 Quratul Ain1,2Matija Cevc3Tatiana Marusic4Jaka Sikonja5,6Fouzia Sadiq2Ursa Sustar4Matej Mlinaric4Jernej Kovac4Hijab Batool7Mohammad Iqbal Khan2,8Katarina Trebusak Podkrajsek4,6Barbara Jenko Bizjan4Tadej Battelino4,6Zlatko Fras4,6Muhammad Ajmal1Urh Groselj4,6*
Quratul Ain1,2Matija Cevc3Tatiana Marusic4Jaka Sikonja5,6Fouzia Sadiq2Ursa Sustar4Matej Mlinaric4Jernej Kovac4Hijab Batool7Mohammad Iqbal Khan2,8Katarina Trebusak Podkrajsek4,6Barbara Jenko Bizjan4Tadej Battelino4,6Zlatko Fras4,6Muhammad Ajmal1Urh Groselj4,6*- 1Translational Genomics Laboratory, Department of Biosciences, COMSATS University Islamabad, Islamabad, Pakistan
- 2Directorate of Research, Shifa Tameer-e-Millat University, Islamabad, Pakistan
- 3Division of Medicine, Centre for Preventive Cardiology, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 4Department of Endocrinology, Diabetes, and Metabolic Diseases, University Children’s Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 5Department of Endocrinology, Diabetes and Metabolic Diseases, Division of Medicine, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 6Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 7Department of Clinical Chemistry and Immunology, Chughtai Institute of Pathology, Lahore, Pakistan
- 8Department of Vascular Surgery, Shifa International Hospital, Islamabad, Pakistan
Introduction: Hypertriglyceridemia (HTG) is a complex disorder caused by genetic and environmental factors that frequently results from loss-of-function variants in the gene encoding lipoprotein lipase (LPL). Heterozygous patients have a range of symptoms, while homozygous LPL deficiency presents with severe symptoms including acute pancreatitis, xanthomas, and lipemia retinalis.
Methods: We described the clinical characteristics of three Slovenian patients (an 8-year-old female, an 18-year-old man, and a 57-year-old female) and one Pakistani patient (a 59-year-old male) with LPL deficiency. We performed next-generation sequencing (NGS) targeting all coding exons and intron-exon boundaries of the LPL gene, and Sanger sequencing for variant confirmation. In addition, we performed a systematic literature review of all cases with three identified variants and described their clinical characteristics.
Results: Two Slovenian patients with a heterozygous pathogenic variant NM_000237.3:c.984G>T (p.Met328Ile) were diagnosed within the first three years of life and had triglyceride (TG) values of 16 and 20 mmol/L. An asymptomatic Pakistani patient with TG values of 36.8 mmol/L until the age of 44 years, was identified as heterozygous for a pathogenic variant NM_000237.3:c.724G>A (p.Asp242Asn). His TG levels dropped to 12.7 mmol/L on dietary modifications and by using fibrates. A Slovenian patient who first suffered from pancreatitis at the age of 18 years with a TG value of 34 mmol/L was found to be homozygous for NM_000237.3:c.337T>C (p.Trp113Arg).
Conclusions: Patients with LPL deficiency had high TG levels at diagnosis. Homozygous patients had worse outcomes. Good diet and medication compliance can reduce severity.
1 Introduction
Hypertriglyceridemia (HTG) is a multifaceted lipid metabolism disorder, influenced by genetic and environmental factors. It is estimated that approximately 30% of the global population is affected by HTG i.e. triglycerides (TG) more than 150 mg/dL, with men (37%) being more commonly affected than women (23%) (1). TG levels between 150–199 mg/dL are classified as mild HTG, while levels ranging from 200–999 mg/dL indicate moderate HTG. Severe HTG is characterized by levels between 1000–1999 mg/dL, and very severe HTG is defined as levels ≥2000 mg/dL (2). While only a small percentage (1.1%) of individuals exhibit biallelic or homozygous monogenic HTG, also known as familial chylomicronemia (FCS), the majority of cases (46.3%) are attributed to polygenic HTG while the remaining 52.6% HTG cases are genetically unidentified (3). The majority of FCS cases (around 95%) are associated with specific pathogenic variants of the lipoprotein lipase (LPL) gene, and 5% of FCS cases are linked to variants in four other genes (APOC, APOA5, LMF1, and GPIHBP1), encoding proteins that modulate the activity of LPL (4, 5) Multifactorial chylomicronemia syndrome (MCS) is a common cause of severe hypertriglyceridemia, linked to risks of acute pancreatitis, cardiovascular issues, and non-alcoholic steatohepatitis. Moulin et al. (6) have proposed a diagnostic algorithm to differentiate FCS from MCS. FCS score is calculated based upon certain clinical features of patients i.e., the severe elevation of plasma TGs, resistance to standard TG-lowering therapies, young age at onset, absence of secondary factors, and a history of acute pancreatitis episodes. If the score is 10 or higher, FCS is highly probable; if it’s 9 or lower, FCS diagnosis is unlikely, and if it’s 8 or lower, FCS is highly unlikely (6).
LPL is located on vascular endothelial cells in adipose tissue, the heart, and muscles, hydrolyzing TG-rich lipoproteins, such as chylomicrons and very low-density lipoproteins (VLDL), facilitating their clearance from the blood (7). To date, 250 distinct variants have been reported in the LPL gene, among these the most common being missense/nonsense pathogenic variants (8, 9). Diagnosis of LPL deficiency can be challenging due to variations in biochemical parameters and often overlapping phenotypes with other etiologically different HTGs (10).
LPL deficiency (OMIM #238600) is characterised by the accumulation of TG-rich lipoproteins (11). Individuals with partial LPL deficiency, due to heterozygous variants, have reduced LPL enzyme activity. This leads to elevated postprandial TG levels and an increased risk of pancreatitis and cardiovascular disease. While, patients with homozygous LPL variants experience complete LPL deficiency, resulting in no LPL enzyme activity. This leads to severe HTG and associated complications such as pancreatitis, abdominal pain, eruptive xanthomas, and hepatosplenomegaly, which are unresponsive to standard treatments (6, 12, 13).
The primary objective of our current study is to comprehensively characterise the genetic and clinical features of four patients diagnosed with LPL deficiency, from both Slovenia and Pakistan. This will provide insight into how these variants may contribute to the development and progression of LPL deficiency. By comparing the clinical profiles of these patients, we seek to identify influences on the disease presentation, severity, and associated complications. Additionally, we conducted a systematic review of the existing literature to further explore the clinical characteristics of patients, associated with identified LPL genetic variants, in our study. We aim to improve the understanding of the intricate interplay between genetics and clinical manifestations of LPL deficiency, ultimately contributing to improved disease management.
2 Materials and methods
2.1 Study design and cohort description
In this retrospective observational study, we examined the medical health records of four unrelated patients diagnosed with HTG. Patients 1, 2, and 4 are an 8-year-old female and an 18-year-old male, and 57 years old female were managed at the University Medical Centre Ljubljana, Slovenia, while patient 3 aged 59 years old male was managed at a tertiary care hospital in Pakistan.
2.2 Compliance with ethics guidelines
The study was approved by the National Medical Ethics Committee of the Republic of Slovenia (number 0120–14/2017/5, and 0120–273/2019/9) and by the institutional review board and ethics Committee of Shifa Tameer-e-Millat University Islamabad, Pakistan (033–523-2019). The principles of the Declaration of Helsinki were followed in this study. All adult patients or their caregivers for child patients provided written informed consent before inclusion in the study.
2.3 Genetic testing
Genetic analysis was conducted at the University Children’s Hospital, Ljubljana, Slovenia, following a previously established protocol in Slovenia (14).
Genomic DNA was isolated from a venous blood sample using a Flexigene kit (Qiagen, Germany). To target the dyslipidemia-associated genes’ coding regions and intron-exon borders, xGen® Lockdown® NGS Probes were employed. Next-generation sequencing (NGS) was performed using the MiSeq Reagent Kit (Illumina, United States) on a MiSeq sequencer, following the manufacturer’s protocol.
The following genes were included in the analysis: ABCA1, ABCG5, ABCG8, ALMS1, APOA1, APOA5, APOB, APOC2, APOC3, APOE, CREB3L3, GPIHBP1, LDLR, LDLRAP1, LIPA, LMF1, LPL, and PCSK9. candidate pathogenic variants identified through NGS following previously described protocol (14) were validated using Sanger sequencing.
2.4 Systematic literature review
Following the PRISMA reporting guidelines, we performed a systematic review to summarize the data about all patients with either one of the three LPL variants (c.984G>T, c.337T>C, and c.724G>A) from our study on 24th March 2023. Our search focused on the Medline database, using the search term “lipoprotein lipase,” which resulted in the identification of 14,053 records. To ensure the relevance and quality of the included studies, two independent reviewers carefully reviewed the titles, abstracts, and full-length text of the identified articles. These inclusion criteria comprised human-based studies published in English, specifically reporting on LPL variants related to our research, and providing clinical data, particularly TGs, along with information on pathogenic variants (Figure 1). From this comprehensive screening process, we selected only eight records that met our predetermined inclusion criteria.

Figure 1 PRISMA flow diagram for systematic literature review.
In addition, we also searched through HGMD and Franklin databases to identify additional reports that were not found in Pubmed.
3 Results
Patient 1 was incidentally discovered in a female at the age of 3 years, during systemic bacterial infection, with her initial recorded TG level being 16.8 mmol/L, indicative of primary HTG. The patient was found to have a heterozygous pathogenic variant NM_000237.3:c.984G>T (p.Met328Ile) in exon 6 of the LPL gene. Following dietary modifications, including a low-fat diet, omega-3 fatty acid supplementation, and avoidance of high-carbohydrate foods, patient 1’s TG levels significantly decreased to 1.9 mmol/L at the last check-up. Table 1 provides a summary of patients’ characteristics. The patient had no family history of dyslipidemia or cardiovascular disease.
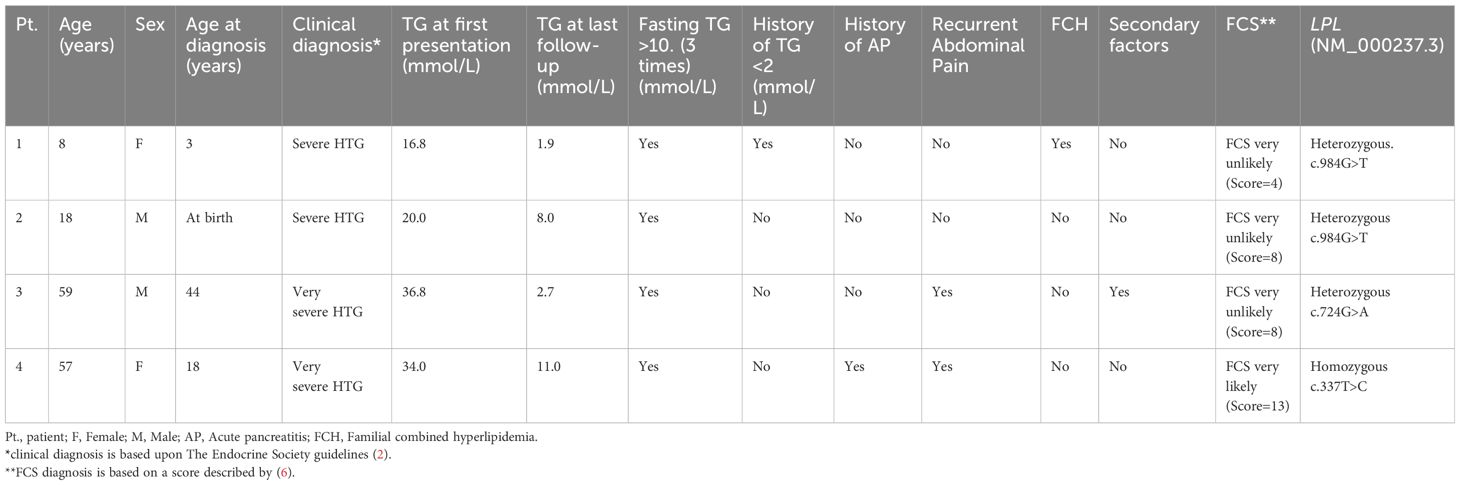
Table 1 Demographic and clinical details of presented patients.
Patient 2 was a male was born at 37 weeks with a body weight of 2930 g and required mechanical support due to meconium aspiration. His plasma TG levels were 20 mmol/L during the neonatal period, and genetic analysis of the patient revealed a heterozygous pathogenic variant in the LPL gene, specifically NM_000237.3:c.984G>T (p.Met328Ile). Dietary restrictions including breast milk were maintained, resulting in a last recorded TG level of 8 mmol/L. The patient’s mother also exhibited high TG levels (11 mmol/L). The calculated FCS score was 8 (Table 1).
Patient 3, a 44-years male was diagnosed with HTG with a TG value of 36.8 mmol/L. The patient underwent genetic testing, which confirmed a heterozygous pathogenic variant in exon 5 of LPL: NM_000273:c.724G>A (p.Asp242Asn). Treatment with gemfibrozil 600 mg, and diet significantly reduced TG levels to 2.7 mmol/L. The patient also had type 2 diabetes and chronic kidney disease. It was noted that patient 3’s brothers also exhibited TG levels above 12 mmol/L.
Patient 4, a female, experienced her first episode of pancreatitis at 18 years old. Upon investigation, a TG level of 34 mmol/L was revealed. The FCS score of the patient was 13. Genetic testing of the patient revealed a homozygous variant in LPL NM_000237.3:c.337T>C (p.Trp113Arg). Despite various attempts, patient 4 continued to experience persistent HTG due to non-adherence to a strict low-fat diet. This led to seven episodes of AP and the development of multiple pancreatic cysts, which required surgical intervention. Treatment with fenofibrate and omega-3 fatty acids, along with the adoption of an appropriate diet, significantly lowered TG levels to 11 mmol/L at the last visit. Her father also suffered from HTG and experienced a heart attack at the age of 65 years.
3.1 Comparison of cases from our study and from systematic literature review
We have comprehensively reviewed previous studies on the LPL c.984G>T pathogenic variant, as presented in Table 2. Homozygous LPL variants resulted in elevated TG levels (65.6 mmol/L) and severe clinical complications, such as hepatosplenomegaly and AP, whereas heterozygous patients showed an average TG value of 11.4 mmol/L. On the other hand, patients with compound heterozygous (CHET) variants had the highest average TG levels (163.7 mmol/L).
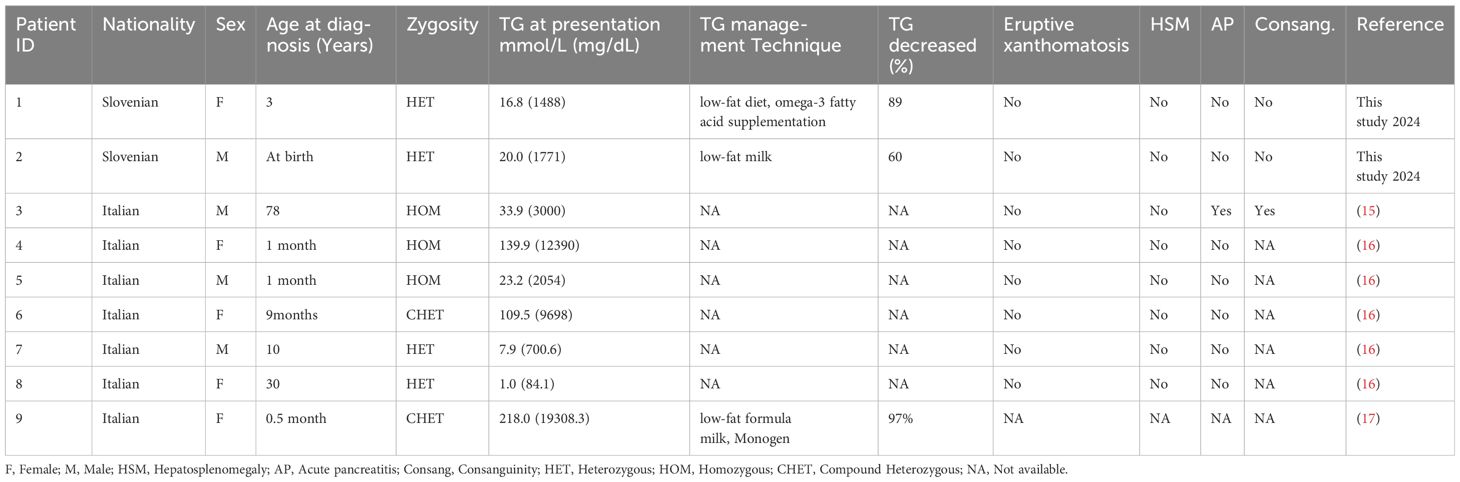
Table 2 Previously reported individuals with pathogenic LPL variants c.984G>T with their clinical details.
Homozygous patients with pathogenic LPL variants c.337T>C had a higher likelihood of experiencing acute pancreatitis, while compound heterozygous and heterozygous patients seemed to experience milder or no complications. The average TG values were highest for homozygous patients (161.3 mmol/L), followed by heterozygous patients (48.4 mmol/L). The only reported case of CHET patients with c.337T>C variant had a TG level of 39.7 mmol/L (Table 3).
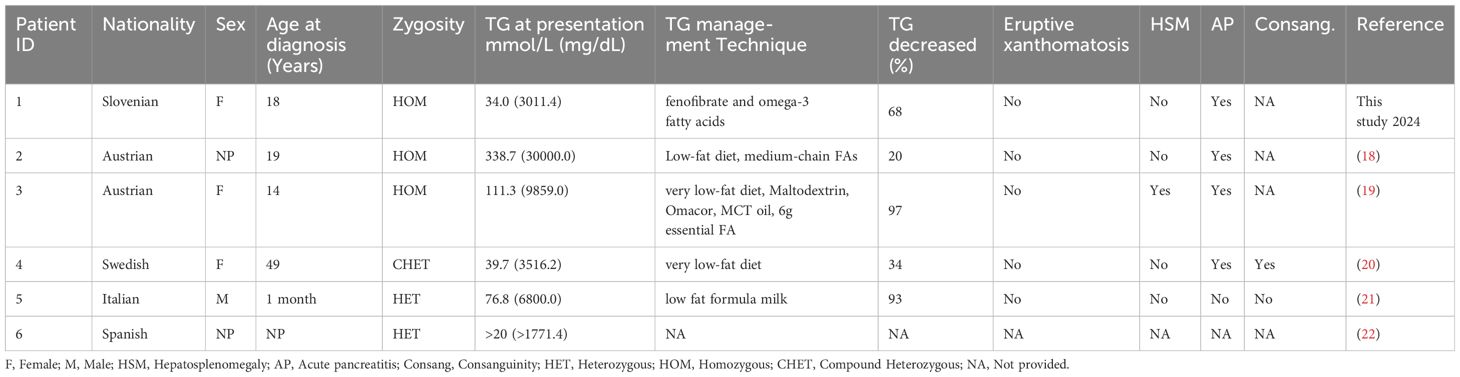
Table 3 Previously reported individuals with pathogenic LPL variants c.337T>C with their clinical details.
Comprehensive analysis of the baseline TG levels at the time of diagnosis and TG levels after implementing TG management techniques showed a significant reduction in the TG levels among all of the patients. The study further revealed that the use of low-fat food in adult patients and low-fat milk in infants led to a significant decrease in TG levels.
However, we could not find enough studies focused on the LPL variant c.724G>A. A study reporting the variant in the Argentinian population was identified, but it didn’t meet our review criteria (23).
4 Discussion
The present study reports four patients who had pathogenic variants in the LPL gene, encoding LPL, a pivotal enzyme in the regulation of TG metabolism (24). Any pathogenic variation in the LPL gene results in the reduction or absence of LPL activity, resulting in higher TG levels in plasma, which in turn leads to the accumulation of TGs in various tissues resulting in pancreatitis, eruptive xanthomas, and hepatosplenomegaly (25). Severe and very severe HTG increase the risk of pancreatitis, while mild or moderate HTG may be a risk factor for cardiovascular disease (2).
We have reported two Slovenian patients with c.984G>T (p.Met328Ile), one Slovenian patient with c.337T>C (p.Trp113Arg), and one Pakistani patient with c.724G>A (p.Asp242Asn) variants in the LPL gene. All patients included in this study presented with HTG when diagnosed. Similar results were obtained in different studies conducted, where LPL pathogenic variants were associated with HTG (9, 26–30). Severe HTG is the third most common reason for developing AP following gallstones and alcohol abuse (31). There is no exact TG threshold level that causes AP, but the risk steadily rises as TG levels increase (32). Patient 4 from our study with c.337T>C (p.Trp113Arg) homozygous LPL variant presented with recurrent pancreatitis. Patient 4 exhibited a severe phenotype attributed to FCS, confirmed by genetic testing and a high FCS score. Similar to our findings various studies have been reported to link this pathogenic variant with pancreatitis (19–21).
HTG and inadequate glycemic control elevate the likelihood of chronic kidney disease (CKD) in individuals with type 2 diabetes mellitus (33). The presence of nephrotic syndrome is associated with a significant reduction in both the abundance and activity of the LPL protein (34). Patient 3 had type II diabetes mellitus and chronic kidney disease, along with HTG, indicating the MCS. An analysis of 18 genes in our NGS panel did not identify any other pathogenic or VUS variants. However, considering the severity of HTG and the coexistence of diabetes and CKD, it is likely that additional genetic variants in other genes may be contributing to the patient’s condition. It is recommended to conduct further genetic testing beyond the current gene panel to identify these potential contributors.
For LPL deficiency patients, diet change, and lifestyle modifications are considered as the first line of treatment. Medications like fibrates are prescribed which increase lipoprotein lipase activity, reducing VLDL levels in the liver (2, 35, 36). Keeping plasma TG below 11.30 mmol/L can help prevent serious complications like severe pancreatitis, which ultimately reduces the high mortality and morbidity rate. In our patients, the implementation of a fat-free diet, lifestyle modification, and medication resulted in a sharp decline in TG levels. Unfortunately, due to resource limitations and the scope of our study, we could not include parental data, which would have provided valuable insights into genetic inheritance patterns. NGS has certain limitations, such as missing large deletions, duplications, or deep intronic mutations, so there is the possibility that one of our heterozygous patients might have an undetected variant, resulting in compound heterozygosity. We analysed LPL variants in Pakistani and Slovenian, complete, and partial LPL deficient populations, providing a deeper understanding of genotype-phenotype relationships for better diagnostics and treatment options. The cases reported in this manuscript contribute to the knowledge, base for precision medicine in the future. In summary, our research reveals that pathogenic variations in the LPL gene are clinically relevant and linked to the severity of HTG.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The study was approved by the National Medical Ethics Committee of the Republic of Slovenia (number 0120-94 14/2017/5, and 0120-273/2019/9) and by the institutional review board and ethics Committee of Shifa Tameer-95 e-Millat University Islamabad, Pakistan (033-523-2019). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
QA: Writing – review & editing, Writing – original draft. MC: Writing – review & editing, Writing – original draft. TM: Writing – review & editing, Writing – original draft. JS: Writing – review & editing, Writing – original draft. FS: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal Analysis, Data curation, Conceptualization. US: Writing – review & editing, Writing – original draft. MM: Writing – review & editing, Writing – original draft, Data curation. JK: Writing – review & editing, Investigation. HB: Writing – review & editing, Data curation. MK: Writing – review & editing, Supervision, Data curation. KT: Writing – review & editing, Supervision, Resources. BB: Writing – review & editing, Supervision, Methodology. TB: Writing – review & editing, Software, Investigation. ZF: Writing – review & editing, Investigation, Formal Analysis. MA: Writing – review & editing, Supervision. UG: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal Analysis, Data curation, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partly supported by The Slovenian National Research Agency (Grants P3-0343, J3-4116, J3-6800, and J3-6798) and the Higher Education Commission, Pakistan (Grant 20-15760).
Acknowledgments
We would like to thank all the patients and their families for their participation and the nurses for their collaboration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ruiz-García A, Arranz-Martínez E, López-Uriarte B, Rivera-Teijido M, Palacios-Martínez D, Dávila-Blázquez GM, et al. Prevalence of hypertriglyceridemia in adults and related cardiometabolic factors. SIMETAP-HTG study. Clinica e Investigacion en Arterioscler. (2020) 32:242–55. doi: 10.1016/j.arteri.2020.04.001
2. Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, et al. Evaluation and treatment of hypertriglyceridemia: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2012) 97(9):2969–89. doi: 10.1210/jc.2011–3213
3. Dron JS, Wang J, Cao H, McIntyre AD, Iacocca MA, Menard JR, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidology. (2019) 13:80–8. doi: 10.1016/j.jacl.2018.10.006
4. Lewis GF, Xiao C, Hegele RA. Hypertriglyceridemia in the genomic era: A new paradigm. Endocrine Rev. (2015) 36:131–47. doi: 10.1210/er.2014–1062
5. Sadiq F, Hegele RA, Catapano AL, Groselj U. Editorial: rare dyslipidemias. Front Genet. (2023) 14. doi: 10.3389/fgene.2023.1248435
6. Moulin P, Dufour R, Averna M, Arca M, Cefalù AB, Noto D, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an “FCS score. In: Atherosclerosis. Elsevier Ireland Ltd, Ireland (2018). p. 265–72. doi: 10.1016/j.atherosclerosis.2018.06.814
7. Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: Structure, function, regulation, and role in disease. J Mol Med. (2002) 80:753–69. doi: 10.1007/s00109–002-0384–9
8. Stenson, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. (2020) 139:1197–207.
9. Li Y, Hu M, Han L, Feng L, Yang L, Chen X, et al. Case report: next-generation sequencing identified a novel pair of compound-heterozygous mutations of LPL gene causing lipoprotein lipase deficiency. Front Genet. (2022) 13:831133. doi: 10.3389/fgene.2022.831133
10. Hegele RA, Ginsberg HN, Chapman MJ, Nordestgaard BG, Kuivenhoven JA, Averna M, et al. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. (2014) 2(8):655–66. doi: 10.1016/S2213–8587(13)70191–8
11. Chait A, Eckel RH. The chylomicronemia syndrome is most often multifactorial. In: Annals of Internal Medicine. American College of Physicians, United States (2019). p. 626–34. doi: 10.7326/M19–0203
12. Dron JS, Hegele RA. Genetics of hypertriglyceridemia. Front Endocrinol. (2020) 11. doi: 10.3389/fendo.2020.00455
13. Muñiz-Grijalvo O, Diaz-Diaz JL. Familial chylomicronemia and multifactorial chylomicronemia. Clinica e Investigacion en Arterioscler. (2021) 33:56–62. doi: 10.1016/j.arteri.2021.02.011
14. Sustar U, Groselj U, Khan SA, Shafi S, Khan I, Kovac J, et al. A homozygous variant in the GPIHBP1 gene in a child with severe hypertriglyceridemia and a systematic literature review. Front Genet. (2022) 13:983283. doi: 10.3389/fgene.2022.983283
15. Vigna GB, Citroni N, Tarugi P, Fellin R. Familial chylomicronemia syndrome. A sixty year follow-up in two siblings and their kindreds. Nosological and clinical considerations. J Clin Lipidology. (2022) 16:591–5. doi: 10.1016/j.jacl.2022.07.013
16. Rabacchi C, Pisciotta L, Cefalù AB, Noto D, Fresa R, Tarugi P, et al. Spectrum of mutations of the LPL gene identified in Italy in patients with severe hypertriglyceridemia. Atherosclerosis. (2015) 241:79–86. doi: 10.1016/j.atherosclerosis.2015.04.815
17. Bordugo A, Carlin E, Demarini S, Faletra F, Colonna F. A neonate with a “milky” blood. What can it be? In: Archives of Disease in Childhood: Fetal and Neonatal Edition. BMJ Group (United Kingdom), vol. 99. (2014). p. F514. doi: 10.1136/archdischild-2014–305940
18. De Gier C, Skacel G, Walleczek NK, Lischka J, Baumgartner M, Greber-Platzer S. Treatment of volanesorsen in a patient with familial chylmicronaemia syndrome (FCS) due to homozygous c.337T>C(p.TRP113ARG) - mutation and impact of dietary incompliance: A case report. Atherosclerosis. (2022) 355:65. doi: 10.1016/j.atherosclerosis.2022.06.421
19. Thajer A, Skacel G, De Gier C, Greber-Platzer S. The effect of a fat-restricted diet in four patients with familial chylomicronemia syndrome: A long-term follow-up study. Children. (2021) 8:1078. doi: 10.3390/children8111078
20. Caddeo A, Mancina RM, Pirazzi C, Russo C, Sasidharan K, Sandstedt J, et al. Molecular analysis of three known and one novel LPL variants in patients with type I hyperlipoproteinemia. Nutrition Metab Cardiovasc Dis. (2018) 28:158–64. doi: 10.1016/j.numecd.2017.11.003
21. Buonuomo PS, Rabacchi C, Macchiaiolo M, Trenti C, Fasano T, Tarugi P, et al. Incidental finding of severe hypertriglyceridemia in children. Role of multiple rare variants in genes affecting plasma triglyceride. J Clin Lipidology. (2017) 11:1329–37.e3. doi: 10.1016/j.jacl.2017.08.017
22. Rodrigues R, Artieda M, Tejedor D, Martínez A, Konstantinova P, Petry H, et al. Pathogenic classification of LPL gene variants reported to be associated with LPL deficiency. J Clin Lipidology. (2016) 10:394–409. doi: 10.1016/j.jacl.2015.12.015
23. Corral P, Geller AS, Polisecki EY, Lopez GI, Bañares VG, Cacciagiu L, et al. Unusual genetic variants associated with hypercholesterolemia in Argentina. Atherosclerosis. (2018) 277:256–61. doi: 10.1016/j.atherosclerosis.2018.06.009
24. Basu D, Goldberg IJ. Regulation of lipoprotein lipase-mediated lipolysis of triglycerides. In: Current Opinion in Lipidology. Lippincott Williams and Wilkins, United States (2020). p. 154–60. doi: 10.1097/MOL.0000000000000676
25. Packard CJ, Boren J, Taskinen MR. Causes and consequences of hypertriglyceridemia. Front Endocrinol. (2020) 11:252. doi: 10.3389/fendo.2020.00252
26. Jap TS, Jenq SF, Wu YC, Chiu CY, Cheng HM. Mutations in the lipoprotein lipase gene as a cause of hypertriglyceridemia and pancreatitis in Taiwan. Pancreas. (2003) 27:122–6. doi: 10.1097/00006676–200308000–00003
27. Chen TZ, Xie SL, Jin R, Huang ZM. A novel lipoprotein lipase gene missense mutation in Chinese patients with severe hypertriglyceridemia and pancreatitis. Lipids Health Dis. (2014) 13:52. doi: 10.1186/1476–511X-13–52
28. Khovidhunkit W, Charoen S, Kiateprungvej A, Chartyingcharoen P, Muanpetch S, Plengpanich W. Rare and common variants in LPL and APOA5 in Thai subjects with severe hypertriglyceridemia: A resequencing approach. J Clin lipidology. (2016) 10:505–11.e1. doi: 10.1016/J.JACL.2015.11.007
29. Li X, Yang Q, Shi X, Chen W, Pu N, Li W, et al. Compound but non-linked heterozygous p.W14X and p.L279 v LPL gene mutations in a Chinese patient with long-term severe hypertriglyceridemia and recurrent acute pancreatitis. Lipids Health Dis. (2018) 17:1–7. doi: 10.1186/s12944–018-0789–2
30. Chyzhyk V, Brown AS. Familial chylomicronemia syndrome: A rare but devastating autosomal recessive disorder characterised by refractory hypertriglyceridemia and recurrent pancreatitis. Trends Cardiovasc Med. (2020) 30:80–5. doi: 10.1016/j.tcm.2019.03.001
31. Yang AL, McNabb-Baltar J. Hypertriglyceridemia and acute pancreatitis. Pancreatology. (2020) 20:795–800. doi: 10.1016/j.pan.2020.06.005
32. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in Hypertriglyceridemic Pancreatitis-An Update. Journal of Clinical Gastroenterology. Lippincott Williams & Wilkins, United States (2014) doi: 10.1097/01.mcg.0000436438.60145.5a.
33. Migdalis IN, Ioannidis IM, Papanas N, Raptis AE, Sotiropoulos AE, Dimitriadis GD, et al. Hypertriglyceridemia and other risk factors of chronic kidney disease in type 2 diabetes: A hospital-based clinic population in Greece. J Clin Med. (2022) 11:3224. doi: 10.3390/jcm11113224
34. Vaziri ND. Molecular mechanisms of lipid disorders in nephrotic syndrome. Kidney Int. (2003) 63:1964–76. doi: 10.1046/j.1523-1755.2003.00941.x
35. Oh RC. Management of Hypertriglyceridemia (2007). Available online at: www.aafp.org/afp.
Keywords: hypertriglyceridemia, lipoprotein lipase, LPL, lipoprotein lipase deficiency, pancreatitis, case series
Citation: Ain Q, Cevc M, Marusic T, Sikonja J, Sadiq F, Sustar U, Mlinaric M, Kovac J, Batool H, Khan MI, Trebusak Podkrajsek K, Bizjan BJ, Battelino T, Fras Z, Ajmal M and Groselj U (2024) Genetic and clinical characteristics of patients with lipoprotein lipase deficiency from Slovenia and Pakistan: case series and systematic literature review. Front. Endocrinol. 15:1387419. doi: 10.3389/fendo.2024.1387419
Received: 17 February 2024; Accepted: 21 May 2024;
Published: 07 June 2024.
Edited by:
Kayoko Hosaka, Karolinska Institutet (KI), SwedenReviewed by:
Teresa Villarreal-Molina, Instituto Nacional de Medicina Genómica (INMEGEN), MexicoPatryk Lipiński, Maria Sklodowska-Curie Medical Academy, Poland
Copyright © 2024 Ain, Cevc, Marusic, Sikonja, Sadiq, Sustar, Mlinaric, Kovac, Batool, Khan, Trebusak Podkrajsek, Bizjan, Battelino, Fras, Ajmal and Groselj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Urh Groselj, urh.groselj@kclj.si