 Cheng Zhang1†
Cheng Zhang1† Gang Wei
Gang Wei Lin Liu
Lin Liu- 1National Drug Clinical Trial Center, The First Affiliated Hospital of Bengbu Medical University, Bengbu, Anhui, China
- 2Beijing Key Laboratory of Diabetes Research and Care, Department of Endocrinology, Beijing Diabetes Institute, Beijing Tongren Hospital, Capital Medical University, Beijing, China
Background: The causal relationship between familial hypercholesterolemia (FH) and various vitamin deficiencies has not yet been elucidated. Therefore, this study investigated the cause-and-effect relationship between FH and the risk of multiple vitamin deficiencies in humans.
Methods: Mendelian randomization (MR) analysis was performed by extracting six datasets for FH, FH with ischemic heart disease (IHD), and vitamin deficiency (vitamin A, thiamine, other B-group vitamins, and vitamin D) from the FinnGen study, covering a total of 329,115; 316,290; 354,932; 354,949; 355,411 and 355,238 individuals, respectively.
Results: FH was suggestively associated with higher odds of thiamine deficiency [inverse variance weighted odds ratio (ORIVW) 95% confidence interval (CI): 1.62 (1.03, 2.55), P = 0.036] and vitamin D deficiencies [ORIVW CI: 1.35 (1.04, 1.75), P = 0.024], low-density lipoprotein receptor (LDLR) rs112898275 variant, rs11591147 and rs499883 in proprotein convertase subtilisin/kexin 9 (PCSK9), rs9644862 in cyclin-dependent kinase inhibitor 2 B antisense RNA1 (CDKN2B-AS1), and rs142834163 in dedicator of cytokinesis 6 (DOCK6) and rs115478735 in ABO blood group (ABO) strongly influenced the risk of thiamine deficiency, while the rs7412 variant in apolipoprotein E (APOE) mostly influenced the risk of vitamin D deficiency. FH with IHD was suggestively associated with higher odds of vitamin D deficiency (ORIVW, weighted median [WM][95%CI]: 1.31 [1.05, 1.64]; 1.47 [1.10, 1.97]) (P = 0.018; 0.010) without any single significant SNPs observed.
Conclusion: FH was positively associated with increased risks of thiamine and vitamin D deficiencies, revealing a prospective and unfortunate complication of FH.
Introduction
Vitamins are a cluster of organic substances that sustain the growth, reproduction, and homeostasis of the human body (1). They exist as fat-soluble (vitamins A, D, E, and K) and water-soluble (vitamins B and C) vitamin forms (2). Except for vitamin D, which can be synthesized by humans, the other vitamins are obtained from food intake and gut microbiota (2, 3). Vitamin deficiency is common worldwide at all ages (4). Minor vitamin deficiencies are insidious and often overlooked in the clinic, but can have severe negative effects (4). The most common examples of these effects include xerophthalmia, anemia, a high incidence rate of infectious diseases (vitamin A), beriberi (thiamine or vitamin B1), rickets, osteomalacia, and a possible association with increased infectious diseases (vitamin D) (4). Globally, vitamin A deficiency occurs in nearly 30% of children <5 years of age. The morbidity of thiamine deficiency is 20%–90%, and the regional epidemic rate of vitamin D deficiency is 5.5–85.1% (5–7). Vitamin deficiency has become an important public health issue due to its high prevalence and undesirable endpoints (8).
Familial hypercholesterolemia (FH) is a congenital mal-transportation of lipids bearing homozygous or heterozygous family mutated genes, which is marked by a superelevation of plasma low-density lipoprotein cholesterol (LDL-C) levels (9). This overload can genetically accelerate vitamin D deficiency (10). Additionally, it has been reported from observational investigation that vitamins D, B6, and B12 deficiencies occurred in hyperlipidemic patients, with vitamins B6 significantly negatively associated with total cholesterol and non-HDL levels (11). Hence, patients with FH are likely complicated with multiple vitamin deficiencies, spanning the entire course of disease. However, the known Mendelian effect of high LDL-C levels on vitamin D deficiency has significant pleiotropy (10); meanwhile, the observational investigation has the sample size of only 60 (including 40 hyperlipidemic patients and 20 healthy controls), and the subjects of only males from Jordanian (11). Therefore, the Mendelian model of FH can be applied to assess the future risk of multiple vitamin deficiencies to ensure its prevention in the background of FH.
Mendelian randomization (MR) design assessed the possible causal relationship between FH and the risk of multiple vitamin deficiencies, including vitamin A, thiamine, other B-group vitamins, and vitamin D.
Materials and methods
Figures 1, 2 show the graphical abstract and an overview of the study design, respectively. An MR design based on public summary-level data derived from genome-wide association studies (GWASs) was adopted to evaluate the possible causal relationship between FH and ischemic heart disease (IHD) with the risk of multiple vitamin deficiencies (vitamin A, thiamine, other B-group vitamins, and vitamin D).

Figure 1. Graphical abstract of this study.
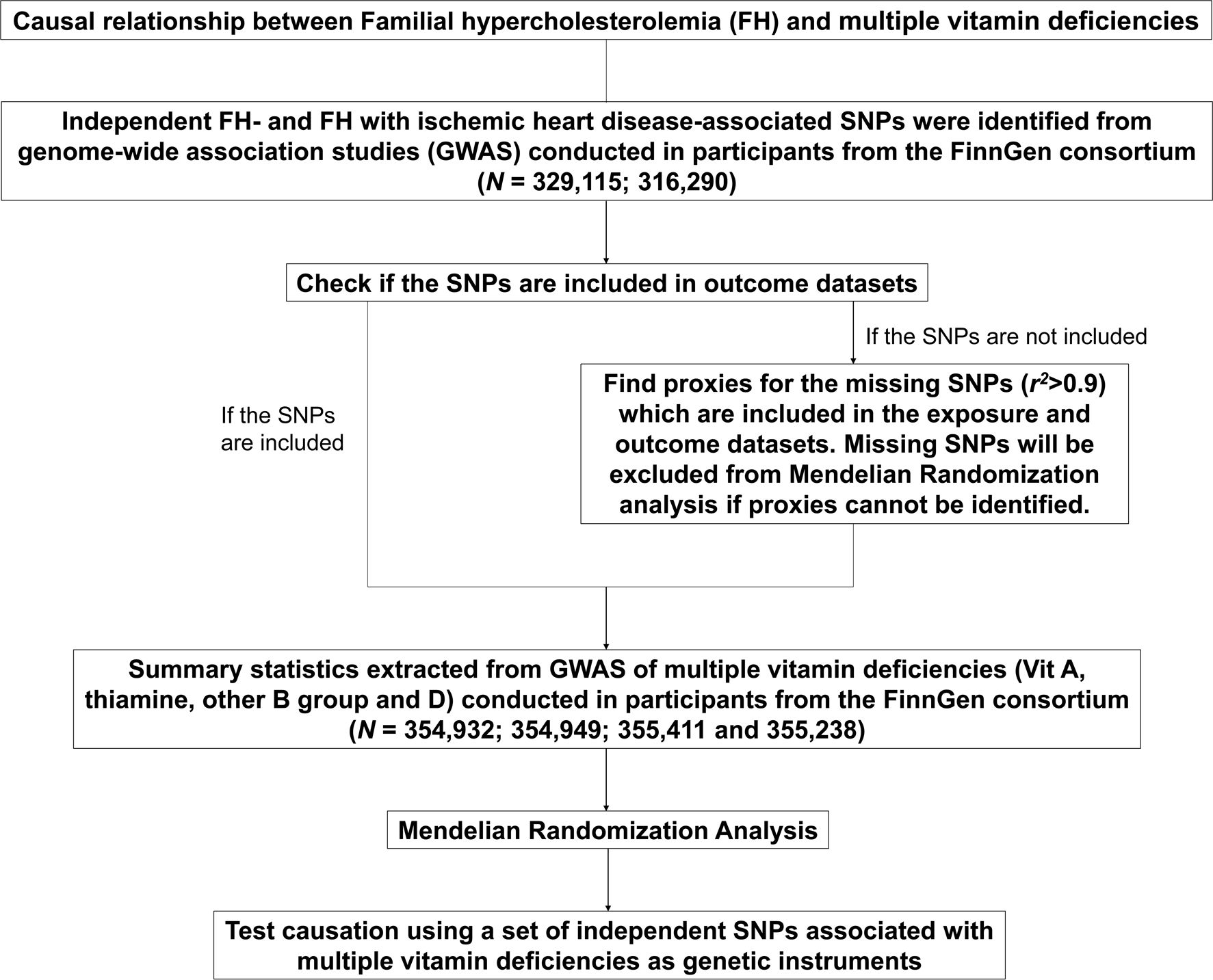
Figure 2. Flowchart of the Mendelian randomization analyses of familial hypercholesterolemia and the risk of multiple vitamin deficiencies. FH, familial hypercholesterolemia.
Instrumental variable selection
Genetic variants of FH (ICD10: E7800) and FH with IHD were obtained from a recently published GWAS of European ancestry (FinnGen) (N = 329,115; 316,290) (Supplementary Table S2). Summary datasets are available on the following websites (https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_E4_FH.gz and https://storage.googleapis.com/finngenpublic-data-r9/summary_stats/finngen_R9_E4_FH_IHD.gz). In the MR analysis, IVs were chosen on the basis of specific criteria. Single nucleotide polymorphisms (SNPs) associated with FH and FH with IHD were selected at a genome-wide significance level of P < 5×10−8. When the r2 of SNPs was < 0.001 within a 10000 kb window, the SNP with a more significant P-value was retained from the analysis. SNPs not available in the vitamin deficiency datasets were either replaced with proxy SNPs in high linkage disequilibrium (LD, r2 > 0.9) or discarded. Finally, palindromic SNPs were discarded based on their allele frequencies.
The use of strong instruments is key to improving the accuracy and efficiency of estimating causal effects in the MR model. However, if the genetic variation is only weakly linked to the exposure variable, it may introduce bias in the estimates of the MR model, which is commonly referred to as weak instrument bias (12). We further assessed the strength of genetic variants by computing the F-statistic (F = β2/SE2) for each SNP, ensuring that the F-statistic exceeded 10 (12, 13). In this study, the minimum F-statistic observed was 30.85, suggesting strong instruments and consequently a low likelihood of bias from weak instruments. Supplementary Table S1 provides an overview of these data.
Outcome data source
Genetic variants of multiple vitamin deficiencies (vitamin A (ICD10: E50), thiamine (ICD10: E51), other B-group vitamins (ICD10: E53), and vitamin D (ICD10: E55)) were obtained from FinnGen (N = 354,932; 354,949; 355,411; and 355,238, respectively). The summary dataset is available on the website (https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_E4_VIT_A_DEF.gz; https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_E4_THIA_DEF.gz; https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_E4_VIT_B_DEF.gz; https://storage.googleapis.com/finngen-public-data-r9/summary_stats/finngen_R9_E4_VIT_D_DEF.gz). Supplementary Table S2 provides detailed information.
Statistical analyses
The MR method was used to explore the causal associations between genetically predicted differences per standard deviation (SD) increases in FH and the risk of vitamin deficiency by reporting odds ratios (ORs). The conventional inverse-variance weighted (IVW) and weighted median (WM) methods were used in the primary analysis (14). Cochran’s Q test using the IVW model was applied to quantify heterogeneity (15). The MR-Egger regression model was used to determine unknown pleiotropic effects. A non-zero intercept from MR-Egger indicates that the IVW estimate may be invalid due to horizontal pleiotropy (16). Sensitivity analysis based on the MR-Egger regression model and leave-one-out sensitivity analysis were performed (16). Furthermore, the PhenoScanner database was searched to assess the association of the selected SNPs with possible pleiotropy at a genome-wide significance level of P <5×10−8 (17, 18). To strengthen the reliability of the results, this study assessed the statistical power of significant associations. This calculation determined the probability of detecting a true effect in this MR study, considering the specified sample size and effect size (19, 20).
All statistical analyses were conducted using R software (R 4.0.5, The R Foundation for Statistical Computing) and the “TwoSampleMR” package (21). The Bonferroni correction is a conservative method for probability thresholding to control the occurrence of false positives. To account for multiple testing, the Bonferroni correction threshold of P-value < 6.25 × 10-3 (0.05/8 [2 exposures and 4 outcomes]) was prespecified. Significance was determined at a P-value < 6.25 × 10-3, while the P-values between 6.25 × 10-3 and 0.05 was considered suggestive (22, 23). For analyses of heterogeneity and pleiotropy, P-value < 0.05 indicated significant (22).
Results
Genetic variants associated with familial hypercholesterolemia
The genetic tool extraction identified 13 SNPs related to FH, and 10 SNPs related to FH with IHD (all P < 5×10−8, r2 < 0.001, 10000 kb) (Supplementary Table S1).
Cause-and-effect relationship of familial hypercholesterolemia with multiple vitamin deficiencies
The primary results of IVW and WM estimated the causal relationship between FH/FH with IHD and multiple vitamin deficiencies. The outcomes of IVW model suggested positive genetic causal associations between FH and deficiencies of thiamine and vitamin D, respectively (OR [95%CI]: 1.62 [1.03, 2.55]; 1.35 [1.04, 1.75]) (P = 0.036; 0.024). Similarly, the outcomes of IVW and WM models suggested positive genetic causal association between FH with IHD and vitamin D deficiency (OR [95%CI]: 1.31 [1.05, 1.64]; 1.47 [1.10, 1.97]) (P = 0.018; 0.010) (Table 1).
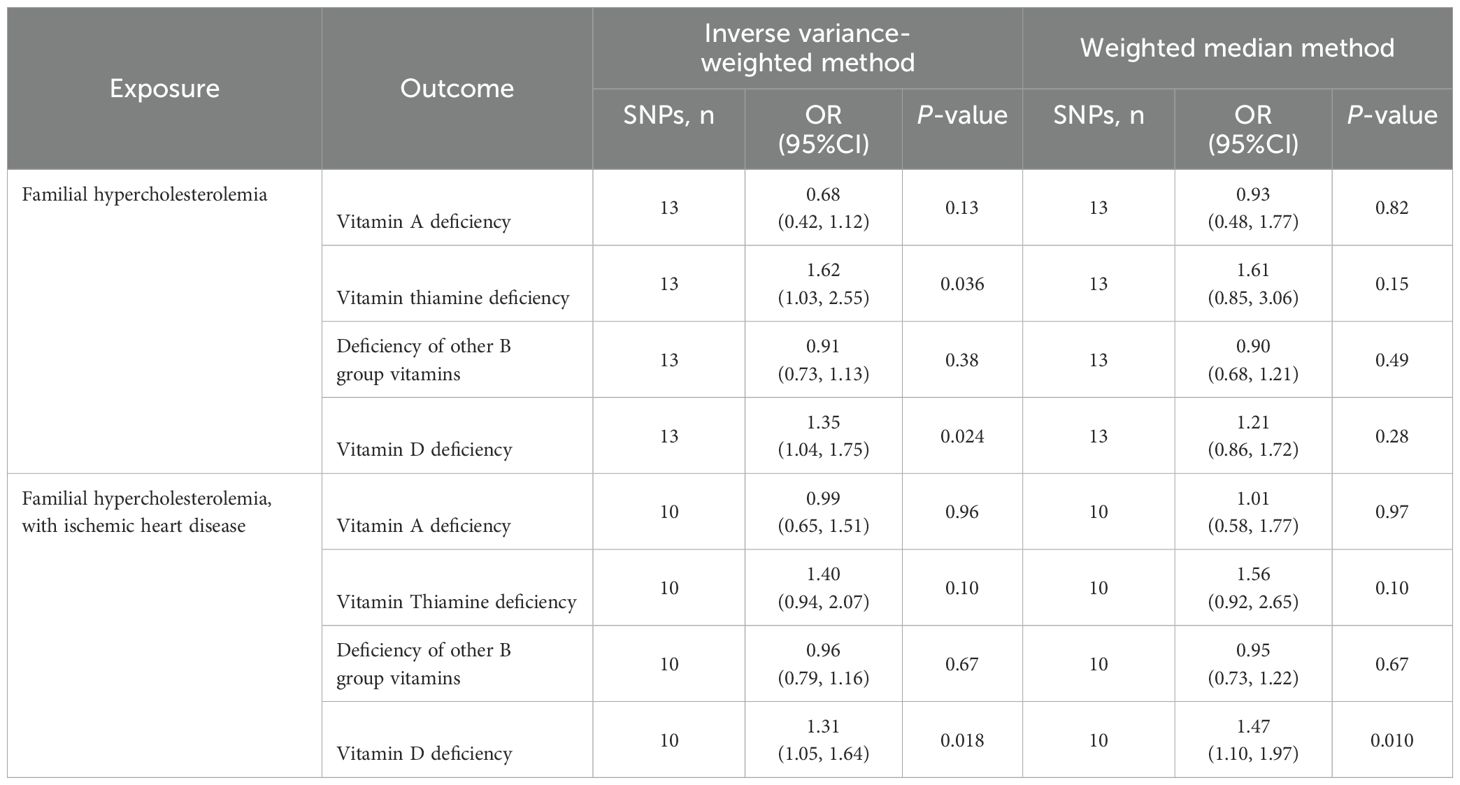
Table 1. Inverse variance-weighted and weighted median analyses of familial hypercholesterolemia and risk of multiple vitamin deficiencies.
In addition, no causal relationships showed between FH and the risks of deficiencies in vitamin A and other B-groups vitamins, and between FH with IHD and the risks of deficiencies in vitamin A, thiamine, and other B-group vitamins (P > 6.25 × 10-3) (Table 1).
Sensitivity analysis of familial hypercholesterolemia with multiple vitamin deficiencies
Sensitivity analysis was conducted on the suggestive exposure-outcome associations obtained to verify their reliability. No significant heterogeneity (P = 0.52) and horizontal pleiotropy (P = 0.40) were observed in the correlation analysis between FH and thiamine deficiency. No significant heterogeneity (P = 0.88) and horizontal pleiotropy (P = 0.28) were observed in the correlation analysis between FH and vitamin D deficiency. No significant heterogeneity (P = 0.55) or horizontal pleiotropy (P = 0.62) was observed in the correlation analysis between FH with IHD and vitamin D deficiencies (Table 2).
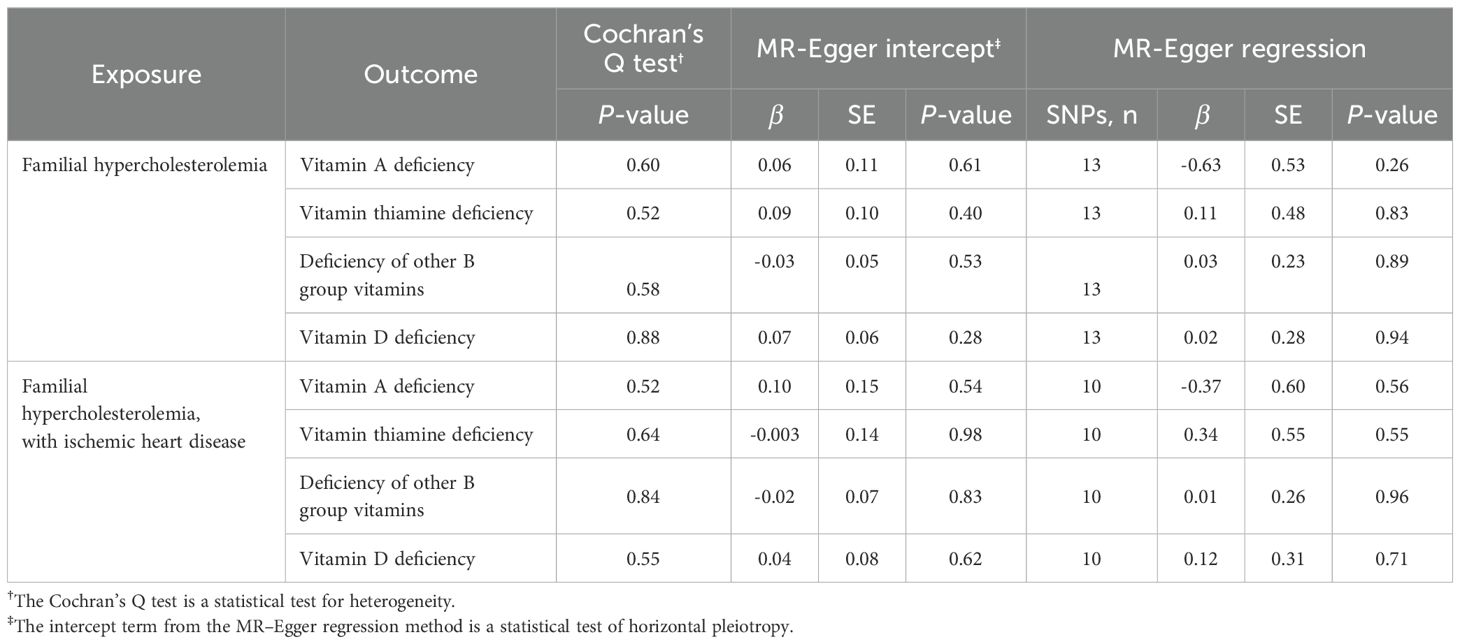
Table 2. Cochran’s Q test, MR–Egger intercept and MR-Egger regression of familial hypercholesterolemia and risk of multiple vitamin deficiencies.
The leave-one-out analysis was used to analyze single instrumental variable influencing the causal effects of FH/FH with IHD on multiple vitamin deficiencies. Leaving rs112898275, rs115478735, rs11591147, rs142834163, rs499883, and rs9644862 out respectively abrogated the correlation of FH with thiamine deficiency (β = -0.32103, 0.141933, -0.50812, 0.508043, 0.144529, 0.115318) (Figure 3A; Supplementary Table S1). Leaving rs7412 out, the correlation of FH with vit D deficiency did not remain (β = -0.42272) (Figure 3B; Supplementary Table S1). However, the correlation between FH with IHD and vitamin D deficiency remained even after removing any single SNPs (Figure 3C).
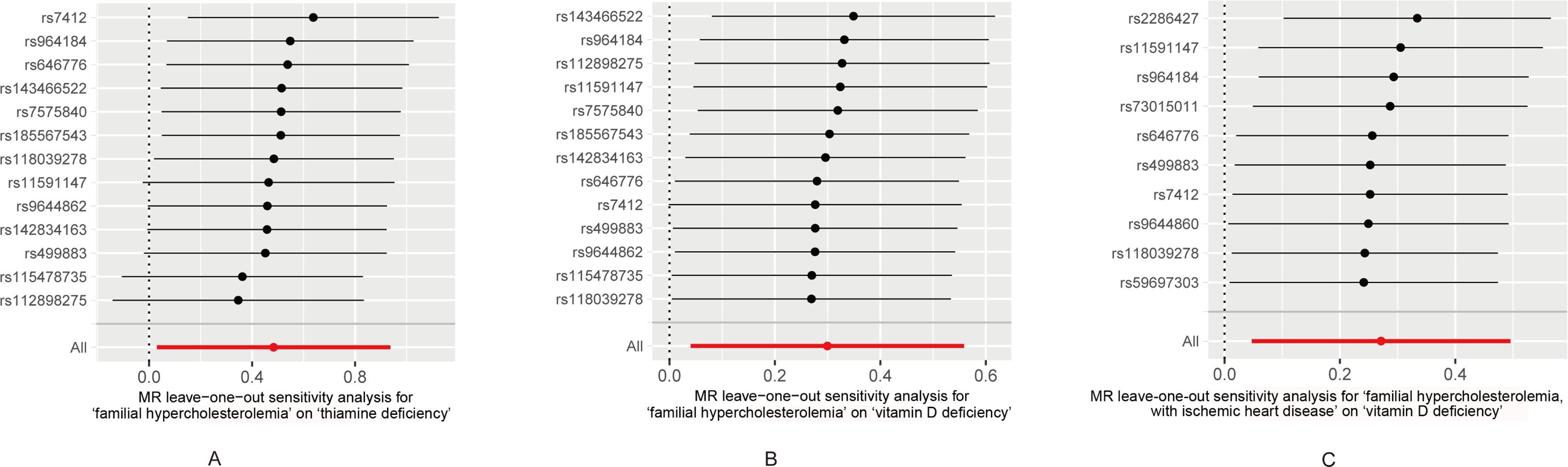
Figure 3. (A) Leave-one-out sensitivity and Mendelian randomization analyses based on the inverse-variance weighted (IVW) model for determining the effects of familial hypercholesterolemia on the risk of thiamine deficiency. (B) Leave-one-out sensitivity and Mendelian randomization analyses based on the inverse-variance weighted (IVW) model for determining the effects of familial hypercholesterolemia on the risk of vitamin D deficiency. (C) Leave-one-out sensitivity and Mendelian randomization analyses based on the inverse-variance weighted (IVW) model for determining the effects of familial hypercholesterolemia with ischemic heart disease on the risk of vitamin D deficiency.
Moreover, the statistical power of FH and thiamine/vitamin D deficiencies were 50%/49%, and FH with IHD and vitamin D deficiency was 49%.
Discussion
This study is the first to evaluate the causal effect of FH on the development of multiple vitamin deficiencies. The results of the MR analysis suggested positive associations between FH and thiamine and vitamin D deficiencies. Unlike FH, FH with IHD was only positively associated with higher odds of vitamin D deficiency. Significant associations were further tested using a leave-one-out sensitivity analysis of the mechanism. The typical signals are depicted in Figure 4.
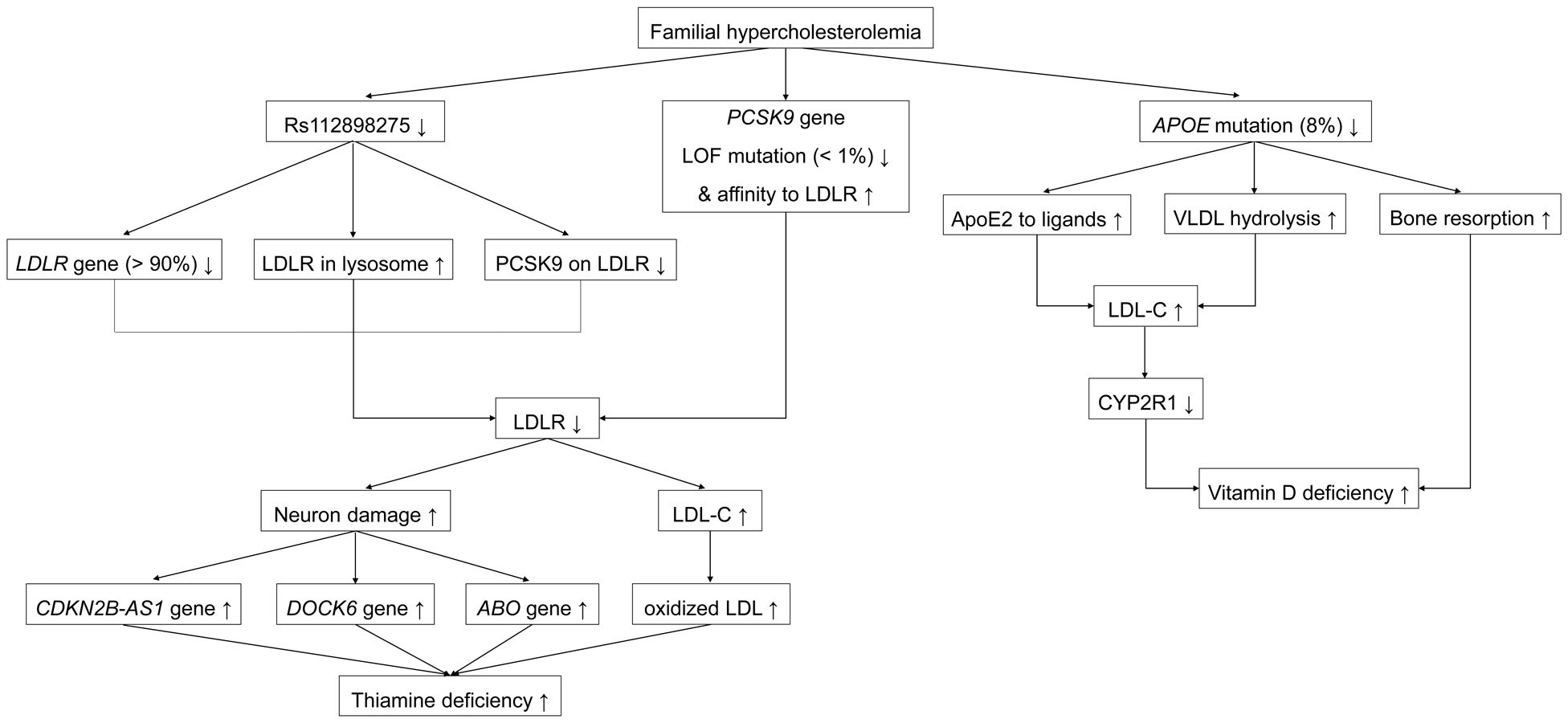
Figure 4. Representative signaling pathways involved in the associations between familial hypercholesterolemia and multiple vitamin deficiencies. ApoE, apolipoprotein E; PCSK9, proprotein convertase subtilisin/kexin 9; CDKN2B-AS1, cyclin-dependent kinase inhibitor 2B antisense RNA1; LOF, loss of function; GOF, gain of function; LDLR, low-density lipoprotein receptor.
Cause-and-effect relationship of familial hypercholesterolemia with thiamine deficiency
Rs112898275 is a SNP in the low-density lipoprotein receptor (LDLR) gene shown in the Phenoscanner database. LDLR is 45 kb long, is located on the short arm of chromosome 19p13.2, and includes 18 exons and 17 introns (24–26). LDLR gives rise to the LDLR protein comprising 860 amino acids harboring a 21-amino acid signal peptide (26). The LDLR protein is processed in the endoplasmic reticulum (ER), where the signal peptide is cleaved; the cleaved protein is then transformed into a mature 160-kDa (839 amino acids) glycoprotein by glycosylation (25). Mature LDLR is a transmembrane receptor located on most cellular surfaces and includes LDL receptor type A (LA) repeats, an epidermal growth factor precursor (EGFP), a transmembrane section, and a cytoplasmic tail with at least one NPxY theme (25, 27). The LA repeats bind to LDL-C on the cell membrane, and EGFP facilitates the combination of LDLR with proprotein convertase subtilisin/kexin 9 (PCSK9) on the hepatocellular surface (primary) and in the trans-Golgi network (TGN) (secondary), and liberation of bound LDL-C in the endosome, whereby LDLR/PCSK9 and released LDL-C are targeted for lysosomal degradation and non-adherent LDLR returns to the cell surface for reutilization (27, 28). LDLR mutagenesis accounts for > 90% of mutations in FH (29). The inverse β-coefficients of rs112898275 lead us to hypothesize that increased LDLR rs112898275 in patients with FH diminishes LDLR reutilization or adhesion to PCSK9.
Rs11591147 and rs499883 are SNPs in the proprotein convertase subtilisin/kexin 9 (PCSK9) gene (30). PCSK9 is located on the small arm of chromosome 1p32 and has a length of 25 kb, 12 exons, and 11 introns (28). The main organ producing PCSK9 is the liver, followed by the kidneys and intestines (28). PCSK9 initially synthetizes the inactive 692-amino acid PCSK9 protein with a signal peptide (amino acids 1–30) (31). The PCSK9 protein is processed in the ER, where the signal peptide is removed and cleavage occurs autocatalytically at the VFAQ152-SIP site (28, 31). The cleaved mature protein is then delivered to the Golgi apparatus in eukaryotic cells (28, 32). Later, when dispersed in plasma, the catalytic domain (amino acids 153–421) of the mature PCSK9 protein links with EGFP (amino acids 314–355) of the LDLR on the cellular surface of liver cells (primary) and in the TGN (secondary) by protein-protein communication (28, 33). LDLR/PCSK9 travels into lysosomes from endosomes for degradation, resulting in inadequate LDLR recycling to the cytomembrane to eliminate LDL-C (25). The two contrary false expressions of PCSK9 include loss-of-function (LOF), which decreases cholesterol, and gain-of-function (GOF), which increases cholesterol levels (34). Among FH mutations, < 1% are GOF mutations in PCSK9; the remaining >99% are non-GOF (29). The rs11591147 (p.Arg46Leu) is located in exon 1 of PCSK9; this LOF-mutation site impairs lysosomal degradation by reducing by 15% the connectivity of PCSK9 to LDLR, thereby combating high LDL-C (25, 28, 32, 35, 36). Meanwhile, rs499883 is an intronic SNP in PCSK9 that positively regulates PCSK9 expression via an unknown mechanism (30). Hence, decreased rs11591147 (p.Arg46Leu) and increased rs499883 in patients with FH leads to increased PCSK9 levels, which strengthens the binding of PCSK9 to LDLR for lysosomal degradation, resulting in insufficient LDLR for recycling.
Rs9644862 is a SNP in the cyclin-dependent kinase inhibitor 2 B antisense RNA1 (CDKN2B-AS1) gene (known as antisense non-coding RNA in the INK4 locus (ANRIL), p15AS, PCAT12, CDKN2BAS, CDKN2B-AS, NCRNA00089) gene (37–39). CDKN2B-AS1 is located on the short arm of human chromosome 9p21.3 and has 21 exons (40, 41). CDKN2B-AS1, a product of CDKN2B-AS1, is a 3.8 kb non-coding RNA (lncRNA) (40, 41). CDKN2B-AS1 protects against neuronal apoptosis by increasing glial cell-derived neurotrophic factor (GDNF) levels and preventing neuronal apoptosis by absorbing micro-RNA (miR)-133 (42).
Rs142834163 is a SNP in the dedicator of cytokinesis 6 (DOCK6) gene, as shown in the Phenoscanner database. DOCK6 (also known as AOS2 or ZIR1) is located on chromosome 19p13.2 and contains 51 exons (43). DOCK6 encodes the 2047 amino acid DOCK6 protein (44). DOCK6 belongs to the DOCK family of guanine nucleotide exchange factors (GEFs), which promote the exchange of guanosine diphosphate (GDP) with guanosine triphosphate (GTP) to regulate Rho GTPase activity (45). DOCK6 has two DOCK homology region (DHR) domains with phospholipid-binding and membrane-targeting activity in DHR-1 and GEF in DHR-2 (46). During neurodevelopment, DOCK6 plays a role in neurite outgrowth, axon growth, and regeneration by exchanging GDP with GTP for ras-related C3 botulinum toxin substrate 1 (RAC1) and cell division cycle 42 (CDC42), which control lamellipodia and filopodia morphology, respectively (46–48).
Rs115478735, a SNP in ABO, is strongly associated with plasma proteins of the immunoglobulin superfamily, including leucine-rich repeat protein 2 (ISLR2) and protein-tyrosine sulfotransferase 2 (TPST2) (49). ISLR2 (also known as LINX) is a type I transmembrane protein and a subset of the leucine-rich repeat and immunoglobulin (LIG) family of proteins comprising five tandem leucine-rich repeats (LRRs), an immunoglobulin (IG) domain, a transmembrane domain, and a short cytoplasmic tail (50). ISLR2 uniquely determines axon extension, guidance, and branching by interacting with Trk receptor tyrosine kinases (RTKs) to regulate their activities (51). ISLR2 interacts with and enhances Rho-kinase activity to rebuild the cytoskeleton during neuronal development (50). TPST2 is a tyrosyl protein sulfotransferase (TPST), a membrane-bound enzyme that catalyzes the protein-tyrosine sulfation in the cellular TGN by transferring a sulfuryl group from 3’-phosphoadenosine 5’-phosphosulfate (PAPS) to tyrosine residues (52). Tyrosine sulfation plays an elusive role in neuronal growth and maintenance (52).
In the central nervous system (CNS), LDLR is predominantly located in neurons, astrocytes, and oligodendrocytes and blocks neuronal pyroptosis by hindering ROS-NLRP3 inflammasome activation (53). In animal models, the absence of LDLR increases ROS generation, vulnerability to amyloid-β (Aβ)-induced neurotoxicity, and caspase-1-dependent gasdermin D (GSDMD) cleavage, and leads to neuronal pyroptosis (53, 54). Besides, shortage of LDLR can also increases blood LDL levels for more LDL oxidation (25, 55). Therefore, patients with FH who lack LDLR naturally develop neuronal dysfunction and elevation of serum oxidized low-density lipoprotein (ox-LDL) levels.
Thiamine is a water-soluble vitamin; Thiamine is present in meat, beef, pork, legumes, whole grains, and nuts; however, milled rice and grains contain small amounts of thiamine because the processing involved in creating these food products removes thiamine. Additionally, food products such as tea, coffee, raw fish, and shellfish contain thiaminases that destroy thiamine (56). Intake thiamine, in cationic form of T+, is hydrolyzed by intestinal phosphatase into free form, following absorbed by small intestine (57, 58). Thiamine diphosphate (TDP), also named as Thiamine pyrophosphate (TPP), which is derived from T+ by thiamine pyrophosphokinase-1 (TPK1), is the main active form of Thiamine, playing as a co-enzyme for glucose, amino acid, and lipid metabolism (57, 58). Thiamine favors neuronal function by serving as a site-specific antioxidant, and promotes energy production by utilizing carbohydrates (59). At the same time, TPP prevents atherosclerosis by reducing macrophage uptake of ox-LDL via antagonizing macrophage P2Y6 receptor (60). In patients with FH, Thiamine may be required to not only repair secondary neuronal damage but also decrease macrophage uptake of excessive ox-LDL, both of which caused by a lack of LDLR. Thiamine deficiency begins when demand exceeds supply, and poor intake intensifies this damage. The effects of decreased rs112898275 and rs11591147, combined with increased rs115478735, rs142834163, rs499883, and rs9644862 on thiamine deficiency requires further investigation.
Cause-and-effect relationship of familial hypercholesterolemia with vitamin D deficiency
Rs7412 is a SNP in the apolipoprotein E (APOE) gene (61). APOE resides on chromosome 19p13.32 and is 3,612 bp in length, including four exons and three introns (62). The liver is the dominant expresser of ApoE, which is less abundant in the brain, spleen, kidneys, gonads, adrenal glands, and macrophages (61). APOE originally generates the 317-amino acid apoE precursor, including an 18-amino acid signal peptide (62). Cleavage of this 18-amino acid signal peptide and glycosylation results in the conversion of the precursor into a mature 299-amino acid, 34,200-kDa protein (61). Apo appears in chylomicrons, very low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, high-density lipoprotein (HDL), and lipoprotein (a) (Lp(a)), and connects to each corresponding receptor to clear them (61). ApoE has a 22-kDa receptor-binding amino-terminal domain (amino acids 1–191) near residues 136–150 and a 10-kDa lipid-binding carboxyl-terminal domain (amino acids 216–299) (63). The three subtypes—apoE2 (Cys112; Cys158), apoE3 (Cys112; Arg158), and apoE4 (Arg112; Arg158)—result from the APOE*ϵ2, APOE*ϵ3 and APOE*ϵ4 alleles, with rs7412 in exon 4 contributing to codon 158 (64–66). ApoE2 from rs7412 (p.Arg176Cys), deforms the salt bridge in the structure to reduce the positive potential in the receptor-binding region, which reduces the affinity of apoE2 to the LDLR, VLDL receptor (VLDLR), and LDLR-related protein (LRP) (61). In contrast, apoE2 rs7412 (p.Arg176Cys) prevents the hydrolysis of triglycerides in VLDL to form VLDL remnants (IDL) as LDL precursors by inhibiting lipoprotein lipase (LPL) on the capillary endothelium (61). This mutation accounts for nearly 8% of the population and results in an LDLR affinity of < 2% of normal, which represents as negativity for β-coefficient with LDL-C in FH, consistent with the findings of this study (67, 68). Thus, decreased rs7412 (p.Arg176Cys) in patients with FH increases LDL-C levels through the joint effects of apoE2 adhesion and VLDL hydrolysis. Moreover, apoE2 is correlated with low bone mass and bone mineral density (BMD), as reflected by biomarkers of high bone resorption, including a reduced serum ratio of osteoprotegerin/receptor activator of nuclear factor kappa B (NFkB) ligand (OPG/RANKL) in men and high serum C-terminus collagen peptide and urinary deoxypyridinoline levels in postmenopausal women (69, 70).
Vitamin D is a fat-soluble vitamin; the two most important members are exogenous vitamin D2 derived from ergosterol in plants and fungi, and endogenous vitamin D3 derived from 7-dehydrocholesterol (7-DHC) in the skin (71). 7-DHC stored in the human epidermis is irradiated with ultraviolet B (UVB) rays (290–315 nm) in sunlight to produce the vitamin D3 precursor. Both vitamins D2 and pre-D3 are synthesized as blood biomarkers of 25 hydroxyvitamin D (25OHD) in the liver, mainly through equal catalysis by 25 hydroxylase (CYP2R1) (71). Subsequently, 25OHD is converted into the active form of 1,25-dihydroxyvitamin D (1,25 (OH)2D) by 25OHD-1α hydroxylase (CYP27B1) or into the inactive form of 24,25-dihydroxyvitamin D (24,25(OH)2D) by 24-hydroxylase (CYP24) in the kidney (71, 72). Circulating 1,25 (OH)2D is transported by vitamin D-binding protein (DBP) to target organ tissues such as the intestine, kidneys, and bones (71). After binding with the vitamin D receptor (VDR) in these tissues, 1,25 (OH)2D regulates the transcription of target genes, thereby playing a classic role in calcium balance (71). An animal model fed a high-fat and high-cholesterol diet showed that the reduced production of vitamin D could be attributed to the alleviated CYP2R1 expression in the liver induced by increasing circulating cholesterol, glucose, and insulin levels (72). Thus, vitamin D deficiency may occur due to elevated LDL-C levels and bone resorption in patients with FH. The effect of rs7412 (p.Arg176Cys) on vitamin D deficiency require further examination.
Advantages and limitations
This study has several advantages. First, this is the first human-based study to analyze the cause-and-effect relationship between FH and the risk of multiple vitamin deficiencies. Second, FH showed significant harmful effects on thiamine and vitamin D deficiencies. Third, the results of the leave-one-out sensitivity analysis confirmed the causal relationships between FH and the risk of thiamine and vitamin D deficiencies. Moreover, the SNPs contributing to thiamine and vitamin D deficiencies in FH were identified. However, this study has several limitations. First, Although Bonferroni correction was used to control the occurrence of false positives in the analysis, type I error may be increased by the sample overlapping of exposures and outcomes, both of which were from Finngen consortium in this study, leading to bias in classic MR methods, thus these findings should be interpreted with caution. Second, the causal association between rs112898275 and thiamine deficiency has not been reported yet. The effect of rs112898275 in LDLR region on thiamine deficiency was hypothesized. The actual contribution of rs112898275 in LDLR region for the association between FH and thiamine deficiency will be studied in the future. Third, MR analysis can only calculate the specific OR value without determining the hazard ratio (HR). Future longitudinal studies should use log-rank tests and Cox regression models for HR. Fourth, FH showed no deficiencies in other neurotropic B, vitamin B6, and vitamin B12 (59), The mechanism, which differs from that of vitamin thiamine deficiency in FH, requires clarification in the future. Fifth, another mutation in FH, ApoB, did not exert an important effect on thiamine and vitamin D deficiencies. Future studies are needed to elucidate this mechanism, which is distinct from those of LDLR and PCSK9. Sixth, only causal associations between FH with IHD and vitamin D deficiency were detected, and the prominent locus was not identified by leave-one-out sensitivity analysis. More comprehensive measures are needed to elucidate profound mechanisms that differ from that of FH. Finally, the FinnGen GWAS database has not yet released data on other vitamins such as vitamins C, E, and K, and folic acid, which will be analyzed in the future.
Conclusion
FH was positively associated with increased risks of thiamine and vitamin D deficiencies, thus revealing a prospective and unfortunate complication of FH.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
This study utilized publicly available data from participant studies that had already received ethical approval from a committee responsible for human experimentation. No additional ethical approval was necessary for this particular study.
Author contributions
CZ: Data curation, Formal analysis, Writing – original draft, Writing – review & editing. GW: Funding acquisition, Writing – original draft, Writing – review & editing. HZ: Writing – original draft, Writing – review & editing. LL: Conceptualization, Data curation, Formal analysis, Methodology, Resources, Software, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the National Natural Science Foundation of China (No. 82370847; 82000804, GW).
Acknowledgments
We thank FinnGen study, all the organizations and participants for the dataset in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1401260/full#supplementary-material
References
1. Bhagavan NV, Ha C-E. Chapter 36 - vitamin metabolism. In: Bhagavan NV, Ha C-E, editors. Essentials of Medical Biochemistry, 2nd ed. Academic Press, San Diego (2015). p. 683–99.
2. Gironés-Vilaplana A, Villaño D, Marhuenda J, Moreno DA, García-Viguera C. Chapter 6 - vitamins. In: Galanakis CM, editor. Nutraceutical and Functional Food Components. Academic Press (2017). p. 159–201. doi: 10.1016/B978-0-12-805257-0.00006-5
3. Zhai Z, Dong W, Sun Y, Gu Y, Ma J, Wang B, et al. Vitamin-microbiota crosstalk in intestinal inflammation and carcinogenesis. Nutrients. (2022) 14:3383. doi: 10.3390/nu14163383
4. Griffiths JK. 144 - vitamin deficiencies. In: Ryan ET, Hill DR, Solomon T, Aronson NE, Endy TP, editors. Hunter’s Tropical Medicine and Emerging Infectious Diseases, Tenth Edition. Elsevier, London (2020). p. 1042–7.
5. Wirth JP, Petry N, Tanumihardjo SA, Rogers LM, McLean E, Greig A, et al. Vitamin A supplementation programs and country-level evidence of vitamin A deficiency. Nutrients. (2017) 9:190. doi: 10.3390/nu9030190
6. Marrs C, Lonsdale D. Hiding in plain sight: modern thiamine deficiency. Cells. (2021) 10:2595. doi: 10.3390/cells10102595
7. Cui A, Zhang T, Xiao P, Fan Z, Wang H, Zhuang Y. Global and regional prevalence of vitamin D deficiency in population-based studies from 2000 to 2022: A pooled analysis of 7.9 million participants. Front Nutr. (2023) 10:1070808. doi: 10.3389/fnut.2023.1070808
8. Darnton-Hill I. Public health aspects in the prevention and control of vitamin deficiencies. Curr Develop Nutr. (2019) 3:nzz075. doi: 10.1093/cdn/nzz075
9. Vaezi Z, Amini A. Familial Hypercholesterolemia. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Afshin Amini declares no relevant financial relationships with ineligible companies. Treasure Island (FL): StatPearls Publishing (2024).
10. Lu Z, Jiao Y, Li J. Higher genetically predicted triglycerides, LDL, and HDL increase the vitamin D deficiency: A mendelian randomization study. Front Nutr. (2022) 9:862942. doi: 10.3389/fnut.2022.862942
11. Al-Qusous MN, Al Madanat WKJ, Mohamed Hussein R. Association of vitamins D, B6, and B12 deficiencies with hyperlipidemia among Jordanian adults. Rep Biochem Mol Biol. (2023) 12:415–24. doi: 10.61186/rbmb.12.3.415
12. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. (2011) 40:755–64. doi: 10.1093/ije/dyr036
13. Wang X, Gao X, Han Y, Zhang F, Lin Z, Wang H, et al. Causal association between serum thyrotropin and obesity: A bidirectional, mendelian randomization study. J Clin Endocrinol Metab. (2021) 106:e4251–e9. doi: 10.1210/clinem/dgab183
14. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40:304–14. doi: 10.1002/gepi.21965
15. Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. (2018) 27:R195–r208. doi: 10.1093/hmg/ddy163
16. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. (2017) 32:377–89. doi: 10.1007/s10654-017-0255-x
17. Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. (2016) 32:3207–9. doi: 10.1093/bioinformatics/btw373
18. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. (2019) 35:4851–3. doi: 10.1093/bioinformatics/btz469
19. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. (2011) 40:740–52. doi: 10.1093/ije/dyq151
20. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. (2013) 42:1497–501. doi: 10.1093/ije/dyt179
21. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. (2018) 7:e34408. doi: 10.7554/eLife.34408
22. Wang M, He X. Mendelian randomization analysis reveals causal associations of inflammatory bowel disease with Spondylarthritis. Gene. (2024) 902:148170. doi: 10.1016/j.gene.2024.148170
23. Ranstam J. Multiple P-values and bonferroni correction. Osteoarthritis Cartilage. (2016) 24:763–4. doi: 10.1016/j.joca.2016.01.008
24. Singh S, Bittner V. Familial hypercholesterolemia–epidemiology, diagnosis, and screening. Curr Atheroscl Rep. (2015) 17:482. doi: 10.1007/s11883-014-0482-5
25. Abifadel M, Boileau C. Genetic and molecular architecture of familial hypercholesterolemia. J Internal Med. (2023) 293:144–65. doi: 10.1111/joim.13577
26. Brautbar A, Leary E, Rasmussen K, Wilson DP, Steiner RD, Virani S. Genetics of familial hypercholesterolemia. Curr Atheroscl Rep. (2015) 17:491. doi: 10.1007/s11883-015-0491-z
27. Jeon H, Blacklow SC. Structure and physiologic function of the low-density lipoprotein receptor. Annu Rev Biochem. (2005) 74:535–62. doi: 10.1146/annurev.biochem.74.082803.133354
28. Awan Z, Baass A, Genest J. Proprotein convertase subtilisin/kexin type 9 (PCSK9): lessons learned from patients with hypercholesterolemia. Clin Chem. (2014) 60:1380–9. doi: 10.1373/clinchem.2014.225946
29. Tricou EP, Morgan KM, Betts M, Sturm AC. Genetic testing for familial hypercholesterolemia in clinical practice. Curr Atheroscl Rep. (2023) 25:197–208. doi: 10.1007/s11883-023-01094-2
30. Brumpton BM, Fritsche LG, Zheng J, Nielsen JB, Mannila M, Surakka I, et al. Variation in serum PCSK9 (Proprotein convertase subtilisin/kexin type 9), cardiovascular disease risk, and an investigation of potential unanticipated effects of PCSK9 inhibition. Circ Genomic Precis Med. (2019) 12:e002335. doi: 10.1161/circgen.118.002335
31. Spolitu S, Dai W, Zadroga JA, Ozcan L. Proprotein convertase subtilisin/kexin type 9 and lipid metabolism. Curr Opin Lipidol. (2019) 30:186–91. doi: 10.1097/mol.0000000000000601
32. Humphries SE, Neely RD, Whittall RA, Troutt JS, Konrad RJ, Scartezini M, et al. Healthy individuals carrying the PCSK9 p.R46L variant and familial hypercholesterolemia patients carrying PCSK9 p.D374Y exhibit lower plasma concentrations of PCSK9. Clin Chem. (2009) 55:2153–61. doi: 10.1373/clinchem.2009.129759
33. Alghamdi RH, O’Reilly P, Lu C, Gomes J, Lagace TA, Basak A. LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (153-421): design, synthesis and biochemical evaluation. Eur J Med Chem. (2015) 92:890–907. doi: 10.1016/j.ejmech.2015.01.022
34. Benjannet S, Hamelin J, Chrétien M, Seidah NG. Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem. (2012) 287:33745–55. doi: 10.1074/jbc.M112.399725
35. Qiu C, Zeng P, Li X, Zhang Z, Pan B, Peng ZYF, et al. What is the impact of PCSK9 rs505151 and rs11591147 polymorphisms on serum lipids level and cardiovascular risk: a meta-analysis. Lipids Health Dis. (2017) 16:111. doi: 10.1186/s12944-017-0506-6
36. Tarasov K, Ekroos K, Suoniemi M, Kauhanen D, Sylvänne T, Hurme R, et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J Clin Endocrinol Metab. (2014) 99:E45–52. doi: 10.1210/jc.2013-2559
37. Pott J, Burkhardt R, Beutner F, Horn K, Teren A, Kirsten H, et al. Genome-wide meta-analysis identifies novel loci of plaque burden in carotid artery. Atherosclerosis. (2017) 259:32–40. doi: 10.1016/j.atherosclerosis.2017.02.018
38. Yuan W, Zhang W, Zhang W, Ruan ZB, Zhu L, Liu Y, et al. New findings in the roles of Cyclin-dependent Kinase inhibitors 2B Antisense RNA 1 (CDKN2B-AS1) rs1333049 G/C and rs4977574 A/G variants on the risk to coronary heart disease. Bioengineered. (2020) 11:1084–98. doi: 10.1080/21655979.2020.1827892
39. Wan P, Su W, Zhuo Y. The role of long noncoding RNAs in neurodegenerative diseases. Mol Neurobiol. (2017) 54:2012–21. doi: 10.1007/s12035-016-9793-6
40. Aguilo F, Di Cecilia S, Walsh MJ. Long non-coding RNA ANRIL and polycomb in human cancers and cardiovascular disease. Curr Topics Microbiol Immunol. (2016) 394:29–39. doi: 10.1007/82_2015_455
41. Razeghian-Jahromi I, Karimi Akhormeh A, Zibaeenezhad MJ. The role of ANRIL in atherosclerosis. Dis Markers. (2022) 2022:8859677. doi: 10.1155/2022/8859677
42. Jiang S, Wang C, Zhu J, Huang X. Regulation of glial cell-derived neurotrophic factor in sevoflurane-induced neuronal apoptosis by long non-coding RNA CDKN2B-AS1 as a ceRNA to adsorb miR-133. Am J Trans Res. (2021) 13:4760–70.
43. Information NCfB. Available online at: https://www.ncbi.nlm.nih.gov/gene/57572. (accessed 27-Nov-2023).
44. Wang Z, Wang X, Guiyu L, Litao Q, Shasha B, Tang X, et al. Novel compound heterozygous mutations of the DOCK6 gene in a familial case of Adams-Oliver syndrome 2. Gene. (2019) 700:65–9. doi: 10.1016/j.gene.2019.03.023
45. Cerikan B, Schiebel E. Mechanism of cell-intrinsic adaptation to Adams-Oliver Syndrome gene DOCK6 disruption highlights ubiquitin-like modifier ISG15 as a regulator of RHO GTPases. Small GTPases. (2019) 10:210–7. doi: 10.1080/21541248.2017.1297882
46. Shi L. Dock protein family in brain development and neurological disease. Commun Integr Biol. (2013) 6:e26839. doi: 10.4161/cib.26839
47. Miyamoto Y, Yamauchi J, Sanbe A, Tanoue A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp Cell Res. (2007) 313:791–804. doi: 10.1016/j.yexcr.2006.11.017
48. Benson CE, Southgate L. The DOCK protein family in vascular development and disease. Angiogenesis. (2021) 24:417–33. doi: 10.1007/s10456-021-09768-8
49. Considine DPC, Jia G, Shu X, Schildkraut JM, Pharoah PDP, Zheng W, et al. Genetically predicted circulating protein biomarkers and ovarian cancer risk. Gynecol Oncol. (2021) 160:506–13. doi: 10.1016/j.ygyno.2020.11.016
50. Abudureyimu S, Asai N, Enomoto A, Weng L, Kobayashi H, Wang X, et al. Essential role of linx/islr2 in the development of the forebrain anterior commissure. Sci Rep. (2018) 8:7292. doi: 10.1038/s41598-018-24064-0
51. Mandai K, Guo T, St Hillaire C, Meabon JS, Kanning KC, Bothwell M, et al. LIG family receptor tyrosine kinase-associated proteins modulate growth factor signals during neural development. Neuron. (2009) 63:614–27. doi: 10.1016/j.neuron.2009.07.031
52. Sherry DM, Murray AR, Kanan Y, Arbogast KL, Hamilton RA, Fliesler SJ, et al. Lack of protein-tyrosine sulfation disrupts photoreceptor outer segment morphogenesis, retinal function and retinal anatomy. Eur J Neurosci. (2010) 32:1461–72. doi: 10.1111/j.1460-9568.2010.07431.x
53. Sun R, Peng M, Xu P, Huang F, Xie Y, Li J, et al. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ischemia/reperfusion injury. J Neuroinflamm. (2020) 17:330. doi: 10.1186/s12974-020-01988-x
54. de Oliveira J, Moreira EL, dos Santos DB, Piermartiri TC, Dutra RC, Pinton S, et al. Increased susceptibility to amyloid-β-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J Alzheimer’s Dis: JAD. (2014) 41:43–60. doi: 10.3233/jad-132228
55. Yoshida H, Kisugi R. Mechanisms of LDL oxidation. Clin Chim Acta. (2010) 411:1875–82. doi: 10.1016/j.cca.2010.08.038
56. Wiley KD, Gupta M. Vitamin B1 (Thiamine) Deficiency. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Mohit Gupta declares no relevant financial relationships with ineligible companies. Treasure Island (FL): StatPearls Publishing (2024).
57. Martel JL, Kerndt CC, Doshi H, Sina RE, Franklin DS, et al. Vitamin B1 (Thiamine) [Updated 2024 jan 31]. In: StatPearls. StatPearls Publishing, Treasure Island (FL (2024).
58. Mrowicka M, Mrowicki J, Dragan G, Majsterek I. The importance of thiamine (vitamin B1) in humans. Biosci Rep. (2023) 43:BSR20230374. doi: 10.1042/bsr20230374
59. Baltrusch S. The role of neurotropic B vitamins in nerve regeneration. BioMed Res Int. (2021) 2021:9968228. doi: 10.1155/2021/9968228
60. Li Y, Zhou M, Li H, Dai C, Yin L, Liu C, et al. Macrophage P2Y6 receptor deletion attenuates atherosclerosis by limiting foam cell formation through phospholipase Cβ/store-operated calcium entry/calreticulin/scavenger receptor A pathways. Eur Heart J. (2024) 45:268–83. doi: 10.1093/eurheartj/ehad796
61. Khalil YA, Rabès JP, Boileau C, Varret M. APOE gene variants in primary dyslipidemia. Atherosclerosis. (2021) 328:11–22. doi: 10.1016/j.atherosclerosis.2021.05.007
62. Forero DA, López-León S, González-Giraldo Y, Dries DR, Pereira-Morales AJ, Jiménez KM, et al. APOE gene and neuropsychiatric disorders and endophenotypes: A comprehensive review. Am J Med Genet Part B Neuropsychiatr Genet. (2018) 177:126–42. doi: 10.1002/ajmg.b.32516
63. Dong LM, Innerarity TL, Arnold KS, Newhouse YM, Weisgraber KH. The carboxyl terminus in apolipoprotein E2 and the seven amino acid repeat in apolipoprotein E-Leiden: role in receptor-binding activity. J Lipid Res. (1998) 39:1173–80. doi: 10.1016/S0022-2275(20)32541-4
64. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. (2019) 51:165–76. doi: 10.1016/j.pathol.2018.11.002
65. Mannila MN, Mahdessian H, Franco-Cereceda A, Eggertsen G, de Faire U, Syvänen AC, et al. Identification of a functional apolipoprotein E promoter polymorphism regulating plasma apolipoprotein E concentration. Arteriosclerosis Thrombosis Vasc Biol. (2013) 33:1063–9. doi: 10.1161/atvbaha.112.300353
66. Li Z, Shue F, Zhao N, Shinohara M, Bu G. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegeneration. (2020) 15:63. doi: 10.1186/s13024-020-00413-4
67. Vanhoye X, Bardel C, Rimbert A, Moulin P, Rollat-Farnier PA, Muntaner M, et al. A new 165-SNP low-density lipoprotein cholesterol polygenic risk score based on next generation sequencing outperforms previously published scores in routine diagnostics of familial hypercholesterolemia. Trans Res. (2023) 255:119–27. doi: 10.1016/j.trsl.2022.12.002
68. Wilson C, Mau T, Weisgraber KH, Wardell MR, Mahley RW, Agard DA. Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure (London England: 1993). (1994) 2:713–8. doi: 10.1016/s0969-2126(00)00072-1
69. Dieckmann M, Beil FT, Mueller B, Bartelt A, Marshall RP, Koehne T, et al. Human apolipoprotein E isoforms differentially affect bone mass and turnover in vivo. J Bone Mineral Res. (2013) 28:236–45. doi: 10.1002/jbmr.1757
70. Souza LS, Rochette NF, Pedrosa DF, Magnago RPL, Filho TBF, Vieira FLH, et al. Role of APOE gene in bone mineral density and incidence of bone fractures in Brazilian postmenopausal women. J Clin Densitometry. (2018) 21:227–35. doi: 10.1016/j.jocd.2017.03.005
71. Bikle D, Christakos S. New aspects of vitamin D metabolism and action - addressing the skin as source and target. Nat Rev Endocrinol. (2020) 16:234–52. doi: 10.1038/s41574-019-0312-5
72. Zhu T, Zhao J, Zhuo S, Hu Z, Ouyang S, Wunier, et al. High fat diet and high cholesterol diet reduce hepatic vitamin D-25-hydroxylase expression and serum 25-hydroxyvitamin D(3) level through elevating circulating cholesterol, glucose, and insulin levels. Mol Nutr Food Res. (2021) 65:e2100220. doi: 10.1002/mnfr.202100220
Keywords: familial hypercholesterolemia, genetic association, human, thiamine deficiency, vitamin D deficiency
Citation: Zhang C, Wei G, Zhou H and Liu L (2024) Causal relationships of familial hypercholesterolemia with the risk of multiple vitamin deficiencies: a Mendelian randomization study. Front. Endocrinol. 15:1401260. doi: 10.3389/fendo.2024.1401260
Received: 03 April 2024; Accepted: 02 October 2024;
Published: 22 October 2024.
Edited by:
Amani Kallel, Ministry of Public Health, TunisiaReviewed by:
Jesus M. Martin-Campos, Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau, SpainXiaopan Li, Shanghai Medical College of Fudan University, China
Copyright © 2024 Zhang, Wei, Zhou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan Zhou, emhvdWh1YW5AYmJtdS5lZHUuY24=; Lin Liu, MzgwODkyN0BxcS5jb20=
†These authors share first authorship