
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 16 September 2024
Sec. Adrenal Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1399847
 Briana N. Cortez1
Briana N. Cortez1 Mickey J. M. Kuo1,2
Mickey J. M. Kuo1,2 Abhishek Jha1
Abhishek Jha1 Mayank Patel1,3
Mayank Patel1,3 Jorge A. Carrasquillo4Tamara Prodanov1Kailah M. Charles1Sara Talvacchio1Alberta Derkyi1
Jorge A. Carrasquillo4Tamara Prodanov1Kailah M. Charles1Sara Talvacchio1Alberta Derkyi1 Frank I. Lin4
Frank I. Lin4 David Taïeb5
David Taïeb5 Jaydira Del Rivero6
Jaydira Del Rivero6 Karel Pacak1*
Karel Pacak1*Few reports have highlighted the rare presence of somatic ATRX variants in clinically aggressive, metastatic pheochromocytoma/paraganglioma (PCC/PGL); however, none have addressed detailed clinical presentation (including biochemistry and imaging) and management of these patients. Here, we address these clinical features and management based on four PCC patients with somatic ATRX variants from our National Institutes of Health PCC/PGL cohort. A total of 192 patients underwent exome sequencing (germline, somatic, or both), and four males were found to have somatic ATRX variants (with additional somatic VHL and FH oncogenic variants in patients 2 and 4, respectively). Per-lesion and per-patient comparisons were performed among functional imaging scans performed at the NIH. Biochemical phenotype and response to systemic treatment were evaluated. This mini-series supports prior studies showing aggressive/metastatic PCC in patients with somatic ATRX variants, as all developed widespread metastatic disease. All four PCC patients presented with noradrenergic biochemical phenotype, and some with significant elevation in 3-methoxytyramine. 18F-FDOPA PET/CT was found to be the superior functional imaging modality, with 100% lesion detection rate when compared to that of 68Ga-DOTATATE, 18F-FDG, 18F-FDA, and 123I-MIBG scans. While patients did not respond to chemotherapy or tyrosine kinase inhibitors, they responded to targeted radiotherapy using high-specific-activity 131I-MIBG (Azedra®) or 177Lu-DOTATATE (Lutathera®).
Pheochromocytomas and paragangliomas (PCCs/PGLs) are neuroendocrine tumors derived from neural crest cells. They produce, metabolize, and secrete catecholamines and their metabolites, metanephrines and 3-methoxytyramine. While PCCs originate from sympathetic paraganglia of the adrenal medulla, PGLs arise from extra-adrenal autonomic nervous system ganglia and their accompanying nerves (1). In 2017, the World Health Organization Classification of Tumors of Endocrine Organs identified all PCC/PGLs as having metastatic potential based on similar histology, thus eliminating the use of “benign” and “malignant” (1).
In PCCs/PGLs, abnormally elevated plasma 3-methoxytyramine levels, large tumor size (mostly over 5–6 cm), and extra-adrenal location all significantly correlate with metastatic disease and consequently with poor prognosis (2–4). Furthermore, pathogenic variants in genes encoding succinate dehydrogenase complex A (SDHA) and B (SDHB), and endothelial PAS domain protein 1/hypoxia-inducible transcription factor 2α (EPAS1/HIF2A) correlate with increased risk of metastasis (5–9). Other risk factors include TERT overexpression, pS100 loss, presence of brown adipose tissue, tumor necrosis, and vascular invasion (10–14). Small studies have shown that somatic ATRX pathogenic variants also correlate with aggressive/metastatic PCC/PGL (7, 15).
Germline ATRX pathogenic variants are associated with the rare neurodevelopmental disorder alpha-thalassemia X-linked intellectual disability syndrome (16). More recently, the role of ATRX in tumor suppression has been described. Somatic ATRX pathogenic variants correlate with initiation and progression of tumors, including pancreatic neuroendocrine tumors, neuroblastoma, and pediatric osteosarcoma (17–20). ATRX maintains chromatin remodeling and telomere maintenance, essential in tumorigenesis and replicative immortality. Telomeres are maintained by telomerase or a telomerase-independent mechanism involving homologous recombination, named alternative lengthening of telomeres (ALT) (21). ATRX and the death domain associated protein (DAXX), work jointly to maintain telomere chromatin structure and replication by adding histone H3.3 into heterochromatin. Telomere stress develops in the absence of ATRX, leading to ALT (22). A pioneering study by Fishbein et al. was the first to report the presence of ATRX variants in clinically aggressive PCC/PGLs. In their discovery and validation cohorts, 9.5% and 12.6% of PCC/PGL samples had somatic ATRX pathogenic variants, respectively (23). Use of telomere fluorescence in situ hybridization (FISH) showed ALT in the presence of ATRX variants. Another study by Job et al. found somatic ATRX variants in 4.8% of their PCC/PGL cohort, which were most frequently seen in the setting of germline SDHB pathogenic variants. Additionally, out of 25 PCC/PGL patients with ALT in their study, 24% had ATRX variants with poorer prognosis and greater metastatic risk (24).
Thus, in this present study, we focused on PCC/PGL patients enrolled in our National Institutes of Health (NIH) IRB-approved protocol (NCT00004847) with somatic ATRX loss-of-function variants to describe their comprehensive evaluation, including biochemistry, genetics, functional imaging, and clinical outcomes.
A male patient with PCC passed at age 71, 11 years after his initial diagnosis. He initially presented in 1999 with diaphoresis, palpitations, left-sided varicocele, and uncontrolled hypertension (HTN) despite pharmacological intervention. An abdominal ultrasound and subsequent computed tomography (CT) scan found a 14 cm left adrenal tumor. Subsequent biochemical testing revealed elevated plasma norepinephrine (2850 pg/mL [<175 pg/mL]). He underwent a left adrenalectomy and nephrectomy. Histopathology reported a well-encapsulated PCC with no invasion into the kidney or surrounding structures. Three years later, he experienced recurrent night sweats, irritable bowel symptoms, and HTN (up to 170/110 mmHg), with elevated plasma normetanephrine (681 pg/mL [<175 pg/mL]) and chromogranin A (CgA) (244 ng/mL [<76 ng/mL]). CT, 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)/CT, 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, and technetium-99m methylene diphosphonate (MDP) bone scintigraphy detected multiple metastatic lesions, including lungs, lymph nodes, and bones. Due to 123I-MIBG-avid metastatic lesions, he began 131I-MIBG radiotherapy with interval improvement in periaortic and multiple abdominal lymph nodes. The following year, widespread metastatic progression was noted, involving the lungs, lymph nodes, and bones. He was referred to our team at the NIH and underwent 18F-fluorodopa (18F-FDOPA) PET/CT, 18F-fluorodopamine (18F-FDA) PET, and anatomic imaging, which confirmed extent of metastatic involvement. Upon our recommendations, he underwent a right middle lobectomy and mediastinal node dissection. Plasma normetanephrine (313 pg/mL [18–122 pg/mL]) and CgA (635 ng/mL [<225 ng/mL]) remained elevated. Due to progressive and extensive metastases, he began treatment with cyclophosphamide, vincristine, and dacarbazine (CVD). He completed 3 cycles, complicated by neutropenia and peripheral neuropathy. Further progression of the disease was observed in the lungs, lymph nodes, and bone, leading to the discontinuation of CVD therapy. He planned to receive radiation therapy to a growing right hilar mass in his home state; however, he was lost to follow-up and the NIH was notified of the patient’s death 2 years later from extensive metastatic disease.
A 28-year-old male initially presented to the emergency room with a pounding headache, abdominal pain, and nausea in 1987 and was admitted to the hospital with hypertensive crisis. Magnetic resonance imaging (MRI) identified a right adrenal mass, for which he underwent a right adrenalectomy. Histopathology reported a PCC measuring 13 cm. He was lost to follow-up, and twenty-six years later, MRI, CT, 18F-FDG, 18F-FDOPA, and 68Ga-DOTA(0)-Tyr(3)-octreotate (68Ga-DOTATATE) PET/CT, and 123I-MIBG scintigraphy revealed multiple tumors in the abdomen, lungs, bones, and an aortocaval mass. Biochemical evaluation showed elevated plasma normetanephrine (1462 pg/mL [18–122 pg/mL]), dopamine (25 pg/mL [0–24 pg/mL]), and CgA (1349 ng/mL [<93 ng/mL]). He subsequently underwent an exploratory laparotomy, open cholecystectomy, and resection of the right aortocaval mass. The following year, he developed recurrence in the right adrenalectomy bed, and progression of metastatic lesions in the lungs, lymph nodes, and bones. One year later, he underwent a resection of a C2 paraspinal mass (Figure 1) and intensity-modulated radiation therapy (IMRT, 5400 cGy) to the right sacral area with shrinkage of the lesion. Due to continued progression, he began Sandostatin® long-acting release (LAR) for 4 months, followed by sunitinib for 5 months. Sunitinib was discontinued due to uncontrolled HTN, flushing, ageusia, and weight loss. Due to progressive disease and recurrence of his C2 paraspinal mass, he was evaluated at the NIH and enrolled in our NCT03206060 protocol: Lu-177-DOTATATE (Lutathera®) in Therapy of Inoperable Pheochromocytoma/Paraganglioma Clinical Trial. He completed 4 cycles of treatment without progression and shrinkage in several lung, liver, and lymph node metastatic lesions. After 2 years, his CgA levels began to rise, and after 3 years, pulmonary progression was seen on CT imaging. He restarted 177Lu-Dotatate and completed 4 additional cycles. Since then, disease remains stable on imaging, and he remains in close follow-up with the NIH.
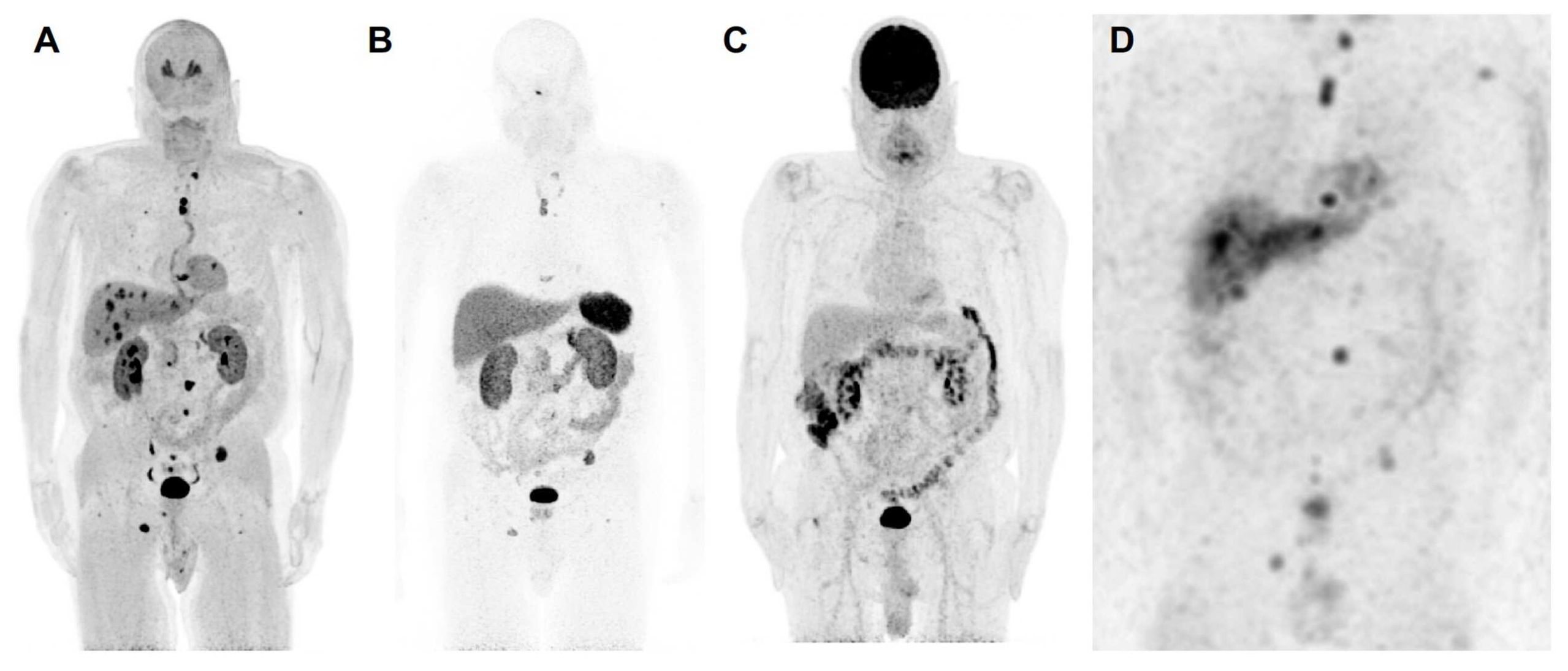
Figure 1. The anterior maximum intensity projection images of 18F-FDOPA positron emission tomography/computed tomography (PET/CT) (A), 68Ga-DOTATATE PET/CT (B), 18F-FDG PET/CT (C), and 123I-MIBG single photon emission computed tomography/computed tomography (SPECT/CT) (D) of a 67-year-old male (patient 3) with a likely oncogenic somatic ATRX variant (c.2018dup, p.Thr674fs) with a history of a previously resected 8.7 cm right pheochromocytoma demonstrates metastatic lesions in lungs, liver, and bones. 18F-FDOPA PET/CT shows superiority in detection of metastatic lesions compared to 68Ga-DOTATATE PET/CT, 18F-FDG PET/CT, and 123I-MIBG SPECT/CT. Liver lesions are detected by 18F-FDOPA PET/CT and 123I-MIBG SPECT/CT and are not seen on 68Ga-DOTATATE PET/CT and 18F-FDG PET/CT. Furthermore, 18F-FDOPA PET/CT detects more bone and lung lesions compared to 68Ga-DOTATATE PET/CT, 18F-FDG PET/CT, and 123I-MIBG SPECT scintigraphy. Based on widespread avidity of metastatic lesions on 123I-MIBG scintigraphy compared to 68Ga-DOTATATE PET/CT, the patient was recommended for high-specific-activity 131I-MIBG (Azedra®) targeted radiotherapy. Additionally, 18F-FDG PET/CT demonstrates diffuse increased uptake in the colon, likely physiologic and not suggestive of malignancy. The 18F-FDA PET/CT, unfortunately, was not performed in this patient. To note, contrast recovery, sensitivity, and spatial resolution of PET/CT imaging is superior to SPECT/CT imaging, and therefore, smaller lesions on PET/CT scans may not be visible on SPECT/CT scans despite adequate uptake.
A male patient with PCC passed at age 68, two years after his initial diagnosis. He initially presented with longstanding HTN, right back pain, pounding headaches, vertigo, fatigue, and multiple subcutaneous tumors. During routine abdominal ultrasound for risk of abdominal aortic aneurysm due to cigarette smoking, an incidental 1.7 x 1.6 x 1.4 cm hypoechoic lesion in the left liver lobe was identified, along with an enlarged paraaortic lymph node. He was evaluated at the NIH with anatomic and functional imaging scans confirming these lesions, along with a large 8.7 x 7 cm right adrenal mass, multiple masses in the liver, lungs, bone, and enlarged mesenteric and retroperitoneal lymph nodes. Histopathology from a liver biopsy specimen showed a well-differentiated neuroendocrine neoplasm, consistent with metastatic PCC. Biochemical evaluation showed elevated plasma normetanephrine (8592 pg/mL [<148 pg/mL]) and metanephrine (1261 pg/mL [<57 pg/mL]). Subsequently, he underwent a laparoscopic right adrenalectomy, with histopathologically confirmed PCC and Ki-67 < 3%. The following year, increasing size and number of metastatic lesions were noted in the lungs, liver, lymph nodes, and bones. Based on the avidity of his metastatic lesions on 123I-MIBG scintigraphy, he planned to start treatment with high-dose iobenguane-131I-MIBG (Azedra®). Approximately one year later, he was admitted to the emergency department experiencing shortness of breath and ultimately passed away due to complications from pneumonia and sepsis. It is believed that his advanced pulmonary metastatic disease played a role in his death.
A male patient with PCC passed at age 64, twenty years after diagnosis. The patient initially presented with progressive HTN, palpitations, flushing, and uncontrolled type 2 diabetes mellitus. He was found to have an elevated vanillylmandelic acid; metanephrines were unavailable. He underwent an abdominal ultrasound and CT scan showing a large left-sided adrenal mass. Due to suspicion of PCC, he underwent a left radical adrenalectomy and nephrectomy without evidence of recurrence until 9 years later. At that time, he developed progressive HTN and uncontrolled hyperglycemia. He was evaluated at the NIH with MRI, CT, 18F-FDG, and 123I-MIBG scintigraphy, which revealed pulmonary, liver, lymph node, peritoneal, and left mesocolon metastasis and recurrence in the left surgical adrenal bed. Biochemical analysis revealed elevated plasma normetanephrine (3307 pg/mL [18–122 pg/mL]), norepinephrine (4949 pg/mL [80–498 pg/mL]), dopamine (59 pg/mL [3–46 pg/mL]), and CgA (1940 ng/mL [≤ 225 ng/mL]). He subsequently underwent resection of left adrenal surgical bed recurrence and metastatic lesions located in the subdiaphragmatic lymph node and mesocolon. Three years later, he presented with worsening HTN and glucose management. Imaging detected pulmonary, liver, lymph node, and bone metastatic lesions. He subsequently underwent exploratory laparotomy with lysis of adhesions and resections of the left periaortic and retropancreatic lymph node masses. Approximately 3 years later, due to progressive disease, he completed 4 cycles of 177Lu-DOTATATE, followed by 14 months of lanreotide injections, and subsequently 3 cycles of CVD, all without stabilization of disease. The patient ultimately succumbed to metastatic disease.
The study protocol was approved by the Intramural Program of the National Institutes of Health, the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board (NCT00004847). Written informed consent was obtained for all clinical, genetic, biochemical, and imaging studies. Our institution complies with all applicable laws, regulations, and policies concerning privacy and confidentiality.
Among NIH study participants without known genetic etiology for a personal history of PCC/PGL, 192 patients underwent exome sequencing (germline, somatic, or both) through the Center for Cancer Research Sequencing Facility. Next-generation sequencing was run on a NovaSeq 6000 S2 using Agilent SureSelect Human All Exon V7 in paired-end sequencing mode. DRAGEN was used for mapping to the reference genome hg38, and variant calling. Germline and somatic variants in published PCC/PGL-associated genes (including SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, RET, NF1, IDH1, KIF1B, HRAS, EPAS1, EGLN1, EGLN2, MAX, TMEM127, FH, BAP1, MDH2, ATRX, DLST, ACO1/IRP1, MAML3, GOT2, and DNMT3A) identified by next-generation sequencing (NGS) were confirmed by targeted Sanger sequencing. After removal of false positive results (those not confirmed by Sanger sequencing) and variants classified as benign or likely benign, four males had somatic putative loss-of-function ATRX variants, including three variants that were classified as likely oncogenic and one as a variant of uncertain significance (VUS), as determined by application of the Standards for the Classification of Pathogenicity of Somatic Variants in Cancer (Oncogenicity) (25) (Table 1). Each of the four patients described in detail here underwent paired exome sequencing (i.e., germline and somatic sequencing with only one tumor used for each patient), and none were found to have germline pathogenic or likely pathogenic variants, nor any suspicious variants of uncertain significance in the genes analyzed. There were no other relevant somatic variants in the genes analyzed that passed confirmation by Sanger sequencing except for those listed explicitly in the Results section. Rare Exome Variant Ensemble Learner (REVEL) scores are derived from the aggregate of multiple computational tools to predict pathogenicity of missense variants as a probability, ranging from 0 to 1 (26).
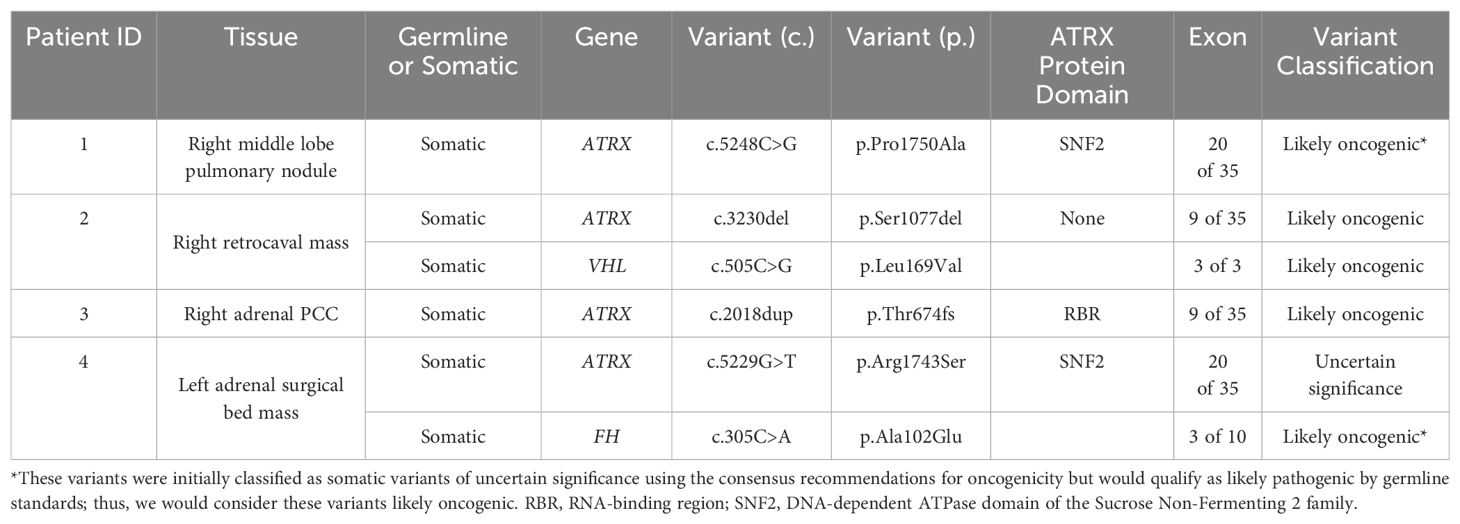
Table 1. Somatic variants identified by exome sequencing of patient tumors.
Biochemical evaluation was based on data sent to and performed at the NIH, which included plasma normetanephrine, norepinephrine, metanephrine, epinephrine, 3-methoxytyramine, dopamine, and chromogranin A. Response to systemic therapies was determined by comparing the last biochemical value pre- and post-treatment.
Biochemical values were compiled throughout patients’ metastatic disease course. Given inconsistencies in laboratory reported reference ranges, fold changes above the upper reference limit (URL) were reported. If the patient’s plasma metanephrine amounted to less than 5% of their total metanephrine, normetanephrine, and 3-methoxytyramine sum, corrected by their respective upper reference limits, they were described as having a noradrenergic biochemical phenotype (27).
Per-lesion and per-patient analyses were performed using the following scans available at the NIH: 18F-FDOPA PET or PET/CT, 18F-FDA PET or PET/CT, 68Ga-DOTATATE PET/CT, 18F-FDG PET/CT, and 123I-MIBG scintigraphy. The duration between functional imaging scans in all patients was less than 3 months except in patient 2, where 18F-FDA was performed about a year prior to other scans. Histologic proof of all metastatic lesions was not feasible. Therefore, a composite of all the imaging studies served as a reference standard for the calculation of detection rates. A lesion was classified as true positive if found to be positive on at least two functional imaging modalities (18F-FDOPA, 18F-FDG, 68Ga-DOTATATE, 18F-FDA, and 123I-MIBG). A positive lesion found on one functional imaging modality and negative on all others was considered a false-positive (Table 2). A patient was considered positive regardless of the number of positive lesions present; counting of metastatic lesions was limited to a maximum of 15 lesions per region (lymph nodes, lungs, mediastinum, liver, and abdomen). Ratios were defined by the number of lesions detected by functional imaging compared to the composite reference standard. McNemar’s test was used to determine significance (p-value <0.05) between 18F-FDOPA and other imaging modalities as it detected all the lesions on imaging comparator.
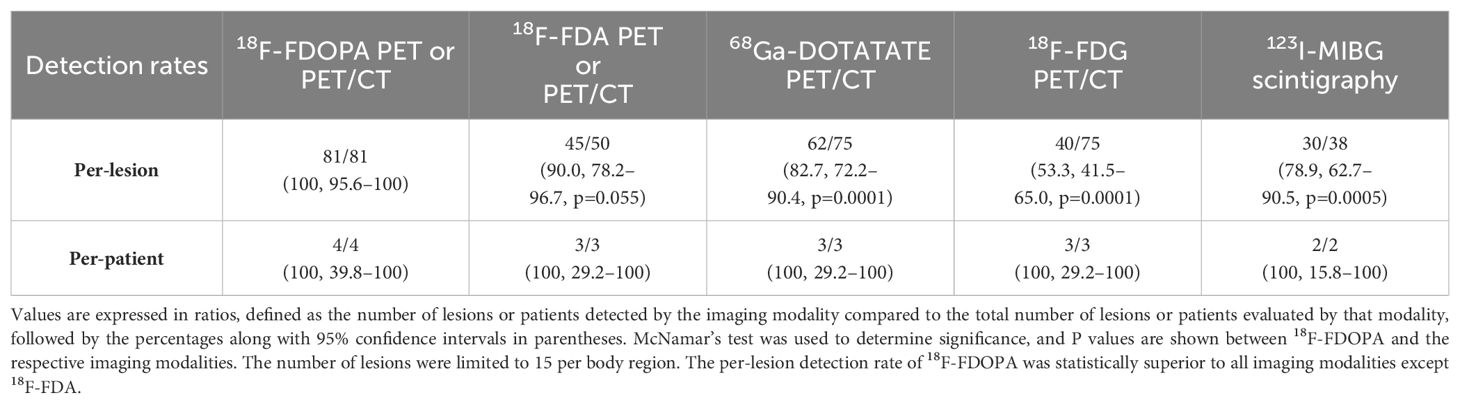
Table 2. Per-lesion and per-patient detection rates of various imaging modalities.
Immunohistochemistry (IHC) for ATRX was performed in tumor specimens from the four patients to assess functional correlation of the protein. A Sigma-Aldrich anti-ATRX antibody (polyclonal, HPA001906, St. Louis, MO) was used at a dilution of 1:200 on unstained slides from formalin fixed paraffin embedded (FFPE) tissue using the Ventana Benchmark XT (Ventana, Tucson, AZ, USA). A Santa Cruz Biotechnology anti-FH antibody (monoclonal, sc-100743, Dallas TX) was used at a dilution of 1:100 on unstained slides from formalin fixed paraffin embedded (FFPE) tissue using the Ventana Benchmark XT (Ventana, Tucson, AZ, USA). Validation of the ATRX staining was performed on the daily clinical laboratory control, the same day these patient tumor samples were stained, by the surgical pathologist on clinical service at the Laboratory of Pathology, National Cancer Institute.
Patient characteristics are summarized in Supplementary Table 1. Four male PCC patients had somatic ATRX (NM_000489.6) variants detected in primary (patient 3) and metastatic lesions (patients 1, 2, and 4). Patients 2 and 4 had additional somatic VHL (NM_000551.4) and FH (NM_000143.4) pathogenic variants, respectively, revealed years after their initial diagnosis when comprehensive genetic analysis was re-initiated at the NIH. Age at initial diagnosis ranged from 28 to 66 years (median=52.5 years). PCC sizes ranged from 8.7 to 14 cm (median=12 cm). Two patients (50%, patients 1 and 4) had left PCC, and the remaining (50%, patients 2 and 3) had right PCC. All patients developed metastatic disease.
Clinical characteristics of primary and metastatic disease are summarized in Supplementary Table 1. Initially, all patients underwent surgical resection of their PCCs. One patient (25%, patient 3) presented with synchronous metastases, in which PCC and distant metastases were found within 6 months, and the remaining three had metachronous metastases (75%, patients 1, 2, and 4). Time between primary resection and development of metastasis in these three patients ranged from 4–26 years (median=9 years). Two patients (50%, patients 2 and 4) had recurrence in their primary adrenalectomy bed ranging between 9–28 years (median=18.5 years). Patient 2 had recurrence of a C2 paraspinal mass 2 years after resection. This patient, the youngest in our cohort, exhibited the most prolonged interval from primary surgical resection to metastatic disease on imaging. However, it is noteworthy that the patient did not adhere to regular follow-up throughout those 26 years.
All patients presented with multiple metastatic lesions to bones, lungs, and lymph nodes. Three patients (75%, patients 2, 3, and 4) had metastases to the liver. Patient 4 (25%) had metastasis to the mesocolon. Those with pulmonary, hepatic, and osseous disease had multiple metastatic lesions in these locations. Two patients (50%, patients 1 and 2) presented with synchronous organ and osseous metastases, while these were metachronous for the remaining two (50%, patients 3 and 4).
Three of four somatic ATRX variants were classified as likely oncogenic. Patients 1 and 4 both had missense variants in exon 20: c.5248C>G (p.Pro1750Ala) and c.5229G>T (p.Arg1743Ser), respectively, both in the ATP-binding SNF2 protein domain (20). REVEL scores for these variants in patients 1 and 4 were 0.94 and 0.709, respectively, providing “strong” and “supporting” levels of computational evidence, respectively, of pathogenicity for these variants (28). Given that the germline classification of the ATRX variant for patient 1 would be considered likely pathogenic, this variant was considered likely oncogenic in the somatic context (25, 29). Of all these somatic ATRX variants, only this one (p.Pro1750Ala) had been reported previously in the Catalogue of Somatic Mutations in Cancer (COSMIC: https://cancer.sanger.ac.uk/cosmic), found in a uterine leiomyosarcoma. Patients 2 and 3 had frameshift truncating variants in exon 9: c.3230del (p.Ser1077del) and c.2018dup (p.Thr674fs), respectively, the latter of which is located in the RBR domain (20). Both of these variants met criteria to be classified as likely oncogenic (Supplementary Table 2).
In addition to each hemizygous somatic ATRX oncogenic variant, two patients (2 and 4) were found to have additional somatic variants: VHL c.505C>G (p.Leu169Val) and FH c.305C>A (p.Ala102Glu), respectively, both of which were classified as likely oncogenic missense variants (Table 1; Supplementary Table 2). None of these variants (in ATRX, VHL, or FH) were present in ClinVar.
ATRX staining for patient 4 tumor sample showed loss of nuclear ATRX in the tumor samples with faintly positive stromal cells serving as an internal control (Figure 2). ATRX staining in tumor samples from patients 2 and 3 failed to display an adequate internal control staining in the stromal cell nuclei, and the tumor sample from patient 1 showed subsets of tumor cell nuclei with ATRX loss and ATRX retention. Samples from patients 1, 2, and 3 were determined to be inadequate for interpretation due to these possible technical concerns in the staining pattern. Loss of FH protein in the tumor cells was confirmed by IHC (Supplementary Figure 1) in patient 4, with validation performed on the same day clinical laboratory control sample.
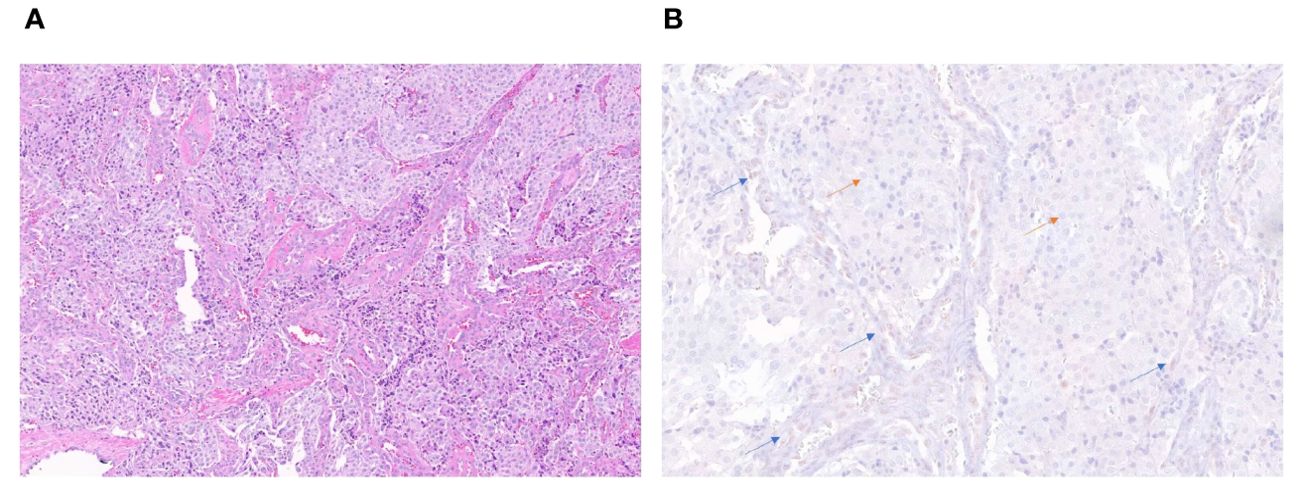
Figure 2. H&E stain (20x) of the periaortic lymph node metastatic lesion in patient 4 (A). Immunohistochemistry stain (40x) showing retained ATRX protein (brown staining) in internal control of endothelial and stromal cells of vessels (blue arrows) and loss of protein in tumor cells (orange arrows) (B).
All patients underwent biochemical analysis, as described above (Supplementary Table 3). However, assessment was not available at regular intervals since patients were largely managed and diagnosed outside the NIH.
Out of four patients, two (patients 1 and 3) had biochemical evaluation at initial diagnosis. Patient 1 had plasma normetanephrine elevation 15.3-fold above the URL. Patient 3 (with synchronous metastases) had elevated plasma normetanephrine, norepinephrine, metanephrine, epinephrine, and dopamine by 64.9-, 1.8-, 17.9-, 5.9-, and 14.6-fold above the URL, respectively.
Biochemistries were followed after metastatic disease was detected. All patients had plasma elevations in normetanephrine (median=1.3-fold above the URL, 0.02–64.9-fold) and norepinephrine (median=1.1-fold above the URL, 0.02–41.9-fold). Additionally, all patients had plasma 3-methoxytyramine elevations (median=4.5-fold above the URL, 1.1–71.8-fold). Three patients (75%, patients 2, 3, and 4) had plasma dopamine elevation (median=0.52-fold above the URL, 0.04–25.5-fold). Two patients (50%, patients 2 and 3) had metanephrine (median = 0.3-fold above the URL, 0.05–18.0-fold) and epinephrine elevations (median = 3.0-fold above the URL, 0.1–5.9-fold). Additionally, all had significant CgA elevation (median= 29.1-fold above the URL, 1.0–204.9-fold).
In summary, at the time of metastatic involvement, all had a noradrenergic biochemical phenotype with significant plasma 3-methoxytyramine elevation, and CgA elevation.
Multiple modalities of anatomical and functional imaging were used to diagnose primary adrenal tumors in our patient cohort. The functional imaging studies are summarized in Supplementary Table 4. Detection was performed by abdominal ultrasound followed by CT in three patients (75%, patient 1, 3, and 4), and MRI in one patient (25%, patient 2).
Functional imaging studies evaluating metastatic disease are summarized in Supplementary Table 4. Of four patients, all (100%) underwent 18F-FDOPA PET (n=1, 25%, patient 1) or PET/CT (n=3, 75%; patients 2–4), and three (75%, patients 2–4) underwent 68Ga-DOTATATE and 18F-FDG PET/CT. Three patients (75%) had 18F-FDA PET (n=1, 33%, patient 1) or PET/CT (n=2, 67%, patients 2 and 4). Finally, two patients had additional 123I-MIBG imaging (50%, patients 2 and 3). 68Ga-DOTATATE PET/CT did not detect liver metastases in patient 3, which were seen on CT, 123I-MIBG scintigraphy, 18F-FDG, and 18F- FDOPA PET/CT.
The total number of lesions identified among all patients was 81. The per-lesion detection rate of 18F-FDOPA PET or PET/CT was 100% (81/81, 95% CI 95.6–100%), which was superior compared to that of 68Ga-DOTATATE PET/CT (82.7% [62/75], 95% CI 72.2–90.4%, p=0.0001), 18F-FDG PET/CT (53.3% [40/75], 95% CI 41.5–65.0%, p=0.0001), and 123I-MIBG scintigraphy (78.9% [30/38], 95% CI 62.7–90.5%, p=0.0005). The difference in per-lesion detection rates between 18F-FDOPA PET/CT and other functional imaging modalities reached statistical significance except for 18F-FDA PET or PET/CT (90% [45/50], 95% CI 78.2–96.7%, p=0.055), which tended towards significance (p=0.055). 18F-FDG PET/CT performed the worst in comparison to all other functional imaging modalities. The per-patient detection rate of all performed functional imaging modalities was 100% (Table 2).
Three patients (patients 1, 3, and 4) have succumbed to metastatic disease. Overall survival from the time of diagnosis ranged from 2 to 26 years with a median of 11 years. One patient (25%, patient 2) is alive 36 years after initial diagnosis with stable disease 2 years after completion of 8 cycles of Lutathera®.
Patients 1, 2, and 4 received various therapies, including cytotoxic chemotherapy, targeted radiotherapies, external beam radiation, and somatostatin analogs. Patient 1 received 131I-MIBG therapy, with mixed response, specifically, shrinkage in one lymph node and progression in pulmonary disease, ultimately requiring surgical pulmonary debulking. Upon continued progression and widespread metastatic disease, he received 3 cycles of CVD with progression on therapy leading to death from metastatic disease.
Patient 2 received IMRT (5400 cGy) to the right sacral area with shrinkage and was also treated with Sandostatin® LAR and sunitinib for widespread organ metastases, during which progression was noted. He subsequently received four cycles of Lutathera®, leading to shrinkage of liver, lung, and lymph node lesions, along with stabilization of other areas for 3 years. Once progression was noted, he restarted Lutathera® for an additional 4 cycles and continues to have stable disease 2 years after treatment.
Lastly, patient 4 received 4 cycles of Lutathera®, followed by lanreotide and CVD, with progression, leading to death from metastatic disease.
Overall, treatment with cytotoxic chemotherapy and a tyrosine kinase inhibitor (TKI) were not effective in stabilizing or shrinking tumors. Systemic radiation with 131I-MIBG and IMRT targeted to sacral metastasis showed response in a periaortic and multiple abdominal lymph nodes (patient 1, 25%) and a sacral osseous metastasis (patient 2, 25%), respectively. Lanreotide treatment in two patients (patients 2 and 4) showed the most durable response with stabilization and shrinkage of tumors in patient 2.
Some reports have described somatic ATRX variants as clinically important risk factors for aggressive/metastatic PCCs/PGLs (23, 24), however, few have described their detailed associations with biochemical phenotypes, functional imaging, or treatment outcomes and survival of patients with these tumors. We identified four patients with somatic ATRX variants and metastatic PCC. Patients 2 and 4 had additional somatic pathogenic variants in VHL and FH, respectively, which are known PCC/PGL susceptibility genes. All four patients were males, who could exhibit greater susceptibility to effects of ATRX loss-of-function variants due to hemizygosity, but based on this small series, we cannot definitively infer biological outcomes based on sex differences. Further investigation should be performed in a greater subset of patients, including females with PCC/PGL found to have oncogenic ATRX variants.
This mini-case series corroborates previous evidence that presence of somatic ATRX variants is associated with aggressive/metastatic PCC. In our cohort, three out of four patients (patient 1, 3, and 4) have succumbed to metastatic disease. Patient 4 harbored a somatic FH pathogenic variant, which is often associated with metastatic disease (1). However, prior seminal studies have shown aggressive/metastatic behavior in the presence of ATRX variants, even in the presence of other well-known aggressive pathogenic variants. For example, Fishbein et al. showed that approximately 67% of their PCC/PGL samples with somatic ATRX pathogenic variants, also harbored SDHB germline pathogenic variants. Telomere FISH revealed that all had ALT activation, and clinically aggressive behavior (23). The aggressive/metastatic nature of PCC with somatic ATRX pathogenic variants suggests a need for heightened frequency of clinical evaluation and development of specific recommendations for patients harboring these variants. Additionally, use of telomere FISH to assess for ALT as a result of oncogenic ATRX variants may be helpful in clinical assessment in these patients when observing tumor specimens by histopathology as was done in Fishbein et al. (23), but additional studies are needed to validate its clinical utility.
To date, biochemical phenotypes associated with ATRX variants have not been well reported. All four patients presented with a noradrenergic biochemical phenotype with significant elevations in 3-methoxytyramine, indicating substantial dopamine production. Interestingly, CgA was also substantially elevated in all patients, especially with metastatic disease. Patients 2 and 4 harbored variants in PCC/PGL cluster 1 genes, VHL and FH, respectively, which have been known to express the noradrenergic biochemical phenotype (30). Thus, while we cannot definitively assert that ATRX variants are driving the noradrenergic phenotype, tumors with somatic ATRX variants in this study were associated with this phenotype and with elevation in 3-methoxytyramine. Future analysis in a larger cohort of patients should be performed to further uncover the impact of ATRX variants on biochemical phenotype.
Our small cohort of PCCs with ATRX variants also provides insight on functional imaging modalities in these patients. 18F-FDOPA PET/CT was shown to be superior to all other functional imaging modalities, with 100% lesion detection in each patient as compared to the reference standard. 18F-FDG PET/CT performed the worst in these patients, with a per-lesion detection of only 53.3%. Thus, 18F-FDOPA PET/CT seems to be the imaging modality of choice for these patients, similar to patients having pathogenic variants in cluster 1b or 2 genes or those with sporadic PCCs (31–35). It is important to note that while patient 2, with a pathogenic VHL variant, already falls into cluster 1b, associated with preferred 18F-FDOPA PET/CT imaging, patient 4, with an FH variant (cluster 1a) may have been appropriately imaged with 68Ga-DOTATATE PET/CT or 18F-FDOPA PET/CT given prior reports (36, 37). These data suggest that the presence of ATRX variants favors 18F-FDOPA PET/CT as the imaging modality of choice.
While PCCs/PGLs are highly heritable tumors that show considerable heterogeneity (38), none of these patients were found to have germline variants in PCC/PGL susceptibility genes. A substantial proportion of these tumors are also due to somatic variants in PCC/PGL susceptibility genes, including many of the same genes implicated in heritable disease (NF1, SDHx, VHL, and RET), but also somatic (or somatic mosaic) variants in genes, including HRAS, FGFR1, EPAS1, H3F3A, and ATRX (39, 40). As our understanding of the molecular biology of these tumors improves, knowledge of the underlying genetic pathogenesis may inform molecular targeted therapies, as demonstrated in a patient with an EPAS1 gain-of-function mutation (41). Therefore, genetic testing for both germline as well as somatic etiologies is crucial for these tumors, as suggested by other experts previously (42). Additionally, multi-omic comparisons between primary and metastatic sites in patients may be useful in forming a deeper understanding of the fundamental biology of tumor evolution in future cohorts.
The five-year survival for those with metastatic PCC/PGL is 37%, and male biological sex and presence of synchronous metastases are associated with higher mortality (43), even though median overall survival is approximately 116.0 months (44). In our cohort, two patients presented with synchronous metastases. Most presented with large PCC (median=12 cm), and therefore, such large tumors could further contribute to metastatic disease as described previously (2, 43). PCC is a tumor type known to have better clinical outcomes (45), however our PCC patients developed metastatic disease and ultimately died, thus suggesting that the presence of ATRX variants and perhaps a large tumor size, rather than the primary location, contributed to adverse outcomes. While two patients had additional pathogenic VHL or FH variants, these have been described to have a lesser impact on metastatic disease (1, 46). However, since all patients with ATRX variants presented with aggressive/metastatic disease despite initially presenting with PCC (low metastatic risk) and VHL or FH variants (low-moderate metastatic risk), we speculate, as others, that ATRX variants contribute to metastatic risk (23). Future prospective studies with a larger cohort of patients could deepen our understanding of the nature of ATRX variants in PCC/PGL when compared to appropriately matched controls without these variants.
Therapeutic outcomes cannot be definitively established in this small study; however, it seems that these patients do not respond to CVD or TKIs but could respond (at least partially) to somatostatin analogs and systemic radiotherapy using either 131I-MIBG (Azedra®) or 177Lu-DOTATATE (Lutathera®), based on relative avidities of metastatic lesions for 123I-MIBG scintigraphy or 68Ga-DOTATATE, respectively (47–49). In summary, here we present four male PCC patients with somatic ATRX variants presenting with metastatic/aggressive disease. While this case series cannot come to definite conclusions due to its limited size, we found that these patients presented mainly with a noradrenergic biochemical phenotype, elevated 3-methoxytyramine levels, and large tumors. 18F-FDOPA PET/CT performed best on per-lesion detection, and systemic radiotherapy using 131I-MIBG (Azedra®) or 177Lu-DOTATATE (Lutathera®) may be considered as a form of therapy in these patients with aggressive/metastatic disease, as they led to limited therapeutic response and stabilization of disease compared to other treatment options.
Original datasets from the patients are available in the publicly accessible repository dbGaP with accession number phs002405.v2.p1.
The studies involving humans were approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
BC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Writing – original draft, Writing – review & editing. MK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, Software, Validation. AJ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing, Validation. MP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. JC: Formal analysis, Investigation, Methodology, Writing – review & editing, Validation. TP: Data curation, Writing – review & editing. KC: Data curation, Writing – review & editing. ST: Data curation, Writing – review & editing. AD: Data curation, Writing – review & editing. FL: Writing – review & editing, Formal analysis, Investigation, Methodology, Validation. DT: Writing – review & editing, Validation. JR: Writing – review & editing, Validation. KP: Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the National Institutes of Health (grant number Z1AHD008735). This work was supported by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Authors’ (KP, MK, AJ, MP, JC, TP, KC, ST, AD, FL, DT, JR) contribution to the Work was done as part of the Authors’ official duties as NIH employees and is a Work of the United States Government. Therefore, copyright may not be established in the United States, 17 U.S.C. § 105. If the Publisher intends to disseminate the Work outside of the U.S., the Publisher may secure copyright to the extent authorized under the domestic laws of the relevant country, subject to a paid-up, nonexclusive, irrevocable worldwide license to the United States in such copyrighted work to reproduce, prepare derivative works, distribute copies to the public and perform publicly and display publicly the work, and to permit others to do so.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1399847/full#supplementary-material
1. Pacak K. New biology of pheochromocytoma and paraganglioma. Endocr Pract. (2022) 28:1253–69. doi: 10.1016/j.eprac.2022.09.003
2. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. (2012) 48:1739–49. doi: 10.1016/j.ejca.2011.07.016
3. Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for Malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. (2011) 96:717–25. doi: 10.1210/jc.2010-1946
4. Zelinka T, Musil Z, Dušková J, Burton D, Merino MJ, Milosevic D, et al. Metastatic pheochromocytoma: does the size and age matter? Eur J Clin Invest. (2011) 41:1121–8. doi: 10.1111/j.1365-2362.2011.02518.x
5. Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. (2005) 23:8812–8. doi: 10.1200/JCO.2005.03.1484
6. Bechmann N, Moskopp ML, Ullrich M, Calsina B, Wallace PW, Richter S, et al. HIF2α supports pro-metastatic behavior in pheochromocytomas/paragangliomas. Endocr Relat Cancer. (2020) 27:625–40. doi: 10.1530/ERC-20-0205
7. Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma: a systematic review and individual patient meta-analysis. Endocr Relat Cancer. (2019) 26:539–50. doi: 10.1530/ERC-19-0024
8. Jha A, de Luna K, Balili CA, Millo C, Paraiso CA, Ling A, et al. Clinical, diagnostic, and treatment characteristics of SDHA-related metastatic pheochromocytoma and paraganglioma. Front Oncol. (2019) 9:53. doi: 10.3389/fonc.2019.00053
9. Patel M, Jha A, Ling A, Chen CC, Millo C, Kuo MJM, et al. Performances of functional and anatomic imaging modalities in succinate dehydrogenase A-related metastatic pheochromocytoma and paraganglioma. Cancers (Basel). (2022) 14(16):3886. doi: 10.3390/cancers14163886
10. Dwight T, Flynn A, Amarasinghe K, Benn DE, Lupat R, Li J, et al. TERT structural rearrangements in metastatic pheochromocytomas. Endocr Relat Cancer. (2018) 25:1–9. doi: 10.1530/ERC-17-0306
11. Mei L, Khurana A, Al-Juhaishi T, Faber A, Celi F, Smith S, et al. Prognostic factors of Malignant pheochromocytoma and paraganglioma: A combined SEER and TCGA databases review. Horm Metab Res. (2019) 51:451–7. doi: 10.1055/a-0851-3275
12. Abdul Sater Z, Jha A, Hamimi A, Mandl A, Hartley IR, Gubbi S, et al. Pheochromocytoma and paraganglioma patients with poor survival often show brown adipose tissue activation. J Clin Endocrinol Metab. (2020) 105:1176–85. doi: 10.1210/clinem/dgz314
13. Pierre C, Agopiantz M, Brunaud L, Battaglia-Hsu SF, Max A, Pouget C, et al. COPPS, a composite score integrating pathological features, PS100 and SDHB losses, predicts the risk of metastasis and progression-free survival in pheochromocytomas/paragangliomas. Virchows Arch. (2019) 474:721–34. doi: 10.1007/s00428-019-02553-5
14. Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, et al. Prognosis of Malignant pheochromocytoma and paraganglioma (MAPP-prono study): A european network for the study of adrenal tumors retrospective study. J Clin Endocrinol Metab. (2019) 104:2367–74. doi: 10.1210/jc.2018-01968
15. Comino-Méndez I, Tejera ÁM, Currás-Freixes M, Remacha L, Gonzalvo P, Tonda R, et al. ATRX driver mutation in a composite Malignant pheochromocytoma. Cancer Genet. (2016) 209:272–7. doi: 10.1016/j.cancergen.2016.04.058
16. León NY, Harley VR. ATR-X syndrome: genetics, clinical spectrum, and management. Hum Genet. (2021) 140:1625–34. doi: 10.1007/s00439-021-02361-5
17. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. (2011) 331:1199–203. doi: 10.1126/science.1200609
18. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. (2012) 483:589–93. doi: 10.1038/nature10910
19. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. (2014) 7:104–12. doi: 10.1016/j.celrep.2014.03.003
20. Valenzuela M, Amato R, Sgura A, Antoccia A, Berardinelli F. The multiple facets of ATRX protein. Cancers (Basel). (2021) 13(19):2211. doi: 10.3390/cancers13092211
21. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. (2010) 11:319–30. doi: 10.1038/nrg2763
22. Li F, Deng Z, Zhang L, Wu C, Jin Y, Hwang I, et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. (2019) 38:e96659. doi: 10.15252/embj.201796659
23. Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. (2015) 6:6140. doi: 10.1038/ncomms7140
24. Job S, Draskovic I, Burnichon N, Buffet A, Cros J, Lépine C, et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. (2019) 25:760–70. doi: 10.1158/1078-0432.CCR-18-0139
25. Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet Med. (2022) 24:986–98. doi: 10.1016/j.gim.2022.01.001
26. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
27. Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. (2005) 51:735–44. doi: 10.1373/clinchem.2004.045484
28. Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/j.ajhg.2022.10.013
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
30. Eisenhofer G, Pamporaki C, Lenders JWM. Biochemical assessment of pheochromocytoma and paraganglioma. Endocr Rev. (2023) 44:862–909. doi: 10.1210/endrev/bnad011
31. Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. (2019) 46:2112–37. doi: 10.1007/s00259-019-04398-1
32. Carrasquillo JA, Chen CC, Jha A, Ling A, Lin FI, Pryma DA, et al. Imaging of pheochromocytoma and paraganglioma. J Nucl Med. (2021) 62:1033–42. doi: 10.2967/jnumed.120.259689
33. Taïeb D, Jha A, Treglia G, Pacak K. Molecular imaging and radionuclide therapy of pheochromocytoma and paraganglioma in the era of genomic characterization of disease subgroups. Endocr Relat Cancer. (2019) 26:R627–r52. doi: 10.1530/ERC-19-0165
34. Jha A, Patel M, Carrasquillo JA, Ling A, Millo C, Saboury B, et al. Sporadic primary pheochromocytoma: A prospective intraindividual comparison of six imaging tests (CT, MRI, and PET/CT using (68)Ga-DOTATATE, FDG, (18)F-FDOPA, and (18)F-FDA). AJR Am J Roentgenol. (2022) 218:342–50. doi: 10.2214/AJR.21.26071
35. Taïeb D, Jha A, Guerin C, Pang Y, Adams KT, Chen CC. 18F-FDOPA PET/CT Imaging of MAX-Related Pheochromocytoma. J Clin Endocrinol Metab. (2018) 103(4):1574-82. doi: 10.1210/jc.2017-02324
36. Imperiale A, Pepponi M, Poterszman N, Jha A, Collen C, Brumaru D, et al. Molecular imaging phenotyping of germline fumarate hydratase (FH) pathogenic variant-positive metastatic pheochromocytoma. Eur J Nucl Med Mol Imaging. (2023) 50:3155–6. doi: 10.1007/s00259-023-06261-w
37. Nambuba J, Därr R, Janssen I, Bullova P, Adams KT, Millo C, et al. Functional imaging experience in a germline fumarate hydratase mutation–positive patient with pheochromocytoma and paraganglioma. AACE Clin Case Rep. (2016) 2:e176–e81. doi: 10.4158/EP15759.CR
38. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. (2014) 14:108–19. doi: 10.1038/nrc3648
39. Burnichon N, Buffet A, Parfait B, Letouzé E, Laurendeau I, Loriot C, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. (2012) 21:5397–405. doi: 10.1093/hmg/dds374
40. Crona J, Delgado Verdugo A, Maharjan R, Stålberg P, Granberg D, Hellman P, et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab. (2013) 98:E1266–71. doi: 10.1210/jc.2012-4257
41. Kamihara J, Hamilton KV, Pollard JA, Clinton CM, Madden JA, Lin J, et al. Belzutifan, a potent HIF2α Inhibitor, in the Pacak-Zhuang syndrome. N Engl J Med. (2021) 385:2059–65. doi: 10.1056/NEJMoa2110051
42. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. (2017) 13:233–47. doi: 10.1038/nrendo.2016.185
43. Hamidi O, Young WF Jr., Gruber L, Smestad J, Yan Q, Ponce OJ, et al. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin Endocrinol (Oxf). (2017) 87:440–50. doi: 10.1111/cen.13434
44. Li H, Abbas KS, Abdelazeem B, Xu Y, Lin Y, Wu H, et al. A predictive nomogram for early death in pheochromocytoma and paraganglioma. Front Oncol. (2022) 12:770958. doi: 10.3389/fonc.2022.770958
45. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. (2021) 43:199–239. doi: 10.1210/endrev/bnab019
46. Cui Y, Zhou Y, Gao Y, Ma X, Wang Y, Zhang X, et al. Novel alternative tools for metastatic pheochromocytomas/paragangliomas prediction. J Endocrinol Invest. (2024) 47:1191–203. doi: 10.1007/s40618-023-02239-5
47. Jha A, Taïeb D, Carrasquillo JA, Pryma DA, Patel M, Millo C, et al. High-specific-activity-(131)I-MIBG versus (177)Lu-DOTATATE targeted radionuclide therapy for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. (2021) 27:2989–95. doi: 10.1158/1078-0432.CCR-20-3703
48. Carrasquillo JA, Chen CC, Jha A, Pacak K, Pryma DA, Lin FI. Systemic radiopharmaceutical therapy of pheochromocytoma and paraganglioma. J Nucl Med. (2021) 62:1192–9. doi: 10.2967/jnumed.120.259697
Keywords: case report, pheochromocytoma, ATRX, prognosis, imaging
Citation: Cortez BN, Kuo MJM, Jha A, Patel M, Carrasquillo JA, Prodanov T, Charles KM, Talvacchio S, Derkyi A, Lin FI, Taïeb D, Del Rivero J and Pacak K (2024) Case Series: ATRX Variants in Four Patients with Metastatic Pheochromocytoma. Front. Endocrinol. 15:1399847. doi: 10.3389/fendo.2024.1399847
Received: 12 March 2024; Accepted: 10 June 2024;
Published: 16 September 2024.
Edited by:
Carl D. Malchoff, University of Connecticut Health Center, United StatesReviewed by:
Daniela Regazzo, University Hospital of Padua, ItalyCopyright © 2024 Cortez, Kuo, Jha, Patel, Carrasquillo, Prodanov, Charles, Talvacchio, Derkyi, Lin, Taïeb, Del Rivero and Pacak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karel Pacak, a2FyZWxAbWFpbC5uaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.