 Thao Nguyen1*†
Thao Nguyen1*† Zehra Ordulu2*†
Zehra Ordulu2*† Sunaina Shrestha2Urja Patel3Paul L. Crispen4Lisa Brown5Sara M. Falzarano2,4,6Hans K. Ghayee3,7Juan Pablo Perdomo Rodriguez3
Sunaina Shrestha2Urja Patel3Paul L. Crispen4Lisa Brown5Sara M. Falzarano2,4,6Hans K. Ghayee3,7Juan Pablo Perdomo Rodriguez3- 1Department of Medicine, University of Florida, Gainesville, FL, United States
- 2Department of Pathology, Immunology and Laboratory Medicine, University of Florida College of Medicine, Gainesville, FL, United States
- 3Division of Endocrinology, University of Florida, Gainesville, FL, United States
- 4Department of Urology, University of Florida, Gainesville, FL, United States
- 5Department of Genetics, University of Florida, Gainesville, FL, United States
- 6Health Cancer Center, University of Florida, Gainesville, FL, United States
- 7Division of Endocrinology, Malcom Randall Veterans Affairs (VA) Medical Center, Gainesville, FL, United States
Background: Paragangliomas (PGL) are rare neuroendocrine tumors derived from the autonomic nervous system paraganglia. Urinary bladder paragangliomas (UBPGL) originate from the sympathetic neurons of the urinary bladder wall and represent 0.7% of all paragangliomas and <0.05% of all bladder tumors. PGL and UBPGL can be associated with SDHB, SDHD, NF1, and VHL gene variants, with the most common germline alterations found in SDHB and VHL.
Case report: We report a case of a 42-year-old woman who presented with menorrhagia/hematuria, uterine leiomyomas, as well as cardiac and bladder masses. The cardiac mass was favored to be a myxoma based on clinical findings, while the bladder mass was diagnosed as UBPGL. A novel SDHB mutation (c.642G>A, p Q214Q), detected in the UBPGL, was proven to be somatic. Although this variant was seemingly synonymous, it was predicted to have a loss of function due to the splice site effect, which was further supported by the immunohistochemical loss of SDHB.
Conclusion: This case highlights the challenges of diagnosing an extremely rare entity, bladder paraganglioma, with an emphasis on the multidisciplinary approach to navigate various clinical and imaging findings that may initially be misleading. In addition, a novel loss of function SDHB variant that could have been overlooked as a synonymous variant is herein reported, while also illustrating the importance of both germline and somatic mutation testing.
Introduction
Paragangliomas (PGLs) are rare neuroendocrine tumors derived from extra-adrenal chromaffin cells (1). The incidence of paragangliomas is often described together with the incidence of pheochromocytomas (PCCs), which is approximately 0.6 cases per 100,000 person-years (2). PGLs located in the neck and skull are usually parasympathetic and nonfunctional while those located in the thorax, abdomen, and pelvis tend to be sympathetic and hypersecretory (1). Urinary bladder paragangliomas (UBPGL) are extremely rare tumors originating from the sympathetic neurons of the urinary bladder wall, which represent 0.7% of all PGLs and <0.05% of all bladder tumors (3). UBPGLs may manifest with catecholamine excess, ranging from 55% to 91% of the cases (4–6) Diagnosing non-functioning UBPGLs can be challenging, as they may only manifest as hematuria or be detected incidentally from imaging studies. Consequently, up to two-thirds of UBPGLs are diagnosed following surgery or a biopsy (7).
Risk factors for metastatic UBPGLs include high levels of catecholamine excess, young age, and large tumor size (7).PGLs and UBPGLs can be associated with mutations involving SDHB, SDHD, VHL, and NF1 (2). The former two genes, particularly SDHB are the most commonly mutated genes in patients with germline mutations (7). Previous research has illustrated that pathogenic SDHB variants in PGLs increase the risk of metastatic disease (7, 8).
Case presentation
A 42-year-old female with menometrorrhagia secondary to presumed uterine leiomyomas presented for hysterectomy evaluation with her gynecologist. The patient had a Foley catheter placed due to urinary retention and hematuria, for which she underwent cystoscopic examination at an outside facility. A bladder mass was identified and biopsied, revealing a paraganglioma. She was thus referred to our institution for further evaluation of the bladder mass.
A CT intravenous pyelogram revealed an enhancing 3.7 cm bladder mass centered near the left ureterovesical junction without hydronephrosis (Figure 1A), as well as a hyperdense cardiac mass on the atrial septum, incompletely evaluated. A subsequent pelvic and abdominal MRI highlighted a well-defined heterogeneous soft tissue mass in the anterior inferior aspect of the bladder abutting the urethra and vagina, measuring 3.8x3.2x3.0 cm, that demonstrated a mild hyperintense signal on T2-weighted images (Figure 1B) as well as an isointense signal on T1-weighted images. This also showed a dominant 7.4 cm intramural leiomyoma in the anterior myometrial wall with a physiologic left ovarian cyst and no other cystic solid adnexal lesions. A transurethral resection of the bladder mass (TURBT) was scheduled.
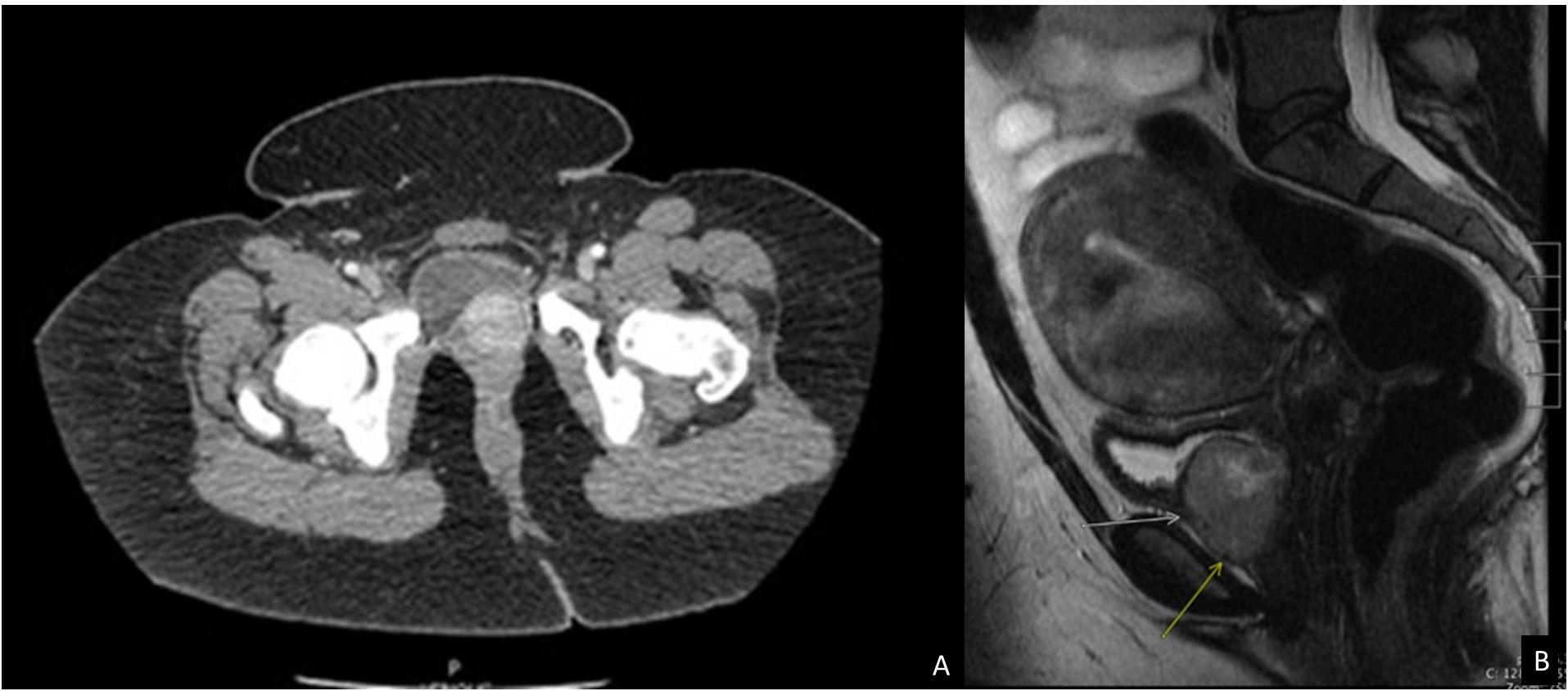
Figure 1 Imaging studies of the patient. (A) CT Intravenous pyelogram demonstrating an enhancing 3.7 cm bladder mass centered near the left ureterovesical junction without hydronephrosis or lymphadenopathy. (B) MRI pelvis and abdomen showing a well-defined 3.8 cm mass (arrows) in the anterior inferior aspect of the bladder abutting the urethra and vagina with a mild hypertense signal on T2 weighted image.
In the meanwhile, based on the initial pathology results, the patient was also referred to endocrinology. Upon further interview at the endocrinology clinic, the patient reported excessive bleeding, both during her period and between her periods (menometrorrhagia), as well as episodes of spotting between menses. Bleeding episodes varied in quantity and quality; it is possible that the hematuria could have been masked by the abundant menometrorrhagia. She also reported fatigue and shortness of breath, which was attributed to severe iron deficiency secondary to abnormal uterine bleeding. Moreover, she complained of mild abdominal pain but denied episodic headaches, sweating, tremors, palpitations, anxiety, fevers, dysuria, or recreational drug use. The patient had no other past medical history or concomitant medication except for multivitamin supplements. Her family history was significant for stroke in her grandparents and father. There was no known family history of endocrine tumors.
Initial blood pressure and heart rate were 110/80 mmHg and 70–80 beats per minute respectively. However, ambulatory blood pressure monitoring revealed occasional values of 160/90 mmHg. At night, her blood pressures did not decrease. Of note, blood pressures were not monitored post-micturition. The physical exam revealed normal heart rate and rhythm with no murmur, rub, or gallop.
Further workup showed a slightly elevated plasma metanephrine level of 74 pg/mL (normal <57 pg/mL) with normal plasma normetanephrine and serum chromogranin levels. In her 24-hour urine studies, the levels of dopamine (564 mcg/24h; normal 52–480 mcg/24h) and metanephrine (305 mcg/24h; normal 58- 203 mcg/24h) were slightly elevated, while the normetanephrine level was normal.
There were no signs of pheochromocytoma or abdominal paragangliomas on abdominal MRI. To further evaluate synchronous paraganglioma, 68Ga-DOTATATE-PET/CT scan was performed before surgery and showed no evidence of metastatic disease, and no uptake of the hyperdense cardiac mass. Unfortunately, bladder visualization was limited due to the excreted tracer. Of note, both abdominal MRI and PET scans revealed suspicious breast nodules, however, biopsy showed fibroadenomatoid changes consistent with sclerosing adenosis with no evidence of malignancy.
To further characterize the cardiac mass, an echocardiogram was performed, which did not show the mass seen earlier on the CT scan. Further workup with cardiac MRI showed a well-circumscribed mass in the interatrial septum measuring 2.0 cm x 1.7 cm, with heterogeneous pattern in the late gadolinium enhancement sequences. A cardiologist was consulted for the left atrial mass, who concluded that it was most likely benign atrial myxoma based on MRI findings. No further treatment, including anticoagulation, was recommended.
To prevent an intraoperative hypertensive crisis, given occasional hypertension on ambulatory blood pressure monitoring, the patient received alpha blockade with doxazosin 2mg daily, which was up titrated to target blood pressure <130/80 mmHg supine and systolic blood pressure 90–110 mmHg upright, and to eliminate all occasionally high values. Her final dose before surgery was 8 mg twice daily. She then underwent TURBT for UBPGL resection. Doxazosin was continued post-operatively due to an elevated blood pressure reading. However, she decided to go home immediately after the surgery rather than stay overnight for monitoring. Therefore, she was discharged with doxazosin. Ten days later, she was seen by her cardiologist for a follow-up regarding her cardiac mass, and her dose was reduced. The patient had multiple rescheduled and missed appointments with endocrinologists. When she eventually followed up with endocrinology at three-month post-procedure, her dose was completely discontinued.
On pancystoscopy, the bladder mass appeared as a large (greater than 5 cm) endophytic mass of the left lateral and anterior walls that extended to the bladder neck. The gross specimen consisted of multiple irregularly and rectangularly shaped fragments of soft pinkish tissue that in total measured 7.0x6.0x1.0 cm in aggregate. Histologic sections showed again fragments of paraganglioma, composed of variably sized nests of eosinophilic cells with round to oval nuclei mostly arranged in a classic Zellballen pattern of growth, separated by a delicate fibrovascular network. No comedonecrosis or vascular invasion was identified. Immunohistochemical stains showed strong diffuse immunoreactivity with synaptophysin, chromogranin, and GATA-3, with negative keratin (AE1/AE3) and p63 stains (Figure 2). The ki-67 labeling index was greater than 3% (7.5% on average, as calculated over 1000 cells from hot spots in a representative section of the tumor). SDHB immunohistochemistry testing showed loss of stain in the tumor cells, with appropriately staining internal controls, represented by the endothelial and stromal cells (Figure 3). Overall, a diagnosis of primary UBPGL was made based on clinicopathologic findings.
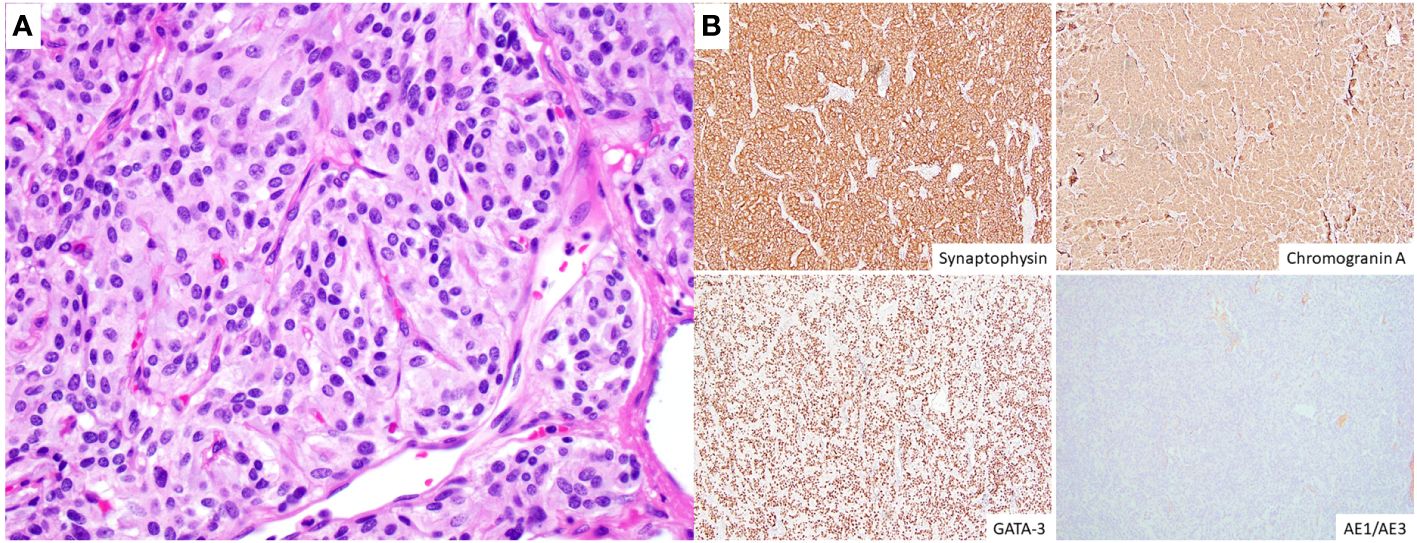
Figure 2 Histologic and immunohistochemical findings. (A) High magnification image (H&E, original magnification 400x) illustrating the tumor cells arranged in irregular nests surrounded by a fine capillary network, and showing moderate to abundant, eosinophilic cytoplasm, round to oval nuclei with finely granular (“salt and pepper”) chromatin and inconspicuous to absent nucleoli. (B) The tumor cells (immunohistochemistry, original magnification 100x) show diffuse cytoplasmic expression of synaptophysin and chromogranin A and nuclear immunoreactivity for GATA-3, with negative keratin (AE1/AE3) stains.
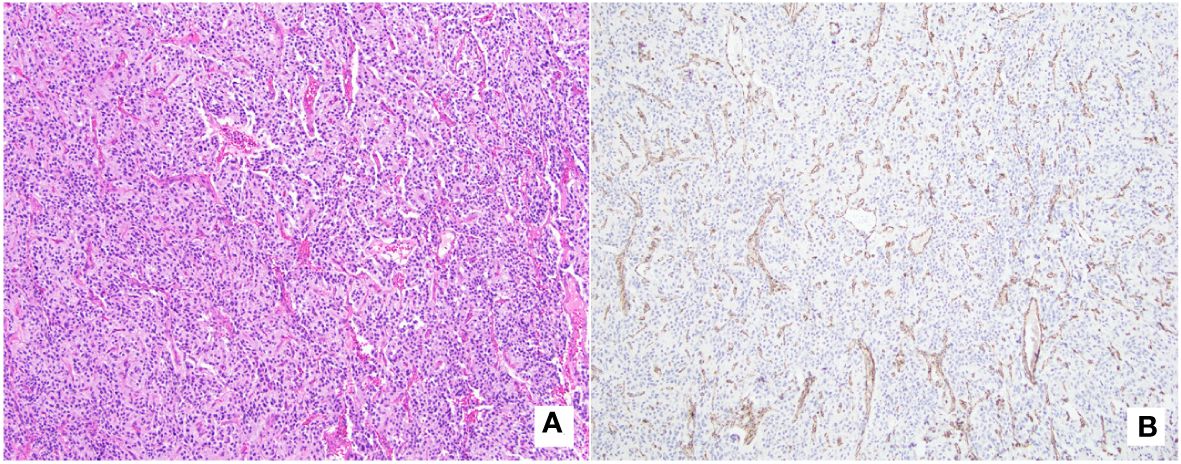
Figure 3 SDHB-immunochemistry with matching histology. (A) (H&E, original magnification 100x): Tumor is composed of cells with abundant eosinophilic cytoplasm arranged in a variable sized nested pattern, with associated thin capillary network. (B) (SDHB immunostain, original magnification 100x): Immunohistochemistry testing shows loss of SDHB protein expression in tumor cells, with appropriately staining internal control (endothelial cells outlining the vascular spaces, and stromal cells).
Both germline and tumor genetic testing were undertaken as part of the clinical work-up. The germline testing was performed via the Ambry Genetics Custom-Next Cancer Panel (consisting of 85 genes) with RNA Insight and revealed a variant of unknown significance (VUS) in MET (c.816G>C, pQ272H). The tumor sample was sequenced by using our institution’s clinically validated 700-gene next-generation sequencing (NGS) panel known as UF Health Pathlabs Gatorseq700 NGS Screening Panel to evaluate for mutations and copy number variants. Briefly, genomic DNA extracted from the tumor was amplified using the GatorSeq700 NGS Panel and sequenced on the Novaseq 6000 to high uniform depth (targeting 500x coverage by non-PCR duplicate read pairs with >99% of exons at coverage >100x). Sequence data were processed using the genomic analysis application DRAGEN (enrichment version 3.9.5) with UCSC hg19-altaware as the reference genome. The mutation nomenclature was based on the convention recommended by the Human Genome Variation Society and interpretation was performed per clinical guidelines (9, 10). Tumor sequencing revealed a seemingly synonymous SDHB mutation (c.642G>A; p.Q214Q) with a deletion in the entire short arm of chromosome 1 spanning SDHB (Figure 4) and no evidence of loss of heterozygosity for the MET VUS. In silico analysis by multiple computational prediction tools supported the deleterious effect of the novel SDHB variant (c.642G>A; p.Q214Q), favoring a donor loss (see Supplementary Data).
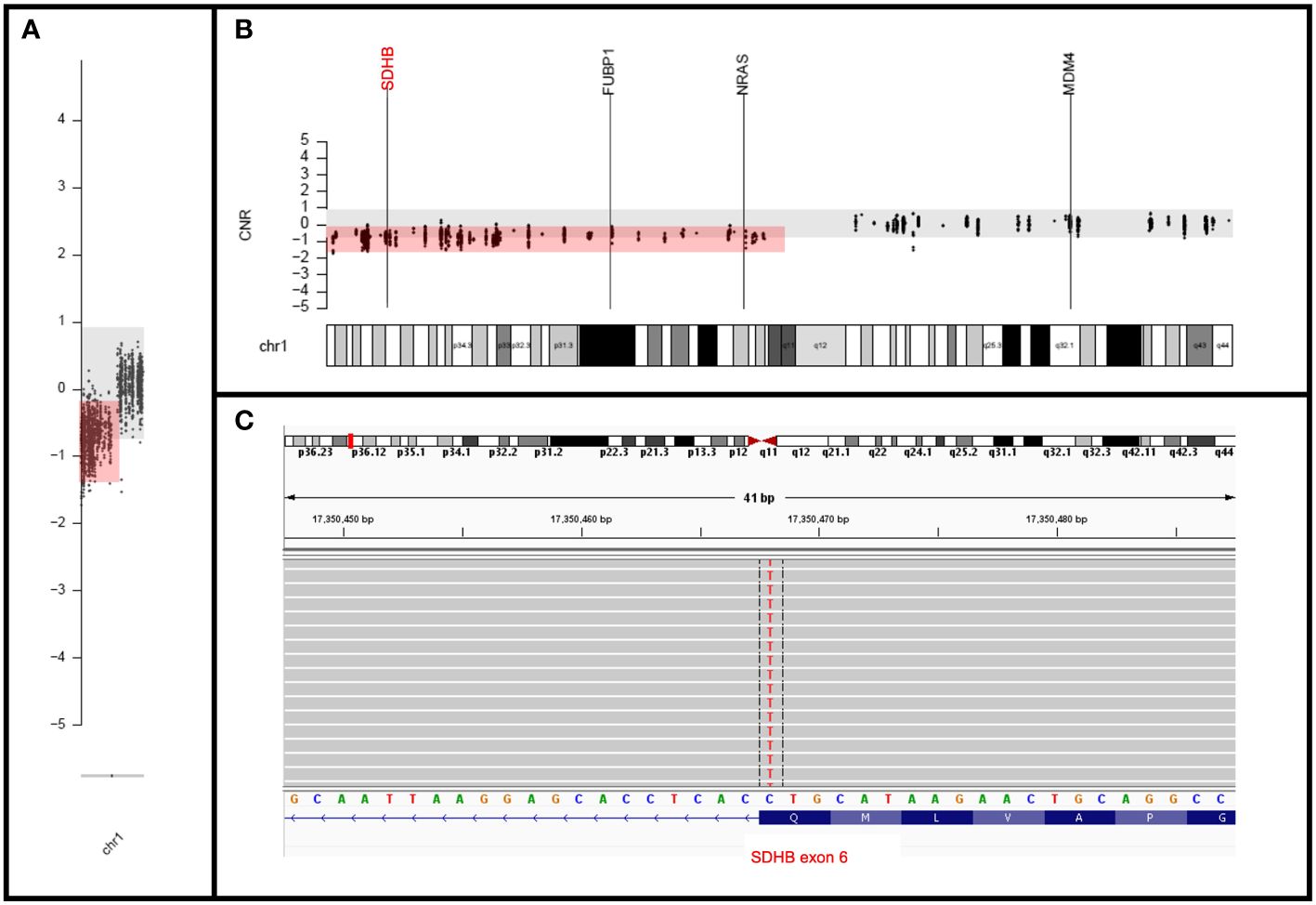
Figure 4 Molecular findings. (A, B) Copy number plots showing chromosome (A) and band level (B) loss of entire short arm of chromosome 1 with deletion of SDHB. (C) IGV view of the mutation in the last nucleotide of the SDHB exon 6 (negatively oriented gene) at the splice site.
Subsequently, the patient underwent a repeated bladder cystoscopy and an additional TURBT, which was negative for any residual tumor. Unfortunately, the patient has been lost to follow-up. We have contacted her in order to stress the importance of follow-up and to inquire about her blood pressure, menometrorrhagia, and repeated biochemical profiling.
Discussion
Herein, we present a unique UBPGL case that illustrates the importance of a multidisciplinary team approach to navigate the clinical and imaging findings of this rare entity that was diagnosed during the workup of menometrorrhagia. In addition, we report a novel loss of function variant in SDHB which may be challenging to interpret given that there is no change in the predicted amino acid transcription if one solely focuses on the nucleotide changes in the codon (c.642G>A p.Q214Q).
PCCs are rare neuroendocrine tumors deriving from chromaffin cells of the adrenal glands, and approximately 10% of these neoplasms are in the extra-adrenal sites and are referred to as PGLs. Among them, UBPGLs are one of the rarest forms with only a couple of hundred cases reported in the literature (3, 5). They can manifest with a range of symptoms, although they may remain clinically silent. Between 30–53% of UBPGLs present with symptoms of catecholamine excess triggered by micturition, 35–47% present with hematuria, while about 3–10% are discovered on imaging incidentally (4–6). Up to 45% of the UBPGLs may be non-functioning (4–6). When there is clinical suspicion for a UBPGL, evaluation includes a cystoscopy and a CT scan of the abdomen and pelvis (11). In a retrospective study by Zhang et al. (12) looking at imaging characteristics of 16 UBPGL cases (9 of which were female patients), 13 patients underwent CT scans, which all exhibited slight hypoattenuation and moderate to marked enhancement of the bladder mass. There was only one case with leiomyomas on imaging, but it was unclear if that patient was symptomatic (12). No other reported cases of concurrent leiomyomas were noted in the literature. A multicentric study that investigated 110 patients diagnosed with UBPGL showed that only 37% were diagnosed prior to biopsy based on more characteristic symptoms (7). Overall, these studies highlight that PGLs may not be considered in the differential diagnosis of bladder masses during the initial workup.
Likewise, in our patient’s case, the history of leiomyomas and lack of significant symptoms of catecholamine excess made it challenging to initially consider a UBPGL in the differential diagnosis. Moreover, the slight increase in metanephrine level did not reach the level of significance (11) and could have been related to pain at the time. Considering the typical biochemical profile of paragangliomas against our findings, we concluded that our patient likely had a nonfunctioning bladder paraganglioma. Our case demonstrates that common tumors with expected symptoms, such as leiomyomas that present with abnormal uterine bleeding, may obscure findings of rare entities. The case also serves to encourage the clinicians to obtain detailed clinical histories and perform adequate work-up of incidental findings with a broad differential diagnosis.
Patients with UBPGLs present at a median age of 50 years and with a median tumor size of 2 cm (7). Hereditary PGLs manifest at an earlier age (approximately 15 years younger than average) and often present as multiple tumors (2, 7). Hereditary PGLs most commonly show germline mutations in SDHB and VHL (13, 14) while other genes such as FH (15) or MET (16) may rarely be involved. Studies have shown a strong correlation between loss of SDHB immunohistochemistry expression and SDHx mutations, with sensitivity and specificity both greater than 80% (15). Therefore, routine SDHB immunohistochemistry testing with PCCs or PGL tumors is a highly effective and rather inexpensive surrogate marker for SDHx mutations, thus representing a valuable screening tool for determining the necessity of germline testing in these tumors. The World Health Organization (WHO) classification of endocrine tumors considers SDHB immunohistochemistry “essential” in the histopathologic diagnosis of parasympathetic (most head and neck) PGLs and “desirable” in sympathetic PGLs (17). Our patient was relatively young and had multiple organ masses based on imaging. In addition, her UBPGL’s pathology had a loss of SDHB expression. Although these findings may initially raise the potential for germline syndrome, she had only one histologically confirmed PGL from her urinary bladder. Her breast lesions were biopsy proven to be non-malignant and her cardiac tumor was favored to be a myxoma based on MRI and PET findings. The immunohistochemical loss of SDHB was accompanied by the tumor NGS testing finding of a novel SDHB variant (c.642G>A; p.Q214Q), however, germline testing excluded this variant as being hereditary. Of note, there was a germline MET variant (c.816G>C, pQ272H) which was interpreted by the reference laboratory and clinical team as a VUS (also supported by ClinVar entries (18) Variant ID: 1401743).
Overall, despite the initial findings pointing towards a hereditary syndrome, a thorough workup by multidisciplinary teams, including additional germline and somatic (tumor) molecular testing, favored this tumor to be a sporadic UBPGL with a novel somatic SDHB variant. Even in cases of SDH-deficient neoplasia where no germline mutation was found, surveillance and further follow-up for other SDH-deficient neoplasms are still recommended (19). Even though, a germline pathogenic variant was not found in our patient, future follow-up with 68Ga-DOTATATE-PET/CT will be pursued, to continue to monitor for metastases and other neoplasms, based on other risk factors present in this case, including the young (41 years) age at presentation, the relatively large UBPGL size (3.8 cm on imaging), and the tumorigenic mutation in SDHB.
The SDHB (c.642G>A; p.Q214Q) variant itself raised a molecular diagnostic challenge in this case due to its seemingly synonymous change in the protein nomenclature. However, it occurred at the last nucleotide of exon 6 (Figure 4), which was predicted to result in a splice site effect (20) in this gene with loss of function variants considered pathogenic. This amino acid position is highly conserved across species and this variant has not been observed in population databases. Different mutations at the same nucleotide position have been reported as likely pathogenic; these include c.642G>T (2 ClinVar entries, Variation ID: 480788) and c.642G>C (reported in a patient with a malignant paraganglioma) (21) and listed in Human Gene Mutation Database (CM065460). In addition, the deleterious effect of this variant was further supported by in silico analysis by multiple prediction tools (see Supplementary Material). Lastly, the WHO for Genetic Tumour Syndromes recommends performing SDHB immunohistochemistry for evaluation of a VUS in SDHB, with a loss implying pathogenicity (19).
In combination with the patient’s tumor histology and the loss of immunohistochemical expression of SDHB, this somatic variant is ultimately interpreted as a likely pathogenic somatic variant (confirmed by germline testing) and the potential driver of this presumed sporadic tumor. Of note, there was a loss of heterozygosity of SDHB by deletion of the entire short arm of chromosome 1, which is a common mechanism for biallelic inactivation in the setting of somatic mutations, first described in this tumor type by van Nederveen et al. (22).
Surgical intervention for UBPGLs is typically personalized to the patient due to the lack of prospective research and clear criteria for malignancy prediction. It is known that there are no absolute histologic criteria or single biomarkers to reliably predict the biological behavior of PGLs or PCCs, and multi-parameter scoring systems have been proposed. The GAPP (Grading system for Adrenal Pheochromocytoma and Paraganglioma) (23) and modified GAPP (24) have shown predictive value to varying degrees, and they may be particularly useful to “rule out” potentially aggressive behavior rather than “rule it in” for risk stratification. The current patient’s tumor characteristics would add up to a GAPP score of 4 (“intermediate risk”). However, the risk stratification also depends on extra-adrenal location, patient age, number of tumors, and evidence of metastasis. A comprehensive approach should include clinical, biochemical, molecular, and pathological assessments (25) Short-term safety for procedures like transurethral resection and cystectomies is documented, yet long-term outcome data remains limited. Usually, UBPGLs are initially treated with either cystectomy or TURBT (7). Repeated surgery is sometimes required, especially in those of younger age (<5 years old) and large tumor size (>1 cm). Patients with incomplete resection or higher tumor stages (‗ T3) are at higher risk of recurrence, metastases, and death when compared to those with lower stages (26). Although the prognosis is usually good, about 8% of patients present with synchronous metastases, and 22% of patients develop metachronous metastases. These patients tend to be young, have a large UBPGL size, and have a high degree of catecholamine excess (4). Our patient was asymptomatic with negative repeated bladder resection at 3 months; however, our study is limited due to a lack of long-term follow-up.
This unique case of a UBPGL showcases multiple layers of diagnostic challenges starting with the patient’s initial abnormal uterine bleeding and leiomyoma masking the symptoms of this rare entity. The patient’s relatively young age, additional findings throughout the clinical workup of other masses at different sites, and the loss of SDHB immunohistochemistry in the UBPGL raised the potential for a germline syndrome, which was then excluded with germline and tumor genetic testing. Lastly, the somatic SDHB variant highlights the importance of positional effects at splice sites in “seemingly synonymous” variants in molecular diagnostics. Overall, a comprehensive approach combining multiple layers of data, from clinical history to molecular findings with multidisciplinary teamwork, is essential for diagnosing rare and challenging cases like ours.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
TN: Writing – original draft, Writing – review & editing. ZO: Writing – review & editing, Investigation, Supervision, Formal Analysis, Conceptualization. SS: Writing – review & editing. UP: Investigation, Writing – review & editing. PC: Investigation, Writing – review & editing. LB: Investigation, Writing – review & editing. SF: Investigation, Writing – review & editing, Resources. HG: Conceptualization, Investigation, Supervision, Validation, Writing – review & editing, Project administration, Resources. JP: Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We acknowledge the collaboration of pathology, genetics, endocrinology, and urology departments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1386285/full#supplementary-material
References
1. Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. (2001) 86:5210–6. doi: 10.1210/jcem.86.11.8034
2. Neumann HPH, Young WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
3. Purnell S, Sidana A, Maruf M, Grant C, Agarwal PK. Genitourinary paraganglioma: demographic, pathologic, and clinical characteristics in the surveillance, epidemiology, and end results (SEER) database (2000–2012). Urol Oncol. (2017) 35:457.e9–457.e14. doi: 10.1016/j.urolonc.2017.02.006
4. Beilan JA, Lawton A, Hajdenberg J, Rosser CJ. Pheochromocytoma of the urinary bladder: a systematic review of the contemporary literature. BMC Urol. (2013) 13:22. doi: 10.1186/1471–2490-13–22
5. Li M, Xu X, Bechmann N, Pamporaki C, Jiang J, Propping S, et al. Differences in clinical presentation and management between pre- and postsurgical diagnoses of urinary bladder paraganglioma: is there clinical relevance? A systematic review. World J Urol. (2022) 40:385–90. doi: 10.1007/s00345-021-03851-x
6. Park S, Kang SY, Kwon GY, Kwon JE, Kim SK, Kim JY, et al. Clinicopathologic characteristics and mutational status of succinate dehydrogenase genes in paraganglioma of the urinary bladder: A multi-institutional Korean study. Arch Pathol Lab Med. (2016) 141:671–7. doi: 10.5858/arpa.2016-0403-OA
7. Yu K, Ebbehøj AL, Obeid H, Vaidya A, Else T, Wachtel H, et al. Presentation, management, and outcomes of urinary bladder paraganglioma: results from a multicenter study. J Clin Endocrinol Metab. (2022) 107:2811–21. doi: 10.1210/clinem/dgac427
8. Su T, Yang Y, Jiang L, Xie J, Zhong X, Wu L, et al. SDHB immunohistochemistry for prognosis of pheochromocytoma and paraganglioma: A retrospective and prospective analysis. Front Endocrinol (Lausanne). (2023) 14:1121397. doi: 10.3389/fendo.2023.1121397
9. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagn. (2017) 19:4–23. doi: 10.1016/j.jmoldx.2016.10.002
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014–1498
12. Zhang J, Bai X, Yuan J, Zhang X, Xu W, Ye H, et al. Bladder paraganglioma: CT and MR imaging characteristics in 16 patients. Radiol Oncol. (2021) 56:46–53. doi: 10.2478/raon-2021-0055
13. de Tersant M, Généré L, Freyçon C, Villebasse S, Abbas R, Barlier A, et al. Pheochromocytoma and paraganglioma in children and adolescents: experience of the French society of pediatric oncology (SFCE). J Endocr Soc. (2020) 4:bvaa039. doi: 10.1210/jendso/bvaa039
14. Virgone C, Andreetta M, Avanzini S, Chiaravalli S, De Pasquale D, Crocoli A, et al. Pheochromocytomas and paragangliomas in children: Data from the Italian Cooperative Study (TREP). Pediatr Blood Cancer. (2020) 67:e28332. doi: 10.1002/pbc.28332
15. Udager AM, Magers MJ, Goerke DM, Vinco ML, Siddiqui J, Cao X, et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum Pathol. (2018) 71:47–54. doi: 10.1016/j.humpath.2017.10.013
16. Toledo RA, Qin Y, Cheng ZM, Gao Q, Iwata S, Silva GM, et al. Recurrent mutations of chromatin remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin Cancer Res. (2016) 22:2301–10. doi: 10.1158/1078-0432.CCR-15-1841
17. WHO Classification of Tumours Editorial Board. Who Classification of Tumours: Endocrine and Neuroendocrine Tumours. 5 ed. Lyon (France: International Agency for Research on Cancer (2022).
18. VCV001401743.8 - ClinVar - NCBI. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/1401743/?oq=1401743&m=NM_000245.4(MET):c.816G%3EC%20(p.Gln272His).
19. WHO Classification of Tumours Editorial Board. Who Classification of Tumours: Genetic Tumour Syndromes Website beta version. International Agency for Research on Cancer (2024). Available at: https://publications.iarc.fr. cited 2024 03/31/2024.
20. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. (2002) 3:285–98. doi: 10.1038/nrg775
21. Jochmanova I, Wolf KI, King KS, Nambuba J, Wesley R, Martucci V, et al. SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. J Cancer Res Clin Oncol. (2017) 143:1421–35. doi: 10.1007/s00432-017-2397-3
22. van Nederveen FH, Korpershoek E, Lenders JWM, de Krijger RR, Dinjens WNM. Somatic SDHB mutation in an extraadrenal pheochromocytoma. N Engl J Med. (2007) 357:306–8. doi: 10.1056/NEJMc070010
23. Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. (2014) 21:405–14. doi: 10.1530/ERC-13-0494
24. Koh JM, Ahn SH, Kim H, Kim BJ, Sung TY, Kim YH, et al. Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma. PLoS One. (2017) 12:e0187398. doi: 10.1371/journal.pone.0187398
25. Kimura N, Takekoshi K, Naruse M. Risk stratification on pheochromocytoma and paraganglioma from laboratory and clinical medicine. J Clin Med. (2018) 7:242. doi: 10.3390/jcm7090242
Keywords: paraganglioma, bladder paraganglioma, SDHB, c.642G>A, p.Q214Q, leiomyoma
Citation: Nguyen T, Ordulu Z, Shrestha S, Patel U, Crispen PL, Brown L, Falzarano SM, Ghayee HK and Perdomo Rodriguez JP (2024) Case report: A novel somatic SDHB variant in a patient with bladder paraganglioma. Front. Endocrinol. 15:1386285. doi: 10.3389/fendo.2024.1386285
Received: 14 February 2024; Accepted: 22 May 2024;
Published: 07 June 2024.
Edited by:
Bojana Popovic, University of Belgrade, SerbiaReviewed by:
Madson Almeida, University of São Paulo, BrazilKatarzyna Ziemnicka, Poznan University of Medical Sciences, Poland
Chunzhang Yang, National Cancer Institute (NIH), United States
Copyright © 2024 Nguyen, Ordulu, Shrestha, Patel, Crispen, Brown, Falzarano, Ghayee and Perdomo Rodriguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thao Nguyen, VGhhby5uZ3V5ZW5AbWVkaWNpbmUudWZsLmVkdQ==; Zehra Ordulu, bW9yZHVsdXNhaGluQHVmbC5lZHU=
†These authors share first authorship