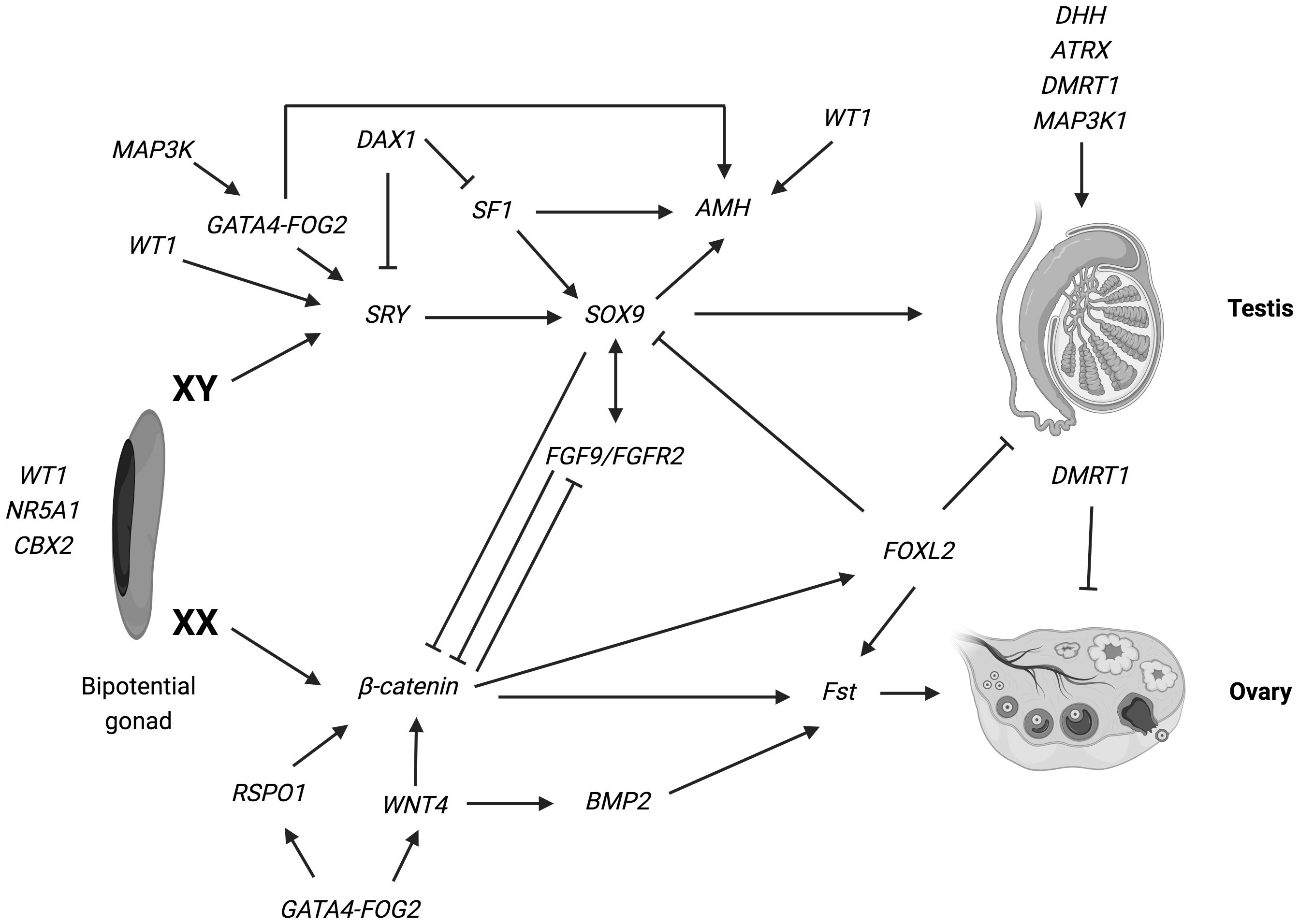
Figure 1 Overview of the genetic pathways involved in gonadal determination. Arrows indicate activation of a downstream target. Lines ending in bars indicate repression of a downstream target. The genes identified as vital for the development of the bi-potential gonad include: nuclear receptor subfamily 5, group A, member 1 (NR5A1); Wilms’ tumor suppressor 1 (WT1) and chromobox homolog 2 (CBX2) (ref (2, 6, 8–10, 28–30). Various genes have been implicated in the pathways leading the bipotential indifferent gonad either into an ovary or a testis. In XY: SRY determine an increase of Sox9 expression, which then stimulate Fgf9 expression. Both Fgf9 and SOX9 suppress the female specific pathway, especially β-catenin and WNT4, supporting testis specific program. Numerous other genes such as WT1, DAX1, AMH, MAP3K1 and DMRT1 are necessary for the development and maintenance of testicular gonad. In XX: SRY is absent and specific genes are involved in ovarian development: WNT4 and RSPO1 have a synergic role on the activation of β-catenin, that suppress the SOX9/Fgf9 testicular pathway. Moreover, WNT4, RSPO1 and FOXL2 active Fst (follistatin) expression (ref. 17–19). Figure adapted from Tevosian SG. Genetic control of ovarian development. Sex Dev. 2013;7 (1–3):33–45 (ref.17); Eggers S, Ohnesorg T, Sinclair A. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol. 2014;10 (11):673–683 (ref.18); Ohnesorg T, Vilain E, Sinclair AH. The genetics of disorders of sex development in humans. Sex Dev. 2014;8 (5):262–272 (ref.19). Created with BioRender.com.
Modification of genetic pathways, due to loss of function of the above listed genes, at this early stage of human gestation, may lead to DSD (31–40). In Table 1 we have summarized current understanding on genes involved in genital ridge development and their pathogenetic role in 46,XX individuals.
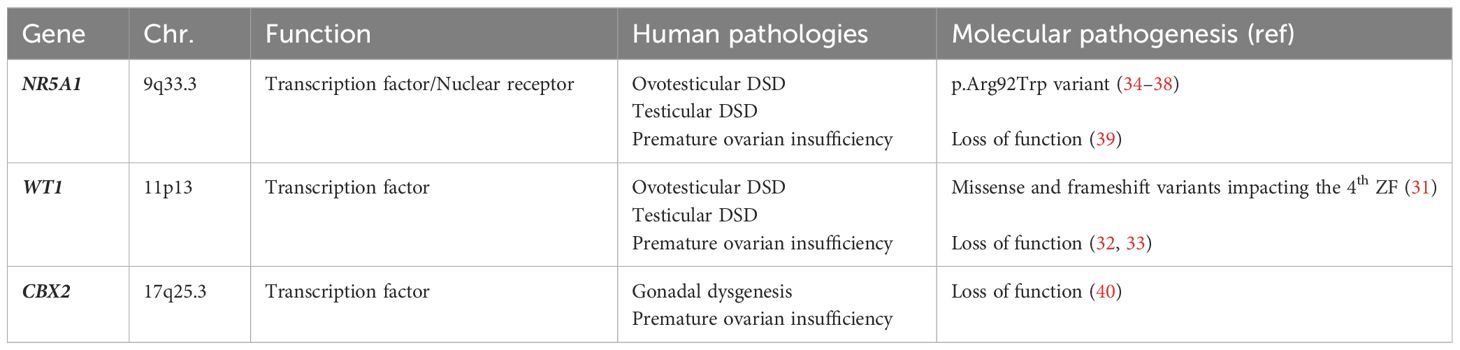
Table 1 Genes involved in genital ridge development and their pathogenetic role in 46,XX DSD.
Until the beginning of differentiation at 6th-7th week of gestation, there are no structural differences between XX and XY gonadal primordium (7). In 46,XX fetuses, the gonads remain undifferentiated for a longer period of time compared to 46,XY, probably due to a later expression of the specific ovarian differentiation pathways (9, 17, 30).
The two main roles of the ovary are the production of steroid hormones and the generation of mature oocyte. For these reasons, ovarian differentiation is characterized by two milestones: (i) the entry of XX germ cells into meiosis and (ii) follicle formation (41, 42). Primordial cells differentiate into oogonia from week 8; at week 11-12, germ cells enter into the first meiosis and primordial follicles develop. At week 15, the primary follicles develop, with the differentiation of theca cells. By the time of birth, the majority of the oocytes in the human ovary are contained in primary follicles, and a few in primordial follicles; the number of secondary or tertiary antral follicles is very small (42).
Although the female regulatory cascade still lacks a ‘master’ regulator as an equivalent of SRY gene in males, it came to light that specific pathways, involving different genes and transcription factors, such as the wingless-type MMTV integration site family, member 4 (WNT4), R-spondin1 (RSPO1), Forkhead box L2 (FOXL2), Gata4/6 and Fog2 and CTNNB1/beta catenin, are vital for promoting ovarian differentiation and oocyte maturation and maintenance (8–10).
Components of both male and female pathways antagonize each other to promote development of either testes or ovaries (Figure 1).
Changes on these specific pathways, altering this perfect antagonism of ‘battle of sexes’, determine several spectrum of DSD conditions, with variable phenotypes (43). In Table 2 we have summarized current knowledge on genes involved in gonadal development and their pathogenetic role in 46,XX DSD (41, 44–51).
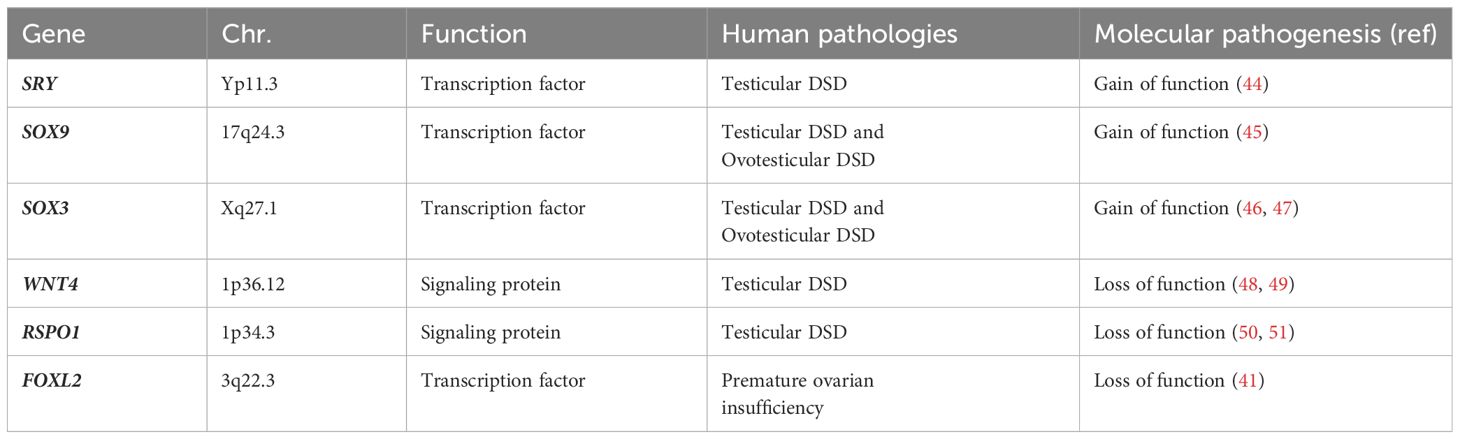
Table 2 Genes involved in gonadal development and their pathogenetic role in 46,XX DSD.
2.2 Internal genitalia differentiation
The internal genitalia are similar in both sexes until 9th week of gestation, when they will differentiate into specific male or female ducts, depending, in turn, by gonadal differentiation. At 5th of gestation, two pairs of ducts, derived from the intermediate mesoderm, begin to develop in fetus: the Wolff ducts, which eventually lead to the epididymis, vas deferens and seminal vesicles) and Mullerian ducts (lateral and parallel to Wolff), which eventually lead to the uterus, fallopian tubes, cervix and the upper third of the vagina (52).
After gonadal differentiation, in case of testis development, synthesis of testosterone by Leydig cells from 9-10th of gestation and anti-mullerian hormone (AMH) by Sertoli cells from 12th week of gestation, determine the regression of Mullerian ducts and the stabilization of Wolff ducts. On the contrary, in females, the absence of elevated androgens and AMH determine the regression of Wolff ducts and development of Mullerian structures (42, 52).
During the embryogenesis, in XX individuals, a maldevelopment of Mullerian ducts may occur, determining the Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, that will be discussed later in detail.
2.3 External genitalia differentiation
Development of external genitalia depends on androgen-independent and androgen-dependent two different pathways. The first phase begins at 5th week of gestation, with the formation of different structures, identical in both sexes: the genital tubercle, located just cranial to the midline opening of the urogenital ostium; the urogenital folds, laterally to the ostium; and the genital swelling/labioscrotal folds, laterally to the urogenital folds. Formation of these structures occurs identically in males and females, during the so-called ambisexual stage of external genitalia development, a hormone-independent phase, from 5th to 12th week of gestation (53).
After this first phase, in males, the androgen production determines the elongation of genital tubercle to a penis, the development of the penile urethra from the urogenital folds and the fusion of labioscrotal folds in the midline to form the scrotum. In females, the absence of elevated amount of androgens determine that genital tubercle differentiate into the clitoris, the urogenital ostium remains open with the subsequent development of the anterior urethra ostium and posterior vagina ostium, the urogenital folds become the labia minora and the labioscrotal folds become the major labia (53, 54).
In XX individuals, fetuses exposure to androgens between 8th and 15th week of gestation determine virilization of external genital with several degree of ‘masculinization’, as in the cases of 46,XX newborns with atypical genitalia, due to gonadal maldevelopment or steroidogenesis defects. These conditions will be discussed below in detail.
3 46,XX Differences of Sex Development
According to the Chicago Consensus (11), recently revised in a European Consensus Statement (12), 46,XX Differences of Sex Development (DSD) include a wide spectrum of conditions due to atypical gonadal development or altered hormonal secretion; in the same group are also classified patients with atypical differentiation of Mullerian structures, affected by the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH).
Disorders/Differences of gonadal development include testicular DSD, ovotesticular DSD and primary ovarian insufficiency. Disorders/Differences of androgen excess comprise the different forms of Congenital Adrenal Hyperplasia (not discussed in this review), Aromatase deficiency and Maternal causes of androgen excess, usually Luteoma.
In the next paragraphs we will summarize the most relevant data on diagnosis, clinical aspects and management of these rare diseases, outside CAH.
3.1 46,XX DSD due to disorders/differences of gonadal development
3.1.1 46,XX testicular DSD
It is characterized by the presence of testes in 46,XX individuals, with concomitant absence of Mullerian derivatives. In 60s A. de la Chapelle identified these subjects as ‘XX males with male phenotype, male psychosexual gender identity, gonads differentiated as testes without macroscopic and microscopic evidence of ovarian tissue and absence of female internal genitalia’ (55).
3.1.1.1 Genetic
The prevalence of this disease is estimated as 1:20.000 (56) and in the majority of cases (almost 90%), the pathogenetic cause is the translocation of SRY gene to the X chromosome or more rarely to an autosome (56). 46,XX testicular DSD SRY negative, may be due to an increased expression of genetic pathways involved in testicular differentiation and/or an insufficient expression of genetic pathways involved in ovarian differentiation (43) (Table 2).
3.1.1.2 Clinical aspects, puberty, sex hormone replacement therapy and possibility of fertility
From a clinical point of view, affected patients usually present with normal virilized male external genitalia; in other cases, newborns may present with atypical genitalia (46–48, 50, 51, 57–63) (Table 3). In patients with normal male external genitalia, diagnoses may be delayed until adult life when they could be referred to an endocrinologist because of infertility. Youngest boys may also be referred to a pediatric endocrinologist because of puberty delay or testicular hypoplasia (43) (Table 3). It has been speculated that at pubertal age, the absence of Y chromosome and genetic pathway needed for testicular maintenance, determine a progressive germ cell loss, azoospermia and consequent reduced testicular volume (64).
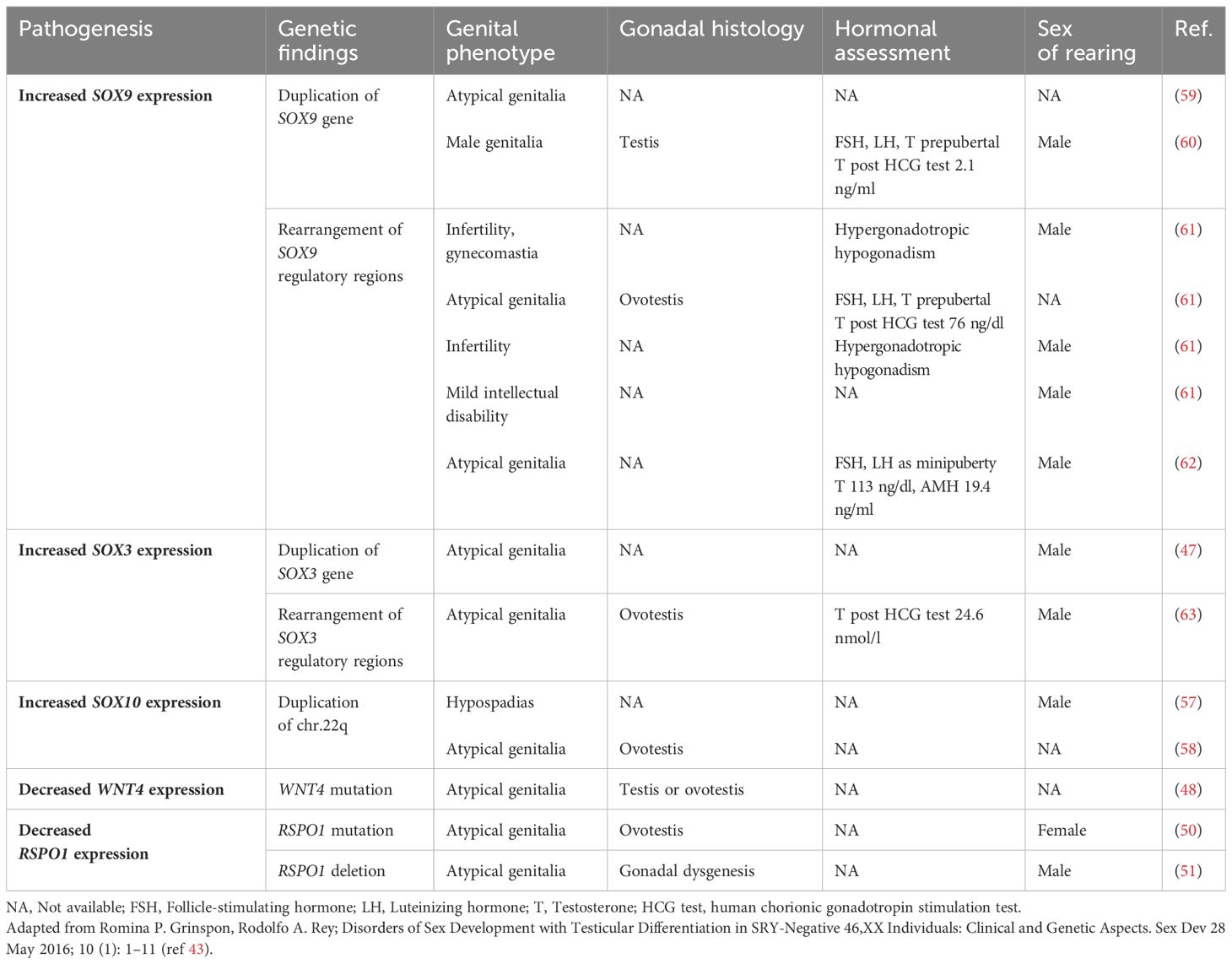
Table 3 Genetics, clinical phenotypes and hormonal assessment of SRY-negative 46,XX testicular and ovotesticular DSD.
In these patients, gender identity is almost always male, but they would need testosterone replacement therapy starting from young adult age (43). It was suggested that levels of testosterone may be normal during adolescence, but decreased in adulthood (65), however, few data have been published so far. Moreover, no studies on treatment to induce or sustain puberty in male patients with gonadal dysgenesis are available in literature (66). Hormone replacement therapy (HRT), if needed, consists of Testosterone treatment, with formulations and therapeutic schemes recommended in International clinical practice guidelines (66–68), that we have summarized in Table 4.
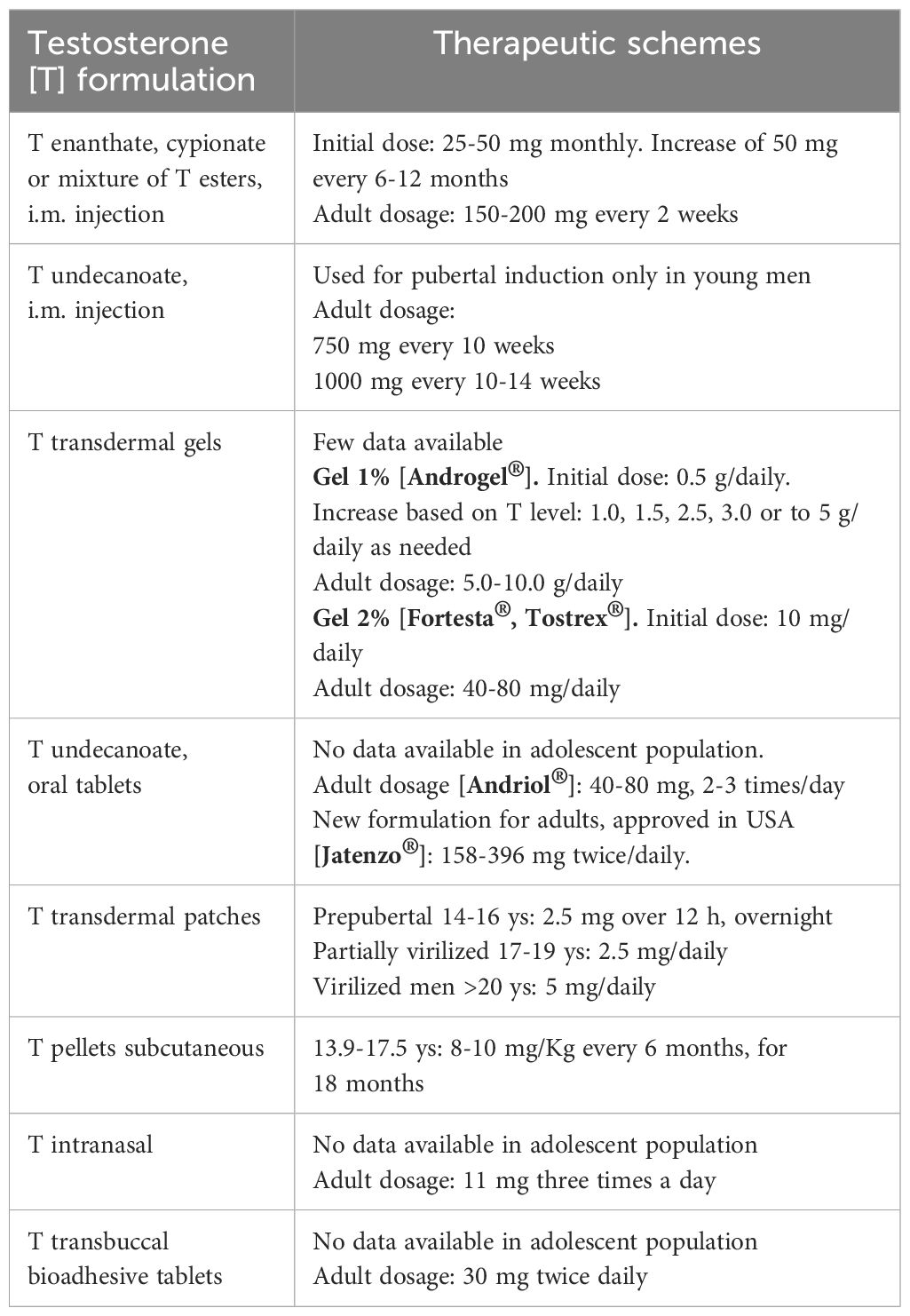
Recently, Chen et al. retrospectively revised a population of 144 males with 46,XX DSD (71 SRY positive, 15 SRY negative), analyzing clinical characteristics and assisted reproductive technology (ART) outcomes (69). The mean age of patients included in the study was 29.06 ± 4.5 yrs. Greater than 90% of cases had elevated levels of follicle-stimulating hormone and luteinizing hormone, almost 63% of cases had low testosterone values. The mean volume (95% CI) of left and right testis was 2.16 (1-82-2.49) and 2.16 (1.83-2.49) ml, respectively: 143 patients had bilateral atrophic testes with testicular volume less than 15 ml on both sides. Among patients with normal ejaculatory function, azoospermia was found in the totality of cases, all presenting the deletion of AZFa, AZFb and AZFc regions. Fertility was achieved in 87 patients, through ART: the live birth rates using artificial insemination (AID) or in vitro fertilization (IVF) with donor spermatozoa was 18.11 and 58.09% respectively, as already described in other retrospective studies of ART (70).
3.1.2 46,XX Ovotesticular DSD
The differential diagnosis between ovotesticular and testicular DSD is based on histological analysis. It requires the concomitant existence of testicular tissue (seminiferous tubules) and ovarian tissue (follicles containing oocytes) that can be found in each of the two gonads (bilateral ovotestis), just in one of the two gonads (unilateral ovotestis) with the other one normally differentiated as testis or ovary, or one testis in one side and ovary on the other (lateral ovotestis). The most common for is the unilateral (ovotestis/ovary) in 34% of cases, followed by bilateral ovotestis in 29% of cases, lateral (ovary/testis) in 25% of cases and unilateral (ovotestis/testis) in 12% of cases (71).
3.1.2.1 Genetic
Contrary to Testicular DSD, 90% of patients are SRY negative. However, similar to testicular DSD, also in these cases, a genetic imbalance of testis and ovarian pathways determining an increased expression of pro-testis genes and an insufficient expression of pro-ovarian/anti-testis genes, results crucial for the onset of these diseases (43, 72) (Table 3).
3.1.2.2 Clinical aspects, puberty, sex hormone replacement therapy and possibility of fertility
The clinical presentation is highly variable, ranging from a normal male phenotype or mild/severe hypospadias to a female presentation with genital tubercle hypertrophy. Less virilized patients may also present Mullerian derivatives (46–48, 50, 51, 57–63) (Table 3).
Sex assignment represents an important challenge for clinicians, and the multidisciplinary team together with patients and families, will apply clinical and ethical considerations discussed in the Consensus Statement (12), to ensure the best quality of life of each affected individual (14, 73–75). These state that the following principles should guide clinical decisions: minimizing physical and psychosocial risks, preserving the potential for fertility and satisfying sexual relations in adolescence and adulthood, leaving options open for the future if necessary, respecting the parents’ wishes, beliefs and sociocultural tradition, when possible, to guarantee the best options for a healthy life (76) (health is a state of complete physical, mental and social well-being and not merely the absence of disease or infirmity; WHO, 1948).
When the sex may not be easily defined at birth, more time is needed to determine the gender identity and the hormonal assessment of patients, in terms of androgen or estrogen gonadal production, waiting until pubertal age. Patients and families will be followed by a specialized DSD multidisciplinary team at each stage of life (12).
The risk of tumor development in the testicular portion it has been reported to be low, probably due to the absence of Y chromosome (77); the ovarian tissue, before puberty, remain quiescent. For these reasons, in view of the unpredictable gender outcome and in line with the recent recommendations of human rights principles, the irreversible gonadectomy should be avoided in any child with OT- DSD and postponed at pubertal age, after discussion with patients theirselves and families (78).
At the onset of puberty, gonadectomy would be needed, to avoid undesirable gonadal hormone production. However, surgical irreversible procedure should be performed until patient’s gender identity is confirmed by the psychological and psychiatric team. When gender identity is still uncertain, temporary treatment with GnRH analogs has been proposed, becoming a well-established clinical practice in DSD nowadays (78, 79), as for young adolescents with gender dysphoria (80).
Few studies have reported pubertal and fertility outcomes in OT-DSD so far, analyzing just small cohorts (79, 81). Some XX-OT boys may start and develop puberty spontaneously, other may need HRT to induce puberty or maintain testosterone level through adulthood (78, 80). As previously discussed, the absence of Y chromosome and genetic pathway needed for testicular maintenance, generally determine a progressive germ cell loss and azoospermia in male individuals (78). Female OT-DSD patients, who have normally developed ovarian tissue, may have physiological pubertal development and regular cyclic menstruation (77, 78, 80).
Several uneventful pregnancies, spontaneous or induced through ART, have been reported (78, 79, 82–85).
Hormone replacement therapy, if needed, consists of Testosterone or Estrogen treatment, depending on gender assignment, with formulations and therapeutic schemes recommended in International guidelines (66).
As previously discussed, no studies on treatment to induce or sustain puberty in male patients with gonadal dysgenesis are available in literature (66). Therapeutic approaches with Testosterone, standardized in clinical practice, have been summarized in Table 4.
On the contrary, looking at clinical studies on treatment to induce or sustain puberty in females with gonadal dysgenesis, several randomized trials and cohort studies have been compared (66); however, the majority of these, have brought patients diagnosed with Turner syndrome, primary ovarian failure or transgender women (66, 86, 87). Further specific studies, focusing on DSD patients, are needed to evaluate the best therapeutic approach ensuring a good quality of life on a long-term follow-up. In Table 5 we have summarized the most common preparations and relative therapeutic schemes that can be used for pubertal induction and maintenance in DSD girls with hypogonadism.

Table 5 Estrogen and progesterone preparations for pubertal induction and maintenance in girls with hypogonadism (ref 66, 85).
3.1.3 Premature ovarian insufficiency (POI)
According to the ESHRE guidelines, Premature ovarian insufficiency (POI) is defined as a clinical syndrome characterized by loss of ovarian activity before the age of 40 years, determining menstrual disturbance (amenorrhea or oligomenorrhea) with raised gonadotrophins and low estradiol (88).
3.1.3.1 Genetic
A wide range of different etiological causes may determine POI disease, including genetic, autoimmune, metabolic, infectious and iatrogenic factors (41, 89).
3.1.3.2 Clinical aspects, puberty, sex hormone replacement therapy and possibility of fertility
The clinical presentation of POI is highly heterogeneous as it can be associated with gonadal dysgenesis and consequent absence of spontaneous pubertal development, primary amenorrhea or secondary amenorrhea due to anticipated depletion of the ovarian reserve before 40 years of age.
Patients with autoimmune or genetic disorders, which it is known to have an increased risk to develop a POI (such as X chromosome defects, BPES, AIRE and other), may benefit from fertility preservation at young age with ovarian tissue or egg freezing (90). In the same way, the evidence of pathogenetic variants in adult women already diagnosed with POI, could be extremely useful for female relatives, who can be precociously referred to specialized team of endocrinologists and gynecologists.
Hormone replacement therapy consists of Estrogen and Progesterone treatments, with formulations and therapeutic schemes recommended in International guidelines (66), summarized in Table 5.
3.2 46,XX DSD due to atypical differentiation of Mullerian structures
3.2.1 Mayer-Rokitansky-Küster-Hauser syndrome
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, also referred to as Müllerian agenesis or aplasia, is a congenital disorder caused by embryologic underdevelopment of the Müllerian duct and characterized by agenesis of the uterus, the cervix and the upper two-thirds of the vagina in otherwise phenotypically 46,XX females. The ovaries, considering their different embryogenesis (as mentioned previously), are typically normal in morphology and function (91).
MRKH syndrome has an estimated incidence of about 1:4.000 to 1:5.000 female live births (92) and it is classified among the most severe uterine malformation by the “European Society of Human Reproduction and Embryology” and the “European Society for Gynaecological Endoscopy” classification. It represents the second most common cause of primary amenorrhea after ovarian insufficiency, reported in ~ 16% of female with primary amenorrhea (93).
3.2.1.1 Genetic
The etiology of MRKH syndrome remains unclear. Environmental and genetic causes that may interfere during the embryonic development have been proposed (91, 94). However, currently, the pathogenesis remains not completely understood and the majority of cases do not have a molecular diagnosis. Also, the inheritance is not completely clear: the identification of monozygotic twins discordant for MRKH syndrome (95) and the absence of genital malformations in biological children of women with MRKH syndrome born from surrogate mothers (96), support the sporadic nature of the disease; however, familial cases have been described (97) indicating that, at least in a subgroup of patients, MRKH syndrome may be an inherited disorder.
The most reported genes implicated in the pathogenesis of MRKH syndrome have been WNT4, LHX1 (LIM homebox protein 1) and HNF1B (hepatocyte nuclear factor-1B) (98). In particular, LHX1 and HNF1B, are considered important transcription factors regulating Mullerian ducts development (98). Mutations in LHX1 (99) and deletions at 17q12, encompassing LHX1 and HNF1B, have been detected in patients with MRKH syndrome (100, 101); mutations in the HNF1B gene have been detected in a familial case, in which two out of four female mutation carriers were affected by uterine malformations (102).
3.2.1.2 Clinical aspects, puberty, sex hormone replacement therapy and possibility of fertility
Physical examination of patient with MRKH syndrome reveals female external genitalia with short blind-ending vagina. Patients usually reach puberty at the physiological time, showing normal development of secondary sex characteristics and do not need HRT. The most frequent reason for referral to the endocrinologist or gynecologist is primary amenorrhea, and the median age at first presentation has been reported to be 17.5 years (92). In rare cases, patients may be referred at younger age for ‘incidental’ evidence of uterus agenesis at abdomen ultrasound or abdominal surgery performed for other reasons.
The role of an expert multidisciplinary team (MDT) is essential for the diagnosis and management of the disease.
Pelvic ultrasound (US) is considered the first line diagnostic tool, demonstrating the absence of uterus but the presence of normal bilateral ovaries; pelvic magnetic resonance imaging (MRI) is the gold standard for diagnosis, showing the Müllerian structures in detail, with a better resolution compared to the US (103, 104). One of the most common condition that may be confused with MRKH syndrome is Complete Androgen Insensitivity Syndrome (CAIS), however with a 46,XY karyotype, so the importance of MDT with a great experience on DSD management.
The evaluation of the presence of concomitant congenital extragenital anomalies is essential in patients with MRKH syndrome: in some individuals, uterine maldevelopment may be associated with urological abnormalities or other malformations (91). For this reason, in literature, it is possibly to classify the MRKH syndrome into the type I, (isolated or typical) and type II, where additional extragenital malformation are documented, involving mainly the kidneys and the axial skeleton and less frequently heart and hearing (91).
In European patients with MRKH syndrome, the proportion of type II was reported to be 43.5-54.4% (92, 105), while in Chinese patients was reported to be 30.4% (106). This discrepancy may be explained by ethnic differences. Renal malformations are the most frequent extragenital abnormalities, occurring in ~ 30–40% in European cohorts (92, 105). Unilateral renal agenesis is the most frequent anomaly accounting about half of all renal malformations associated with MRKH syndrome; it is often associated with complete absence of the ipsilateral Müllerian duct which suggests a close relationship between early kidney and Müllerian duct development. Other renal malformations include pelvic kidney, duplex kidney, and horseshoe kidney (92).
Anomalies of the skeleton are the second most frequent extragenital manifestations affecting about ~10–40% of patients (92). Skeletal anomalies typically involve the axial skeleton (scoliosis, Klippel-Feil anomaly, hemivertebrae, rib aplasia) and more rarely the extremities.
Cardiac abnormalities are reported in < 5% of patients (pulmonary valve stenosis, atrial septal defect) (92). Hearing impairment, including both sensorineural and conductive hearing, is generally reported in < 5% of patient, but is not routinely examined (92).
The coexistence of Müllerian aplasia with unilateral renal aplasia/ectopic kidney and cervicothoracic somite dysplasia is called Müllerian aplasia, renal aplasia, and cervicothoracic somite dysplasia syndrome (MURCS) (107).
Caring of patients with MRKH syndrome require a multidisciplinary team consisting of expert gynecologists, surgeons, physiologists and sexologists, playing a key role at diagnosis and long-term follow-up (12).
The creation of a new functional neovagina represents one of the most important intervention to discuss with patients and families. In the last decades, different surgical and non-surgical treatment strategies have been suggested for vaginal reconstruction.
The American College of Obstetricians and Gynecologists (ACOG) has recommended self-dilation therapy using vaginal dilatators as first line approach in most patients, based on the high overall success rate, safeness with low complication rate and reduced operating costs than surgery (108). Adverse effects reported with dilation include urinary complaints, bleeding and pain. Patients should receive a psychological support and encouraged to start dilation when she feels emotionally and physically ready (109).
ACOG recommend that surgery should be reserved for those patients experiencing failure with dilation, choosing among different surgical approaches and techniques. A comprehensive literature review on the management of vaginal agenesis has been conducted by Callens et al. discussing outcome, advantages and disadvantages of the different procedures (110). However, discuss with patients and families the possibility of different therapeutical approaches is absolutely important, highlighting that also the surgical option require a postoperative dilation to ensure a satisfactory long-term outcome.
Currently, patients affected by MRKH syndrome may experience motherhood adopting a child or, in some Countries also with a surrogate pregnancy, transferring the embryo derived from their own oocytes and partner’s sperm (111, 112); very recently, the uterus transplantation technique has also been reported (113). Since the first live birth after uterus transplantation achieved by the Swedish team (113), more than 80 procedures have been performed around the globe and at least 40 children were born (114).
Despite recent scientific progress, further studies are needed to improve quality of life of these patients and reduce psychological discomforts (115, 116).
3.3 46,XX DSD due to androgen excess
3.3.1 Aromatase deficiency
Aromatase deficiency (AroD) is a rare genetic condition that is caused by mutations in the CYP19A1 gene, located on the long arm of chromosome 15 (15q21.1) (16). It is an autosomal recessive disorder, first described by Shozu et al. in 1991 (117); then, almost 40 cases have been described, due to several pathogenetic variations in the CYP19A1 gene (118).
Aromatase is the enzyme that catalyzes the synthesis of estrogens from androgens. The three main precursors are androstenedione, testosterone, and 16-α-hydroxy dehydroepiandrosterone sulfate, catalyzed into estrone, estradiol, and estriol, respectively. During pregnancy, 16OH-dehydroepiandrosterone sulfate (16OH-DHEAS) arising from fetal liver hydroxylation of fetal adrenal DHEAS represents an important substrate for placental aromatase and subsequent estriol production (119).
3.3.1.1 Genetic
In human, the CYP19A1 gene and its product aromatase are expressed in the ovary, testis, placenta, adipose tissue, skin, and the brain. The size of the aromatase gene is greater than 123 kb, and its tissue-specific expression is regulated by the use of tissue-specific promoters involving alternative splicing (120). Moreover, CYP19 gene expression is regulated by several hormones and factors that differ markedly between tissues. Thus, a strict control over tissue-specific expression is needed for proper regulation of estrogen synthesis during fetal development and post-natal life. The biological importance of the aromatase complex is related not only to its role in the synthesis of estrogens, but also to its potential influence on the balance of the androgen-estrogen ratio in different tissues (118).
3.3.1.2 Clinical aspects, puberty, sex hormone replacement therapy
Clinical phenotype in patients affected by AroD is quite variable, depending on the enzymatic activity (121). In most 46,XX newborn diagnosed with aromatase deficiency, atypical genitalia with various degrees of masculinization of the external genitalia have been described (16, 118, 121). A typical aspect that could alert to the possibility of diagnosis is the maternal virilization during gestation, that progressively disappear after delivery.
During infancy and childhood, there are usually no symptoms of aromatase deficiency. However, it has been reported that basal and stimulated gonadotropin levels (LH, FSH) remain significantly high during the infancy and childhood period, resulting in a higher risk of follicular ovarian cysts (122, 123). It has been speculated that a prolonged effect of androgens or lack of estrogens during gestation, persisting into infancy, might result in an irreversible incorrect maturation of the GnRH pulse generator (123).
In adolescent girls, AroD may determine several conditions such as puberty delay, hypergonadotropic hypogonadism or primary amenorrhea, due to estrogen deficiency; moreover, clinical signs of hyperandrogenism may also be present (16). In affected individuals, regular follow-up is needed to avoid long-term consequences of hypoestrogenism.
To date there is no consensus on the appropriate age, type and dosage of estrogen replacement therapy and the usefulness of starting low-dose estrogen treatment from infancy in affected females.
Further retrospective and prospective studies on large population are needed to define the best management of these patients.
4 Conclusions
The 46,XX DSD group includes a wide spectrum of conditions, with different etiopathogenesis. Even if the majority of cases are caused by CAH, it is crucial to know that other rarer disorders exist, in order to make a correct and preferable early diagnosis. Patients should be referred to a specialized Centre, where a trained multidisciplinary team (MDT) could manage these children/adolescents (and families), from diagnosis to adulthood. Support group may have a key role, together with MDT, helping families to not fell alone, sharing parents’ and patients’ experiences. While many patients fare well and have a good quality of life, other individuals have expressed a sense of anxiety and discomfort about the DSD condition or have reported poor quality of life. For these reasons is important to improve our knowledge on diagnosis, management and long-term prognosis of these individuals, organizing international collaborative studies focusing on more debate aspects, such as clinical care, psychosocial development and psychosocial adaptation.
Author contributions
MS: Conceptualization, Writing – original draft, Writing – review & editing. SM: Writing – original draft. CB: Writing – original draft. GR: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hughes IA. Minireview: sex differentiation. Endocrinology. (2001) 142:3281–7. doi: 10.1210/endo.142.8.8406
2. Bashamboo A, McElreavey K. Mechanism of sex determination in humans: insights from disorders of sex development. Sex Dev. (2016) 10:313–25. doi: 10.1159/000452637
3. Valenzuela N. Sexual development and the evolution of sex determination. Sex Dev. (2008) 2:64–72. doi: 10.1159/000129691
4. Shimada K. Sex determination and sex differentiation. Avian Poult Biol Rev. (2002) 13:1–14. doi: 10.3184/147020602783698449
5. She ZY, Yang WX. Molecular mechanisms involved in mammalian primary sex determination. J Mol Endocrinol. (2014) 53:R21–37. doi: 10.1530/JME-14-0018
6. Eggers S, Sinclair A. Mammalian sex determination—insights from humans and mice. Chromosom Res. (2012) 20:215–38. doi: 10.1007/s10577-012-9274-3
7. Makiyan Z. Studies of gonadal sex differentiation. Organogenesis. (2016) 12:42–51. doi: 10.1080/15476278.2016.1145318
8. Biason-Lauber A. The battle of the sexes: human sex development and its disorders. Results Probl Cell Differ. (2016) 58:337–82. doi: 10.1007/978-3-319-31973-5_13
9. Lucas-Herald AK, Bashamboo A. Gonadal development. Endocr Dev. (2014) 27:1–16. doi: 10.1159/000363608
10. Bashamboo A, McElreavey K. Human sex-determination and disorders of sex-development (DSD). Semin Cell Dev Biol. (2015) 45:77–83. doi: 10.1016/j.semcdb.2015.10.030
11. Hughes IA, Houk CP, Ahmed SF, Lee PA, ESPE Consensus Group, Group EC. Consensus statement on management of intersex disorders. Arch Dis Child. (2006) 91:554–63. doi: 10.1136/adc.2006.098319
12. Cools M, Nordenström A, Robeva R, Hall J, Westerveld P, Flück C, et al. Caring for individuals with a difference of sex development (DSD): a Consensus Statement. Nat Rev Endocrinol. (2018) 14:415–29. doi: 10.1038/s41574-018-0010-8
13. Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. Global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr. (2016) 85:158–80. doi: 10.1159/000442975
14. Ahmed SF, Bryce J, Hiort O. International networks for supporting research and clinical care in the field of disorders of sex development. Endocr Dev. (2014) 27:284–92. doi: 10.1159/000363676
15. Pasterski V, Mastroyannopoulou K, Wright D, Zucker KJ, Hughes IA. Predictors of posttraumatic stress in parents of children diagnosed with a disorder of sex development. Arch Sex Behav. (2014) 43:369–75. doi: 10.1007/s10508-013-0196-8
16. Baronio F, Ortolano R, Menabò S, Cassio A, Baldazzi L, Natale Di V, et al. 46,XX DSD due to androgen excess in monogenic disorders of steroidogenesis: genetic, biochemical, and clinical features. Int J Mol Sci. (2019) 20:4605. doi: 10.3390/ijms20184605
17. Tevosian SG. Genetic control of ovarian development. Sex Dev. (2013) 7:33–45. doi: 10.1159/000339511
18. Eggers S, Ohnesorg T, Sinclair A. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol. (2014) 10:673–83. doi: 10.1038/nrendo.2014.163
19. Ohnesorg T, Vilain E, Sinclair AH. The genetics of disorders of sex development in humans. Sex Dev. (2014) 8:262–72. doi: 10.1159/000357956
20. Jost A. The age factor in the castration of male rabbit fetuses. Proc Soc Exp Biol Med. (1947) 66:302. doi: 10.3181/00379727-66-16071
21. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. (1990) 346:240–4. doi: 10.1038/346240a0
22. Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman DE. WT1 mutations contribute to abnormal genital system development and hereditary Wilms’ tumour. Nature. (1991) 353:431–4. doi: 10.1038/353431a0
23. Muscatelli F, Strom TM, Walker AP, Zanaria E, Récan D, Meindl A, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. (1994) 372:672–6. doi: 10.1038/372672a0
24. Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. (1994) 79:1111–20. doi: 10.1016/0092-8674(94)90041-8
25. Sekido R, Lovell-Badge R. Genetic control of testis development. Sex Dev. (2012) 7:21–32. doi: 10.1159/000342221
26. McElreavey K, Vilain E, Cotinot C, Payen E, Fellous M. Control of sex determination in animals. Eur J Biochem. (1993) 218:769–83. doi: 10.1111/j.1432-1033.1993.tb18432.x
27. Goodfellow PN, Lovell-Badge R. SRY and sex determination in mammals. Annu Rev Genet. (1993) 27:71–92. doi: 10.1146/annurev.ge.27.120193.000443
28. Yang Y, Workman S, Wilson MJ. The molecular pathways underlying early gonadal development. J Mol Endocrinol. (2019) 62:R47–64. doi: 10.1530/JME-17-0314
29. Hart D, Rodríguez Gutiérrez D, Biason-Lauber A. CBX2 in DSD: the quirky kid on the block. Sex Dev. (2022) 16:162–70. doi: 10.1159/000522164
30. Zaytouni T, Efimenko EE, Tevosian SG. GATA transcription factors in the developing reproductive system. Adv Genet. (2011) 76:93–134. doi: 10.1016/B978-0-12-386481-9.00004-3
31. Eozenou C, Gonen N, Touzon MS, Jorgensen A, Yatsenko SA, Fusee L, et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. Proc Natl Acad Sci. (2020) 117:13680–8. doi: 10.1073/pnas.1921676117
32. Wang Y, Chen Q, Zhang F, Yang X, Shang L, Ren S, et al. Whole exome sequencing identified a rare WT1 loss-of-function variant in a non-syndromic POI patient. Mol Genet Genomic Med. (2022) 10:e1820. doi: 10.1002/mgg3.1820
33. Wang H, Li G, Zhang J, Gao F, Li W, Qin Y, et al. Novel WT1 missense mutations in han chinese women with premature ovarian failure. Sci Rep. (2015) 5:13983. doi: 10.1038/srep13983
34. Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. (2016) 25:3446–53. doi: 10.1093/hmg/ddw186
35. Baetens D, Stoop H, Peelman F, Todeschini A-L, Rosseel T, Coppieters F, et al. NR5A is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med. (2017) 19:367–76. doi: 10.1038/gim.2016.118
36. Igarashi M, Takasawa K, Hakoda A, Kanno J, Takada S, Miyado M, et al. Identical NR5A1 missense mutations in two unrelated 46,XX individuals with testicular tissues. Hum Mutat. (2017) 38:39–42. doi: 10.1002/humu.23116
37. Knarston IM, Robevska G, van den Bergen JA, Eggers S, Croft B, Yates J, et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum Mutat. (2019) 40:207–16. doi: 10.1002/humu.23672
38. Swartz JM, Ciarlo R, Guo MH, Abrha A, Weaver B, Diamond DA, et al. A 46,XX ovotesticular disorder of sex development likely caused by a steroidogenic factor-1 (NR5A1) variant. Horm Res Paediatr. (2016) 87:191–5. doi: 10.1159/000452888
39. Lourenço D, Brauner R, Lin L, Perdigo De A, Weryha G, Muresan M, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. (2009) 360:1200–10. doi: 10.1056/NEJMoa0806228
40. Bouazzi L, Sproll P, Eid W, Biason-Lauber A. The transcriptional regulator CBX2 and ovarian function: a whole genome and whole transcriptome approach. Sci Rep. (2019) 9:17033. doi: 10.1038/s41598-019-53370-4
41. Elzaiat M, Todeschini AL, Caburet S, Veitia RA. The genetic make-up of ovarian development and function: the focus on the transcription factor FOXL2. Clin Genet. (2017) 91:173–82. doi: 10.1111/cge.12862
42. Kurilo LF. Oogenesis in antenatal development in man. Hum Genet. (1981) 57:86–92. doi: 10.1007/BF00271175
43. Grinspon RP, Rey RA. Disorders of sex development with testicular differentiation in SRY-negative 46,XX individuals: clinical and genetic aspects. Sex Dev. (2016) 10:1–11. doi: 10.1159/000445088
44. Terribile M, Stizzo M, Manfredi C, Quattrone C, Bottone F, Giordano DR, et al. 46,XX testicular disorder of sex development (DSD): a case report and systematic review. Medicina (Kaunas). (2019) 55:371. doi: 10.3390/medicina55070371
45. Croft B, Ohnesorg T, Sinclair AH. The role of copy number variants in disorders of sex development. Sex Dev. (2017) 12:19–29. doi: 10.1159/000481896
46. Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. (2011) 121:328–41. doi: 10.1172/JCI42580
47. Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bägli DJ, Thomas P, et al. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet. (2012) 158A:1759–64. doi: 10.1002/ajmg.a.35390
48. Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. (2008) 82:39–47. doi: 10.1016/j.ajhg.2007.08.005
49. Ragitha TS, Sunish KS, Gilvaz S, Daniel S, Varghese PR, Raj S, et al. Mutation analysis of WNT4 gene in SRY negative 46,XX DSD patients with Mullerian agenesis and/or gonadal dysgenesis- An Indian study. Gene. (2023) 861:147236. doi: 10.1016/j.gene.2023.147236
50. Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. (2008) 29:220–6. doi: 10.1002/humu.20665
51. Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, et al. R-spondin1 is essential in sex determination, skin differentiation and Malignancy. Nat Genet. (2006) 38:1304–9. doi: 10.1038/ng1907
52. Mullen RD, Behringer RR. Molecular genetics of müllerian duct formation, regression and differentiation. Sex Dev. (2014) 8:281–96. doi: 10.1159/000364935
53. Blaschko SD, Cunha GR, Baskin LS. Molecular mechanisms of external genitalia development. Differentiation. (2012) 84:261–8. doi: 10.1016/j.diff.2012.06.003
54. Baskin L, Derpinghaus A, Cao M, Sinclair A, Li Y, Overland M, et al. Hot spots in fetal human penile and clitoral development. Differentiation. (2020) 112:27–38. doi: 10.1016/j.diff.2019.11.001
55. de la Chapelle A, Hortling H, Niemi M, Wennström J. XX sex chromosomes in a human male. Acta Med Scand. (1964) 175:25–38. doi: 10.1111/j.0954-6820.1964.tb04630.x
56. Alkhzouz C, Bucerzan S, Miclaus M, Mirea AM, Miclea D. 46,XX DSD: developmental, clinical and genetic aspects. Diagnostics (Basel). (2021) 11:1379. doi: 10.3390/diagnostics11081379
57. Seeherunvong T, Perera EM, Bao Y, Benke PJ, Benigno A, Donahue RP, et al. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet Part A. (2004) 127A:149–51. doi: 10.1002/ajmg.a.20630
58. Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet. (1999) 85:2–4. doi: 10.1002/(SICI)1096-8628(19990702)85:1%3C2::AID-AJMG2%3E3.0.CO
59. Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet. (1999) 87:349–53. doi: 10.1002/(SICI)1096-8628(19991203)87:4%3C349::AID-AJMG13%3E3.0.CO
60. Lee GM, Ko JM, Shin CH, Yang SW. A Korean boy with 46,XX testicular disorder of sex development caused by SOX9 duplication. Ann Pediatr Endocrinol Metab. (2014) 19:108–12. doi: 10.6065/apem.2014.19.2.108
61. Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. (2015) 23:1025–32. doi: 10.1038/ejhg.2014.237
62. Qian Z, Grand K, Freedman A, Nieto MC, Behlmann A, Schweiger BM, et al. Whole genome sequencing identifies a cryptic SOX9 regulatory element duplication underlying a case of 46,XX ovotesticular difference of sexual development. Am J Med Genet Part A. (2021) 185:2782–8. doi: 10.1002/ajmg.a.62373
63. Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab. (2015) 100:E815–820. doi: 10.1210/jc.2014-4383
64. Huang WJ, Yen PH. Genetics of spermatogenic failure. Sex Dev. (2008) 2:251–9. doi: 10.1159/000152041
65. Velasco G, Savarese V, Sandorfi N, Jimenez SA, Jabbour S. 46,XX SRY-positive male syndrome presenting with primary hypogonadism in the setting of scleroderma. Endocr Pract. (2011) 17:95–8. doi: 10.4158/EP10184.CR
66. Nordenström A, Ahmed SF, van den Akker E, Blair J, Bonomi M, Brachet C, et al. Pubertal induction and transition to adult sex hormone replacement in patients with congenital pituitary or gonadal reproductive hormone deficiency: an Endo-ERN clinical practice guideline. Eur J Endocrinol. (2022) 186:G9–49. doi: 10.1530/EJE-22-0073
67. Stancampiano MR, Lucas-Herald AK, Russo G, Rogol AD, Ahmed SF. Testosterone therapy in adolescent boys: the need for a structured approach. Horm Res Paediatr. (2019) 92:215–28. doi: 10.1159/000504670
68. Bhasin S, Yialamas MA, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, et al. Testosterone therapy in men with hypogonadism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:1715–44. doi: 10.1210/jc.2018-00229
69. Chen T, Tian L, Wu F, Xuan X, Ma G, Tang R, et al. Clinical and genetic analysis in males with 46,XX disorders of sex development: A reproductive centre experience of 144 cases. Andrologia. (2019) 51:e13232. doi: 10.1111/and.13232
70. Gerkowicz SA, Crawford SB, Hipp HS, Boulet SL, Kissin DM, Kawwass JF. Assisted reproductive technology with donor sperm: national trends and perinatal outcomes. Am J Obstet Gynecol. (2018) 218:421.e1–421.e10. doi: 10.1016/j.ajog.2017.12.224
71. Vilain E. Chapter 14A - The genetics of ovotesticular differences of sex development. In: Maria I, editor. New, Genetic Steroid Disorders, 2nd ed. Academic Press (2023). p. 285–8. doi: 10.1016/B978-0-12-821424-4.00004-6
72. McElreavey K, Bashamboo A. Monogenic forms of DSD: an update. Horm Res Paediatr. (2023) 96:144–68. doi: 10.1159/000521381
73. Schweizer K, Brunner F, Gedrose B, Handford C, Richter-Appelt H. Coping with diverse sex development: treatment experiences and psychosocial support during childhood and adolescence and adult well-being. J Pediatr Psychol. (2017) 42:504–19. doi: 10.1093/jpepsy/jsw058
74. Siminoff LA, Sandberg DE. Promoting shared decision making in disorders of sex development (dsd): decision aids and support tools. Horm Metab Res. (2015) 47:335–9. doi: 10.1055/s-0035-1545302
75. Gillam LH, Hewitt JK, Warne GL. Ethical principles for the management of infants with disorders of sex development. Horm Res Paediatr. (2010) 74:412–8. doi: 10.1159/000316940
76. Bertelloni S, Tyutyusheva N, Valiani M, D’Alberton F, Baldinotti F, Caligo MA, et al. Disorders/Differences of sex development presenting in the newborn with 46,XY karyotype. Front Pediatr. (2021) 9:627281. doi: 10.3389/fped.2021.627281
77. Looijenga LHJ, Hersmus R, de Leeuw BH, Stoop H, Cools M, Oosterhuis JW, et al. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol Metab. (2010) 24:291–310. doi: 10.1016/j.beem.2009.10.002
78. Syryn H, Van De Vijver K, Cools M. Ovotesticular Difference of Sex Development: Genetic background, histological features, and clinical management. Horm Res Paediatr. (2023) 96:180–9. doi: 10.1159/000519323
79. Lambert S, Peycelon M, Samara-Boustani D, Hyon C, Dumeige L, Peuchmaur M, et al. SRY-negative 46,XX testicular/ovotesticular DSD: long-term outcomes and early blockade of gonadotropic axis. Clin Endocrinol (Oxf). (2021) 94:667–76. doi: 10.1111/cen.14389
80. Hembree WC, Cohen-Kettenis PT, Gooren L, Hannema SE, Meyer WJ, Murad MH, et al. Endocrine treatment of gender-dysphoric/gender-incongruent persons: an endocrine society* clinical practice guideline. J Clin Endocrinol Metab. (2017) 102:3869–903. doi: 10.1210/jc.2017-01658
81. Verkauskas G, Jaubert F, Lortat-Jacob S, Malan V, Thibaud E, Nihoul-Fékété C. The long-term followup of 33 cases of true hermaphroditism: a 40-year experience with conservative gonadal surgery. J Urol. (2007) 177:726–31. doi: 10.1016/j.juro.2006.10.003
82. Matsuda S, Kuwabara Y, Kato R, Nakao K, Yonezawa M, Ono S, et al. Successful in vitro fertilization pregnancy and delivery by an infertile woman with ovotesticular disorder of sex development: A Case Report. J Nippon Med Sch. (2023) 90:240–4. doi: 10.1272/jnms.JNMS.2023_90-202
83. Minowada S, Fukutani K, Hara M, Shinohara M, Kamioka J, Isurugi K, et al. Childbirth in true hermaphrodite. Eur Urol. (2017) 10:414–5. doi: 10.1159/000463848
84. Słowikowska-Hilczer J, Hirschberg AL, Claahsen-van der Grinten H, Reisch N, Bouvattier C, Thyen U, et al. Fertility outcome and information on fertility issues in individuals with different forms of disorders of sex development: findings from the dsd-LIFE study. Fertil Steril. (2017) 108:822–31. doi: 10.1016/j.fertnstert.2017.08.013
85. Schultz BAH, Roberts S, Rodgers A, Ataya K. Pregnancy in true hermaphrodites and all male offspring to date. Obstet Gynecol. (2009) 113:534–6. doi: 10.1097/AOG.0b013e3181866456
86. Klein KO, Phillips SA. Review of hormone replacement therapy in girls and adolescents with hypogonadism. J Pediatr Adolesc Gynecol. (2019) 32:460–8. doi: 10.1016/j.jpag.2019.04.010
87. Swee DS, Javaid U, Quinton R. Estrogen replacement in young hypogonadal women—transferrable lessons from the literature related to the care of young women with premature ovarian failure and transgender women. Front Endocrinol (Lausanne). (2019) 10:685. doi: 10.3389/fendo.2019.00685
88. European Society for Human Reproduction and Embryology (ESHRE) Guideline Group on POI, Webber L, Davies M, Anderson R, Bartlett J, Braat D, Cartwright B, et al. ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. (2016) 31:926–37. doi: 10.1093/humrep/dew027
89. Rossetti R, Ferrari I, Bonomi M, Persani L. Genetics of primary ovarian insufficiency. Clin Genet. (2017) 91:183–98. doi: 10.1111/cge.12921
90. La Marca A, Mastellari E. Fertility preservation for genetic diseases leading to premature ovarian insufficiency (POI). J Assist Reprod Genet. (2021) 38:759–77. doi: 10.1007/s10815-021-02067-7
91. Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. (2020) 15:214. doi: 10.1186/s13023-020-01491-9
92. Herlin M, Bjørn A-MB, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: a nationwide registry-based study. Hum Reprod. (2016) 31:2384–90. doi: 10.1093/humrep/dew220
93. Timmreck LS, Reindollar RH. Contemporary issues in primary amenorrhea. Obstet Gynecol Clin North Am. (2003) 30:287–302. doi: 10.1016/s0889-8545(03)00027-5
94. Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development. (2010) 137:1407–20. doi: 10.1242/dev.024166
95. Duru UA, Laufer MR. Discordance in Mayer-von Rokitansky- Küster -Hauser syndrome noted in monozygotic twins. J Pediatr Adolesc Gynecol. (2009) 22:e73–75. doi: 10.1016/j.jpag.2008.07.012
96. Petrozza JC, Gray MR, Davis AJ, Reindollar RH. Congenital absence of the uterus and vagina is not commonly transmitted as a dominant genetic trait: outcomes of surrogate pregnancies. Fertil Steril. (1997) 67:387–9. doi: 10.1016/S0015-0282(97)81927-9
97. Herlin M, Højland AT, Petersen MB. Familial occurrence of Mayer–Rokitansky–Küster–Hauser syndrome: a case report and review of the literature. Am J Med Genet. (2014) 164:2276–86. doi: 10.1002/ajmg.a.36652
98. Triantafyllidi VE, Mavrogianni D, Kalampalikis A, Litos M, Roidi S, Michala L. Identification of genetic causes in Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: A systematic review of the literature. Children (Basel). (2022) 9:961. doi: 10.3390/children9070961
99. Ledig S, Brucker S, Barresi G, Schomburg J, Rall K, Wieacker P. Frame shift mutation of LHX1 is associated with Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome. Hum Reprod. (2012) 27:2872–5. doi: 10.1093/humrep/des206
100. Bernardini L, Gimelli S, Gervasini C, Carella M, Baban A, Frontino G, et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis. (2009) 4:25. doi: 10.1186/1750-1172-4-25
101. Nik-Zainal S, Strick R, Storer M, Huang N, Rad R, Willatt L, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Müllerian aplasia. J Med Genet. (2011) 48:197–204. doi: 10.1136/jmg.2010.082412
102. Williams LS, Demir Eksi D, Shen Y, Lossie AC, Chorich LP, Sullivan ME, et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. (2017) 108:145–51. doi: 10.1016/j.fertnstert.2017.05.017
103. Lermann J, Mueller A, Wiesinger E, Häberle L, Brucker S, Wallwiener D, et al. Comparison of different diagnostic procedures for the staging of malformations associated with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. (2011) 96:156–9. doi: 10.1016/j.fertnstert.2011.04.051
104. Preibsch H, Rall K, Wietek BM, Brucker SY, Staebler A, Claussen CD, et al. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. Eur Radiol. (2014) 24:1621–7. doi: 10.1007/s00330-014-3156-3
105. Kapczuk K, Iwaniec K, Friebe Z, Kędzia W. Congenital malformations and other comorbidities in 125 women with Mayer-Rokitansky-Küster-Hauser syndrome. Eur J Obstet Gynecol Reprod Biol. (2016) 207:45–9. doi: 10.1016/j.ejogrb.2016.10.014
106. Chen N, Pan H, Luo G, Wang P, Xie Z, Hua K, et al. Clinical characteristics of 1,055 Chinese patients with Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide multicentric study. Fertil Steril. (2021) 116:558–65. doi: 10.1016/j.fertnstert.2021.02.033
107. Kumar S, Sharma S. MURCS (Müllerian duct aplasia–renal agenesis–cervicothoracic somite dysplasia): a rare cause of primary amenorrhoea. Oxford Med Case Rep. (2016) 2016:73–5. doi: 10.1093/omcr/omw022
108. Committee Opinion: no. 562: müllerian agenesis: diagnosis, management, and treatment. Obstet Gynecol. (2013) 121:1134–7. doi: 10.1097/01.AOG.0000429659.93470.ed
109. Heller-Boersma JG, Schmidt UH, Keith Edmonds D. Psychological distress in women with uterovaginal agenesis (Mayer-Rokitansky-Küster-Hauser Syndrome, MRKH). Psychosomatics. (2009) 50:277–81. doi: 10.1176/appi.psy.50.3.277
110. Callens N, De Cuypere G, De Sutter P, Monstrey S, Weyers S, Hoebeke P, et al. An update on surgical and non-surgical treatments for vaginal hypoplasia. Hum Reprod Update. (2014) 20:775–801. doi: 10.1093/humupd/dmu024
111. White PM. Commercialization, altruism, clinical practice: seeking explanation for similarities and differences in californian and canadian gestational surrogacy outcomes. Women’s Heal Issues. (2018) 28:239–50. doi: 10.1016/j.whi.2018.01.004
112. Hodson N, Townley L, Earp BD. Removing harmful options: the law and ethics of international commercial surrogacy. Med Law Rev. (2019) 27:597–622. doi: 10.1093/medlaw/fwz025
113. Brännström M, Johannesson L, Dahm-Kähler P, Enskog A, Mölne J, Kvarnström N, et al. First clinical uterus transplantation trial: a six-month report. Fertil Steril. (2014) 101:1228–36. doi: 10.1016/j.fertnstert.2014.02.024
114. Sousa C, Carton I, Jaillard S, Cospain A, Lavillaureix A, Timoh KN, et al. Mayer-Rokitansky-Küster-Hauser syndrome patients’ interest, expectations and demands concerning uterus transplantation. J Gynecol Obstet Hum Reprod. (2023) 52:102674. doi: 10.1016/j.jogoh.2023.102674
115. Bean EJ, Mazur T, Robinson AD. Mayer-Rokitansky- Küster-Hauser Syndrome: sexuality, psychological effects, and quality of life. J Pediatr Adolesc Gynecol. (2009) 22:339–46. doi: 10.1016/j.jpag.2008.11.006
116. Weijenborg PTM, Kluivers KB, Dessens AB, Kate-Booij MJ, Both S. Sexual functioning, sexual esteem, genital self-image and psychological and relational functioning in women with Mayer–Rokitansky–Küster–Hauser syndrome: a case–control study. Hum Reprod. (2019) 34:1661–73. doi: 10.1093/humrep/dez130
117. Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. (1991) 72:560–6. doi: 10.1210/jcem-72-3-560
118. Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res. (2009) 72:321–30. doi: 10.1159/000249159
119. Belgorosky A, Rivarola MA. Physiology and pathophysiology of estrogens: lessons from pediatric patients with complete aromatase deficiency. Endocrinologist. (2004) 14:93–100. doi: 10.1097/01.ten.0000123567.27319.e2
120. Means G, Mahendroo M, Corbin C, Mathis JM, Powell FE, Mendelson CR, et al. Structural analysis of the gene encoding human aromatase cytochrome P-450, the enzyme responsible for estrogen biosynthesis. J Biol Chem. (1989) 264:19385–91. doi: 10.1016/S0021-9258(19)47313-4
121. Lin L, Ercan O, Raza J, Burren CP, Creighton SM, Auchus RJ, et al. Variable Phenotypes associated with aromatase (cyp19) insufficiency in humans. J Clin Endocrinol Metab. (2007) 92:982–90. doi: 10.1210/jc.2006-1181
122. Conte FA, Grumbach MM, Ito Y, Fisher CR, Simpson ER. A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab. (1994) 78:1287–92. doi: 10.1210/jcem.78.6.8200927
123. Belgorosky A, Pepe C, Marino R, Guercio G, Saraco N, Vaiani E, et al. Hypothalamic-pituitary-ovarian axis during infancy, early and late prepuberty in an aromatase-deficient girl who is a compound heterocygote for two new point mutations of the cyp19 gene. J Clin Endocrinol Metab. (2003) 88:5127–31. doi: 10.1210/jc.2003-030433
Keywords: 46, XX DSD, gonadal differentiation, atypical genitalia, gonadal dysgenesis, ovotestis, Mayer-Rokitansky-Küster-Hauser syndrome, aromatase deficiency
Citation: Stancampiano MR, Meroni SLC, Bucolo C and Russo G (2024) 46,XX Differences of Sex Development outside congenital adrenal hyperplasia: pathogenesis, clinical aspects, puberty, sex hormone replacement therapy and fertility outcomes. Front. Endocrinol. 15:1402579. doi: 10.3389/fendo.2024.1402579
Received: 17 March 2024; Accepted: 22 April 2024;
Published: 22 May 2024.
Edited by:
Antonio Balsamo, Alma Mater Studiorum - University Hospital S.Orsola Malpighi, ItalyReviewed by:
Ralitsa Robeva, Medical University - Sofia, BulgariaLaura Audí, Vall d’Hebron University Hospital, Spain
Copyright © 2024 Stancampiano, Meroni, Bucolo and Russo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianna Rita Stancampiano, c3RhbmNhbXBpYW5vLm1hcmlhbm5hcml0YUBoc3IuaXQ=