 Zihan Li
Zihan Li Ya-yi Jiang
Ya-yi Jiang Caiyi Long1,2
Caiyi Long1,2 Xi Peng
Xi Peng Yueheng Pu
Yueheng Pu Rensong Yue
Rensong Yue
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 10 May 2024
Sec. Neuroendocrine Science
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1393253
This article is part of the Research TopicStrategies to Overcome Metabolic Syndrome and Related DiseasesView all 16 articles
Metabolic syndrome (MetS) and cognitive dysfunction pose significant challenges to global health and the economy. Systemic inflammation, endocrine disruption, and autoregulatory impairment drive neurodegeneration and microcirculatory damage in MetS. Due to their unique anatomy and function, astrocytes sense and integrate multiple metabolic signals, including peripheral endocrine hormones and nutrients. Astrocytes and synapses engage in a complex dialogue of energetic and immunological interactions. Astrocytes act as a bridge between MetS and cognitive dysfunction, undergoing diverse activation in response to metabolic dysfunction. This article summarizes the alterations in astrocyte phenotypic characteristics across multiple pathological factors in MetS. It also discusses the clinical value of astrocytes as a critical pathologic diagnostic marker and potential therapeutic target for MetS-associated cognitive dysfunction.
MetS is a syndrome of multiple metabolic disorders that seriously jeopardize cardiovascular health. MetS leads to myocardial metabolic, hemodynamic, and microcirculatory dysfunction by activating the sympathetic nervous system, the renin-angiotensin system, and pro-inflammatory adipokines (1, 2). Combining several recognized diagnostic criteria, the main diagnostic features of MetS include abdominal obesity, dyslipidemia, hyperglycemia, insulin resistance (IR), and elevated blood pressure (3–10). The multivessel risk factors of MetS jeopardize the cerebral vasculature and reduce cerebral perfusion while accelerating neuronal cell senescence and degeneration (11, 12). Evidence from diverse studies supports the association of MetS with vascular dementia and Alzheimer’s disease dementia (13). Various components of MetS have been found in cross-sectional and longitudinal studies to cause decreases in learning memory, attention, visuospatial and executive functions, and processing speed (14, 15). A 15-year follow-up analysis of 176,000 non-demented participants found that MetS led to a 12% increased risk of developing all-cause dementia (16).
Astrocytes are the primary glial cells and are essential for maintaining brain homeostasis. Astrocytes endfeet envelop neurons and the cerebral vasculature, linking cerebrovascular nutrient uptake transport to high oxygen- and sugar-dependent synaptic activity. Astrocytes shape synapses and their surrounding microenvironment, regulate cerebrovascular structure and perfusion, and influence neuroinflammation. Astrocytes become “reactive astrocytes” when stimulated by metabolic changes (e.g., glucose and lipid metabolism) (17). Reactive astrocytes are traditionally thought to have a double-edged role in cytotoxicity and neuroprotection (18). Cytotoxicity of astrocytes is defined as driving pathologic progression through the release of toxic factors such as inflammatory cytokines. Neuroprotective effects are usually heavy in ischemic injury, and reactive astrocytes promote vascular repair and remodeling. Escartin et al. (19) have pointed out the shortcomings of this binary division in recent years based on transcriptomic studies, suggesting that heterogeneity of reactive astrocytes should be emphasized.
Chronic low-level inflammatory states, peripherally and centrally, and systemic IR, are critical in the MetS, leading to cognitive dysfunction (20). A more detailed understanding of the underlying molecular mechanisms of MetS-related cognitive dysfunction will facilitate the development of new approaches to prevention and treatment. MetS-related nutritional and hormonal changes can significantly alter blood metabolic signaling, thereby regulating astrocytes’ responsive activation and specific genomic programs and functional transitions (17). Astrocyte activation is often considered an adaptive mechanism for metabolic adaptation and relief of neuronal stress, but it can also have multifaceted effects on cognitive function and metabolic homeostasis (21). However, persistent astrocyte proliferation and neurotoxic phenotype are essential causes of neuroinflammatory spread and chronicity (18, 22). Identifying the activation state of these astrocytes and the associated molecular mechanisms may provide new targets for treating MetS-related cognitive dysfunction (23).
In this review, we overview the multifaceted role of astrocytes in MetS-related cognitive impairment. Recent discoveries on astrocyte subpopulations and their regulation of cognitive and metabolic functions are highlighted.
Neuroimaging changes associated with MetS have been observed in clinical studies, including reduced gray matter volume, cerebral white matter microstructural changes, cerebral atrophy, and lacunar cerebral infarcts (24, 25). Reduced resting-state functional connectivity between MetS-related vascular risk factors and multiple higher-order cognitive function-related neural networks (26). MetS leads to cerebrovascular injury and neurodegenerative lesions through complex mechanisms that ultimately produce altered cognitive function (27) (Figure 1).
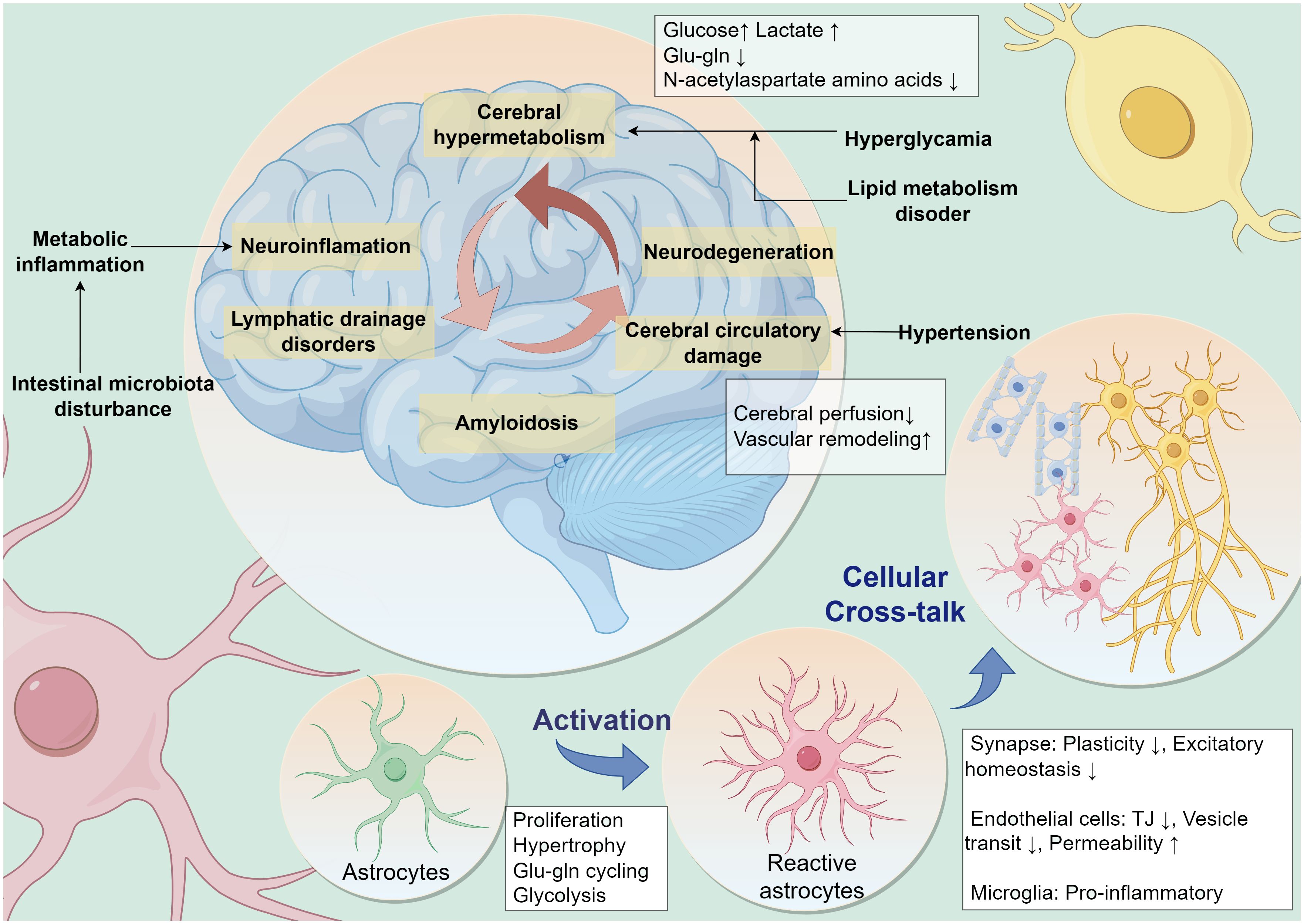
Figure 1 MetS leads to cognitive dysfunction through multiple pathologic factors. Multiple peripheral metabolic disorders in MetS, such as persistent hyperglycemic states, disorders of lipid metabolism, hypertension, metabolic inflammation, and disturbances in intestinal microbiota, are contributing to neurocognitive decline. Neuropathology in the brain leads to a vicious cycle of cognitive decline caused by neuroinflammation, cerebral microcirculatory dysfunction, impaired glial lymphatic system drainage, and accumulation of pathologic proteins. Astrocytes are potentially central to this vicious cycle. Metabolic stress pressure drives reactive activation of astrocytes and influences their interactive dialog with surrounding cells. Glu-gln, glutamate-glutamine; TJ, Tight Junction.
MetS components such as low-density lipoprotein (LDL), high-density lipoprotein (HDL), hypertension, and advanced glycation end products (AGEs) accumulation all contribute to cerebral atherosclerosis, accelerating white matter damage, lacunar microinfarcts, and microhemorrhages (28, 29). The cerebral circulatory system has the capacity for adaptive regulation to maintain cerebral perfusion in fluctuating arterial blood pressure. MetS contributes to decreased cerebral microvascular density and blood flow, impairing cerebrovascular responsive autoregulation and blood flow reserve functions (30, 31). Excessive or unstable arterial blood pressure levels reduce cerebrovascular autoregulation, leading to cerebral hypoperfusion and neurodegenerative pathologies (32) and amyloid-β (Aβ) protein deposition by mechanical stretch (33). Stimulation by high blood pressure arterial wall shear stress increases vascular smooth muscle cell hypertrophy and proliferation, leading to vascular remodeling (34, 35). High-fat diet(HFD) rats were more sensitive to ischemia-reperfusion injury. Compared to normal diet rats, they exhibited more significant decreases in cerebral blood flow (CBF) and elevated levels of oxidative stress (36).
Oxidative stress and mitochondrial malfunction in MetS also promote neuroinflammation and neurodegeneration (37). High levels of circulating inflammatory markers are a common feature of MetS and cognitive dysfunction (38, 39). Ectopic fat accumulation, fatty inflammation (40), and disruption of intestinal flora (41) during the MetS disease process lead to a chronic low-grade inflammatory state. High levels of circulating pro-inflammatory cytokines in MetS disrupt and cross the blood-brain barrier (BBB) into the brain, thereby activating astrocytes and microglia to trigger a neuroinflammatory response, leading to a critical mechanism of MetS-related cognitive dysfunction (42, 43). Peripheral inflammation leads to neuroinflammation and oxidative stress, impairing energy supply to synaptic mitochondria (44).
Astrocytes serve as a communication bridge between the central and peripheral nervous systems. They are essential components of the neurovascular unit and communicate extensively with neurons, endothelial cells, and glia via dendritic structures (45). The complex and diverse dynamic network of astrocytes is the anatomical basis for their maintenance of metabolic and immune homeostasis of the brain. The astrocyte network integrates nutrient metabolic signals and interacts with hypothalamic functional neurons to exert central feedback regulation of appetite regulation, glucose sensing, and other systemic metabolism (46–48).
Synaptic plasticity and presynaptic vesicle release in neurons are the mechanisms underlying working memory. Astrocytes are a new target for improving cognitive function, forming glial isolates that wrap 50-60% of brain synapses and regulate synaptic plasticity through glycolytic energy supply and release of multiple neuroactive substances (e.g., glutamate, ATP, adenosine, and D-serine) (49). Astrocytes regulate the neurosynaptic microenvironment with their unique glial-isolated structure and immunometabolic properties (50, 51). Perisynaptic astrocyte processes (PAPs) constitute the glial segregation of the synaptic gap. Astrocytes regulate glutamate concentration through glutamate transporter subtype 1(GLT-1) and deliver lactate through monocarboxylate transporters1 and 4 (MCT1, MCT4). Aging and atrophy of PAPs are accompanied by a decline in glutamate clearance and a decrease in Ca2+ events, and excess glutamic acid spillage in the synaptic gap will activate N-methyl-D-aspartic acid (NMDA) receptors to reduce the amplitude of the neuron’s long-term potentiation (LTP) (52). Furthermore, astrocytes control synaptic plasticity by producing neurotrophic factors, releasing and clearing neurotransmitters, and regulating ion levels in the extracellular environment (53).
In addition to regulating synapses, astrocytes regulate the electrical activity of neuronal networks by releasing gliotransmitters such as ATP, glutamate, and Ca2+ wave oscillations (54). Computational model studies of working memory suggest that astrocytes can store traces of neuronal activation in information processing (55). In contrast, memory extraction depends on astrocytes’ modulation of spiking neuron network connections (56). Astrocytes influence memory performance through states of conscious vigilance and basal arousal. Sustained neuronal firing in the hippocampus induces astrocytic γ-aminobutyric acid G protein-coupled receptor signals that control the oscillatory activity of the θ and γ oscillations of the hippocampal neuronal network (57).
Astrocytes coordinate interneuronal network projections between the hippocampus and cortex, thus participating in memory consolidation and storage. Astrocytes modulate communication between hippocampal CA1 and cingulate cortex to promote memory consolidation and retention (58). Dysfunctional cell division in astrocytes impairs hippocampal-prefrontal theta synchronization (59).
The neural axons and telopods of astrocytes wrap around the cerebral vascular system to detect nutrients and metabolic hormones in the arterial blood that enters the brain (60). Astrocytes form an intercellular communication network with each other through connexins, bridging the intravascular nutrient supply with the energy demand of neuronal activity. The astrocyte network enables the propagation and sharing of small molecule nutrient metabolic signals at the cellular network level (61). Astrocytes are the primary cells for glycogen storage and glycolysis in the brain. They shape the fundamental pattern of brain energy metabolism by coupling with neuronal oxidative phosphorylation (62, 63). Astrocytes can bridge the transient energy demands of neurosynaptic activity with the supply of circulating fuel (64).
Astrocytes store lipid droplets (LDs) by taking up excess fatty acids (FAs) from neurons via rich acid transport protein and lipid transport protein, making them critical sites for fatty acid oxidation. Astrocytes take up FAs produced by neuronal metabolism and help highly active neurons relieve lipid oxidative stress through astrocyte consumption of LDs for ATP production via NMDA receptor-mediated mitochondrial β-oxidation (65).
Recent studies have shown that astrocyte function is equally dependent on mitochondria and oxidative phosphorylation (66). Mitochondrial disorders in astrocytes affect brain oxidative phosphorylation metabolism and contribute to forming metabolic stresses such as reactive oxygen species (ROS) (67). Astrocytes can exert neuroprotective effects by delivering mitochondria to neurons. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) and mGluR5(metabotropic glutamate receptor 5) modulate the mitochondrial network of astrocyte cells and produce positive Ca2+ signaling ion conductance to synapses (68). Restoring mitochondrial biogenesis in astrocytes may be a therapeutic target for neuropsychiatric disorders with impaired synapse formation.
The endfoot of astrocytes envelop the endothelium of the cerebral microcirculation. Astrocytes rapidly regulate CBF to meet neurocognitive energy demands, ensuring nutrient and oxygen delivery (69). Arachidonic acid, prostaglandin E2, and nitric oxide produced by astrocytes connect directly with small smooth muscle cells in the arteries (70). Additionally, astrocytes monitor cerebral perfusion pressure and regulate blood flow by modulating the diameter of cerebral arteries through their unique anatomical location and pressure-sensitive membrane structures (71). The end-foot of astrocytes abutting endothelial cells expresses the transient potential receptor vanilloid 4 (TRPV4) and is influenced by transmural pressure in penetrating arterioles and blood flow levels (72).
The endfoot of astrocytes with tight junction (TJ) proteins are the basic structures that comprise the BBB (73). Ablating astrocytes increases BBB permeability and impairs repair (74). Astrocytes regulate the TJ structure through the vascular permeability factor, matrix metalloproteinase (75). Astrocyte endfoot around blood vessels are tightly connected by connexins 30 (Cx30) and connexins 43 (Cx43), which allow ion exchange in the peri-endothelial astrocyte network. Deletion of these connexins weakens the BBB, leading to its opening under increased hydrostatic vascular pressure (76). Physiologically, astrocytes Cx30 and Cx43 are involved in memory formation (77). Astrocyte Cx43 controls the rate of synaptic vesicle release to regulate presynaptic function, controls glutamate levels and allows glutamine release to maintain synaptic transmission (78). Astrocyte-specific Cx30 and Cx43 double knockouts lead to widespread activation of astrocytes and microglia, significant suppression of neuronal excitability, excitatory synaptic transmission in hippocampal CA1 region, and decrease of spatial learning and memory (79). In addition, astrocytes secrete a variety of vasoactive substances, among which angiopoietin-1 (ANG-1), sonic hedgehog (SHH), and insulin-like growth factor-1 (IGF-1) protect the BBB. In contrast, vascular endothelial growth factors (VEGF), matrix metalloproteinases (MMP), nitric oxide, glutamate, and endothelin-1 lead to structural damage of the BBB (80). Damage to astrocyte structures reduces BBB permeability, thus allowing pathogens and toxins to enter the central nervous system (81). Peripheral inflammatory factor across BBB and drives the pro-inflammatory phenotype of glial cells. This is a crucial pathway by which systemic inflammation triggers neuroinflammation (82).
Astrocytes undergo adaptive activation both anatomically morphologically and functionally as they respond to and maintain homeostasis in the brain’s internal environment. Both clinical and animal studies have found that disorders of glucolipid metabolism and hypertension can induce reactive activation of brain astrocytes and overexpression of glial fibrillary acidic protein (GFAP) (83). Astrocytes, as reactive cells, are regulated by complex factors of circulating origin. Different types of stimuli induce specific reactive changes. In addition, sex was an essential variable in the analysis of MetS and cognitive dysfunction, and there was sex- and age-related heterogeneity in the altered responsiveness of astrocytes (84). The prevalence of MetS and related complications is higher in men than in women at ages younger than 50 years. The risk of MetS and associated complications in women exceeds that of men, with a decline in estrogen levels after menopause (85, 86). Recent reports suggest that astrocyte numbers, differentiation, and function differ between the sexes. Sex differences in reactive astrocytes are responsible for the emergence of sex differences in neuroendocrine regulation and cognitive function. Astrocytes isolated from female rats were more resistant to cell death induced by hypoxia, palmitic acid (PA), and lipopolysaccharide (LPS) than male astrocytes (87–89).
Increased numbers and morphological hypertrophy of reactive astrocytes in the brain have been observed in several animal models of hypertension (Table 1). 26-week-old SHR showed increased numbers and areas of immunoreactive positive astrocytes in the prefrontal cortex, occipital cortex and significantly higher numbers of GFAP immunoreactive positive astrocytes in the hippocampal region, compared to the same-week-old WKY rats (94). In the chronic hypertension model induced by 8 weeks of AngII infusion, a linear positive correlation between astrocyte morphology and elevated arterial blood pressure proliferated in cerebral white matter (95). Astrocyte endfoot are in contact with cerebral blood vessels, directly sensing circulating hemodynamic changes and releasing vasoactive substances to modulate slight arterial tone to maintain CBF independent of blood pressure fluctuations (96). Astrocyte reactive activation accompanied by transient potential receptor vanilloid 4 (TRPV4) activation was observed in the hippocampus of an AngII 28-day injection-induced mouse model of chronic hypertension. In this study, astrocyte TRPV4 mediated an increase in spontaneous Ca2+ events within microdomains, which enhanced parenchymal arteriole tone and decreased cognitive function (90).
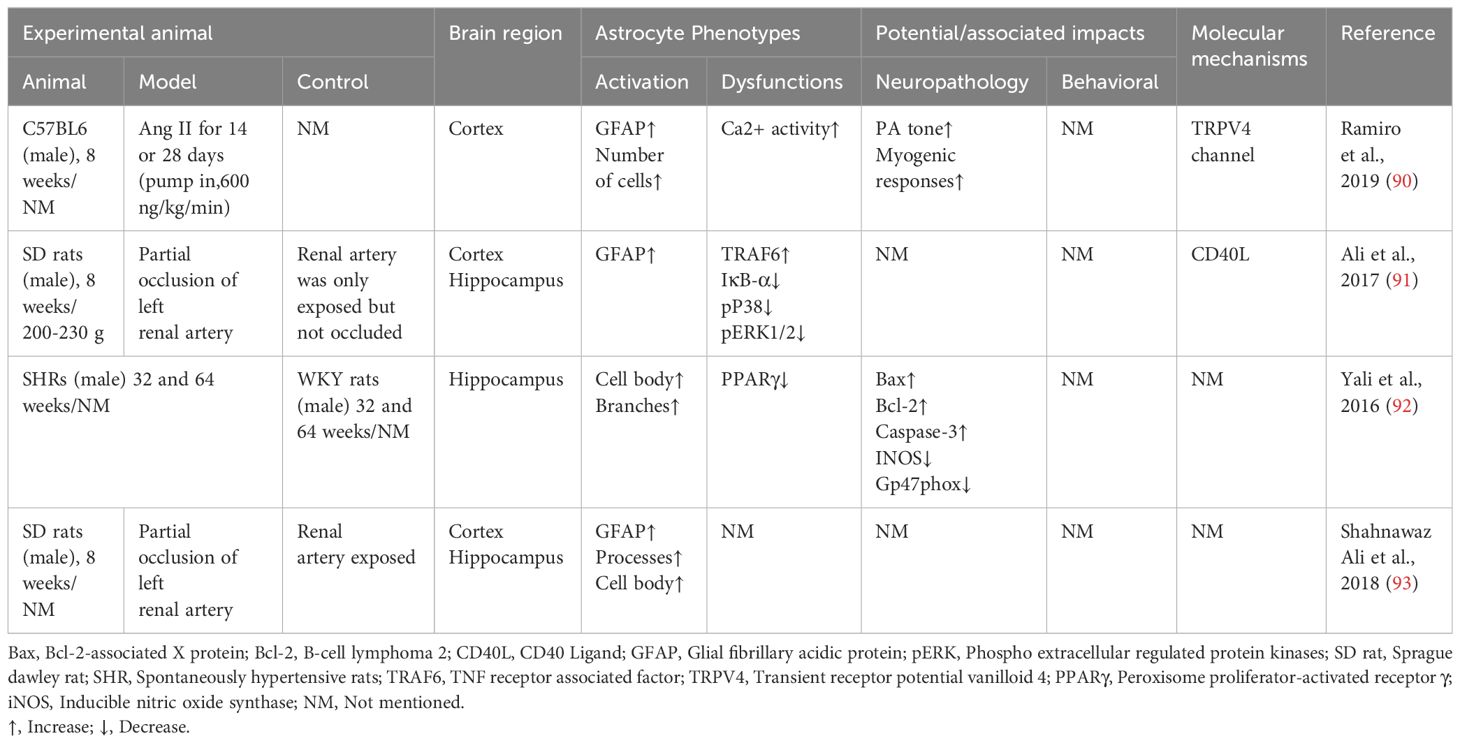
Table 1 Effects of hypertension on astrocyte pathology and cognitive functions.
Studies in hypertensive humans and hypertensive rat models have shown that an overactive brain renin-angiotensin system (RAS), which leads to oxidative stress and neuroinflammation in several brain regions, including the brainstem cardiovascular centers and the hippocampus (97, 98). The overactive brain RAS in hypertension also contributes to cognitive dysfunction and exacerbates hypertension through sympathetic excitation. In several experimental and genetic models of hypertension, including spontaneously hypertensive rats (SHR) (99, 100) and desoxycorticosterone acetate salt hypertensive rats (101), hyperactivity of the central RAS was observed, especially increased levels of angiotensin II (AngII), angiotensin III (AngIII) and angiotensin II receptor type 1 (AT1R). In vitro studies have shown the expression of AT1R and AT2R on human astrocytoma cell lines (102)and primary cerebral cortex astrocytes (103). Therefore, it has been suggested that astrocytes may play a role in neuroinflammation and oxidative stress caused by AngII and AngIII in the brain RAS (102). Studies conducted on primary astrocytes isolated from SHR have shown that AngII causes the secretion of IL-6 from astrocytes through the activation of NF-κB/ROS and overexpressing cyclooxygenase 2 via astrocyte AT1R (104, 105). In primary rat astrocytes derived from SD rats, AngIII targets AT1R to activate extracellular regulated protein kinases (ERK)1/2 MAP kinases and c-Jun N-terminal kinase (JNK) phosphorylation to promote astrocyte proliferation (106).
Astrocyte activation and proliferation in the hippocampus and hypothalamus have been observed in HFD-induced obese rat models (Table 2) (114–117).
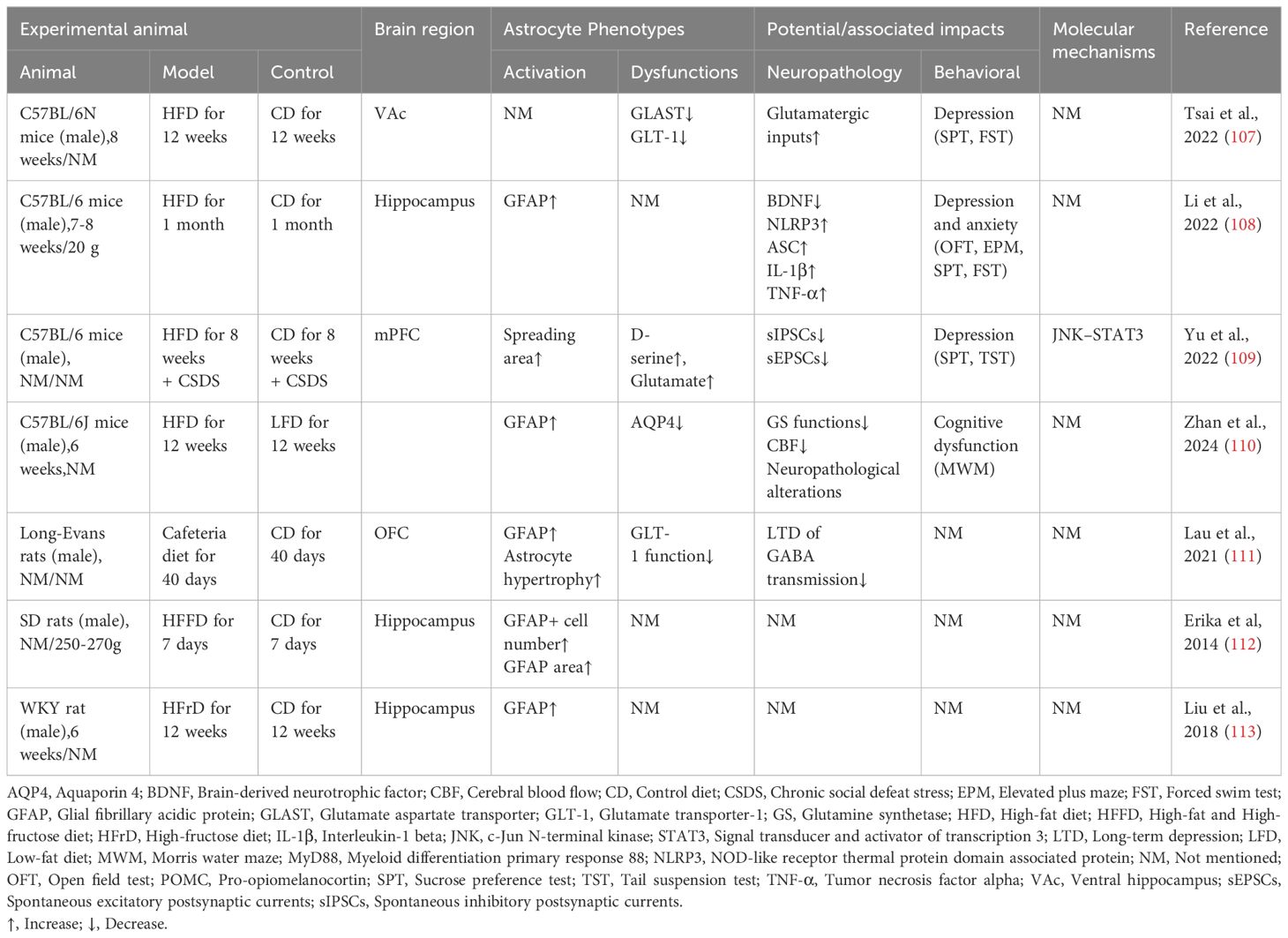
Table 2 Effects of hyperlipidemia and obesity on astrocyte pathology and cognitive functions.
HFD-induced obesity also affects astrocyte lipid oxidation and mitochondrial metabolism. Fatty acid β-oxidation (FAO) in the brain occurs mainly in astrocytes. Astrocytes store overloaded free fatty acids as LDs, which are used via FAO to energize neurons and protect them from lipotoxic damage. Obesity leads to the accumulation of astrocyte misfolded proteins, induced endoplasmic reticulum stress, and thus crosstalk with neurodegenerative (21). Obesity decreases fatty acid oxidation in hypothalamic astrocytes, leading to disturbed mitochondrial dynamics (118). In the hippocampus of mice raised on a HFD for 1 month, astrocytes’ lipid and cholesterol content was elevated, accompanied by an increase in the number of secondary branches and lobules of neural protrusions (119). In vitro studies have found that saturated and unsaturated fatty acids have opposite regulatory effects on astrocyte lipoprotein lipase (LPL). TGs and palmitic acid decrease LPL expression and oleic acid elevate LPL. HFD-induced elevation of LPL in hypothalamic astrocytes of obese rats increases the accumulation of LDs. It impairs glycolytic metabolism, impairing glucose tolerance, increases food intake, and aggravates obesity (120).
In addition, obesity and pathologic fat accumulation lead to decreased function of astrocyte-neuron crosstalk, in which high serum levels of leptin inhibit astrocyte excitatory amino acid transporter protein (EAAT) expression and promote sympathetic overactivation (121). Obesity impairs glutamate clearance from the synaptic gap by astrocytes and attenuates the endogenous cannabinoid pathway and the synaptic plasticity it mediates in vertebral neurons in the orbitofrontal cortex (111). Astrocytes present a compensatory neuroprotective effect in the early stages of lipid metabolism disorders and are progressively bettered by chronic stressful pressures. It was found that 8 weeks of HFD induced astrocyte proliferation and limited neuronal damage by releasing heat shock protein 70 (HSP70) and ciliary neurotrophic factor (CNTF). In contrast, the compensatory neuroprotective effect of astrocytes was depleted after 20 consecutive weeks of HFD (122).
High blood glucose levels in both in vivo and ex vivo studies resulted in reactive activation of astrocytes accompanied by changes in metabolic processes (Table 3). The astrocyte glycolytic effect is two-sided, with increased glycolytic flux supplying neurons with energy and antioxidants (133), while glycolysis also provides energetic support for inflammatory responses (134). A comprehensive review of astrocyte glycolysis in cellular metabolic immunity is lacking, but some studies have suggested that it undergoes the same progression from compensation to decompensation as immune cells (135). Multiple in vitro studies have found increased glycogen content and glycolytic activity in astrocytes chronically exposed to high glucose (136). In a 1H NMR-based metabonomic approach study, an increase in glucose uptake, glycolytic activity lactate release, and downregulation of TCA cycling activity were found in astrocytes after 72 hours of high-glucose exposure (137). High glucose promotes glucose uptake and glycogen storage in primary astrocytes but reduces maximal respiratory and glycolytic reserve capacity (138). It is suggested that high glucose leads to an increase in astrocyte glucose metabolic flux, but the efficiency of cellular energy utilization is reduced, making it more vulnerable to stressful pressures.
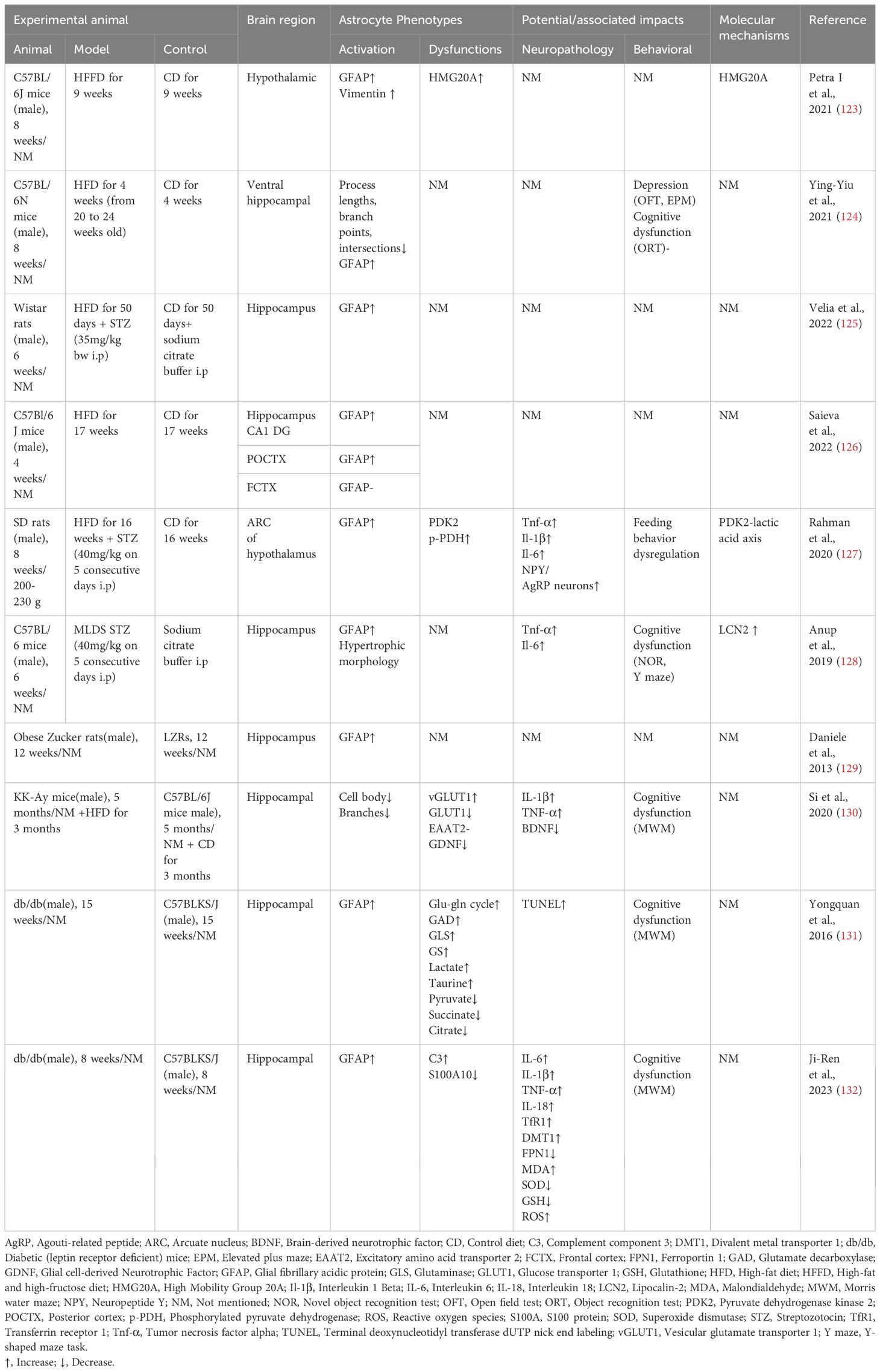
Table 3 Effects of hyperglycemia on astrocyte pathology and cognitive functions.
Astrocyte metabolic plasticity has a double-edged role, feeding the inflammatory immune response process and acting as a buffer against metabolic stress. The astrocyte pentose phosphate pathway and glutathione levels increase with blood glucose, reduce ROS production, and protect neurons from oxidative stress damage (139). In vitro metabolomics studies have found that astrocytes produce and transport more lactate in high-sugar environments, which may work to enhance astrocyte-neuron lactate shuttling (137).
High glucose leads to increased secretion of multiple pro-inflammatory factors by astrocytes, leading to neuroinflammation (140). High glucose increases the expression and secretion of pro-inflammatory cytokines IL-6 and IL-8 in human primary astrocytes and U-118MG astrocytoma cells via STAT-3 (141). Hyperglycemia induces enlargement of astrocytes in the hippocampus and is linked to peripheral recruitment of leukocytes to the cerebrovascular system (142). High glucose exacerbates neuroinflammation via ROS/mitogen-activated protein kinase (MAPK)/NF-κB, ERK, and JNK pathways by upregulating matrix metalloproteinase-9 expression in rat brain astrocytes (143, 144). The toll-like receptor (TLR) of astrocytes serves as an essential target of the innate immune system, and high glucose promotes neuroinflammation and altered cellular metabolism via the TLR/MAPK/PPARs pathway (145).
Astrocyte responsiveness to high glucose affects microcirculatory endothelial barrier structure. Hyperglycemia induces increased secretion of VEGF protein in astrocytes, impairment of gap junctional Cx43 and Cx30 proteins, and reduced transendothelial cell electrical resistance (TEER), which are critical factors for reduced BBB permeability (146, 147). Sustained hyperglycemia induces the non-enzymatic glycosylation of various proteins and the resulting formation of advanced glycation endproducts (AGEs), which mediate the development of diabetic complications by targeting the receptor of advanced glycation endproducts (RAGE). Primary astrocytes from mice cultured with high glucose showed increased expression of immune complement C3 and decreased synaptic number, suggesting that high glucose promotes synaptic phagocytosis of the complement pathway in astrocytes. In this study, the RAGE-p38MAPK-NF-κB pathway was a vital upstream of the synaptic phagocytosis promoted by high glucose in astrocytes (148).
The hippocampus is a major brain region involved in memory functions, and its synaptic plasticity activities of vesicular and ionic channel activity depend on continuous energy support and are susceptible to nutrient metabolism (149). The release of gliotransmitters in astrocytes modulates neural theta oscillations between the dorsal hippocampus and prefrontal cortex, which are involved in memory formation and storage (59). Morphologic, immunologic, and metabolic alterations in astrocytes mediate the contribution of multiple factors to the development of cognitive dysfunction in MetS (Figure 2).
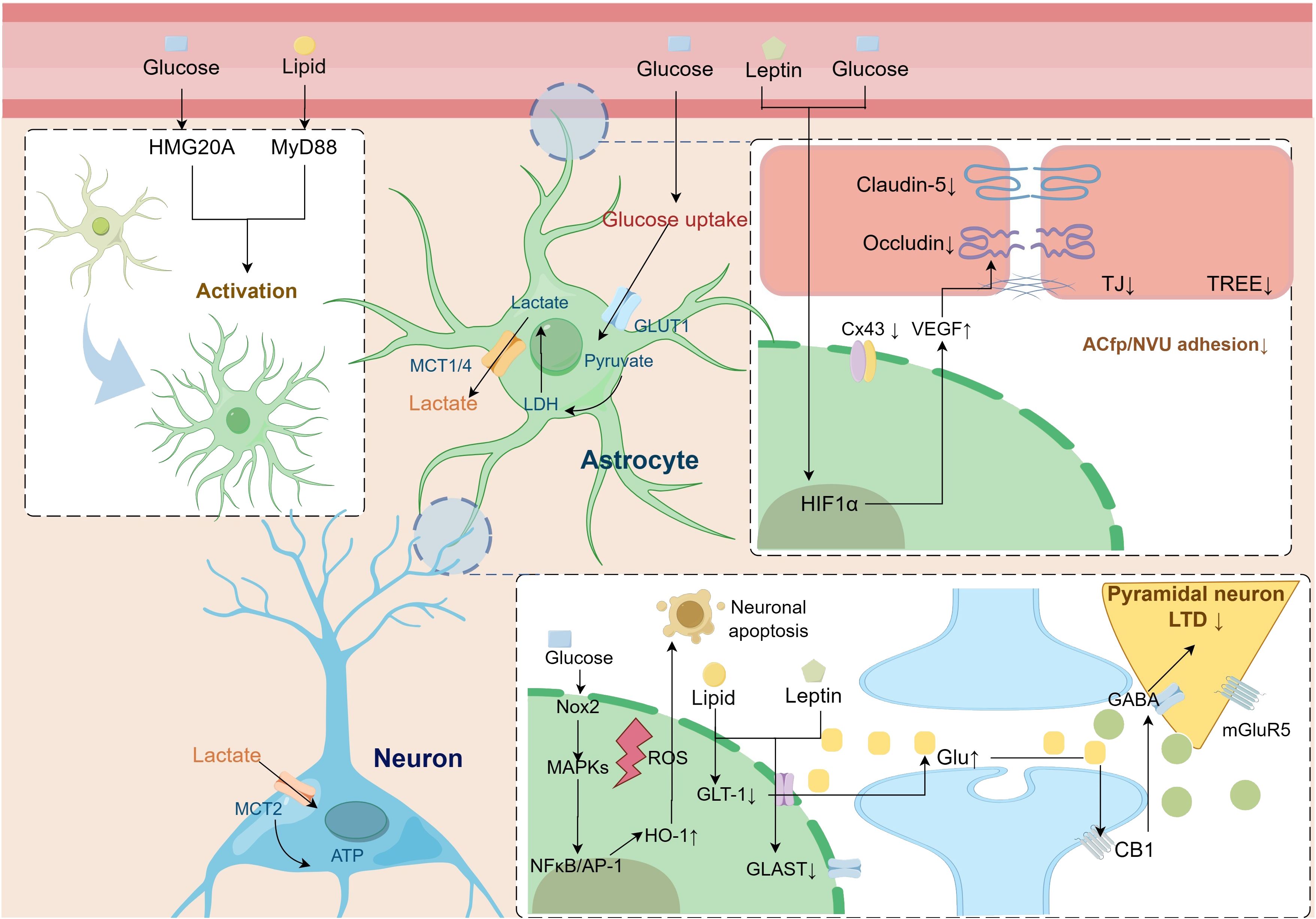
Figure 2 Pathologic alterations of astrocytes in metabolic syndrome leading to cognitive dysfunction. Astrocytes undergo reactive activation driven by a disturbed metabolic state. Activation of the astrocyte HIF-1α pathway secretes multiple factors, including VEGF, that impair BBB-linked proteins, leading to increased blood-brain barrier permeability. At the same time, the lipid, glucose, and amino acid metabolism coupling pathway between astrocytes and neurons is impaired, leading to an imbalance in neuronal excitability and decreased synaptic plasticity. AP-1, activator protein-1; CB1, cannabinoid 1; Cx43, connexin 43; GABA, γ-aminobutyric acid; GLAST, glutamate aspartate transporter; Glu, glutamate; GLUT1, glucose transporter type 1; GLT-1, glutamate transporter subtype I; HIF1-α, hypoxia-inducible factor1-α; HMG20A, high mobility group domain protein 20A; HO-1, heme oxygenase-1; LDH, lactate dehydrogenase; LTD, long-term depression; MAPK, mitogen-activated protein kinase; MCT, monocarboxylate transporter; mGluR5, metabotropic glutamate receptor 5; MyD88, myeloid differentiation primary response 88; NFκB, nuclear Factor-κB; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; TJ, tight Junction; TREE, trans-endothelial/epithelial electrical resistance; VEGF, vascular endothelial growth factor.
In MetS, astrocytes undergo reactive changes in cell morphology, as demonstrated by increased cell proliferation and hypertrophy, neural protrusion density and axon length. It was found that glial segregation and TJ barrier structures constituted by astrocytes proliferating their endfoot can limit neuroinflammatory damage (73). The morphologic plasticity of astrocytes is compatible with their functional transformation to limit the spread of inflammation and support nerve regeneration (150).
The altered morphology of astrocytes leads to impaired communication with other cells in the neural vascular unit, which in turn impairs the barrier immunity and energy substance uptake function of the BBB (151). Both in vivo and ex vivo studies have shown that a hyperglycemic state impairs gap junction communication in astrocytes, inducing swelling of the astrocyte endfoot and detachment from the endothelial cell basement membrane (146). Astrocyte endfoot processes (ACfp) from the neurovascular unit (NVU) were observed in the prefrontal cortex of female diabetic db/db mice. Loss of ACfp/NVU adhesion has been suggested as a potential mechanism contributing to impaired cognitive function in diabetes (152). Obesity induces reactive proliferation of astrocytes, which in turn induces structural remodeling of the neuroglial interface in multiple brain regions and alters the immune and transport functions of the BBB (153, 154). HFD-induced glial proliferation of astrocytes affects the BBB structure in the arcuate nucleus region. It makes it more difficult for neuropeptide Y (NPY) and proopiomelanocortin (POMC) neuronal cytosomes and dendrites in this region to reach the vasculature (155). Obesity is accompanied by increased serum leptin levels, which activate hypoxia-inducible factor 1-alpha (HIF-1α)-VEGF signaling in hypothalamic astrocytes, thereby inducing structural remodeling of the glial interface (156). Astrocytes isolated from stroke-prone spontaneously hypertensive rats (SHRSP) cause TJ damage and high resistance in endothelial cells by secreting large amounts of lactate (157).
Several studies have found that the overall neural processes of astrocytes are shortened in states of metabolic dysregulation, diminishing their modulation of synapses. A HFD for 12 weeks induced an increase in GFAP expression in the rat hippocampus but in turn impaired the length of neural protrusions in astrocytes, as well as the expression of the proteins glutamate aspartate transporter (GLAST), GLT-1, and Cx43, which are associated with synaptic plasticity (124, 158, 159). Chronic overnutrition leads to the shortening of the central neural protrusions of astrocytes through upregulation of the IκB kinase β (IKKβ)/nuclear factor-κB (NF-κB) pathway, which in turn affects their glutamate uptake in the synaptic gap and modulation of synaptic excitability (160).
Astrocytes are thought to be critical regulators of neuroinflammation (161). Peripheral immune signaling drives glial cell immune function switching as a potential mechanism for systemic inflammation to trigger neuroinflammation (82). Astrocytes play a vital role in developing and expanding neuroinflammation by interacting with various central nervous system in situ immune cells, including microglia and T cells (162, 163). Moreover, astrocytes drive perivascular leukocyte recruitment to the brain by secreting C-C motif chemokine ligand 2 (CCL2) and C-X-C motif chemokine ligand 10 (CXCL10) (163, 164). GFAP, S100 calcium-binding protein B (S100B), monoamine oxidase-B (MAO-B), chitinase-3-like protein 1 (YKL-40), and D-serine were used as biomarkers to assess the reactivity and proliferation intensity of astrocytes in tissues, cerebrospinal fluid, and blood (165, 166). High glucose activation of the complement 3 (C3) pathway in astrocytes can lead indirectly and directly to synaptic loss. C3 secreted by astrocytes is able to interact with microglial component 3a (C3a) receptors to modulate synaptic phagocytosis in microglia (167). Reduced secretion of complement factor C3/C3a in high glucose-treated primary astrocytes leads to synaptic protein damage and cognitive dysfunction (168).
As previously described, multiple factors in MetS, such as peripheral metabolic glucolipid metabolism disorders and metabolic inflammation, can drive astrocyte reactive activation. Several studies have demonstrated that enhanced GFAP immunoblotting is observed for T2DM disease states lasting 2-4 weeks, whereas GFAP expression is significantly reduced in more extended weekly studies (169, 170). HFD induces increased hippocampal GFAP expression in rats, which is associated with neuroinflammation, microvascular damage, and subsequent cognitive dysfunction (171, 172). The IKKβ/NF-κB of astrocytes is an essential pathway for HFD-induced hypothalamic inflammation. Knockdown of IKKβ in astrocytes can improve HFD-induced hypothalamic neuroinflammation, insulin resistance status, and glucolipid metabolism (173). HFD induces upregulation of hypothalamic potassium inwardly rectifying channel subfamily J member 2 Gene (Kcnj2), Complement 4b (C4b) and discoidin domain receptor 1 (Ddr1) and co-localizes with GFAP, and is therefore considered an early marker of obesity and diabetes-related cognitive dysfunction (174). Secretion of inflammatory factors by astrocytes is associated with synaptic loss. In neuron-astrocyte co-culture cell studies, LPS increased astrocyte secretion of inflammatory factors and correlated with decreased neuronal synaptophysin (SYN) (175).
MetS leads to the activation of astrocytes, thereby affecting their regulatory role in cognition and behavior. Reactive astrocytes are a potential therapeutic target for ameliorating vascular and neurodegeneration-related cognitive dysfunction (176, 177). Targeting the neurotoxic phenotype of reactive astrocytes alleviates cognitive-behavioral alterations induced by MetS-related factors (169).
Reactive astrocytes respond to metabolic stress by reprogramming metabolic processes and exerting various adaptive compensatory effects to maintain neuronal energy supply (178, 179). Various components of MetS can act directly on metabolic processes such as glycolysis and mitochondrial metabolism in astrocytes. Astrocyte metabolism progresses towards dysregulation under metabolic stress, with mitochondrial malfunction, energy failure, and oxidative stress, which can affect the energy supply of neurosynapses and impaired lymphatic efflux of Aβ proteins (180). Astrocyte glycolysis, gluconeogenesis, and lipid metabolism are plastic to undergo reprogramming during MetS metabolic stress to maintain neuronal energy homeostasis (181). Excessive chronic stressful pressure leads to a compensatory decrease in astrocyte energy metabolism, which may be impaired by promoting cerebral insulin resistance, decreased glucose uptake, and oxidative stress (66).
Central leptin signaling activation in HFD rats reduces astrocytic ghrelin transporter protein and EAAT1 and EAAT2 in the arcuate nucleus of the hypothalamus, resulting in decreased ghrelin uptake and reduced glutamine synthesis (121). Chronic lipid exposure-induced ectopic lipid loading in astrocytes leads to reduced insulin-induced protein kinase B (AKT) phosphorylation and dysregulated glycogen metabolism (182). The metabolomic study showed that selenium amino acid metabolism, urea cycle, and glutamate metabolism were up-regulated in human astrocytes in a palmitic acid-induced lipotoxic environment for several amino acid metabolic pathways (183). Several tricarboxylic acid cycle intermediates, such as succinate and citrate were reduced, glutamine synthetase was increased, and glutaminase and glutamic acid decarboxylase decreased in the hippocampal region of db/db mice (131). During physiological states, lipid synthesis and metabolism in astrocytes regulate hippocampal synapse development and function. Diminishing of sterol regulatory element-binding protein 2 (SREBP2) cleavage-activating protein (SCAP) resulted in lower levels of the synaptosome associated protein 25(SNAP-25) and reduced numbers of synaptic vesicles in the hippocampus of mice (184). Diabetes mellitus leads to impaired brain cholesterol synthesis and reduced synapse number by reducing the transcription factor SREBP2 (185).
Metabolic reprogramming of astrocytes is also thought to be an adaptive change in response to central insulin resistance (186). Astrocytes express insulin receptor and insulin-like growth factor 1 (IGF1), which regulate glucose transporter type 1 (GLUT1) expression to take up circulating glucose (187). In another study, IR knockdown in astrocytes was found to impair tyrosine phosphorylation of Munc18c, reduce ATP cytokinesis, and subsequently lead to reduced neuronal dopamine release and depressive-like behavior (188).
The process of reactive activation of astrocytes is accompanied by a metabolic paradigm shift (189). Activation and maintenance of reactive astrocytes depend on a continuous supply of energy from glycolytic metabolism. The downright inflammatory response of astrocytes to LPS is accompanied by elevated glycolytic flux and elevated activity of critical metabolic enzymes, such as 6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase isoform 3 (PFKFB3) (190). 2-DG glycolysis inhibitor and glycogen phosphorylase inhibitor intervened to regulate the astrocyte glycolysis process and significantly attenuated LPS-induced cytokine release and NF-κB phosphorylation (134). Inhibition of pyruvate dehydrogenase kinase-2(PDK2) in hypothalamic astrocytes of diabetic rats inhibited cellular glycolysis and its inflammatory activation, thus reducing hypothalamic inflammation as well as lactate levels and reversing the increase in food intake (127). The peroxisome proliferator-activated receptor (PPAR) pathway is one of the critical pathways of astrocyte immunometabolism, among which PPARγ stimulates glucose and glutamate uptake and lactate release from astrocytes. At the same time, PPARα induces fatty acid β-oxidation in the presence of impaired glucose metabolism (191). Regulators of aerobic glycolysis, such as HIF-1α and AMPK in astrocytes, are affected by inflammation (192). Nicotinamide phosphoribosyltransferase (NAMPT)-dependent nicotinamide adenine dinucleotide (NAD+) upregulation in astrocytes provides energy for cellular activation and drives transcriptional inflammatory program rearrangements, and inhibition of endogenous NAD+ synthesis impairs astrocyte transcriptional responses to LPS/Interferon-γ(IFNγ) stimulation and attenuates activation-associated neuroinflammation (193).
In summary, it has been elaborated that astrocytes are susceptible to morphologically and functionally responsive changes in metabolically disturbed environments. Reactive astrocytes are involved in pathological processes such as neuroinflammation, energy metabolic homeostasis, and cross-barrier transport of nutrients and amyloid in MetS-associated cognitive dysfunction (165, 194).
Astrocytes express and secrete a variety of specific molecular substances during pathological processes, which are considered promising targets for the development of early screening with humoral or imaging biomarkers (195). Several studies and meta-analyses have found a strong association between astrocyte biomarkers and cognitive decline (166). In a clinical study of 121 older adults, cerebrospinal fluid and plasma levels of GFAP and YKL-40 were shown to relate to Aβ and tau pathology and to mediate hippocampal volume atrophy (196).
Proteomic studies have identified increased levels of the metabolism-related proteins lactate dehydrogenase B-chain (LDHB), pyruvate kinase (PKM), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in M4 Astrocyte in cerebrospinal fluid, which can be used as a biomarker for the early diagnosis of cognitive impairment (197). In addition, at the level of genetic material, astrocyte-derived extracellular vesicles (EVs) and a variety of miRNAs therein are thought to potentially become biomarkers for neurodegenerative diseases (198). Increased secretion of miR-141-3p and miR-30d is detected in primary human astrocytes activated by stimulation with the neuroinflammatory factor IL-1β (199). Regarding imaging, astrocyte metabolic levels and associated metabolites are visualized and analyzed by PET/CT imaging with 11C-acetate and 18F-fluorodeoxyglucose (18F-FDG). In a study, astrocyte acetate hypermetabolism and neuronal glucose hypometabolism were used as a visual diagnostic strategy for early diagnosis of Alzheimer’s disease (200). In addition, machine learning research is an emerging tool for constructing diagnostic models. In a study, automata theory was used to build a diagnostic computational model to monitor the astrocyte metabolic end-product lactate, thereby characterizing the level of glycogen metabolism in the brain (201).
Astrocytes have been progressively recognized as potential biomarkers for the development of cognitive impairment. At the same time, these markers are highly expressed in MetS and its related components. A joint examination of astrocytes in both diseases is still lacking. However, as reactive cells, their immunometabolic flexibility and sensitivity make them valuable as potential diagnostic markers of MetS-associated cognitive disorders.
Conventional treatments have focused on neuron-focused mechanistic interventions, but drug development and clinical translation have been limited. It is currently considered effective in improving the homeostasis of synapses and their adjacent microenvironments through systems biology and multi-targeted therapeutic approaches. The supportive role and pathologic criticality of neuroglia for neuronal function are gradually being demonstrated. Astrocytes have been proposed at multiple levels as potential targets for drug development to ameliorate central neuropathy and restore cognitive function (202, 203). Based on the altered functional and molecular pathways in astrocytes in MetS, targeting their biological processes may have therapeutic value.
Astrocytes have a central role in neuroinflammation (204). Several studies have targeted toll-like receptor proteins (TLRs) (205), NF-κB, and the transcription factor NF-E2-related factor 2 (Nrf2) (206) in astrocytes, thereby limiting their cell proliferation and neurotoxicity (207). In addition, the repair of astrocyte TJ protein structures resulted in the amelioration of BBB permeability damage. In vitro and in vivo studies showed that inhibition of astrocyte Cx43 hemichannel opening prevented astrocyte proliferation (astrogliosis) and improved BBB permeability (208). Clomipramine is a classical tricyclic antidepressant. Epoxomicin is a natural selective proteasome inhibitor and has an anti-inflammatory effect. Both drugs have been attempted as inhibitors of intermediate filament proteins and vimentin associated with astrogliosis, thereby facilitating the limitations on nerve regeneration imposed by persistent, excessive glial proliferation (209).
Astrocytic metabolic plasticity allows astrocytes to act as critical cells in maintaining homeostasis (210), with their glycolytic metabolism and derived metabolites, such as lactate and serine, providing energy support to synapses and maintaining homeostasis of neural excitability (211). Therefore, regulation of astrocyte metabolism has also been recognized as a potential therapeutic target (212). The astrocyte glycolytic metabolite L-lactate and secreted vesicles have also been identified as potential targets in neurological disorders (213). Antidiabetic drugs have been found to improve brain glucose uptake by targeting astrocytes. Metformin, which crosses the BBB, increases glucose consumption and lactate release in astrocytes (214). Metformin treatment normalized GLUT-1 expression in STZ-induced diabetic rats and partially restored hippocampal glucose uptake and transport (215). Notably, glucagon-like peptide-1 (GLP-1) receptor agonists have been shown preclinically in small pilot trials to improve cerebral glucose metabolism and functional connectivity (216, 217). Liraglutide (an analog of GLP-1) improves cognitive function by enhancing astrocyte-promoted aerobic glycolysis and alleviating OXPHOS activation to maintain neuronal support (133).
In addition, promoting the transformation of astrocytes into neurons or other glial cells is a potential but controversial therapeutic modality. Several drug tools for astrocyte-specific delivery have been developed, including pluripotent stem cell therapies (218) and effective viral vectors (219) to control astrocyte-specific gene expression. Some studies have used lineage-tracing strategies to target astrocytes in vivo for transformation into neurons (220). Other studies have attempted to reprogram astrocyte lineage cells into oligodendrocyte cells by targeting Sox2 and Sox10. This method could relieve astrocyte glial scarring and promote myelin regeneration of neural axons (221–223).
MetS causes multiple disorders that worsen in the peripheral circulation and affect the brain, which is considered its target organ for generating metabolic stress damage. As a result, MetS accelerates cognitive decline through the acceleration of neurodegeneration and cerebral circulatory disturbances. Astrocytes change their metabolic and immune phenotypes in response to peripheral metabolic stressors, leading to early compensatory regulation of local neurological microenvironmental homeostasis. However, this compensation is lost when the stressors become too much, leading to worsened neuroinflammation. Astrocytes interact with a wide range of cells in the vascular, neural unit to influence BBB permeability and glial lymphatic system drainage functions and also form structures with neural synapses known as tripartite synapses, which play diverse and complex regulatory roles in neural circuit modulation.
The sensitive metabolic and functional plasticity of astrocytes makes them potential targets for improving the maintenance of brain energy metabolism and sustaining synaptic energy support. Also their cellular markers with specific functional proteins are also being developed as diagnostic markers for cognitive disorders. However, the complex and challenging nature of targeting astrocytes by transgenic techniques still poses a challenge due to the rich diversity of astrocytes and their overlap with other CNS cell genetic lineages (224). Contradictory findings in basic research cast doubt on the transdifferentiation capacity of astrocytes (225, 226). Additionally, there is still caution in clinical development regarding immunogenicity mapping in viral manipulation and the potential off-target risk of transgenic manipulation. Nevertheless, this cell implantation strategy could potentially enable endogenous neuronal regeneration in the future.
LZ: Writing – original draft, Writing – review & editing. JY-Y: Writing – original draft, Writing – review & editing. LC: Formal Analysis, Investigation, Validation, Writing – review & editing. XP: Data curation, Formal Analysis, Funding acquisition, Methodology, Writing – review & editing. TJ: Funding acquisition, Resources, Validation, Visualization, Writing – review & editing. PY: Software, Writing – review & editing. YR: Funding acquisition, Resources, Software, Visualization, Writing – review & editing. PQ:.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors’ work in this area is supported by the Natural Science Foundation of China (82274486); Science and Technology Research Special project of Sichuan Provincial; Department (2022YFS0382); YR’s TCM studio.
Figures were created with Figdraw.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lambert GW, Straznicky NE, Lambert EA, Dixon JB, Schlaich MP. Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther. (2010) 126:159–72. doi: 10.1016/j.pharmthera.2010.02.002
2. Tune JD, Goodwill AG, Sassoon DJ, Mather KJ. Cardiovascular consequences of metabolic syndrome. Transl Res. (2017) 183:57–70. doi: 10.1016/j.trsl.2017.01.001
3. Lemieux I, Després JP. Metabolic syndrome: past, present and future. Nutrients. (2020) 12:3501. doi: 10.3390/nu12113501
4. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. doi: 10.1007/s11906-018-0812-z
5. World Health O. Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications: Report of a Who Consultation. Part 1, Diagnosis and Classification of Diabetes Mellitus. Geneva: World Health Organization (1999).
6. Third report of the national cholesterol education program (Ncep) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult treatment panel iii) final report. Circulation. (2002) 106:3143–421. doi: 10.1161/circ.106.25.3143
7. Einhorn D, Reaven GM, Cobin RH, Ford E, Ganda OP, Handelsman Y, et al. American college of endocrinology position statement on the insulin resistance syndrome. Endocr Pract. (2003) 9:237–52. doi: 10.4158/EP.9.S2.5
8. Alberti KG, Zimmet P, Shaw J. The metabolic syndrome–a new worldwide definition. Lancet. (2005) 366:1059–62. doi: 10.1016/s0140-6736(05)67402-8
9. Lone S, Lone K, Khan S, Pampori RA. Assessment of metabolic syndrome in kashmiri population with type 2 diabetes employing the standard criteria’s given by who, ncepatp iii and idf. J Epidemiol Glob Health. (2017) 7:235–9. doi: 10.1016/j.jegh.2017.07.004
10. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; american heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation. (2009) 120:1640–5. doi: 10.1161/circulationaha.109.192644
11. Fakih W, Zeitoun R, AlZaim I, Eid AH, Kobeissy F, Abd-Elrahman KS, et al. Early metabolic impairment as a contributor to neurodegenerative disease: mechanisms and potential pharmacological intervention. Obes (Silver Spring). (2022) 30:982–93. doi: 10.1002/oby.23400
12. Livingston JM, McDonald MW, Gagnon T, Jeffers MS, Gomez-Smith M, Antonescu S, et al. Influence of metabolic syndrome on cerebral perfusion and cognition. Neurobiol Dis. (2020) 137:104756. doi: 10.1016/j.nbd.2020.104756
13. Kim YJ. Different effects of metabolic syndrome according to dementia type. Alzheimer’s Dementia. (2020) 16:e042370. doi: 10.1002/alz.042370
14. Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. Jama. (2004) 292:2237–42. doi: 10.1001/jama.292.18.2237
15. Kazlauskaite R, Janssen I, Wilson RS, Appelhans BM, Evans DA, Arvanitakis Z, et al. Is midlife metabolic syndrome associated with cognitive function change? The study of women’s health across the nation. J Clin Endocrinol Metab. (2020) 105:e1093–105. doi: 10.1210/clinem/dgaa067
16. Qureshi D, Collister J, Allen NE, Kuźma E, Littlejohns T. Association between metabolic syndrome and risk of incident dementia in uk biobank. Alzheimers Dement. (2024) 20:447–58. doi: 10.1002/alz.13439
17. Xiong X-Y, Tang Y, Yang Q-W. Metabolic changes favor the activity and heterogeneity of reactive astrocytes. Trends Endocrinol Metab. (2022) 33:390–400. doi: 10.1016/j.tem.2022.03.001
18. Ding ZB, Song LJ, Wang Q, Kumar G, Yan YQ, Ma CG. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regener Res. (2021) 16:1702–10. doi: 10.4103/1673-5374.306064
19. Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
20. Arnoriaga-Rodríguez M, Fernández-Real JM. Microbiota impacts on chronic inflammation and metabolic syndrome - related cognitive dysfunction. Rev Endocr Metab Disord. (2019) 20:473–80. doi: 10.1007/s11154-019-09537-5
21. Martin-Jiménez CA, García-Vega Á, Cabezas R, Aliev G, Echeverria V, González J, et al. Astrocytes and endoplasmic reticulum stress: A bridge between obesity and neurodegenerative diseases. Prog Neurobiol. (2017) 158:45–68. doi: 10.1016/j.pneurobio.2017.08.001
22. Uddin MS, Lim LW. Glial cells in Alzheimer’s disease: from neuropathological changes to therapeutic implications. Ageing Res Rev. (2022) 78:101622. doi: 10.1016/j.arr.2022.101622
23. Giovannoni F, Quintana FJ. The role of astrocytes in cns inflammation. Trends Immunol. (2020) 41:805–19. doi: 10.1016/j.it.2020.07.007
24. Alfaro FJ, Gavrieli A, Saade-Lemus P, Lioutas VA, Upadhyay J, Novak V. White matter microstructure and cognitive decline in metabolic syndrome: A review of diffusion tensor imaging. Metabolism. (2018) 78:52–68. doi: 10.1016/j.metabol.2017.08.009
25. Kotkowski E, Price LR, Franklin C, Salazar M, Woolsey M, DeFronzo RA, et al. A neural signature of metabolic syndrome. Hum Brain Mapp. (2019) 40:3575–88. doi: 10.1002/hbm.24617
26. Rashid B, Poole VN, Fortenbaugh FC, Esterman M, Milberg WP, McGlinchey RE, et al. Association between metabolic syndrome and resting-state functional brain connectivity. Neurobiol Aging. (2021) 104:1–9. doi: 10.1016/j.neurobiolaging.2021.03.012
27. Kouvari M, D’Cunha NM, Travica N, Sergi D, Zec M, Marx W, et al. Metabolic syndrome, cognitive impairment and the role of diet: A narrative review. Nutrients. (2022) 14:333. doi: 10.3390/nu14020333
28. van Sloten TT, Sedaghat S, Carnethon MR, Launer LJ, Stehouwer CDA. Cerebral microvascular complications of type 2 diabetes: stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. (2020) 8:325–36. doi: 10.1016/S2213-8587(19)30405-X
29. Borshchev YY, Uspensky YP, Galagudza MM. Pathogenetic pathways of cognitive dysfunction and dementia in metabolic syndrome. Life Sci. (2019) 237:116932. doi: 10.1016/j.lfs.2019.116932
30. Bendlin B, Willette A, Carlsson C, Johnson S, Davenport N, Birdsill A, et al. O3-06-06: metabolic syndrome in middle-aged adults is associated with brain perfusion deficits. Alzheimer’s Dementia. (2012) 8:P442–P. doi: 10.1016/j.jalz.2012.05.1176
31. Efimova I, Efimova N, Lishmanov Y. Cerebral blood flow and cognitive function in patients with metabolic syndrome: effect of antihypertensive therapy. J Clin Hypertension. (2014) 16:900–6. doi: 10.1111/jch.12435
32. Hughes TM, Lockhart SN, Suerken CK, Jung Y, Whitlow CT, Bateman JR, et al. Hypertensive aspects of cardiometabolic disorders are associated with lower brain microstructure, perfusion, and cognition. J Alzheimers Dis. (2022) 90:1589–99. doi: 10.3233/jad-220646
33. Avolio A, Kim MO, Adji A, Gangoda S, Avadhanam B, Tan I, et al. Cerebral haemodynamics: effects of systemic arterial pulsatile function and hypertension. Curr Hypertens Rep. (2018) 20:20. doi: 10.1007/s11906-018-0822-x
34. Schiffrin EL. How structure, mechanics, and function of the vasculature contribute to blood pressure elevation in hypertension. Can J Cardiol. (2020) 36:648–58. doi: 10.1016/j.cjca.2020.02.003
35. Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, et al. Downregulation of tmem16a calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. (2012) 125:697–707. doi: 10.1161/circulationaha.111.041806
36. Obadia N, Lessa MA, Daliry A, Silvares RR, Gomes F, Tibiriçá E, et al. Cerebral microvascular dysfunction in metabolic syndrome is exacerbated by ischemia-reperfusion injury. BMC Neurosci. (2017) 18:67. doi: 10.1186/s12868-017-0384-x
37. Jha SK, Jha NK, Kumar D, Ambasta RK, Kumar P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in neurodegeneration. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1132–46. doi: 10.1016/j.bbadis.2016.06.015
38. Kipinoinen T, Toppala S, Rinne JO, Viitanen MH, Jula AM, Ekblad LL. Association of midlife inflammatory markers with cognitive performance at 10-year follow-up. Neurology. (2022) 99:e2294–e302. doi: 10.1212/wnl.0000000000201116
39. Pedersen KM, Cordua S, Hasselbalch HC, Ellervik C. The association between circulating inflammatory markers and metabolic syndrome: A general population study. Blood. (2018) 132:4305. doi: 10.1182/blood-2018-99-120356
40. Caprio S, Perry R, Kursawe R. Adolescent obesity and insulin resistance: roles of ectopic fat accumulation and adipose inflammation. Gastroenterology. (2017) 152:1638–46. doi: 10.1053/j.gastro.2016.12.051
41. Boulangé CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. (2016) 8:42. doi: 10.1186/s13073-016-0303-2
42. Więckowska-Gacek A, Mietelska-Porowska A, Wydrych M, Wojda U. Western diet as a trigger of Alzheimer’s disease: from metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res Rev. (2021) 70:101397. doi: 10.1016/j.arr.2021.101397
43. Khan MSH, Hegde V. Obesity and diabetes mediated chronic inflammation: A potential biomarker in Alzheimer’s disease. J Pers Med. (2020) 10:42. doi: 10.3390/jpm10020042
44. Crispino M, Trinchese G, Penna E, Cimmino F, Catapano A, Villano I, et al. Interplay between peripheral and central inflammation in obesity-promoted disorders: the impact on synaptic mitochondrial functions. Int J Mol Sci. (2020) 21:5964. doi: 10.3390/ijms21175964
45. Aten S, Kiyoshi CM, Arzola EP, Patterson JA, Taylor AT, Du Y, et al. Ultrastructural view of astrocyte arborization, astrocyte-astrocyte and astrocyte-synapse contacts, intracellular vesicle-like structures, and mitochondrial network. Prog Neurobiol. (2022) 213:102264. doi: 10.1016/j.pneurobio.2022.102264
46. Nampoothiri S, Nogueiras R, Schwaninger M, Prevot V. Glial cells as integrators of peripheral and central signals in the regulation of energy homeostasis. Nat Metab. (2022) 4:813–25. doi: 10.1038/s42255-022-00610-z
47. García-Cáceres C, Balland E, Prevot V, Luquet S, Woods SC, Koch M, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci. (2019) 22:7–14. doi: 10.1038/s41593-018-0286-y
48. Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci. (2014) 17:908–10. doi: 10.1038/nn.3725
49. Dallérac G, Rouach N. Astrocytes as new targets to improve cognitive functions. Prog Neurobiol. (2016) 144:48–67. doi: 10.1016/j.pneurobio.2016.01.003
50. Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology. (2019) 27:663–77. doi: 10.1007/s10787-019-00580-x
51. Zhang H, Zheng Q, Guo T, Zhang S, Zheng S, Wang R, et al. Metabolic reprogramming in astrocytes results in neuronal dysfunction in intellectual disability. Mol Psychiatry. (2022). doi: 10.1038/s41380-022-01521-x
52. Popov A, Brazhe A, Denisov P, Sutyagina O, Li L, Lazareva N, et al. Astrocyte dystrophy in ageing brain parallels impaired synaptic plasticity. Aging Cell. (2021) 20:e13334. doi: 10.1111/acel.13334
53. Santello M, Toni N, Volterra A. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci. (2019) 22:154–66. doi: 10.1038/s41593-018-0325-8
54. Wade JJ, McDaid LJ, Harkin J, Crunelli V, Kelso JA. Bidirectional coupling between astrocytes and neurons mediates learning and dynamic coordination in the brain: A multiple modeling approach. PloS One. (2011) 6:e29445. doi: 10.1371/journal.pone.0029445
55. Tsybina Y, Kastalskiy I, Krivonosov M, Zaikin A, Kazantsev V, Gorban AN, et al. Astrocytes mediate analogous memory in a multi-layer neuron–astrocyte network. Neural Comput Appl. (2022) 34:9147–60. doi: 10.1007/s00521-022-06936-9
56. Gordleeva SY, Tsybina YA, Krivonosov MI, Ivanchenko MV, Zaikin AA, Kazantsev VB, et al. Modeling working memory in a spiking neuron network accompanied by astrocytes. Front Cell Neurosci. (2021) 15:631485. doi: 10.3389/fncel.2021.631485
57. Perea G, Gómez R, Mederos S, Covelo A, Ballesteros JJ, Schlosser L, et al. Activity-dependent switch of gabaergic inhibition into glutamatergic excitation in astrocyte-neuron networks. Elife. (2016) 5:e20362. doi: 10.7554/eLife.20362
58. Kol A, Adamsky A, Groysman M, Kreisel T, London M, Goshen I. Astrocytes contribute to remote memory formation by modulating hippocampal-cortical communication during learning. Nat Neurosci. (2020) 23:1229–39. doi: 10.1038/s41593-020-0679-6
59. Sardinha VM, Guerra-Gomes S, Caetano I, Tavares G, Martins M, Reis JS, et al. Astrocytic signaling supports hippocampal-prefrontal theta synchronization and cognitive function. Glia. (2017) 65:1944–60. doi: 10.1002/glia.23205
60. Marina N, Turovsky E, Christie IN, Hosford PS, Hadjihambi A, Korsak A, et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia. (2018) 66:1185–99. doi: 10.1002/glia.23283
61. Cooper ML, Pasini S, Lambert WS, D’Alessandro KB, Yao V, Risner ML, et al. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc Natl Acad Sci U.S.A. (2020) 117:18810–21. doi: 10.1073/pnas.2009425117
62. Welcome MO, Mastorakis NE. Emerging concepts in brain glucose metabolic functions: from glucose sensing to how the sweet taste of glucose regulates its own metabolism in astrocytes and neurons. Neuromol Med. (2018) 20:281–300. doi: 10.1007/s12017-018-8503-0
63. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
64. Dienel GA. Astrocytic energetics during excitatory neurotransmission: what are contributions of glutamate oxidation and glycolysis? Neurochem Int. (2013) 63:244–58. doi: 10.1016/j.neuint.2013.06.015
65. Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. (2019) 177:1522–35.e14. doi: 10.1016/j.cell.2019.04.001
66. Shen Z, Li ZY, Yu MT, Tan KL, Chen S. Metabolic perspective of astrocyte dysfunction in Alzheimer’s disease and type 2 diabetes brains. BioMed Pharmacother. (2023) 158:114206. doi: 10.1016/j.biopha.2022.114206
67. Gollihue JL, Norris CM. Astrocyte mitochondria: central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res Rev. (2020) 59:101039. doi: 10.1016/j.arr.2020.101039
68. Zehnder T, Petrelli F, Romanos J, De Oliveira Figueiredo EC, Lewis TL Jr., Déglon N, et al. Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. (2021) 35:108952. doi: 10.1016/j.celrep.2021.108952
69. Marina N, Christie IN, Korsak A, Doronin M, Brazhe A, Hosford PS, et al. Astrocytes monitor cerebral perfusion and control systemic circulation to maintain brain blood flow. Nat Commun. (2020) 11:131. doi: 10.1038/s41467-019-13956-y
70. Gordon GRJ, Mulligan SJ, MacVicar BA. Astrocyte control of the cerebrovasculature. Glia. (2007) 55:1214–21. doi: 10.1002/glia.20543
71. Cohen-Salmon M, Slaoui L, Mazaré N, Gilbert A, Oudart M, Alvear-Perez R, et al. Astrocytes in the regulation of cerebrovascular functions. Glia. (2021) 69:817–41. doi: 10.1002/glia.23924
72. Kim KJ, Ramiro Diaz J, Iddings JA, Filosa JA. Vasculo-neuronal coupling: retrograde vascular communication to brain neurons. J Neurosci. (2016) 36:12624–39. doi: 10.1523/jneurosci.1300-16.2016
73. Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, et al. Astrocytic tight junctions control inflammatory cns lesion pathogenesis. J Clin Invest. (2017) 127:3136–51. doi: 10.1172/jci91301
74. Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S. Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia. (2021) 69:436–72. doi: 10.1002/glia.23908
75. Pivoriūnas A, Verkhratsky A. Astrocyte-endotheliocyte axis in the regulation of the blood-brain barrier. Neurochem Res. (2021) 46:2538–50. doi: 10.1007/s11064-021-03338-6
76. Ezan P, André P, Cisternino S, Saubaméa B, Boulay AC, Doutremer S, et al. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. (2012) 32:1457–67. doi: 10.1038/jcbfm.2012.45
77. He JT, Li XY, Yang L, Zhao X. Astroglial connexins and cognition: memory formation or deterioration? Biosci Rep. (2020) 40:BSR20193510. doi: 10.1042/bsr20193510
78. Cheung G, Chever O, Rollenhagen A, Quenech’du N, Ezan P, Lübke JHR, et al. Astroglial connexin 43 regulates synaptic vesicle release at hippocampal synapses. Cells. (2023) 12:3501. doi: 10.3390/cells12081133
79. Hösli L, Binini N, Ferrari KD, Thieren L, Looser ZJ, Zuend M, et al. Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep. (2022) 38:110484. doi: 10.1016/j.celrep.2022.110484
80. MiChinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int J Mol Sci. (2019) 20:571. doi: 10.3390/ijms20030571
81. Osipova ED, Semyachkina-Glushkovskaya OV, Morgun AV, Pisareva NV, Malinovskaya NA, Boitsova EB, et al. Gliotransmitters and cytokines in the control of blood-brain barrier permeability. Rev Neurosci. (2018) 29:567–91. doi: 10.1515/revneuro-2017-0092
82. Hashioka S, Wu Z, Klegeris A. Glia-driven neuroinflammation and systemic inflammation in Alzheimer’s disease. Curr Neuropharmacol. (2021) 19:908–24. doi: 10.2174/1570159x18666201111104509
83. Treviño S, Díaz A, González-López G, Guevara J. Differential biochemical-inflammatory patterns in the astrocyte-neuron axis of the hippocampus and frontal cortex in wistar rats with metabolic syndrome induced by high fat or carbohydrate diets. J Chem Neuroanat. (2022) 126:102186. doi: 10.1016/j.jchemneu.2022.102186
84. Chowen JA, Argente-Arizón P, Freire-Regatillo A, Argente J. Sex differences in the neuroendocrine control of metabolism and the implication of astrocytes. Front Neuroendocrinol. (2018) 48:3–12. doi: 10.1016/j.yfrne.2017.05.003
85. Pucci G, Alcidi R, Tap L, Battista F, Mattace-Raso F, Schillaci G. Sex- and gender-related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: A review of the literature. Pharmacol Res. (2017) 120:34–42. doi: 10.1016/j.phrs.2017.03.008
86. Jaber SM, Bordt EA, Bhatt NM, Lewis DM, Gerecht S, Fiskum G, et al. Sex Differences in the Mitochondrial Bioenergetics of Astrocytes but Not Microglia at a Physiologically Relevant Brain Oxygen Tension. Neurochem Int. (2018) 117:82–90. doi: 10.1016/j.neuint.2017.09.003
87. Santos-Galindo M, Acaz-Fonseca E, Bellini MJ, Garcia-Segura LM. Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol Sex Differ. (2011) 2:7. doi: 10.1186/2042-6410-2-7
88. Chanana V, Tumturk A, Kintner D, Udho E, Ferrazzano P, Cengiz P. Sex differences in mouse hippocampal astrocytes after in-vitro ischemia. J Vis Exp. (2016) 116:53695. doi: 10.3791/53695
89. Hidalgo-Lanussa O, González Santos J, Barreto GE. Sex-specific vulnerabilities in human astrocytes underpin the differential impact of palmitic acid. Neurobiol Dis. (2024) 195:106489. doi: 10.1016/j.nbd.2024.106489
90. Diaz JR, Kim KJ, Brands MW, Filosa JA. Augmented astrocyte microdomain ca(2+) dynamics and parenchymal arteriole tone in angiotensin ii-infused hypertensive mice. Glia. (2019) 67:551–65. doi: 10.1002/glia.23564
91. Bhat SA, Goel R, Shukla R, Hanif K. Platelet cd40l induces activation of astrocytes and microglia in hypertension. Brain Behav Immun. (2017) 59:173–89. doi: 10.1016/j.bbi.2016.09.021
92. Li Y, Liu J, Gao D, Wei J, Yuan H, Niu X, et al. Age-related changes in hypertensive brain damage in the hippocampi of spontaneously hypertensive rats. Mol Med Rep. (2016) 13:2552–60. doi: 10.3892/mmr.2016.4853
93. Bhat SA, Goel R, Shukla S, Shukla R, Hanif K. Angiotensin receptor blockade by inhibiting glial activation promotes hippocampal neurogenesis via activation of wnt/B-catenin signaling in hypertension. Mol Neurobiol. (2018) 55:5282–98. doi: 10.1007/s12035-017-0754-5
94. Sabbatini M, Tomassoni D, Amenta F. Hypertensive brain damage: comparative evaluation of protective effect of treatment with dihydropyridine derivatives in spontaneously hypertensive rats. Mech Ageing Dev. (2001) 122:2085–105. doi: 10.1016/s0047-6374(01)00318-9
95. Levit A, Cheng S, Hough O, Liu Q, Agca Y, Agca C, et al. Hypertension and pathogenic happ independently induce white matter astrocytosis and cognitive impairment in the rat. Front Aging Neurosci. (2020) 12:82. doi: 10.3389/fnagi.2020.00082
96. Turovsky EA, Braga A, Yu Y, Esteras N, Korsak A, Theparambil SM, et al. Mechanosensory signaling in astrocytes. J Neurosci. (2020) 40:9364–71. doi: 10.1523/jneurosci.1249-20.2020
97. Su C, Xue J, Ye C, Chen A. Role of the central renin−Angiotensin system in hypertension (Review). Int J Mol Med. (2021) 47:95. doi: 10.3892/ijmm.2021.4928
98. Elsaafien K, de Kloet AD, Krause EG, Sumners C. Brain angiotensin type-1 and type-2 receptors in physiological and hypertensive conditions: focus on neuroinflammation. Curr Hypertens Rep. (2020) 22:48. doi: 10.1007/s11906-020-01062-0
99. Healy DP, Printz MP. Angiotensinogen levels in the brain and cerebrospinal fluid of the genetically hypertensive rat. Hypertension. (1985) 7:752–9. doi: 10.1161/01.hyp.7.5.752
100. Huang BS, Leenen FH. Brain “Ouabain” and angiotensin ii in salt-sensitive hypertension in spontaneously hypertensive rats. Hypertension. (1996) 28:1005–12. doi: 10.1161/01.hyp.28.6.1005
101. Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. (2013) 113:1087–96. doi: 10.1161/circresaha.113.301811
102. O’Connor AT, Clark MA. Astrocytes and the renin angiotensin system: relevance in disease pathogenesis. Neurochem Res. (2018) 43:1297–307. doi: 10.1007/s11064-018-2557-0
103. Park MH, Kim HN, Lim JS, Ahn JS, Koh JY. Angiotensin ii potentiates zinc-induced cortical neuronal death by acting on angiotensin ii type 2 receptor. Mol Brain. (2013) 6:50. doi: 10.1186/1756-6606-6-50
104. O’Connor AT, Clark MA. Angiotensin ii induces cyclooxygenase 2 expression in rat astrocytes via the angiotensin type 1 receptor. Neuropeptides. (2019) 77:101958. doi: 10.1016/j.npep.2019.101958
105. Gowrisankar YV, Clark MA. Angiotensin ii induces interleukin-6 expression in astrocytes: role of reactive oxygen species and nf-Kb. Mol Cell Endocrinol. (2016) 437:130–41. doi: 10.1016/j.mce.2016.08.013
106. Clark MA, Nguyen C, Tran H. Angiotensin iii induces C-jun N-terminal kinase leading to proliferation of rat astrocytes. Neurochem Res. (2012) 37:1475–81. doi: 10.1007/s11064-012-0738-9
107. Tsai SF, Hsu PL, Chen YW, Hossain MS, Chen PC, Tzeng SF, et al. High-fat diet induces depression-like phenotype via astrocyte-mediated hyperactivation of ventral hippocampal glutamatergic afferents to the nucleus accumbens. Mol Psychiatry. (2022) 27:4372–84. doi: 10.1038/s41380-022-01787-1
108. Li Y, Cheng Y, Zhou Y, Du H, Zhang C, Zhao Z, et al. High fat diet-induced obesity leads to depressive and anxiety-like behaviors in mice via ampk/mtor-mediated autophagy. Exp Neurol. (2022) 348:113949. doi: 10.1016/j.expneurol.2021.113949
109. Yu G, Cao F, Hou T, Cheng Y, Jia B, Yu L, et al. Astrocyte reactivation in medial prefrontal cortex contributes to obesity-promoted depressive-like behaviors. J Neuroinflamm. (2022) 19:166. doi: 10.1186/s12974-022-02529-4
110. Zhan M, Liu X, Xia X, Yang Y, Xie Y, Zhang L, et al. Promotion of Neuroinflammation by the Glymphatic System: A New Insight into Ethanol Extracts from Alisma Orientale in Alleviating Obesity-Associated Cognitive Impairment. Phytomedicine. (2024) 122:155147. doi: 10.1016/j.phymed.2023.155147
111. Lau BK, Murphy-Royal C, Kaur M, Qiao M, Bains JS, Gordon GR, et al. Obesity-induced astrocyte dysfunction impairs heterosynaptic plasticity in the orbitofrontal cortex. Cell Rep. (2021) 36:109563. doi: 10.1016/j.celrep.2021.109563
112. Calvo-Ochoa E, Hernández-Ortega K, Ferrera P, Morimoto S, Arias C. Short-term high-fat-and-fructose feeding produces insulin signaling alterations accompanied by neurite and synaptic reduction and astroglial activation in the rat hippocampus. J Cereb Blood Flow Metab. (2014) 34:1001–8. doi: 10.1038/jcbfm.2014.48
113. Liu WC, Wu CW, Tain YL, Fu MH, Hung CY, Chen IC, et al. Oral pioglitazone ameliorates fructose-induced peripheral insulin resistance and hippocampal gliosis but not restores inhibited hippocampal adult neurogenesis. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:274–85. doi: 10.1016/j.bbadis.2017.10.017
114. Kim N, Lee J, Song HS, Oh YJ, Kwon MS, Yun M, et al. Kimchi intake alleviates obesity-induced neuroinflammation by modulating the gut-brain axis. Food Res Int. (2022) 158:111533. doi: 10.1016/j.foodres.2022.111533
115. Jin S, Kim KK, Park BS, Kim DH, Jeong B, Kang D, et al. Function of astrocyte myd88 in high-fat-diet-induced hypothalamic inflammation. J Neuroinflamm. (2020) 17:195. doi: 10.1186/s12974-020-01846-w
116. Mongin AA. Astrocytes on “Cholesteroids”: the size- and function-promoting effects of a high-fat diet on hippocampal astroglia. Acta Physiol (Oxf). (2022) 236:e13859. doi: 10.1111/apha.13859
117. González-García I, García-Cáceres C. Hypothalamic astrocytes as a specialized and responsive cell population in obesity. Int J Mol Sci. (2021) 22:6176. doi: 10.3390/ijms22126176
118. Varela L, Kim JG, Fernández-Tussy P, Aryal B, Liu ZW, Fernández-Hernando C, et al. Astrocytic lipid metabolism determines susceptibility to diet-induced obesity. Sci Adv. (2021) 7:eabj2814. doi: 10.1126/sciadv.abj2814
119. Popov A, Brazhe N, Fedotova A, Tiaglik A, Bychkov M, Morozova K, et al. A high-fat diet changes astrocytic metabolism to promote synaptic plasticity and behavior. Acta Physiol (Oxf). (2022) 236:e13847. doi: 10.1111/apha.13847
120. Gao Y, Layritz C, Legutko B, Eichmann TO, Laperrousaz E, Moullé VS, et al. Disruption of lipid uptake in astroglia exacerbates diet-induced obesity. Diabetes. (2017) 66:2555–63. doi: 10.2337/db16-1278
121. Liu X, Zheng H. Leptin-mediated sympathoexcitation in obese rats: role for neuron-astrocyte crosstalk in the arcuate nucleus. Front Neurosci. (2019) 13:1217. doi: 10.3389/fnins.2019.01217
122. Dalvi PS, Chalmers JA, Luo V, Han DY, Wellhauser L, Liu Y, et al. High fat induces acute and chronic inflammation in the hypothalamus: effect of high-fat diet, palmitate and tnf-A on appetite-regulating npy neurons. Int J Obes (Lond). (2017) 41:149–58. doi: 10.1038/ijo.2016.183
123. Lorenzo PI, Martin Vazquez E, López-Noriega L, Fuente-Martín E, Mellado-Gil JM, Franco JM, et al. The metabesity factor hmg20a potentiates astrocyte survival and reactive astrogliosis preserving neuronal integrity. Theranostics. (2021) 11:6983–7004. doi: 10.7150/thno.57237
124. Lam YY, Tsai SF, Chen PC, Kuo YM, Chen YW. Pioglitazone rescues high-fat diet-induced depression-like phenotypes and hippocampal astrocytic deficits in mice. BioMed Pharmacother. (2021) 140:111734. doi: 10.1016/j.biopha.2021.111734
125. Cassano V, Tallarico M, Armentaro G, De Sarro C, Iannone M, Leo A, et al. Ranolazine attenuates brain inflammation in a rat model of type 2 diabetes. Int J Mol Sci. (2022) 23:16160. doi: 10.3390/ijms232416160
126. Saieva S, Taglialatela G. Near-infrared light reduces glia activation and modulates neuroinflammation in the brains of diet-induced obese mice. Sci Rep. (2022) 12:10848. doi: 10.1038/s41598-022-14812-8
127. Rahman MH, Bhusal A, Kim JH, Jha MK, Song GJ, Go Y, et al. Astrocytic pyruvate dehydrogenase kinase-2 is involved in hypothalamic inflammation in mouse models of diabetes. Nat Commun. (2020) 11:5906. doi: 10.1038/s41467-020-19576-1
128. Bhusal A, Rahman MH, Lee IK, Suk K. Role of hippocampal lipocalin-2 in experimental diabetic encephalopathy. Front Endocrinol (Lausanne). (2019) 10:25. doi: 10.3389/fendo.2019.00025
129. Tomassoni D, Nwankwo IE, Gabrielli MG, Bhatt S, Muhammad AB, Lokhandwala MF, et al. Astrogliosis in the brain of obese zucker rat: A model of metabolic syndrome. Neurosci Lett. (2013) 543:136–41. doi: 10.1016/j.neulet.2013.03.025
130. Shi S, Yin HJ, Li J, Wang L, Wang WP, Wang XL. Studies of pathology and pharmacology of diabetic encephalopathy with kk-ay mouse model. CNS Neurosci Ther. (2020) 26:332–42. doi: 10.1111/cns.13201
131. Zheng Y, Yang Y, Dong B, Zheng H, Lin X, Du Y, et al. Metabonomic profiles delineate potential role of glutamate-glutamine cycle in db/db mice with diabetes-associated cognitive decline. Mol Brain. (2016) 9:40. doi: 10.1186/s13041-016-0223-5
132. An JR, Liu JT, Gao XM, Wang QF, Sun GY, Su JN, et al. Effects of liraglutide on astrocyte polarization and neuroinflammation in db/db mice: focus on iron overload and oxidative stress. Front Cell Neurosci. (2023) 17:1136070. doi: 10.3389/fncel.2023.1136070
133. Zheng J, Xie Y, Ren L, Qi L, Wu L, Pan X, et al. Glp-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol Metab. (2021) 47:101180. doi: 10.1016/j.molmet.2021.101180
134. Robb JL, Hammad NA, Weightman Potter PG, Chilton JK, Beall C, Ellacott KLJ. The metabolic response to inflammation in astrocytes is regulated by nuclear factor-kappa B signaling. Glia. (2020) 68:2246–63. doi: 10.1002/glia.23835
135. Robb JL, Morrissey NA, Weightman Potter PG, Smithers HE, Beall C, Ellacott KLJ. Immunometabolic changes in glia - a potential role in the pathophysiology of obesity and diabetes. Neuroscience. (2020) 447:167–81. doi: 10.1016/j.neuroscience.2019.10.021
136. Li W, Roy Choudhury G, Winters A, Prah J, Lin W, Liu R, et al. Hyperglycemia alters astrocyte metabolism and inhibits astrocyte proliferation. Aging Dis. (2018) 9:674–84. doi: 10.14336/ad.2017.1208
137. Wang D, Zhao L, Zheng H, Dong M, Pan L, Zhang X, et al. Time-dependent lactate production and amino acid utilization in cultured astrocytes under high glucose exposure. Mol Neurobiol. (2018) 55:1112–22. doi: 10.1007/s12035-016-0360-y
138. Staricha K, Meyers N, Garvin J, Liu Q, Rarick K, Harder D, et al. Effect of high glucose condition on glucose metabolism in primary astrocytes. Brain Res. (2020) 1732:146702. doi: 10.1016/j.brainres.2020.146702
139. Takahashi S, Izawa Y, Suzuki N. Astroglial pentose phosphate pathway rates in response to high-glucose environments. ASN Neuro. (2012) 4:e00078. doi: 10.1042/an20120002
140. Abdyeva A, Kurtova E, Savinkova I, Galkov M, Gorbacheva L. Long-term exposure of cultured astrocytes to high glucose impact on their lps-induced activation. Int J Mol Sci. (2024) 25:1122. doi: 10.3390/ijms25021122
141. Bahniwal M, Little JP, Klegeris A. High glucose induces pro-inflammatory phenotype in human astrocytes and enhances neurotoxicity. FASEB J. (2013) 27:691. doi: 10.1096/fasebj.27.1_supplement.691.8
142. Wanrooy BJ, Kumar KP, Wen SW, Qin CX, Ritchie RH, Wong CHY. Distinct contributions of hyperglycemia and high-fat feeding in metabolic syndrome-induced neuroinflammation. J Neuroinflamm. (2018) 15:293. doi: 10.1186/s12974-018-1329-8
143. Hsieh HL, Wang HH, Wu WB, Chu PJ, Yang CM. Transforming growth factor-B1 induces matrix metalloproteinase-9 and cell migration in astrocytes: roles of ros-dependent erk- and jnk-nf-Kb pathways. J Neuroinflamm. (2010) 7:88. doi: 10.1186/1742-2094-7-88
144. Hsieh HL, Lin CC, Hsiao LD, Yang CM. High glucose induces reactive oxygen species-dependent matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Mol Neurobiol. (2013) 48:601–14. doi: 10.1007/s12035-013-8442-6
145. Chistyakov DV, Azbukina NV, Astakhova AA, Polozhintsev AI, Sergeeva MG, Reiser G. Toll-like receptors control P38 and jnk mapk signaling pathways in rat astrocytes differently, when cultured in normal or high glucose concentrations. Neurochem Int. (2019) 131:104513. doi: 10.1016/j.neuint.2019.104513
146. Gandhi GK, Ball KK, Cruz NF, Dienel GA. Hyperglycaemia and diabetes impair gap junctional communication among astrocytes. ASN Neuro. (2010) 2:e00030. doi: 10.1042/an20090048
147. Garvin J, Semenikhina M, Liu Q, Rarick K, Isaeva E, Levchenko V, et al. Astrocytic responses to high glucose impair barrier formation in cerebral microvessel endothelial cells. Am J Physiol Regul Integr Comp Physiol. (2022) 322:R571–r80. doi: 10.1152/ajpregu.00315.2020
148. Zhao Y, Luo C, Chen J, Sun Y, Pu D, Lv A, et al. High glucose-induced complement component 3 up-regulation via rage-P38mapk-nf-Kb signalling in astrocytes: in vivo and in vitro studies. J Cell Mol Med. (2018) 22:6087–98. doi: 10.1111/jcmm.13884
149. Kirschen GW, Kéry R, Ge S. The hippocampal neuro-glio-vascular network: metabolic vulnerability and potential neurogenic regeneration in disease. Brain Plast. (2018) 3:129–44. doi: 10.3233/bpl-170055
150. Lawal O, Ulloa Severino FP, Eroglu C. The role of astrocyte structural plasticity in regulating neural circuit function and behavior. Glia. (2022) 70:1467–83. doi: 10.1002/glia.24191
151. Mills WA 3rd, Woo AM, Jiang S, Martin J, Surendran D, Bergstresser M, et al. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat Commun. (2022) 13:1794. doi: 10.1038/s41467-022-29475-2
152. Hayden MR. Hypothesis: astrocyte foot processes detachment from the neurovascular unit in female diabetic mice may impair modulation of information processing-six degrees of separation. Brain Sci. (2019) 9:83. doi: 10.3390/brainsci9040083
153. Stranahan AM. Visceral adiposity, inflammation, and hippocampal function in obesity. Neuropharmacology. (2022) 205:108920. doi: 10.1016/j.neuropharm.2021.108920
154. de Paula GC, Brunetta HS, Engel DF, Gaspar JM, Velloso LA, Engblom D, et al. Hippocampal function is impaired by a short-term high-fat diet in mice: increased blood-brain barrier permeability and neuroinflammation as triggering events. Front Neurosci. (2021) 15:734158. doi: 10.3389/fnins.2021.734158
155. Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Sotonyi P, Shanabrough M, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U.S.A. (2010) 107:14875–80. doi: 10.1073/pnas.1004282107
156. Gruber T, Pan C, Contreras RE, Wiedemann T, Morgan DA, Skowronski AA, et al. Obesity-associated hyperleptinemia alters the gliovascular interface of the hypothalamus to promote hypertension. Cell Metab. (2021) 33:1155–70.e10. doi: 10.1016/j.cmet.2021.04.007
157. Yamagata K, Tagami M, Nara Y, Fujino H, Kubota A, Numano F, et al. Faulty induction of blood-brain barrier functions by astrocytes isolated from stroke-prone spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. (1997) 24:686–91. doi: 10.1111/j.1440-1681.1997.tb02113.x
158. Tsai SF, Wu HT, Chen PC, Chen YW, Yu M, Wang TF, et al. High-fat diet suppresses the astrocytic process arborization and downregulates the glial glutamate transporters in the hippocampus of mice. Brain Res. (2018) 1700:66–77. doi: 10.1016/j.brainres.2018.07.017
159. Cano V, Valladolid-Acebes I, Hernández-Nuño F, Merino B, del Olmo N, Chowen JA, et al. Morphological changes in glial fibrillary acidic protein immunopositive astrocytes in the hippocampus of dietary-induced obese mice. NeuroReport. (2014) 25:819–22. doi: 10.1097/WNR.0000000000000180
160. Zhang Y, Reichel JM, Han C, Zuniga-Hertz JP, Cai D. Astrocytic process plasticity and ikkβ/nf-Kb in central control of blood glucose, blood pressure, and body weight. Cell Metab. (2017) 25:1091–102.e4. doi: 10.1016/j.cmet.2017.04.002
161. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. (2020) 9:42. doi: 10.1186/s40035-020-00221-2
162. Williams JL, Manivasagam S, Smith BC, Sim J, Vollmer LL, Daniels BP, et al. Astrocyte-T cell crosstalk regulates region-specific neuroinflammation. Glia. (2020) 68:1361–74. doi: 10.1002/glia.23783
163. Bhusal A, Afridi R, Lee WH, Suk K. Bidirectional communication between microglia and astrocytes in neuroinflammation. Curr Neuropharmacol. (2023) 21:2020–9. doi: 10.2174/1570159x21666221129121715
164. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
165. Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, Zimmer ER. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med. (2019) 25:77–95. doi: 10.1016/j.molmed.2018.11.006
166. Bruna B, João Pedro F-S, Lucas Uglione da R, Stephen FC, Elena R-V, Agneta N, et al. Astrocyte biomarkers in Alzheimer disease. Neurology. (2021) 96:e2944. doi: 10.1212/WNL.0000000000012109
167. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/jneurosci.2117-15.2016
168. Du M, Jiang T, He S, Cheng B, Zhang X, Li L, et al. Sigma-1 receptor as a protective factor for diabetes-associated cognitive dysfunction via regulating astrocytic endoplasmic reticulum-mitochondrion contact and endoplasmic reticulum stress. Cells. (2023) 12:197. doi: 10.3390/cells12010197
169. Meng F, Fu J, Zhang L, Guo M, Zhuang P, Yin Q, et al. Function and therapeutic value of astrocytes in diabetic cognitive impairment. Neurochem Int. (2023) 169:105591. doi: 10.1016/j.neuint.2023.105591
170. Afsari ZH, Renno WM, Abd-El-Basset E. Alteration of glial fibrillary acidic proteins immunoreactivity in astrocytes of the spinal cord diabetic rats. Anat Rec (Hoboken). (2008) 291:390–9. doi: 10.1002/ar.20678
171. Bondan E, Cardoso C, Martins MF, Otton R. Obesity in rats causes hippocampal astrogliosis. J Comp Pathol. (2019) 166:148. doi: 10.1016/j.jcpa.2018.10.158
172. Buckman LB, Thompson MM, Moreno HN, Ellacott KL. Regional astrogliosis in the mouse hypothalamus in response to obesity. J Comp Neurol. (2013) 521:1322–33. doi: 10.1002/cne.23233
173. Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP. Astrocyte ikkβ/nf-Kb signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab. (2017) 6:366–73. doi: 10.1016/j.molmet.2017.01.010
174. Lin L, Basu R, Chatterjee D, Templin AT, Flak JN, Johnson TS. Disease-associated astrocytes and microglia markers are upregulated in mice fed high fat diet. Sci Rep. (2023) 13:12919. doi: 10.1038/s41598-023-39890-0
175. Wasilewski D, Villalba-Moreno ND, Stange I, Glatzel M, Sepulveda-Falla D, Krasemann S. Reactive astrocytes contribute to Alzheimer’s disease-related neurotoxicity and synaptotoxicity in a neuron-astrocyte co-culture assay. Front Cell Neurosci. (2021) 15:739411. doi: 10.3389/fncel.2021.739411
176. Saggu R, Schumacher T, Gerich F, Rakers C, Tai K, Delekate A, et al. Astroglial nf-kb contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. (2016) 4:76. doi: 10.1186/s40478-016-0350-3
177. Sadick JS, Liddelow SA. Don’t forget astrocytes when targeting Alzheimer’s disease. Br J Pharmacol. (2019) 176:3585–98. doi: 10.1111/bph.14568
178. Henn RE, Noureldein MH, Elzinga SE, Kim B, Savelieff MG, Feldman EL. Glial-neuron crosstalk in health and disease: A focus on metabolism, obesity, and cognitive impairment. Neurobiol Dis. (2022) 170:105766. doi: 10.1016/j.nbd.2022.105766
179. Chen Z, Yuan Z, Yang S, Zhu Y, Xue M, Zhang J, et al. Brain energy metabolism: astrocytes in neurodegenerative diseases. CNS Neurosci Ther. (2023) 29:24–36. doi: 10.1111/cns.13982
180. Zyśk M, Beretta C, Naia L, Dakhel A, Påvénius L, Brismar H, et al. Amyloid-B Accumulation in human astrocytes induces mitochondrial disruption and changed energy metabolism. J Neuroinflamm. (2023) 20:43. doi: 10.1186/s12974-023-02722-z
181. Morita M, Ikeshima-Kataoka H, Kreft M, Vardjan N, Zorec R, Noda M. Metabolic plasticity of astrocytes and aging of the brain. Int J Mol Sci. (2019) 20:941. doi: 10.3390/ijms20040941
182. Heni M, Eckstein SS, Schittenhelm J, Böhm A, Hogrefe N, Irmler M, et al. Ectopic fat accumulation in human astrocytes impairs insulin action. R Soc Open Sci. (2020) 7:200701. doi: 10.1098/rsos.200701
183. Castellanos DB, Martín-Jiménez CA, Pinzón A, Barreto GE, Padilla-González GF, Aristizábal A, et al. Metabolomic analysis of human astrocytes in lipotoxic condition: potential biomarker identification by machine learning modeling. Biomolecules. (2022) 12:986. doi: 10.3390/biom12070986
184. van Deijk AF, Camargo N, Timmerman J, Heistek T, Brouwers JF, Mogavero F, et al. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia. (2017) 65:670–82. doi: 10.1002/glia.23120
185. Ferris HA, Perry RJ, Moreira GV, Shulman GI, Horton JD, Kahn CR. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc Natl Acad Sci U.S.A. (2017) 114:1189–94. doi: 10.1073/pnas.1620506114
186. Cao Y, Wang G, Li Z. The effects of antidiabetic drugs on cerebral metabolism in mice with central insulin resistance. Diabetes. (2018) 67:247–LB. doi: 10.2337/db18-247-LB
187. Fernandez AM, Hernandez-Garzón E, Perez-Domper P, Perez-Alvarez A, Mederos S, Matsui T, et al. Insulin regulates astrocytic glucose handling through cooperation with igf-I. Diabetes. (2017) 66:64–74. doi: 10.2337/db16-0861
188. Cai W, Xue C, Sakaguchi M, Konishi M, Shirazian A, Ferris HA, et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J Clin Invest. (2018) 128:2914–26. doi: 10.1172/jci99366
189. Mitra S, Banik A, Saurabh S, Maulik M, Khatri SN. Neuroimmunometabolism: A new pathological nexus underlying neurodegenerative disorders. J Neurosci. (2022) 42:1888–907. doi: 10.1523/jneurosci.0998-21.2022
190. Vizuete AFK, Fróes F, Seady M, Zanotto C, Bobermin LD, Roginski AC, et al. Early effects of lps-induced neuroinflammation on the rat hippocampal glycolytic pathway. J Neuroinflamm. (2022) 19:255. doi: 10.1186/s12974-022-02612-w
191. Iglesias J, Morales L, Barreto GE. Metabolic and inflammatory adaptation of reactive astrocytes: role of ppars. Mol Neurobiol. (2017) 54:2518–38. doi: 10.1007/s12035-016-9833-2
192. Bolaños JP. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J Neurochem. (2016) 139 Suppl 2:115–25. doi: 10.1111/jnc.13486
193. Meyer T, Shimon D, Youssef S, Yankovitz G, Tessler A, Chernobylsky T, et al. Nad(+) metabolism drives astrocyte proinflammatory reprogramming in central nervous system autoimmunity. Proc Natl Acad Sci U.S.A. (2022) 119:e2211310119. doi: 10.1073/pnas.2211310119
194. Galea E, Weinstock LD, Larramona-Arcas R, Pybus AF, Giménez-Llort L, Escartin C, et al. Multi-transcriptomic analysis points to early organelle dysfunction in human astrocytes in Alzheimer’s disease. Neurobiol Dis. (2022) 166:105655. doi: 10.1016/j.nbd.2022.105655
195. Bolsewig K, Hok-A-Hin YS, Sepe FN, Boonkamp L, Jacobs D, Bellomo G, et al. A panel of novel astrocytic and synaptic biomarkers in serum and csf for the differential diagnosis of frontotemporal dementia. Alzheimer’s Dementia. (2021) 17:e051338. doi: 10.1002/alz.051338
196. Ferrari-Souza JP, Ferreira PCL, Bellaver B, Tissot C, Wang YT, Leffa DT, et al. Astrocyte biomarker signatures of amyloid-B and tau pathologies in Alzheimer’s disease. Mol Psychiatry. (2022) 27:4781–9. doi: 10.1038/s41380-022-01716-2
197. Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. (2020) 26:769–80. doi: 10.1038/s41591-020-0815-6
198. Wang X, Yang H, Liu C, Liu K. A new diagnostic tool for brain disorders: extracellular vesicles derived from neuron, astrocyte, and oligodendrocyte. Front Mol Neurosci. (2023) 16:1194210. doi: 10.3389/fnmol.2023.1194210
199. Gayen M, Bhomia M, Balakathiresan N, Knollmann-Ritschel B. Exosomal micrornas released by activated astrocytes as potential neuroinflammatory biomarkers. Int J Mol Sci. (2020) 21:2312. doi: 10.3390/ijms21072312
200. Giannoni P, Arango-Lievano M, Neves ID, Rousset MC, Baranger K, Rivera S, et al. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol Dis. (2016) 88:107–17. doi: 10.1016/j.nbd.2016.01.001
201. Selvakumar R, Muhammad MR. Diagnostic computational model for neuronal disorder through glycogen metabolism in astrocytes. Int J Comput Sci Math. (2018) 9:232–9. doi: 10.1504/IJCSM.2018.093151
202. Gorshkov K, Aguisanda F, Thorne N, Zheng W. Astrocytes as targets for drug discovery. Drug Discovery Today. (2018) 23:673–80. doi: 10.1016/j.drudis.2018.01.011
203. Stogsdill JA, Harwell CC, Goldman SA. Astrocytes as master modulators of neural networks: synaptic functions and disease-associated dysfunction of astrocytes. Ann N Y Acad Sci. (2023) 1525:41–60. doi: 10.1111/nyas.15004
204. Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in cns inflammation. Neuron. (2020) 108:608–22. doi: 10.1016/j.neuron.2020.08.012
205. Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. (2003) 43:281–91. doi: 10.1002/glia.10256
206. Nakano-Kobayashi A, Canela A, Yoshihara T, Hagiwara M. Astrocyte-targeting therapy rescues cognitive impairment caused by neuroinflammation via the nrf2 pathway. Proc Natl Acad Sci U.S.A. (2023) 120:e2303809120. doi: 10.1073/pnas.2303809120
207. Klegeris A. Targeting neuroprotective functions of astrocytes in neuroimmune diseases. Expert Opin Ther Targets. (2021) 25:237–41. doi: 10.1080/14728222.2021.1915993
208. De Bock M, De Smet M, Verwaerde S, Tahiri H, Schumacher S, Van Haver V, et al. Targeting gliovascular connexins prevents inflammatory blood-brain barrier leakage and astrogliosis. JCI Insight. (2022) 7:e135263. doi: 10.1172/jci.insight.135263
209. de Pablo Y, Chen M, Möllerström E, Pekna M, Pekny M. Drugs targeting intermediate filaments can improve neurosupportive properties of astrocytes. Brain Res Bull. (2018) 136:130–8. doi: 10.1016/j.brainresbull.2017.01.021
210. Barros LF, Schirmeier S, Weber B. The astrocyte: metabolic hub of the brain. Cold Spring Harb Perspect Biol. (2024). doi: 10.1101/cshperspect.a041355
211. Beard E, Lengacher S, Dias S, Magistretti PJ, Finsterwald C. Astrocytes as key regulators of brain energy metabolism: new therapeutic perspectives. Front Physiol. (2021) 12:825816. doi: 10.3389/fphys.2021.825816
212. Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. (2018) 19:235–49. doi: 10.1038/nrn.2018.19
213. Horvat A, Vardjan N, Zorec R. Targeting astrocytes for treating neurological disorders: carbon monoxide and noradrenaline-induced increase in lactate. Curr Pharm Des. (2017) 23:4969–78. doi: 10.2174/1381612823666170622112734
214. Hohnholt MC, Blumrich EM, Waagepetersen HS, Dringen R. The antidiabetic drug metformin decreases mitochondrial respiration and tricarboxylic acid cycle activity in cultured primary rat astrocytes. J Neurosci Res. (2017) 95:2307–20. doi: 10.1002/jnr.24050
215. Pilipenko V, Narbute K, Pupure J, Langrate IK, Muceniece R, Kluša V. Neuroprotective potential of antihyperglycemic drug metformin in streptozocin-induced rat model of sporadic Alzheimer’s disease. Eur J Pharmacol. (2020) 881:173290. doi: 10.1016/j.ejphar.2020.173290
216. Monney M, Jornayvaz FR, Gariani K. Glp-1 receptor agonists effect on cognitive function in patients with and without type 2 diabetes. Diabetes Metab. (2023) 49:101470. doi: 10.1016/j.diabet.2023.101470
217. Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In Alzheimer’s disease, 6-month treatment with glp-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci. (2016) 8:108. doi: 10.3389/fnagi.2016.00108
218. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
219. Colin A, Faideau M, Dufour N, Auregan G, Hassig R, Andrieu T, et al. Engineered lentiviral vector targeting astrocytes in vivo. Glia. (2009) 57:667–79. doi: 10.1002/glia.20795
220. Wang L-L, Serrano C, Zhong X, Ma S, Zou Y, Zhang C-L. Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell. (2021) 184:5465–81.e16. doi: 10.1016/j.cell.2021.09.005
221. Farhangi S, Dehghan S, Totonchi M, Javan M. In vivo conversion of astrocytes to oligodendrocyte lineage cells in adult mice demyelinated brains by sox2. Mult Scler Relat Disord. (2019) 28:263–72. doi: 10.1016/j.msard.2018.12.041
222. Liu X, Li C, Li J, Xie L, Hong Z, Zheng K, et al. Egf signaling promotes the lineage conversion of astrocytes into oligodendrocytes. Mol Med. (2022) 28:50. doi: 10.1186/s10020-022-00478-5
223. Zare L, Baharvand H, Javan M. In vivo conversion of astrocytes to oligodendrocyte lineage cells using chemicals: targeting gliosis for myelin repair. Regener Med. (2018) 13:803–19. doi: 10.2217/rme-2017-0155
224. Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. (2010) 20:588–94. doi: 10.1016/j.conb.2010.06.005
225. Hoang T, Kim DW, Appel H, Ozawa M, Zheng S, Kim J, et al. Ptbp1 deletion does not induce astrocyte-to-neuron conversion. Nature. (2023) 618:E1–e7. doi: 10.1038/s41586-023-06066-9
Keywords: metabolic syndrome, cognitive dysfunction, astrocytes, immunometabolism, neurodegeneration
Citation: Li Z, Jiang Y, Long C, Peng X, Tao J, Pu Y and Yue R (2024) Bridging metabolic syndrome and cognitive dysfunction: role of astrocytes. Front. Endocrinol. 15:1393253. doi: 10.3389/fendo.2024.1393253
Received: 28 February 2024; Accepted: 25 April 2024;
Published: 10 May 2024.
Edited by:
Lin Zhu, Vanderbilt University Medical Center, United StatesReviewed by:
Carolina Dalmasso, University of Kentucky, United StatesCopyright © 2024 Li, Jiang, Long, Peng, Tao, Pu and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Rensong, c29uZ3Jlbnl1ZUBjZHV0Y20uZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.