 Lindsay T. Fourman1
Lindsay T. Fourman1 Josivan Gomes Lima2
Josivan Gomes Lima2 Vinaya Simha3
Vinaya Simha3 Marco Cappa4Saif Alyaarubi5
Marco Cappa4Saif Alyaarubi5 Renan Montenegro Jr.6
Renan Montenegro Jr.6 Baris Akinci7,8*
Baris Akinci7,8* Ferruccio Santini9
Ferruccio Santini9- 1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
- 2Hospital Universitário Onofre Lopes, Departamento de Clinica Medica, Universidade Federal do Rio Grande do Norte, Natal, Brazil
- 3Division of Endocrinology, Mayo Clinic, Rochester, MN, United States
- 4Research Area for Innovative Therapies in Endocrinopathies Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 5Pediatric Endocrinology, Oman Medical Specialty Board, Muscat, Oman
- 6Brazilian Group for the Study of Inherited and Acquired Lipodystrophies (BRAZLIPO), Clinical Research Unit, Walter Cantidio University Hospital, Federal University of Ceará/Ebserh, Fortaleza, Brazil
- 7Dokuz Eylul University Health Campus Technopark (DEPARK), Dokuz Eylul University, Izmir, Türkiye
- 8Department of Research Programs, Technological Research, Izmir Biomedicine and Genome Center (IBG), Izmir, Türkiye
- 9Obesity and Lipodystrophy Center, Endocrinology Unit, University Hospital of Pisa, Pisa, Italy
Introduction: Lipodystrophy syndromes are rare diseases that can present with a broad range of symptoms. Delays in diagnosis are common, which in turn, may predispose to the development of severe metabolic complications and end-organ damage. Many patients with lipodystrophy syndromes are only diagnosed after significant metabolic abnormalities arise. Prompt action by clinical teams may improve disease outcomes in lipodystrophy syndromes. The aim of the Rapid Action Plan is to serve as a set of recommendations from experts that can support clinicians with limited experience in lipodystrophy syndromes.
Methods: The Rapid Action Plan was developed using insights gathered through a series of advisory meetings with clinical experts in lipodystrophy syndromes. A skeleton template was used to facilitate interviews. A consensus document was developed, reviewed, and approved by all experts.
Results: Lipodystrophy is a clinical diagnosis. The Rapid Action Plan discusses tools that can help diagnose lipodystrophy syndromes. The roles of clinical and family history, physical exam, patient and family member photos, routine blood tests, leptin levels, skinfold measurements, imaging studies, and genetic testing are explored. Additional topics such as communicating the diagnosis to the patients/families and patient referrals are covered. A set of recommendations regarding screening and monitoring for metabolic diseases and end-organ abnormalities is presented. Finally, the treatment of lipodystrophy syndromes is reviewed.
Discussion: The Rapid Action Plan may assist clinical teams with the prompt diagnosis and holistic work-up and management of patients with lipodystrophy syndromes, which may improve outcomes for patients with this rare disease.
1 Introduction
Lipodystrophies are a group of heterogeneous, rare, and irreversible conditions characterized by an absence of subcutaneous fat (1, 2). The pattern of fat loss can either be across the whole body [generalized lipodystrophy (GL)] or in specific areas [partial lipodystrophy (PL)]. Both forms of lipodystrophy syndromes (GL or PL) may be genetic or acquired, leading to four major categories: congenital generalized lipodystrophy (CGL), familial partial lipodystrophy (FPLD), acquired generalized lipodystrophy (AGL), and acquired partial lipodystrophy (APL) (3, 4). Typical characteristics of these four major categories are summarized in Table 1 (5–13). In addition to these classical categories, additional etiologies such as progeroid syndromes, complex genetic syndromes, autoimmune syndromes, and lipodystrophy induced by myeloablative therapy can also manifest with GL or PL.
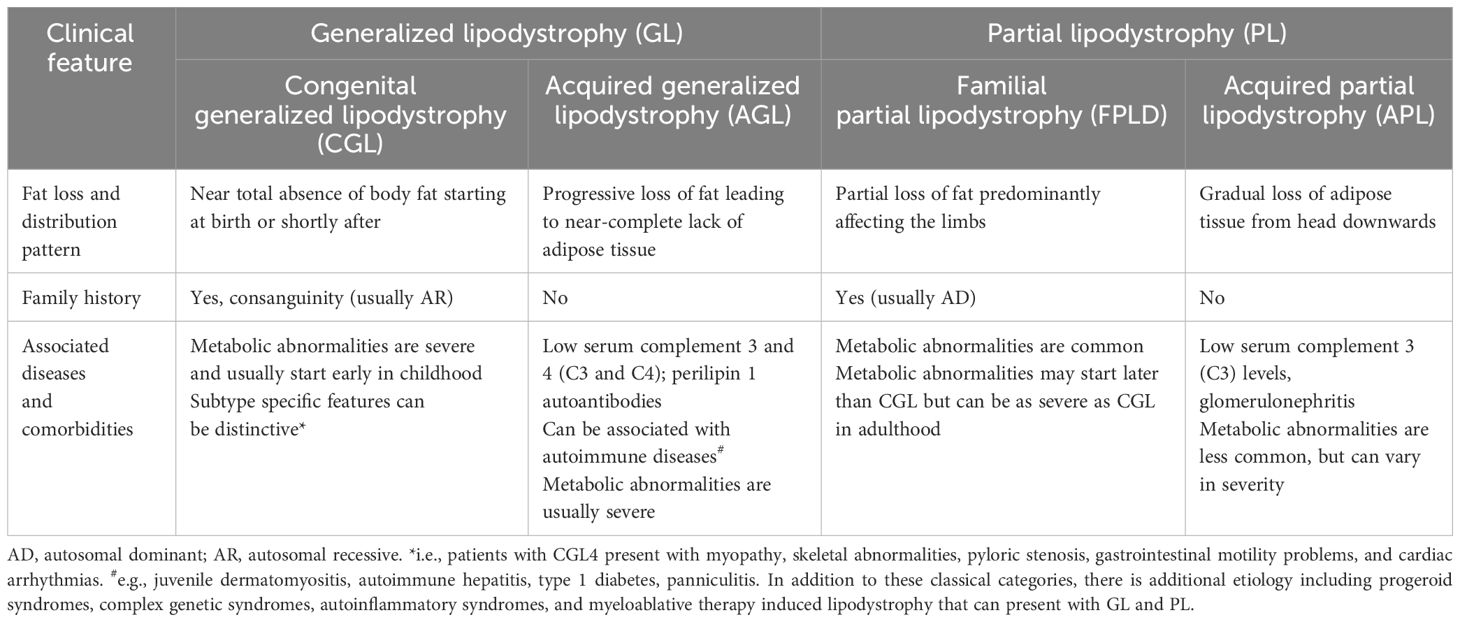
Table 1 Clinical features of four major subtypes of lipodystrophy syndromes.
Patients with lipodystrophy syndromes present with a broad range of symptoms. Clinical characteristics of lipodystrophy syndromes are heterogeneous and may depend on molecular etiology in genetic cases (14). Adipose tissue loss leads to ectopic fat accumulation which, in turn, triggers the development of severe insulin resistance and metabolic disease (15). Reduced leptin secretion contributes to the pathogenesis of lipodystrophy by adversely affecting appetite control, glucose and lipid homeostasis and metabolism (16, 17). In later stages of the disease, symptoms can include severe metabolic abnormalities (e.g., severe insulin resistance and difficult-to-treat diabetes, severe hypertriglyceridemia) and end-organ complications (e.g., non-alcoholic steatohepatitis (NASH), nephropathy, pancreatitis, cardiovascular disorders, neuromuscular system abnormalities) (1, 14, 15, 18–22).
While an international multi-society guideline exists (4) that covers the diagnosis and management of lipodystrophy syndromes, there is still a great amount of variation in the care that patients with lipodystrophy syndromes receive, including the speed with which they receive it. Despite a gradually increasing awareness of lipodystrophy syndromes among clinicians, it is still common for patients to be diagnosed only after they develop severe metabolic abnormalities and organ complications. Delays in diagnosis expose patients to the risk of developing severe metabolic disease and end-organ damage, which have already developed at the time of diagnosis in many cases (19, 23, 24). To address this knowledge gap, we aimed to create a Rapid Action Plan to support clinicians with limited experience in lipodystrophy syndromes by providing expertise from leading clinical teams. The goal of this plan is to reduce the time it takes for patients with lipodystrophy syndromes to receive a comprehensive diagnosis followed by the care and holistic support that they need.
2 Methods
This Rapid Action Plan was developed using insights gathered through a series of advisory meetings with clinical experts in lipodystrophy syndromes. A consensus meeting was held to initiate development of the Rapid Action Plan document at which time a group of international experts in lipodystrophy syndromes (United States, Brazil, Italy, Turkey, and Oman) was invited to discuss the key priorities for clinicians in the first 100 days after seeing a patient with clinical suspicion for the diagnosis. The rationale of the project, its scope and the role of expert contributors were further presented at this meeting. There was broad agreement from experts across this field that additional tools to help clinicians understand key indicators, priority tests and patient follow-up would be valuable. All parties have reviewed and endorsed the steps within this plan. The steps outlined reflect a consensus from experts on what action they would recommend given their experience. It is designed to be a reference tool for clinical teams that can complement national and international guidelines.
A skeleton template was used to facilitate interviews with experts. The first two sections of the skeleton were developed to review general information on lipodystrophy syndromes such as explaining the main subtypes of lipodystrophy. The next section was about the Rapid Action Plan focusing on diagnosing and supporting a patient with a suspicion of lipodystrophy syndromes. The aim of this section was to generate an action plan for clinical teams that can help reach a diagnosis without delay. This section was developed to answer specific questions such as:
● What physical findings and clinical features should raise suspicion for lipodystrophy syndromes?
● What are the other differential diagnoses that are likely to be considered by peers and experts? What is the list of conditions that need to be included in differential diagnosis?
● How can I confirm the diagnosis and screen for complications and comorbidities?
● How can I educate patients about the diagnosis and available support?
Experts were asked to provide information on tools that can support prompt diagnosis and referral. The role of physical signs and clinical history, skinfolds and radiology, laboratory testing, leptin level, and genetic testing was explored. Treatment strategies for lipodystrophy syndromes following metabolic risk stratification were also discussed.
After all interviews were completed, experts were asked for a second round of meetings to review outputs from initial interviews. Based on feedback from experts, revisions were made. A consensus document was developed, reviewed, and approved by all experts.
3 Results
3.1 Diagnosing lipodystrophy syndromes
3.1.1 Physical and metabolic signs
In patients with GL, the condition may be relatively apparent due to absence of fat in the whole body, prominent veins, and increased muscular appearance. However, parents and patients can easily get accustomed to this physical appearance if GL is not diagnosed at birth, and this may delay diagnosis until the development of potentially severe metabolic abnormalities. Children with GL more frequently present with elevated liver enzymes and severe hypertriglyceridemia compared to their counterparts with PL. Abdominal distension and protrusion of the umbilical scar are common and may help with diagnosis.
PL may be more challenging to identify as fat loss is selective. Phenotype also varies according to sex, with men usually presenting with a less prominent fat distribution abnormality and less severe metabolic profile than women. In certain subtypes of lipodystrophy syndromes, such as FPLD type 2 (Dunnigan variety), abnormal fat accumulation, particularly in the face and neck, can be remarkable. Patients with FPLD may exhibit significant muscularity and phlebomegaly in the limbs, which can be remarkable in lower extremities. In pediatric patients, metabolic abnormalities can be absent so adipose tissue distribution should be carefully considered.
Other associated signs and symptoms include insatiable appetite, skin findings (e.g., acanthosis nigricans, hirsutism in women, eruptive xanthomata), skeletal abnormalities (e.g., bone cysts, scoliosis), other organ abnormalities based on molecular etiology (in genetic cases), and autoimmune features (in acquired cases).
There are several disorders that need to be considered in the differential diagnosis of GL and PL. A list of common entities is listed in Table 2 (11, 25).

Table 2 Differential diagnosis of GL and PL.
● Lack of subcutaneous fat is the most critical manifestation.
● Any person with partial or complete lack of subcutaneous fat should be evaluated for the diagnosis of lipodystrophy syndromes.
● Be wary of features of metabolic syndrome in the absence of increased adiposity or high BMI.
● Lipodystrophy syndromes should be considered when metabolic symptoms are disproportionate to body size, including diabetes with high insulin requirements, hypertriglyceridemia, fatty liver disease, or polycystic ovary syndrome (PCOS).
3.1.2 Clinical and family history
Taking a detailed clinical and family history of the patient can help in diagnosing lipodystrophy syndromes and understanding the type of lipodystrophy a patient may have, especially for genetic forms of the disease. The family history should include questions about body shape as well as a history of known (or suspected) metabolic comorbidities or other clinical characteristics. It is important to remember phenotypic differences between men and women as it can be easy to overlook a male relative with lipodystrophy syndromes. Although no other family member with lipodystrophy can be detected in acquired lipodystrophies, it is important to assess for a history of autoimmunity and radiation and rule out HIV infection.
● Collating photos of patients through their lifetime can be very helpful. Sensitively ask for photos of the patient before/after symptoms (and ideally in different states of dress).
● The absence of adipose tissue can be identified at birth or within the first year of life in CGL, while fat loss develops at any time in life in AGL. In FPLD, fat loss can be detected in childhood, but it typically becomes prominent around puberty. Fat loss can start at any time in life in APL (usually in childhood, adolescence, or young adulthood).
● Photos of family members can also be helpful to assess and determine if a suspected lipodystrophy syndrome is genetic.
● Personal or family history of autoimmune diseases can be helpful.
3.1.3 Routine tests
In general, upon initial evaluation of the patient (and at follow-up appointments), routine laboratory tests should be considered to assess diabetes [e.g., fasting glucose levels, glycated hemoglobin (HbA1c)] and lipid abnormalities [fasting serum lipids (especially triglyceride levels)]. Oral glucose tolerance test (OGTT) can be considered on a case-by-case basis. In addition, liver function tests along with hepatic ultrasound and/or liver elastography to identify signs of non-alcoholic fatty liver disease (NAFLD) or hepatic fibrosis are recommended to evaluate hepatic disease. A baseline electrocardiogram (ECG) is recommended. As part of cardiac examination, an echocardiogram can be helpful to detect heart abnormalities (e.g., signs of cardiomyopathy).
● Laboratory tests should be considered to assess for insulin resistance, diabetes, and lipid abnormalities (high triglycerides and low HDL cholesterol are typical findings).
● Liver function tests along with hepatic ultrasound and/or elastography can identify signs of NAFLD and hepatic fibrosis.
3.1.4 Leptin assay
While leptin deficiency is a hallmark in the pathology of lipodystrophy syndromes, consensus among the group was that a given leptin level cannot be used to rule in or out a lipodystrophy diagnosis.
● Low leptin level is supportive but not diagnostic of lipodystrophy syndromes.
● Normal or high leptin levels do not rule out a lipodystrophy diagnosis.
3.1.5 Skinfold measurement
The panel thought that it is important to include objective measures of body composition as part of screening tools to aid in the diagnosis of lipodystrophy syndromes. This is particularly the case in PL where fat loss can be subtle. The panel agreed that skinfold measurement can be useful (particularly alongside other parameters) in the diagnosis of lipodystrophy syndromes, However, it is considered a less precise option when compared to alternative methods for measuring body fat. Where the technology and resources allow, alternatives should be considered before skinfold measurement including dual-energy x-ray absorptiometry (DXA) (see below).
● Mid-thigh skinfold thickness is easy to obtain and can be useful, but it is a rather imprecise option compared with alternatives for measuring body fat. Skin fold thickness values of the anterior thigh <10mm in adult men and <22mm in adult women are supportive information for the diagnosis of GL and FPLD (3).
3.1.6 Radiology
Where available, imaging modalities should be seen as an important objective tool to support the diagnosis of lipodystrophy syndromes. While magnetic resonance imaging (MRI) can be considered a useful tool, the panel noted that it is far less practical and more costly than dual energy X-ray absorptiometry (DXA). If physicians consider it impractical and burdensome to use MRI, it is suggested that this be reserved for research or exceptional circumstances, and that DXA is a suitable, cost-effective and appropriate alternative.
● Where available, imaging modalities should be seen as an important objective tool to support diagnosis of lipodystrophy syndromes.
● DXA is a suitable, cost-effective and appropriate strategy to assess fat quantity and distribution.
● Although clear diagnostic definitions are not established, a very low percentage of body fat may suggest GL, and a Fat Mass Ratio (the ratio between percent of the trunk fat mass and the percent of the lower-limb fat mass) higher than 1.2 in females may suggest FPLD (26).
● MRI is a useful tool to assess body composition but can be less practical and more costly.
3.1.7 Genetic testing
Lipodystrophy in a younger patient or with a positive family history (or consanguinity) may suggest a genetic etiology. Genetic testing in patients with suspected congenital or familial lipodystrophy syndromes can be used to confirm the diagnosis. However, a negative genetic test does not rule out an inherited form of the disease since some genes involved in the pathogenesis of lipodystrophy syndromes have yet to be identified. Furthermore, some forms of genetic lipodystrophy syndromes are polygenic in origin with affected genes not typically included in standard lipodystrophy panels. In patients suspected of lipodystrophy syndromes in whom genetic testing is negative, acquired forms of lipodystrophy syndromes should additionally be considered.
● Preference of the format of genetic testing continues to vary across localities.
● Among the expert panel reviewing this resource, diagnostic genetic panels were generally preferred over single gene testing.
● As caveats to genetic testing, some genes involved in the pathology of lipodystrophy syndromes are yet to be identified and some forms of genetic lipodystrophy syndromes can be polygenic.
● Whole Exome Sequencing (WES) was typically reserved for patients highly suspected of inherited lipodystrophy syndromes for whom the genetic panel did not yield results.
● As technology continues to improve, WES may become increasingly a viable option for routine use.
A diagnostic checklist is presented in Figure 1.

Figure 1 Diagnostic check list.
3.2 Communicating the diagnosis to the patient and families
A diagnosis of lipodystrophy is a life-changing moment and careful consideration must be given as to how best to communicate this to patients and their family members. The following are useful areas to consider on this topic:
• Several discussions may be needed.
Diagnosis is often a relief for people with lipodystrophy syndromes; however, there is often a lot of information to completely understand and it is likely to take several discussions/consultations before the patient is comfortable with the meaning of the diagnosis.
• Include other members of the multidisciplinary team.
Delivering the diagnosis alongside other specialist(s) from the multidisciplinary team that will likely be involved in the management of the patient can be helpful in some cases.
• Discuss proactive management.
The diagnosis can be framed as an opportunity to be proactive with respect to monitoring and managing risk factors for potential comorbidities.
• Distress over irreversible changes to appearance is common.
It is important that the person with lipodystrophy syndromes understands that it is generally not possible to regain lost fat. This can be very distressing and psychological support should be offered. There are also cosmetic options such as reconstruction surgery and fillers that could be considered.
• Consider genetic counselling in relevant cases.
The diagnosis of a heritable form of lipodystrophy syndromes is accompanied by specific considerations, and genetic counselling should be offered.
3.3 Referrals for patients
Referral to an expert center, where there is a multidisciplinary team with expertise in treating lipodystrophy syndromes, is highly recommended by the expert panel. If a nearby expert center does not exist, efforts should be made to consult with specialists who have experience in treating patients with lipodystrophy syndromes.
• Referral to an expert center is highly recommended.
3.4 Screening and monitoring for metabolic dysfunction and organ abnormalities
3.4.1 Insulin resistance, diabetes mellitus, and associated complications
Once a diagnosis of lipodystrophy syndrome is made, it is important to assess patients for insulin resistance, diabetes mellitus, and associated complications. For most patients, following international/national diabetes treatment guidelines is recommended (4, 27, 28). Even if initial laboratory results appear normal, regular monitoring should be conducted.
● Check fasting glucose (consider ordering fasting insulin alongside) as well as HbA1c at least annually.
● Screen patients for complications of insulin resistance and diabetes.
3.4.2 Lipids and pancreatitis
Dyslipidemia (high triglycerides, low HDL-cholesterol) is a major comorbidity of lipodystrophy syndromes which can lead to development of eruptive xanthomas and episodes of acute pancreatitis and is associated with increased cardiovascular risk (20). Following international/national treatment guidelines for hyperlipidemia is generally recommended (4, 29).
● Monitor fasting lipid panel at least annually and repeat with occurrence of abdominal pain or xanthomata.
3.4.3 Liver disease
Liver disease (severe liver steatosis leading to NASH and eventually liver cirrhosis) remains a major cause of mortality in patients with lipodystrophy syndromes and should be carefully monitored (30).
● Monitor liver enzymes at least annually.
● Consider elastography, liver ultrasound, MRI (with Dixon fat) or magnetic resonance elastography at diagnosis, and then every few years or sooner as clinically indicated.
● Liver biopsy is performed as clinically indicated.
3.4.4 Cardiovascular disease
Several forms of lipodystrophy syndromes are associated with heart disease (e.g., cardiomyopathy, arrhythmias, conduction abnormalities, coronary artery disease) due to frequent and severe metabolic complications and underlying molecular etiology (21, 22, 31–36).
● Blood pressure should be measured at every visit.
● Baseline ECG is recommended and should be repeated as clinically indicated.
● Echocardiogram should be considered as clinically indicated.
● Symptoms of coronary artery disease should be assessed carefully, and further testing should be considered based on clinical findings.
● Consider Holter ECG or ECG with exercise test to evaluate for arrhythmias in select patients.
● Risk of sudden death syndrome should not be overlooked in lipodystrophy syndromes.
3.4.5 Kidney disease
Chronic kidney disease (CKD) is a major cause of mortality in lipodystrophy syndromes. CKD is frequent and has an early onset in patients with lipodystrophy syndromes, and thus vigilance is recommended (37).
● Urine protein/creatinine ratio and serum creatinine levels should be assessed at the time of diagnosis and repeated at least annually.
3.4.6 Reproductive system
Reproductive dysfunction such as PCOS, oligo/amenorrhea, reduced fertility, and hirsutism is commonly detected in women with lipodystrophy syndromes. Also, women with lipodystrophy syndromes are theoretically at increased risk for preeclampsia, miscarriage and macrosomia due to poor metabolic control. Early adrenarche, true precocious puberty, or central hypogonadism also may occur in children with GL (1, 15, 38–40).
● Gonadal steroids, gonadotropins, and pelvic ultrasonography can be helpful in identifying reproductive dysfunction.
● Pubertal staging should be performed annually in children.
3.4.7 Hunger and other additional symptoms
Patients with lipodystrophy syndromes, especially GL, are typically hyperphagic due to leptin deficiency (41–45). However, monitoring hunger can be a challenge. While there was no overall consensus on how best to measure hyperphagia, the panel noted that it may be helpful to consider the use of questionnaires such as the Three-Factor Eating Questionnaire (46).
Comorbidities and other impairments of lipodystrophy syndromes that may affect patient quality of life include, but are not limited to, social anxiety and limitations with symptoms or physical appearance leading to inability to work and a reluctance or inability to socialize, chronic pain that can affect a patient’s ability to carry out basic tasks, and fatigue (47, 48).
● Patients with lipodystrophy syndromes, especially GL, typically have hyperphagia due to leptin deficiency which can have implications for patients, family members and caregivers.
● Additional symptoms of lipodystrophy may include anxiety, depression, chronic pain, and fatigue.
3.5 Treating lipodystrophy syndromes
3.5.1 Diet
Generally, patients are advised to follow diets with balanced macronutrient composition (3, 4). Energy-restricted diets improve metabolic abnormalities and may be appropriate in adults. Very-low-fat diets should be used in patients with severe hypertriglyceridemia (triglycerides > 1000 mg/dL). In the absence of severe hypertriglyceridemia, patients should replace refined carbohydrates with unsaturated fat and protein. Alcohol intake should be limited. A dietician should be consulted for specialized dietary needs, especially in infants and young children (3, 4, 49). Overfeeding should be avoided. Medium-chain triglyceride oil formulas can provide energy without raising triglycerides in infants.
● Most patients are recommended to follow a calorically restricted diet with balanced macronutrient composition; however, it may be difficult to control hunger in a leptin deficient state.
● Referral to a dietician is strongly recommended.
● Alcohol and smoking should be avoided.
3.5.2 Exercise
Patients with lipodystrophy syndromes should be encouraged to exercise in the absence of specific contraindications. Patients with subtypes of lipodystrophy syndromes predisposed to cardiomyopathy and/or arrhythmias should undergo cardiac evaluation before initiating an exercise regimen. Contact sports should be avoided in patients with severe hepatosplenomegaly and in patients with CGL who have lytic bone lesions. Referral to an exercise specialist may be appropriate.
● Exercise is encouraged in the absence of specific contraindications.
3.5.3 Role of metreleptin
Metreleptin, a recombinant analog of the human hormone leptin, is an orphan drug used to treat complications of leptin deficiency in lipodystrophy syndromes. In the US, metreleptin is approved as an adjunct to diet as replacement therapy to treat the complications of leptin deficiency in patients with CGL or AGL (50). In Europe and Brazil, metreleptin is indicated as an adjunct to diet as a replacement therapy to treat the complications of leptin deficiency in adults and children 2 years of age and above with confirmed CGL or AGL (51, 52). Metreleptin is also approved for this indication in adults and children 12 years of age and above with confirmed FPLD or APL in whom standard treatments have failed to achieve adequate metabolic control (51). The effects of metreleptin treatment are summarized in Figure 2.
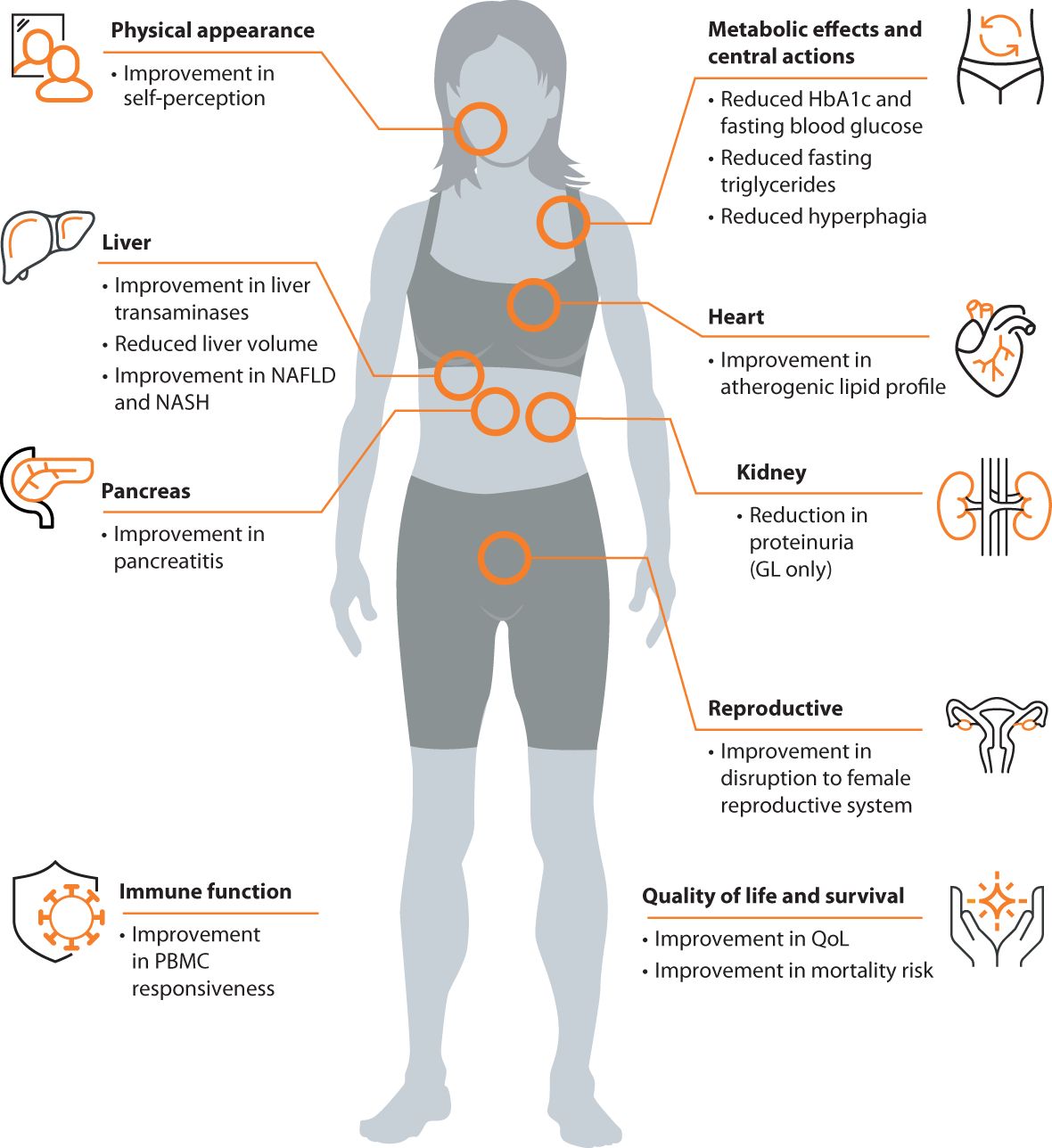
Figure 2 Clinical effects of metreleptin in patients with lipodystrophy syndromes.
● The expert panel concluded that, where available, metreleptin should be used in accordance with local guidelines.
3.5.4 Management of diabetes with standard approaches
In addition to the 2016 multi-society lipodystrophy guideline, international/national diabetes treatment guidelines can be helpful (4, 53). Metformin is the most commonly used agent to improve insulin resistance and glycemic control (3, 4). Thiazolidinediones may improve metabolic complications in PL (4, 54–56) but should be used with caution in GL. While animal data suggest partial reversal of metabolic disturbances in GL mouse models with thiazolidinediones (57), clinical experience in patients with GL is very limited (54, 58, 59). Preliminary data from case reports (60) and a case series indicate sodium-glucose cotransporter 2 (SGLT2) inhibitors can help improve glycemic control among patients with PL (61). Due to severe insulin resistance, many patients with lipodystrophy syndromes will need large amounts of insulin to control their blood glucose. Concentrated insulins should be considered (4, 62). In patients with PL, reduction of body mass index (BMI) may reduce the risk of metabolic comorbidities (63). Glucagon-like peptide-1 (GLP-1) receptor analogues may improve metabolic levels (64, 65). It is not known whether GLP-1 receptor analogues affect fat distribution in lipodystrophy syndromes. Some experts felt that GLP-1 receptor agonists may help reduce localized fat accumulation (e.g., on the abdominal fat) and may also help manage hyperphagia in some cases, although these effects have not been systematically studied in patients with lipodystrophy syndromes. Prior episodes of acute pancreatitis and severely elevated triglyceride levels can be limiting factors to GLP-1 receptor agonist use (66, 67).
● Treat diabetes per guidelines. Maintain oversight but include referral to an endocrinologist as part of management.
3.5.5 Management of lipid abnormalities with standard approaches
Lifestyle modifications are essential in the management of hypertriglyceridemia (see Diet and Exercise sections above). Statins should be used concomitantly with lifestyle interventions in the setting of hypertriglyceridemia, high low-density lipoprotein cholesterol (LDL-C), or diabetes mellitus for cardiovascular risk reduction. Fibrates are commonly used to treat severely elevated triglycerides though physicians should confer with their local prescribing guidelines regarding use in children. Long-chain omega-3 fatty acid use also may be helpful in treating hypertriglyceridemia. Although no studies are available in patients with lipodystrophies, a previous large-scale clinical study (68) reported cardiovascular benefits in patients with fasting triglyceride levels of 135–499 mg/dL following treatment with icosapent ethyl. However, the use of these agents may be limited in patients with lipodystrophies who are at higher risk of developing arrhythmias, as an increased risk of atrial fibrillation has been reported after the use of omega-3 fatty acids, particularly in high doses (69–71).
● Treat dyslipidemia aggressively using lifestyle modification and statins with or without fibrates.
● Cardiovascular disease risk calculators may not necessarily be accurate in patients with lipodystrophy syndromes.
3.5.6 Management of liver disease with standard approaches
The panel agreed that monitoring for liver disease is critical in patients with lipodystrophy. NAFLD may develop early and progress rapidly to NASH and cirrhosis in this population. Weight loss, exercise, and avoidance of excess alcohol are mainstays of treatment per international/national guidelines (3, 4, 72). However, from a patient perspective, significant weight loss can be an undesired outcome in lipodystrophy syndromes, especially in patients with GL.
● Monitor and treat for liver disease per guidelines.
● Consider referral to a hepatologist as part of management.
3.5.7 Management of cardiovascular disease with standard approaches
The panel considers patients with lipodystrophy syndromes to be at high risk of cardiovascular disease and believes that an aggressive risk mitigation and management strategy is needed. In addition to the 2016 multi-society lipodystrophy guidelines (4), the American Diabetes Association (ADA) guideline can be helpful for cardiovascular risk management and hypertension treatment among patients with diabetes (27, 29). Patients with subtypes of lipodystrophy syndromes predisposed to cardiomyopathy or arrhythmia should undergo more detailed cardiac evaluation (32, 33). This is especially important before initiating an exercise regimen.
● Monitor and treat for cardiovascular disease per guidelines.
● Consider referral to a cardiologist as part of management.
● Strongly advise against smoking and assist with smoking cessation.
3.5.8 Management of kidney disease with standard approaches
CKD has a complex background in lipodystrophy syndromes and can be severe and rapidly progressing (37). Depending on the subtype, the etiology of CKD may vary. Although patients with lipodystrophy syndromes may develop CKD in the absence of diabetes, consensus from the group was to treat many of them in a way similar to patients with CKD caused by diabetes (unless CKD is caused by a specific form of kidney disease such as C3-positive membranoproliferative glomerulonephritis) according to international/local guidelines (despite lack of evidence in patients with lipodystrophy syndromes) (73, 74). Angiotensin-converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB) in addition to sodium-glucose cotransporter 2 (SGLT2) inhibitors can be considered if urine protein is elevated (73).
● Monitor and treat for kidney disease per guidelines.
● Consider referral to nephrologist if patient is hypertensive, has elevated creatinine or low estimate glomerular filtration rate (eGFR), or proteinuria.
3.5.9 Management of reproductive health with standard approaches
In patients with low leptin levels, irregular menses may normalize on leptin replacement therapy (40). In patients who are not candidates for or whose menses do not normalize on leptin replacement, if a patient is amenorrhoeic due to a low leptin level and does not improve after leptin treatment, or cannot access leptin treatment, consider hormone replacement therapy– an estrogen patch (less harmful than oral estrogens on triglycerides) and oral progesterone, according to international/local guidelines (4, 75). PCOS in lipodystrophy syndromes is generally managed in line with international guidelines (76). Oral estrogens are generally contraindicated in the presence of severe hypertriglyceridemia (4). Patient attempting pregnancy should be referred to a reproductive clinic.
● Maintain oversight but consider referral to a reproductive endocrinologist or gynecologist as indicated.
4 Discussion
As a rare disease, lipodystrophy syndromes often pose a diagnostic and treatment challenge. In this regard, an international chart review study (19) involving five treatment centers in Brazil, Turkey and the United States has illustrated that patients commonly experience delays of several years before receiving a definitive diagnosis of lipodystrophy syndrome. Although fat loss is known to be prominent at birth in CGL and around puberty in FPLD, first symptoms of lipodystrophy were typically identified during childhood (mean age: 9.2 years) among patients with GL and during early adulthood (mean age: 24.7 years) among those with PL. More importantly, from the time symptoms were first noted, it took an average of 3.1 years in GL and 9.0 years in PL for physicians to diagnose them accurately. Delays in diagnosis and intervention predispose patients with lipodystrophy syndromes to irreversible end-organ damage, and as such comprehensive, multi-disciplinary diagnostic and treatment plans are critically needed. We developed the Rapid Action Plan to enable clinical teams to promptly diagnose and holistically manage patients with lipodystrophy syndromes. This set of guidance from experts is intended to enhance outcomes for individuals afflicted by this disease.
It is important to recognize that lipodystrophy syndromes are heterogeneous, resulting in a wide spectrum of clinical presentations. Nonetheless, regional or generalized absence of subcutaneous fat is the sine qua non of lipodystrophy and should be assessed for on physical exam. Clinical history is also important to differentiate this diagnosis from other possibilities, and to distinguish genetic versus acquired etiologies. Basic laboratory testing and imaging are useful adjuncts to the history and physical exam and can help to risk stratify patients for cardiometabolic complications. Insulin resistance, diabetes mellitus, hypertriglyceridemia, ectopic fat accumulation (e.g., hepatic steatosis), and low leptin level are common characteristics of the disease. However, these features may not be present in patients with all forms of lipodystrophy syndromes and are not specific for the condition.
Several tools can help to verify clinical suspicion for lack of subcutaneous fat. Skinfold measurement is an important part of objective assessment that is portable, fast, and affordable. Specifically, mid-thigh skinfolds with a cut-off of less than 10 mm in adult men and less than 22 mm in adult women (corresponding to approximately the 10th percentile of the US population) are useful to support the diagnosis (3). However, skinfold thicknesses are highly operator-dependent, do not give a comprehensive representation of overall fat distribution, and may be prone to error in patients with low body fat. Thus, imaging modalities are preferred, where available, as discussed below.
Among the most important and practical imaging modality noted by this expert panel was the DXA scan (77–79). Where skinfold measurement provides a simple on-the-spot measurement for body fat, DXA allows for the quantification of not only body fat, but also lean mass over time. Also, fat shadow images, which are color-coded representations highlighting only the fat tissue, can be generated from DXA images to facilitate visualization of fat distribution (80). Body composition can alternatively be assessed by computed tomography (CT) and MRI scans. Among the panel, use of MRI was generally recommended to obtain objective ratios of different fat compartments in patients with for PL (81, 82) and to provide information regarding residual mechanical fat in patients with CGL (83, 84). MRI also can determine the lipid content of tissues, solid organs (such as the liver) (85), and bone marrow fat (84) using the Dixon method or spectroscopy.
Despite the need to be mindful of a ‘false negative’, genetic testing remains an important supportive diagnostic and prognostic tool. A genetic test where the commonly known variants (e.g., LMNA) are negative should not rule out familial or congenital forms of lipodystrophy syndromes, which remain clinical diagnoses. On the other hand, positive genetic testing can be confirmatory of the diagnosis and should prompt a clinician to screen for complications directly or indirectly associated with molecular etiology and to offer genetic testing to affected family members.
Given the progressive nature of lipodystrophy syndromes, regular screening for metabolic abnormalities and end-organ complications is imperative. The comprehensive follow-up algorithm encompasses various components, including detection of insulin resistance, diabetes, complications associated with diabetes, lipid abnormalities, pancreatitis, liver disease, cardiovascular disease, kidney disease, reproductive system abnormalities, and other comorbidities driven by molecular or acquired etiologies. Emerging evidence emphasizes the importance of cardiovascular health in patients with lipodystrophy syndromes. Patients with lipodystrophy syndromes are at risk of coronary artery disease as a result of severe insulin resistance and poor metabolic control (22, 86). It should be noted that patients with LMNA mutations are more likely to develop cardiac disease than others with FPLD (33). Moreover, certain types of lipodystrophy syndromes (e.g., CGL4) are linked to potentially fatal cardiac rhythm changes and prompt diagnosis is therefore vital to minimize risk of sudden cardiac death (31, 32). It is critical to identify and to treat these arrhythmias before they lead to sudden cardiac death.
The current management strategy for lipodystrophy syndromes primarily focuses on metabolic health. Maintaining a controlled diet is key for individuals with lipodystrophy syndromes, although this can be challenging for some patients given a lack of fat and insatiable appetite due to leptin deficiency. Standard medications to treat metabolic disease (e.g., diabetes, hyperlipidemia) can be used with limited efficacy (56, 61, 64, 87). Recombinant leptin, metreleptin, is used as an adjunct to diet as a replacement therapy to treat metabolic complications of lipodystrophy syndromes. The clinical development program for metreleptin includes a pivotal study integrating data from two trials (NIH 991265/20010769; Clinical Trials IDs: NCT00005905 and NCT00025883) and a supportive study (FHA101; Clinical Trials ID: NCT00677313). In patients with GL, the NIH 991265/20010769 study showed a mean absolute reduction of 2.2% in HbA1c levels and a mean relative reduction of 32.1% in plasma triglycerides (88). The supportive study FHA101 yielded consistent efficacy results, albeit with a smaller number of patients (89). Discontinuation of insulin, oral antidiabetic medications, and lipid-lowering therapies was observed in a significant percentage of GL patients after starting metreleptin treatment (88). In patients with PL, metreleptin treatment resulted in significant reductions in HbA1c, fasting triglycerides, and liver volume. A subgroup of patients with baseline HbA1c ≥ 6.5% or plasma triglycerides ≥ 500 mg/dL experienced an absolute reduction of 0.9% in HbA1c and a relative reduction of 37.4% in triglycerides (90). Further studies at the NIH and other treatment centers showed that metreleptin decreased hyperphagia (17), improved insulin sensitivity (91, 92), reduced liver steatosis and improved NASH score on biopsy specimens (30, 93). Improvements in reproductive abnormalities in women (40) and reduction in proteinuria in patients with GL (94, 95) were also observed. While limited, real-life studies have supported the robust effectiveness of metreleptin in GL, whereas the impact of treatment has been heterogeneous in patients with PL. Araujo-Vilar et al. (96) reported a decrease from 11.8% to 6.7% in average HbA1c and a 78% reduction in triglycerides in patients with CGL. In a real-world experience analysis of 53 patients (28 GL and 25 PL) from four countries (France, Spain, Italy, and the UK) (97), one year of metreleptin treatment resulted in a mean reduction of 53% in triglycerides and a 1.9% point decrease in HbA1c in subjects with GL. In patients with PL, the mean percentage reduction in triglycerides was 22% and mean decrease in HbA1c was a 0.5%. A recent multicenter retrospective observational cohort study of 47 patients with lipodystrophy (28 GL and 19 PL) who started metreleptin therapy in France between 2009 and 2020 (98) reported significant improvements in HbA1c (from 8.4% to 6.8%) and fasting triglycerides (from 3.6 mmol/L to 2.2 mmol/L) after one year of treatment with metreleptin, with sustained efficacy thereafter. Additionally, a significant decrease was noted in liver enzymes. However, the impact of treatment was heterogeneous in patients with PL, and overall changes in HbA1c within this subgroup were not significant. Among patients with PL, 61% were responders regarding glucose homeostasis, and 61% were responders regarding hypertriglyceridemia at year 1, with those with more severe metabolic disease and lower leptin at baseline, as well as those with preserved β-cell functions, likely being better responders. Metreleptin has a black box warning for the risk of anti-metreleptin antibodies with neutralizing activity and risk of lymphoma in the US and is available only through a restricted program (50). Metreleptin is subject to additional monitoring in Europe (51). Other potential adverse reactions include hypersensitivity reactions, acute pancreatitis associated with discontinuation of metreleptin, hypoglycemia with concomitant use of insulin and other anti-diabetics, T-cell lymphoma, immunogenicity, and serious and severe infections. Also, data from clinical trials do not support safety and efficacy in patients with HIV-related lipodystrophy. A summary of the reported clinical effects of metreleptin in patients with lipodystrophy syndromes are shown in Figure 2.
Lipodystrophy syndromes not only impacts physical health but can also significantly affect mental well-being and quality of life (47, 48). Therefore, it is essential to assess patients’ psychological health and, where available, patients should be referred to a psychologist or counselor for support. Given that many patients may experience body dysmorphia, referral to a plastic surgeon or aesthetic specialist may be appropriate to address these concerns.
In a rare disease setting, consultation with dedicated specialized centers hold significant potential to reduce or even prevent lengthy diagnostic and treatment journeys. There are several ways to find the nearest specialist or specialist center for lipodystrophy syndromes. Because lipodystrophy specialists can be found via publications, running a PubMed search may be helpful. In Europe, there are specialist centers listed on the European Lipodystrophy Consortium’s (ECLip) website (https://www.eclip-web.org/lipodystrophies/). Also, European Reference Networks (ERN) for rare diseases might be helpful; the endocrinology ERN has a main thematic group on ‘genetic disorder of glucose & insulin homeostasis’ (MTG3) (https://endo-ern.eu/rare-genetic-disorders-of-glucose-insulin-homeostasis/). In the United States (US), the Endocrine Society website hosts a member-exclusive community to connect with specialists (DocMatter tool) (https://www.endocrine.org/membership/endoforum). In the Middle East, there is growing collaboration around lipodystrophy [Arab Society for Pediatric Endocrinology and Diabetes (ASPED)] (https://asped.net/). In Brazil, BrazLipo (https://brazlipo.org/) exists as a reference point for professionals and the public.
In conclusion, the complexity of lipodystrophy syndromes poses a challenge to clinicians with limited expertise in the field. The Rapid Action Plan may prove helpful for clinical teams seeking to promptly diagnose and holistically manage patients with lipodystrophy syndromes. This information has the potential to enhance outcomes for individuals with this rare disease.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
LF: Writing – original draft, Writing – review & editing. JL: Writing – original draft, Writing – review & editing. VS: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing. SA: Writing – original draft, Writing – review & editing. RM: Writing – original draft, Writing – review & editing. BA: Writing – original draft, Writing – review & editing. FS: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Amryt Pharmaceuticals provided funding solely for the research and publication costs associated with the manuscript. Amryt Pharmaceuticals did not provide any funding for authorship of the manuscript. Amryt Pharmaceuticals was not involved in data analysis, in the preparation of the abstract/manuscript nor in the decision to submit it for publication. Amryt Pharmaceuticals is a wholly owned subsidiary of Chiesi Farmaceutici S.p.A.
Acknowledgments
The authors extend their appreciation to Akt Health for not only conducting structured interviews but also for diligently preparing a report based on these interviews.
Conflict of interest
LF serves as a consultant for Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.) and Thera Technologies and receives grant support to her institution from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.). JL received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), is a speaker to Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), Astra Zeneca, NovoNordisk, and Abbott. VS received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.). MC received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.). SA received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.). RM received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.). BA received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), has run projects for and/or served as a consultant, board member, steering committee member, and/or speaker to Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), Alnylam, Regeneron, ThirdRock Ventures, Astra Zeneca, Novonordisk, Boehringer Ingelheim, Sanofi, Bilim Ilac, ARIS, and Servier. FS received a consultancy fee from Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), has run projects for and/or served as a consultant, board member, steering committee member, and/or speaker to Amryt Pharmaceuticals (wholly owned subsidiary of Chiesi Farmaceutici S.p.A.), Novonordisk, Boehringer Ingelheim, Ely Lilly, Pfizer, Bruno Farmaceutici, and BioItalia.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Garg A. Acquired and inherited lipodystrophies. N Engl J Med. (2004) 350:1220–34. doi: 10.1056/NEJMra025261
2. Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol. (2006) 17:162–9. doi: 10.1097/01.mol.0000217898.52197.18
3. Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, et al. The clinical approach to the detection of lipodystrophy - an aace consensus statement. Endocr Pract. (2013) 19:107–16. doi: 10.4158/endp.19.1.v767575m65p5mr06
4. Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The diagnosis and management of lipodystrophy syndromes: A multi-society practice guideline. J Clin Endocrinol Metab. (2016) 101:4500–11. doi: 10.1210/jc.2016–2466
5. Patni N, Garg A. Congenital generalized lipodystrophies–new insights into metabolic dysfunction. Nat Rev Endocrinol. (2015) 11:522–34. doi: 10.1038/nrendo.2015.123
6. Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin North Am. (2016) 45:783–97. doi: 10.1016/j.ecl.2016.06.012
7. Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Med (Baltimore). (2003) 82:129–46. doi: 10.1097/00005792–200303000–00007
8. Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Med (Baltimore). (2004) 83:18–34. doi: 10.1097/01.md.0000111061.69212.59
9. Fernandez-Pombo A, Sanchez-Iglesias S, Cobelo-Gomez S, Hermida-Ameijeiras A, Araujo-Vilar D. Familial partial lipodystrophy syndromes. Presse Med. (2021) 50:104071. doi: 10.1016/j.lpm.2021.104071
10. Hegele RA. Familial partial lipodystrophy: A monogenic form of the insulin resistance syndrome. Mol Genet Metab. (2000) 71:539–44. doi: 10.1006/mgme.2000.3092
11. Vantyghem MC, Balavoine AS, Douillard C, DeFrance F, Dieudonne L, Mouton F, et al. How to diagnose a lipodystrophy syndrome. Ann Endocrinol (Paris). (2012) 73:170–89. doi: 10.1016/j.ando.2012.04.010
12. Corvillo F, Abel BS, Lopez-Lera A, Ceccarini G, Magno S, Santini F, et al. Characterization and clinical association of autoantibodies against perilipin 1 in patients with acquired generalized lipodystrophy. Diabetes. (2023) 72:71–84. doi: 10.2337/db21–1086
13. Ceccarini G, Magno S, Gilio D, Pelosini C, Santini F. Autoimmunity in lipodystrophy syndromes. Presse Med. (2021) 50:104073. doi: 10.1016/j.lpm.2021.104073
14. Akinci B, Meral R, Oral EA. Phenotypic and genetic characteristics of lipodystrophy: pathophysiology, metabolic abnormalities, and comorbidities. Curr Diabetes Rep. (2018) 18:143. doi: 10.1007/s11892–018-1099–9
15. Akinci B, Sahinoz M, Oral E. Lipodystrophy Syndromes: Presentation and Treatment. Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. South Dartmouth (MA: Endotext (2000).
16. Papathanasiou AE, Nolen-Doerr E, Farr OM, Mantzoros CS. Geoffrey harris prize lecture 2018: novel pathways regulating neuroendocrine function, energy homeostasis and metabolism in humans. Eur J Endocrinol. (2019) 180:R59–71. doi: 10.1530/EJE-18–0847
17. McDuffie JR, Riggs PA, Calis KA, Freedman RJ, Oral EA, DePaoli AM, et al. Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J Clin Endocrinol Metab. (2004) 89:4258–63. doi: 10.1210/jc.2003–031868
18. Pedicelli S, de Palma L, Pelosini C, Cappa M. Metreleptin for the treatment of progressive encephalopathy with/without lipodystrophy (Peld) in a child with progressive myoclonic epilepsy: A case report. Ital J Pediatr. (2020) 46:158. doi: 10.1186/s13052–020-00916–2
19. Akinci B, Oral EA, Neidert A, Rus D, Cheng WY, Thompson-Leduc P, et al. Comorbidities and survival in patients with lipodystrophy: an international chart review study. J Clin Endocrinol Metab. (2019) 104:5120–35. doi: 10.1210/jc.2018–02730
20. Simha V, Garg A. Inherited lipodystrophies and hypertriglyceridemia. Curr Opin Lipidol. (2009) 20:300–8. doi: 10.1097/MOL.0b013e32832d4a33
21. Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. (2001) 103:2225–9. doi: 10.1161/01.cir.103.18.2225
22. Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology. (2019) 51:202–12. doi: 10.1016/j.pathol.2018.11.004
23. Araujo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J Endocrinol Invest. (2019) 42:61–73. doi: 10.1007/s40618-018-0887-z
24. Fourman LT, Grinspoon SK. Approach to the patient with lipodystrophy. J Clin Endocrinol Metab. (2022) 107:1714–26. doi: 10.1210/clinem/dgac079
25. Foss-Freitas MC, Akinci B, Luo Y, Stratton A, Oral EA. Diagnostic strategies and clinical management of lipodystrophy. Expert Rev Endocrinol Metab. (2020) 15:95–114. doi: 10.1080/17446651.2020.1735360
26. Valerio CM, Zajdenverg L, de Oliveira JE, Mory PB, Moyses RS, Godoy-Matos AF. Body composition study by dual-energy X-ray absorptiometry in familial partial lipodystrophy: finding new tools for an objective evaluation. Diabetol Metab Syndr. (2012) 4:40. doi: 10.1186/1758–5996-4–40
27. American Diabetes Association Professional Practice C. 4. Comprehensive medical evaluation and assessment of comorbidities: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S52–76. doi: 10.2337/dc24-S004
28. American Diabetes Association Professional Practice C. 6. Glycemic goals and hypoglycemia: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S111–S25. doi: 10.2337/dc24-S006
29. American Diabetes Association Professional Practice C. 10. Cardiovascular disease and risk management: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S179–218. doi: 10.2337/dc24-S010
30. Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. (2005) 41:753–60. doi: 10.1002/hep.20672
31. Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-qt syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (Cgl4) due to ptrf-cavin mutations. PloS Genet. (2010) 6:e1000874. doi: 10.1371/journal.pgen.1000874
32. Akinci G, Alyaarubi S, Patni N, Alhashmi N, Al-Shidhani A, Prodam F, et al. Metabolic and other morbid complications in congenital generalized lipodystrophy type 4. Am J Med Genet A. (2024) 194(6):e63533. doi: 10.1002/ajmg.a.63533
33. Eldin AJ, Akinci B, da Rocha AM, Meral R, Simsir IY, Adiyaman SC, et al. Cardiac phenotype in familial partial lipodystrophy. Clin Endocrinol (Oxf). (2021) 94:1043–53. doi: 10.1111/cen.14426
34. Haque WA, Vuitch F, Garg A. Post-mortem findings in familial partial lipodystrophy, dunnigan variety. Diabetes Med. (2002) 19:1022–5. doi: 10.1046/j.1464-5491.2002.00796.x
35. Yildirim Simsir I, Tuysuz B, Ozbek MN, Tanrikulu S, Celik Guler M, Karhan AN, et al. Clinical features of generalized lipodystrophy in Turkey: A cohort analysis. Diabetes Obes Metab. (2023) 25:1950–63. doi: 10.1111/dom.15061
36. Sanon VP, Handelsman Y, Pham SV, Chilton R. Cardiac manifestations of congenital generalized lipodystrophy. Clin Diabetes. (2016) 34:181–6. doi: 10.2337/cd16–0002
37. Akinci B, Unlu SM, Celik A, Simsir IY, Sen S, Nur B, et al. Renal complications of lipodystrophy: A closer look at the natural history of kidney disease. Clin Endocrinol (Oxf). (2018) 89:65–75. doi: 10.1111/cen.13732
38. Huseman CA, Johanson AJ, Varma MM, Blizzard RM. Congenital lipodystrophy. Ii. Association with polycystic ovarian disease. J Pediatr. (1979) 95:72–4. doi: 10.1016/s0022–3476(79)80087–6
39. Blackwell VC, Salis P, Groves RW, Baldeweg SE, Conway GS. Unwin RJ. Partial lipodystrophy, polycystic ovary syndrome and proteinuria: A common link to insulin resistance? J R Soc Med. (2001) 94:238–40. doi: 10.1177/014107680109400510
40. Oral EA, Ruiz E, Andewelt A, Sebring N, Wagner AJ, Depaoli AM, et al. Effect of leptin replacement on pituitary hormone regulation in patients with severe lipodystrophy. J Clin Endocrinol Metab. (2002) 87:3110–7. doi: 10.1210/jcem.87.7.8591
41. Unger RH. Longevity, lipotoxicity and leptin: the adipocyte defense against feasting and famine. Biochimie. (2005) 87:57–64. doi: 10.1016/j.biochi.2004.11.014
42. Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. (2005) 54:1994–2002. doi: 10.2337/diabetes.54.7.1994
43. Ebihara K, Nakao K. Translational Research of Leptin in Lipodystrophy and Its Related Diseases. In: Nakao K, Minato N, Uemoto S, editors. Innovative Medicine: Basic Research and Development. Tokyo: Springer. (2015). p. 165–75.
44. Aotani D, Ebihara K, Sawamoto N, Kusakabe T, Aizawa-Abe M, Kataoka S, et al. Functional magnetic resonance imaging analysis of food-related brain activity in patients with lipodystrophy undergoing leptin replacement therapy. J Clin Endocrinol Metab. (2012) 97:3663–71. doi: 10.1210/jc.2012–1872
45. Ahima R, Osei SY. Leptin and appetite control in lipodystrophy. J Clin Endocrinol Metab. (2004) 89:4254–7. doi: 10.1210/jc.2004–1232
46. Karlsson J, Persson LO, Sjostrom L, Sullivan M. Psychometric properties and factor structure of the three-factor eating questionnaire (Tfeq) in obese men and women. Results from the swedish obese subjects (Sos) study. Int J Obes Relat Metab Disord. (2000) 24:1715–25. doi: 10.1038/sj.ijo.0801442
47. Calabro PF, Ceccarini G, Calderone A, Lippi C, Piaggi P, Ferrari F, et al. Psychopathological and psychiatric evaluation of patients affected by lipodystrophy. Eat Weight Disord. (2020) 25:991–8. doi: 10.1007/s40519–019-00716–6
48. Demir T, Simsir IY, Tuncel OK, Ozbaran B, Yildirim I, Pirildar S, et al. Impact of lipodystrophy on health-related quality of life: the qualip study. Orphanet J Rare Dis. (2024) 19:10. doi: 10.1186/s13023-023-03004-w
49. Cecchetti C, Belardinelli E, Dionese P, Teglia R, Fazzeri R, MR DA, et al. Is it possible to achieve an acceptable disease control by dietary therapy alone in berardinelli seip type 1? Experience from a case report. Front Endocrinol (Lausanne). (2023) 14:1190363. doi: 10.3389/fendo.2023.1190363
50. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125390s024lbl.pdf.
51. Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/Myalepta.
52. Available online at: https://consultas.anvisa.gov.br/#/bulario/q/?numeroRegistro=175040002.
53. American Diabetes Association Professional Practice C. 9. Pharmacologic approaches to glycemic treatment: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S158–S78. doi: 10.2337/dc24-S009
54. Arioglu E, Duncan-Morin J, Sebring N, Rother KI, Gottlieb N, Lieberman J, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. (2000) 133:263–74. doi: 10.7326/0003–4819-133–4-200008150–00009
55. Agostini M, Schoenmakers E, Beig J, Fairall L, Szatmari I, Rajanayagam O, et al. A pharmacogenetic approach to the treatment of patients with pparg mutations. Diabetes. (2018) 67:1086–92. doi: 10.2337/db17–1236
56. Collet-Gaudillat C, Billon-Bancel A, Beressi JP. Long-term improvement of metabolic control with pioglitazone in a woman with diabetes mellitus related to dunnigan syndrome: A case report. Diabetes Metab. (2009) 35:151–4. doi: 10.1016/j.diabet.2009.01.001
57. Prieur X, Dollet L, Takahashi M, Nemani M, Pillot B, Le May C, et al. Thiazolidinediones partially reverse the metabolic disturbances observed in bscl2/seipin-deficient mice. Diabetologia. (2013) 56:1813–25. doi: 10.1007/s00125–013-2926–9
58. Kumar R, Pilania RK, Bhatia A, Dayal D. Acquired generalised lipodystrophy and type 1 diabetes mellitus in a child: A rare and implacable association. BMJ Case Rep. (2018) 2018:bcr2018225553. doi: 10.1136/bcr-2018–225553
59. Bhatia R, Chennupathi P, Rosenstein ED, Advani S. Spontaneous remission of acquired generalized lipodystrophy presenting in the postpartum period. JCEM Case Rep. (2024) 2:luae009. doi: 10.1210/jcemcr/luae009
60. Nagayama A, Ashida K, Watanabe M, Moritaka K, Sonezaki A, Kitajima Y, et al. Case report: metreleptin and sglt2 inhibitor combination therapy is effective for acquired incomplete lipodystrophy. Front Endocrinol (Lausanne). (2021) 12:690996. doi: 10.3389/fendo.2021.690996
61. Bansal R, Cochran E, Startzell M, Brown RJ. Clinical effects of sodium-glucose transporter type 2 inhibitors in patients with partial lipodystrophy. Endocr Pract. (2022) 28:610–4. doi: 10.1016/j.eprac.2022.03.006
62. Cochran E, Musso C, Gorden P. The use of U-500 in patients with extreme insulin resistance. Diabetes Care. (2005) 28:1240–4. doi: 10.2337/diacare.28.5.1240
63. Koo E, Foss-Freitas MC, Meral R, Ozer M, Eldin AJ, Akinci B, et al. The metabolic equivalent bmi in patients with familial partial lipodystrophy (Fpld) compared with those with severe obesity. Obes (Silver Spring). (2021) 29:274–8. doi: 10.1002/oby.23049
64. Banning F, Rottenkolber M, Freibothe I, Seissler J, Lechner A. Insulin secretory defect in familial partial lipodystrophy type 2 and successful long-term treatment with a glucagon-like peptide 1 receptor agonist. Diabetes Med. (2017) 34:1792–4. doi: 10.1111/dme.13527
65. Oliveira J, Lau E, Carvalho D, Freitas P. Glucagon-like peptide-1 analogues - an efficient therapeutic option for the severe insulin resistance of lipodystrophic syndromes: two case reports. J Med Case Rep. (2017) 11:12. doi: 10.1186/s13256–016-1175–1
66. Cao C, Yang S, Zhou Z. Glp-1 receptor agonists and pancreatic safety concerns in type 2 diabetic patients: data from cardiovascular outcome trials. Endocrine. (2020) 68:518–25. doi: 10.1007/s12020–020-02223–6
67. Sodhi M, Rezaeianzadeh R, Kezouh A, Etminan M. Risk of gastrointestinal adverse events associated with glucagon-like peptide-1 receptor agonists for weight loss. JAMA. (2023) 330:1795–7. doi: 10.1001/jama.2023.19574
68. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. (2019) 380:11–22. doi: 10.1056/NEJMoa1812792
69. Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the strength randomized clinical trial. JAMA. (2020) 324:2268–80. doi: 10.1001/jama.2020.22258
70. Curfman G. Omega-3 fatty acids and atrial fibrillation. JAMA. (2021) 325:1063. doi: 10.1001/jama.2021.2909
71. Bork CS, Myhre PL, Schmidt EB. Do omega-3 fatty acids increase risk of atrial fibrillation? Curr Opin Clin Nutr Metab Care. (2023) 26:78–82. doi: 10.1097/MCO.0000000000000907
72. Cusi K, Isaacs S, Barb D, Basu R, Caprio S, Garvey WT, et al. American association of clinical endocrinology clinical practice guideline for the diagnosis and management of nonalcoholic fatty liver disease in primary care and endocrinology clinical settings: co-sponsored by the american association for the study of liver diseases (Aasld). Endocr Pract. (2022) 28:528–62. doi: 10.1016/j.eprac.2022.03.010
73. American Diabetes Association Professional Practice C. 11. Chronic kidney disease and risk management: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S219–S30. doi: 10.2337/dc24-S011
74. Slinin Y, Ishani A, Rector T, Fitzgerald P, MacDonald R, Tacklind J, et al. Management of hyperglycemia, dyslipidemia, and albuminuria in patients with diabetes and ckd: A systematic review for a kdoqi clinical practice guideline. Am J Kidney Dis. (2012) 60:747–69. doi: 10.1053/j.ajkd.2012.07.017
75. The Hormone Therapy Position Statement of The North American Menopause Society" Advisory P. The 2022 hormone therapy position statement of the north american menopause society. Menopause. (2022) 29:767–94. doi: 10.1097/GME.0000000000002028
76. Teede HJ, Tay CT, Laven JJE, Dokras A, Moran LJ, Piltonen TT, et al. Recommendations from the 2023 international evidence-based guideline for the assessment and management of polycystic ovary syndrome. J Clin Endocrinol Metab. (2023) 108:2447–69. doi: 10.1210/clinem/dgad463
77. Valerio CM, Godoy-Matos A, Moreira RO, Carraro L, Guedes EP, Moises RS, et al. Dual-energy X-ray absorptiometry study of body composition in patients with lipodystrophy. Diabetes Care. (2007) 30:1857–9. doi: 10.2337/dc07–0025
78. Vasandani C, Li X, Sekizkardes H, Adams-Huet B, Brown RJ, Garg A. Diagnostic value of anthropometric measurements for familial partial lipodystrophy, dunnigan variety. J Clin Endocrinol Metab. (2020) 105:2132–41. doi: 10.1210/clinem/dgaa137
79. Ajluni N, Meral R, Neidert AH, Brady GF, Buras E, McKenna B, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). (2017) 86:698–707. doi: 10.1111/cen.13311
80. Meral R, Ryan BJ, Malandrino N, Jalal A, Neidert AH, Muniyappa R, et al. "Fat shadows" from dxa for the qualitative assessment of lipodystrophy: when a picture is worth a thousand numbers. Diabetes Care. (2018) 41:2255–8. doi: 10.2337/dc18–0978
81. Lim K, Haider A, Adams C, Sleigh A, Savage DB. Lipodistrophy: A paradigm for understanding the consequences of "Overloading" Adipose tissue. Physiol Rev. (2021) 101:907–93. doi: 10.1152/physrev.00032.2020
82. Adiyaman SC, Altay C, Kamisli BY, Avci ER, Basara I, Simsir IY, et al. Pelvis magnetic resonance imaging to diagnose familial partial lipodystrophy. J Clin Endocrinol Metab. (2023) 108:e512–e20. doi: 10.1210/clinem/dgad063
83. Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the agpat2 or seipin genes. J Clin Endocrinol Metab. (2003) 88:5433–7. doi: 10.1210/jc.2003–030835
84. Altay C, Secil M, Demir T, Atik T, Akinci G, Ozdemir Kutbay N, et al. Determining residual adipose tissue characteristics with mri in patients with various subtypes of lipodystrophy. Diagn Interv Radiol. (2017) 23:428–34. doi: 10.5152/dir.2017.17019
85. Reeder SB, Sirlin CB. Quantification of liver fat with magnetic resonance imaging. Magn Reson Imaging Clin N Am. (2010) 18:337–57, ix. doi: 10.1016/j.mric.2010.08.013
86. Kinzer AB, Shamburek RD, Lightbourne M, Muniyappa R, Brown RJ. Advanced lipoprotein analysis shows atherogenic lipid profile that improves after metreleptin in patients with lipodystrophy. J Endocr Soc. (2019) 3:1503–17. doi: 10.1210/js.2019–00103
87. Shamsudeen I, Hegele RA. Advances in the care of lipodystrophies. Curr Opin Endocrinol Diabetes Obes. (2022) 29:152–60. doi: 10.1097/MED.0000000000000695
88. Brown RJ, Oral EA, Cochran E, Araujo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. (2018) 60:479–89. doi: 10.1007/s12020–018-1589–1
89. Ajluni N, Dar M, Xu J, Neidert AH, Oral EA. Efficacy and safety of metreleptin in patients with partial lipodystrophy: lessons from an expanded access program. J Diabetes Metab. (2016) 7(3):659. doi: 10.4172/2155–6156.1000659
90. Oral EA, Gorden P, Cochran E, Araujo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine. (2019) 64:500–11. doi: 10.1007/s12020–019-01862–8
91. Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. (2002) 346:570–8. doi: 10.1056/NEJMoa012437
92. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. (2002) 109:1345–50. doi: 10.1172/JCI15001
93. Safar Zadeh E, Lungu AO, Cochran EK, Brown RJ, Ghany MG, Heller T, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol. (2013) 59:131–7. doi: 10.1016/j.jhep.2013.02.007
94. Javor ED, Moran SA, Young JR, Cochran EK, DePaoli AM, Oral EA, et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. J Clin Endocrinol Metab. (2004) 89:3199–207. doi: 10.1210/jc.2003–032140
95. Lee HL, Waldman MA, Auh S, Balow JE, Cochran EK, Gorden P, et al. Effects of metreleptin on proteinuria in patients with lipodystrophy. J Clin Endocrinol Metab. (2019) 104:4169–77. doi: 10.1210/jc.2019–00200
96. Araujo-Vilar D, Sanchez-Iglesias S, Guillin-Amarelle C, Castro A, Lage M, Pazos M, et al. Recombinant human leptin treatment in genetic lipodystrophic syndromes: the long-term spanish experience. Endocrine. (2015) 49:139–47. doi: 10.1007/s12020–014-0450–4
97. Cook K, Stears A, Araujo VD, Santini F, O’Rahilly S, Ceccarini G, et al. Savage DB real-world experience of generalized and partial lipodystrophy patients enrolled in the metreleptin early access program. Endocrine Abstracts. (2019) 63:586. doi: 10.1530/endoabs.63.P586
98. Mosbah H, Vantyghem MC, Nobecourt E, Andreelli F, Archambeaud F, Bismuth E, et al. Therapeutic indications and metabolic effects of metreleptin in patients with lipodystrophy syndromes: real-life experience from a national reference network. Diabetes Obes Metab. (2022) 24:1565–77. doi: 10.1111/dom.14726
Keywords: delay in diagnosis, disease management, lipodystrophy, screening, clinical assessment
Citation: Fourman LT, Lima JG, Simha V, Cappa M, Alyaarubi S, Montenegro Jr. R, Akinci B and Santini F (2024) A rapid action plan to improve diagnosis and management of lipodystrophy syndromes. Front. Endocrinol. 15:1383318. doi: 10.3389/fendo.2024.1383318
Received: 07 February 2024; Accepted: 13 May 2024;
Published: 04 June 2024.
Edited by:
Lin Zhu, Vanderbilt University Medical Center, United StatesReviewed by:
David Araujo-Vilar, University of Santiago de Compostela, SpainSun Manyi, Nankai University, China
Copyright © 2024 Fourman, Lima, Simha, Cappa, Alyaarubi, Montenegro, Akinci and Santini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baris Akinci, YmFyaXNha2luY2ltZEBnbWFpbC5jb20=