
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 08 July 2024
Sec. Pediatric Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1367808
 Mónica Fernández-Cancio1,2*†‡
Mónica Fernández-Cancio1,2*†‡ María Antolín3,4†
María Antolín3,4† María Clemente1,2,5,6Ariadna Campos-Martorell1,5,6Eduard Mogas1,5,6Noelia Baz-Redón1,2Jordi Leno-Colorado3,4Gemma Comas-Armangué3Elena García-Arumí2,3,7
María Clemente1,2,5,6Ariadna Campos-Martorell1,5,6Eduard Mogas1,5,6Noelia Baz-Redón1,2Jordi Leno-Colorado3,4Gemma Comas-Armangué3Elena García-Arumí2,3,7 Laura Soler-Colomer5Núria González-Llorens5
Laura Soler-Colomer5Núria González-Llorens5 Núria Camats-Tarruella1,2Diego Yeste1,2,5,6
Núria Camats-Tarruella1,2Diego Yeste1,2,5,6Introduction: Defects in any thyroid hormone synthesis steps cause thyroid dyshormonogenesis (THD). THD due to thyroglobulin (TG) gene variants is a cause of congenital hypothyroidism (CH) with a wide clinical spectrum, ranging from mild to severe permanent hypothyroidism. We present high-throughput sequencing results of patients with TG variants.
Methods: A CH high-throughput sequencing-panel of the main genes involved in the regulation of thyroid hormonogenesis was performed to identify those TG variants that may be related to patient THD phenotype.
Results: We identified 21 TG gene variants in 19 patients (11.8%) which could explain their phenotype. Ten of those (47.6%) were not previously described. CH was biochemically severe in these 19 patients. Eight of them were reevaluated after one month of discontinuing LT4 treatment and all had severe permanent hypothyroidism. We also identified another 16 patients who presented heterozygous TG variants, of whom, at reevaluation, five had mild permanent and only one had severe permanent hypothyroidisms.
Discussions: In this study, 10 novel and 11 previously reported variants in the TG gene have been identified that could explain the phenotype of 19 patients from non-consanguineous families from a large THD cohort. Although not all these TG gene variants can explain all the patients’ THD phenotypes, some of them had severe or mild permanent hypothyroidism at reevaluation.
Primary congenital hypothyroidism (CH) is the most common endocrine disorder in neonates and infants with an incidence of one in 3,000 newborns (the female to male ratio is 2:1) (1–5) and is also one of the most common preventable causes of intellectual disability. If untreated, CH causes permanent intellectual disability and slow growth. Most CH patients can be detected by newborn screening programs during the first week after birth, when there may be no specific clinical symptoms or signs suggestive of CH.
Genetic classification divides CH into two main categories according to their causes: a) thyroid gland development disorders (the dysembryogenesis or thyroid dysgenesis group), which occurs in 60% of all CH patients or b) defects in any of the steps of thyroid hormone synthesis (the dyshormonogenesis group) in the remaining cases (6, 7). Thyroid dyshormonogenesis (THD) can be caused by different defects in genes involved in thyroid hormone biosynthesis. There are several well-known associated genes, including DUOX1, DUOX2, DUOXA1, DUOXA2, SLC26A4, SLC26A7, SLC5A5, TPO, IYD and TG (3, 6, 8–10).
Dyshormonogenesis due to thyroglobulin (TG) gene variants is a cause of CH with an estimated incidence of 1 in 67,000 to 1 in 100,000 newborns (11–15), although this incidence may be biased and is likely to change with the use of high-throughput sequencing. Their clinical spectrum ranges from euthyroid through mild or severe permanent hypothyroidism with high iodide uptake, negative perchlorate discharge test (PDT), low levels of serum TG, high levels of serum thyroid-stimulating hormone (TSH), and simultaneous low serum total tetraiodothyronine (thyroxine, T4) levels accompanied by low or normal serum triiodothyronine (T3) levels. In untreated patients, TG defects due to loss-of-function variants (frameshift, nonsense and splice site variants) or missense variants involving wild-type cysteine residues and the acetylcholinesterase-homology (ChEL) domain cause persistent thyroid hormone (TH) deficiency, characterized by a severe phenotype resulting in intellectual disability with a large goiter (10).
The human TG gene is a single copy gene of 268 Kb that maps on chromosome 8q24 and is divided into 48 exons, encoding a mRNA sequence of 8,455 nucleotides (nt) (of which 8,304 nt correspond to coding sequences, NCBI: NM_003235.5) (13, 15–19). The monomeric human TG preprotein has a leader peptide of 19 amino acids followed by a 2749-amino acid polypeptide (NP_003226.4) (18). To date, about 230 human TG gene variants associated with THD have been identified (10). Since THD is due to pathogenic variants in TG inherited in an autosomal recessive manner, the patients should be homozygous or compound heterozygous.
In this study, high-throughput sequencing of patients from a Spanish cohort with THD reveal the presence of 19 patients with TG variants. To the best of our knowledge, this cohort study is the largest in Spain with regard to the number of genes analyzed and patients with THD enrolled, about whom an extensive clinical description and a genotype-phenotype correlation have been made.
Patients were diagnosed within the CH Neonatal Screening Program in Catalonia, based on TSH levels, and were treated at the Pediatric Endocrinology Unit of the Vall d’Hebron University Hospital (VUVH) between 2001 and 2022. This study is an ambispective, retrospective and longitudinal study.
All patients, or their responsible adults, provided informed consent for the use of their samples in research studies. Their confidentiality was maintained by assigning a sample code. Our project was approved by the Clinical Research Ethics Committee (CEIC) of the VHUH (PR (AMI) 390/2016).
The inclusion criteria were: serum TSH levels higher than 10 mIU/L (AutoDELFIA®), in a sample drawn at the time of diagnostic confirmation of CH, and with scintigraphic and/or ultrasound studies suggestive of THD. Patients with serum TSH levels below 10 mIU/L, or with thyroid agenesis or ectopia diagnosed by thyroid scintigraphy and/or ultrasound, or with recognized syndromes were excluded. Additional information on the possible existence of thyroid disease in members of the family or associated malformations/diseases was collected in all cases.
Serum TSH levels at confirmation were determined by electrochemiluminescence immunoassay (TSH3-Ultra, ADVIA Centaur®), free T4 (fT4) levels were determined by electrochemiluminescence immunoassay (FT4, ADVIA Centaur®), and TG levels were determined by electrochemiluminescence immunoassay (Elecsys® Tg II-Cobas®).
Permanent or transient CH was determined using results of thyroid function tests after temporary withdrawal of LT4 therapy at approximately 3 years of age. One month after discontinuing levothyroxine (LT4) treatment, TSH and fT4 levels were measured in venous blood sample. Individuals who showed continuous dependency on LT4 were diagnosed with permanent CH. After reevaluation, patients with TSH≥10 mIU/L and fT4<0.8 ng/dL were classified as severe permanent THD and patients with TSH≥10 mIU/L and fT4> 0.8ng/dL as mild permanent THD. Subsequently, these children were then repeatedly evaluated at regular intervals of six months to monitor thyroid function. Those who did not need continuous LT4 therapy were diagnosed with transient CH (TSH<5 mIU/L and fT4>0.8 ng/dL). Reevaluation was not carried out when LT4 requirements (>3.4 mcg/day) and genetic results were suggestive of TG deficiency.
The data describing clinical characteristics and anthropometric neonatal parameters are summarized in Supplementary Table S1.
Peripheral blood DNA was extracted by automatic magnetic extraction (Chemagic™ 360, Perkin-Elmer, Waltham, MA, USA) and DNA concentration was measured by Qubit 2.0 fluorometer (Qubit dsDNA BR Assay; Thermo Fisher, Waltham, MA, USA).
To perform the genetic study, a CH high-throughput sequencing-panel was designed with the GeneRead DNAseq Custom Builder tool from Qiagen (https://www.qiagen.com/en/shop/genes-and-pathways/custom-products/custom-array-products/generead-designer/) which included the main genes involved in the regulation of thyroid hormonogenesis: DUOX2, DUOXA2, IYD, PAX8, TG, TPO, TSHR, SLC26A4 and SLC5A5. This CH panel consisted of 393 amplicons (with an average size of 194 base pairs, and coverage ±10 nt of the exon-intronic junction zone) and allows sequencing of exons and adjacent intronic gene regions of interest, with a total coverage of 96.4% of the target sequence (GeneRead, Qiagen, Hilden, Germany) (20). The GeneRead CH panel was performed on the DNA samples of the patients following the provider’s instructions. In summary, amplification by PCR, indexing and library preparation (NEBNext Ultra DNA library prep kit, New England Biolabs) of the DNA samples were carried out. In those cases of greater clinical complexity, samples were analysed with a customised targeted Seq-Cap EZ HyperCap (NimbleGen) designed to capture the following genes ANO1, CDCA8, DUOX1, DUOX2, DUOXA2, FOXE1, GLIS3, GNAS, IYD, NKX2-1, NKX2-5, PAX8, SLC26A4, SLC5A5, TG, THRB, TPO and TSHR.
Finally, library sequencing was performed in a MiSeq platform (Illumina, San Diego, CA, USA) and bioinformatic analysis of the obtained data was performed with MiSeq Control Software (MCS), MiSeq Reporter (Illumina Inc, San Diego CA, USA), GeneRead SeqVariant Analysis software (Qiagen) and ANNOVAR adapted to our own pipeline. Firstly, overrepresented sequences were trimming using Trimmomatic (21), removing the sequences corresponding to the cut adapters used. Afterwards, sequences were mapped against the GRCh37 reference genome, using BWA (22). Variant calling was performed with Genome Analysis Toolkit (GATK) (23), and the variant annotation was done using ANNOVAR (24). Sanger sequencing was performed to confirm detected candidate variants and to analyze target regions with low coverage (<20).
The classification of the variants was performed following the American College of Medical Genetics and Genomics (ACMG) criteria (https://www.acmg.net/) using Varsome (https://varsome.com/) (25). Prediction of the effect of variants identified in the splicing regions was evaluated by Alamut Visual 2.11 (https://www.interactive-biosoftware.com/es/alamut-visual/). Family co-segregation analysis was carried out, when possible. The nucleotide position in human TG mRNA was designated according to reference sequence (NM_003235.5). Variant nomenclature is according to the guidelines of the Human Genome Variation Society (HGVS) (https://www.hgvs.org/) (26).
The frequency and the functional annotation of the identified variants were checked in public databases (GnomAD, ExAC, 1000 Genome project). Genetic variants including frameshift, missense, nonsense, and splicing-site with a minor allele frequency (MAF) <0.01 identified in known dyshormonogenesis genes were considered for the analysis. If available, segregation analysis results were taken into consideration.
The sequence of TG (NP_003226.4) was aligned with other TG homolog peptide sequences found in the HomoloGene NCBI database (https://www.ncbi.nlm.nih.gov/homologene/).
Results are expressed as mean ± SD for values that followed a normal distribution or median (range) for values that did not follow a normal distribution. Differences for the variables were calculated using non-parametric Kruskal-Wallis test. All analyses were carried out with the statistical program “R” [(R version 3.6.1; 2019-07-05), Copyright (C) 2015 The R Foundation for Statistical Computing], with the support of the Biostatistics Unit of the Vall d’Hebron Research Institute (VHIR).
In this study we patients with TG variants of a Spanish cohort with THD. We identified 21 TG gene variants in 19 patients, which could explain their phenotype (17 compound heterozygotes and 2 homozygotes). Clinical characteristics, hormonal results, scintigraphs, ultrasounds, and perchlorate discharge test (PDT) of patients with THD and TG variants are presented in Table 1. As it is shown, there are no patients in this group with a follow-up classification of transient hypothyroidism; all of them suffered from severe hypothyroidism.
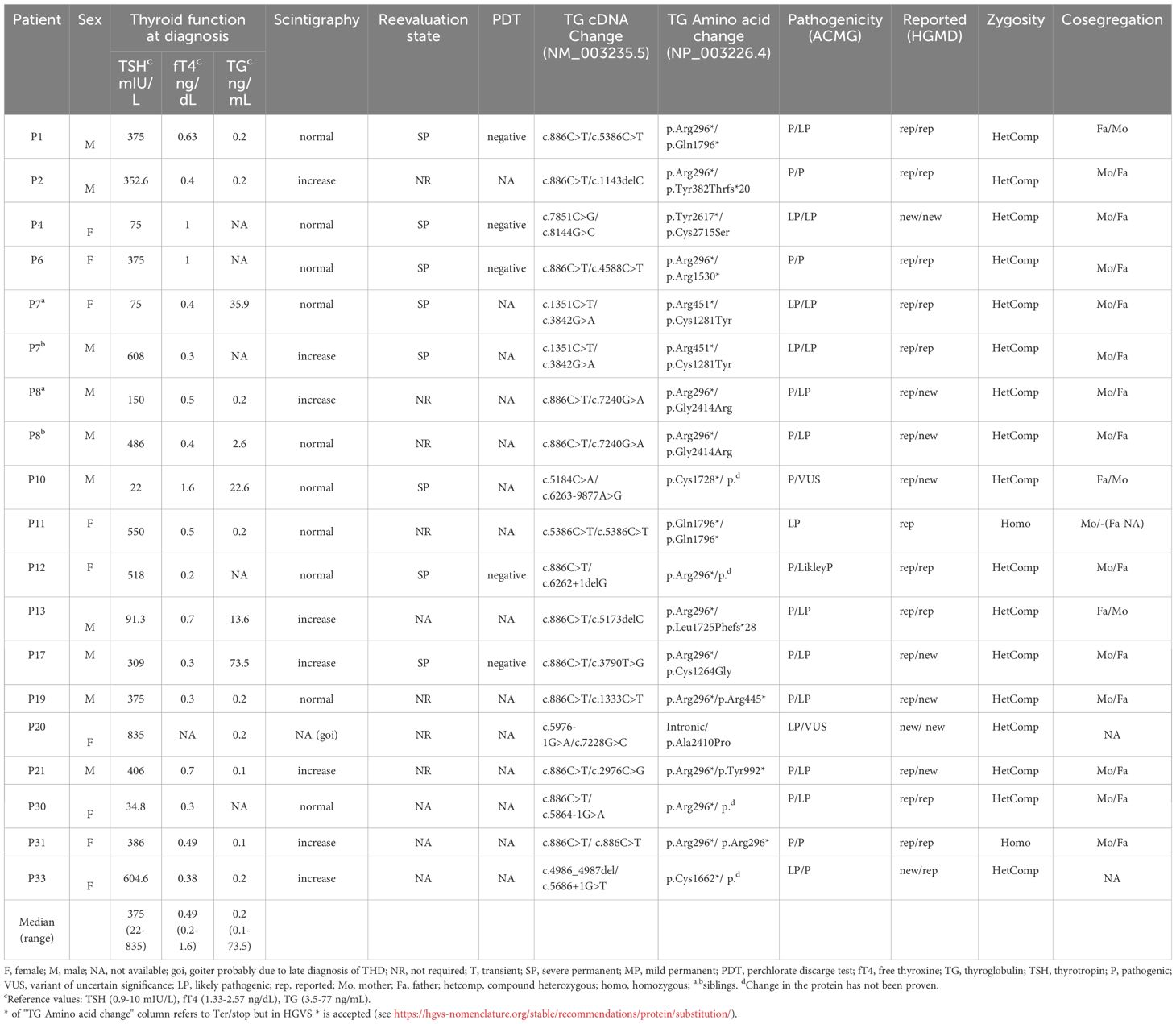
Table 1 Clinical and genetic characteristics of patients with confirmed thyroid dyshormonogenesis diagnosis by variants in the TG gene.
A description of the 21 TG variants detected in our patients is included in Table 2. Four variants were located in the N-terminal domain (NTD), one in the core domain, two in the flap domain, ten in the arm domain and four in the carboxy-terminal domain (CTD). Figure 1 shows the location of variants in the new primary TG structure described recently (10, 27, 28). Ten TG variants (47.6%) had not been previously described in the literature. Of these, six were classified as likely pathogenic and four as variants of uncertain significance (VUS) following ACMG guidelines.
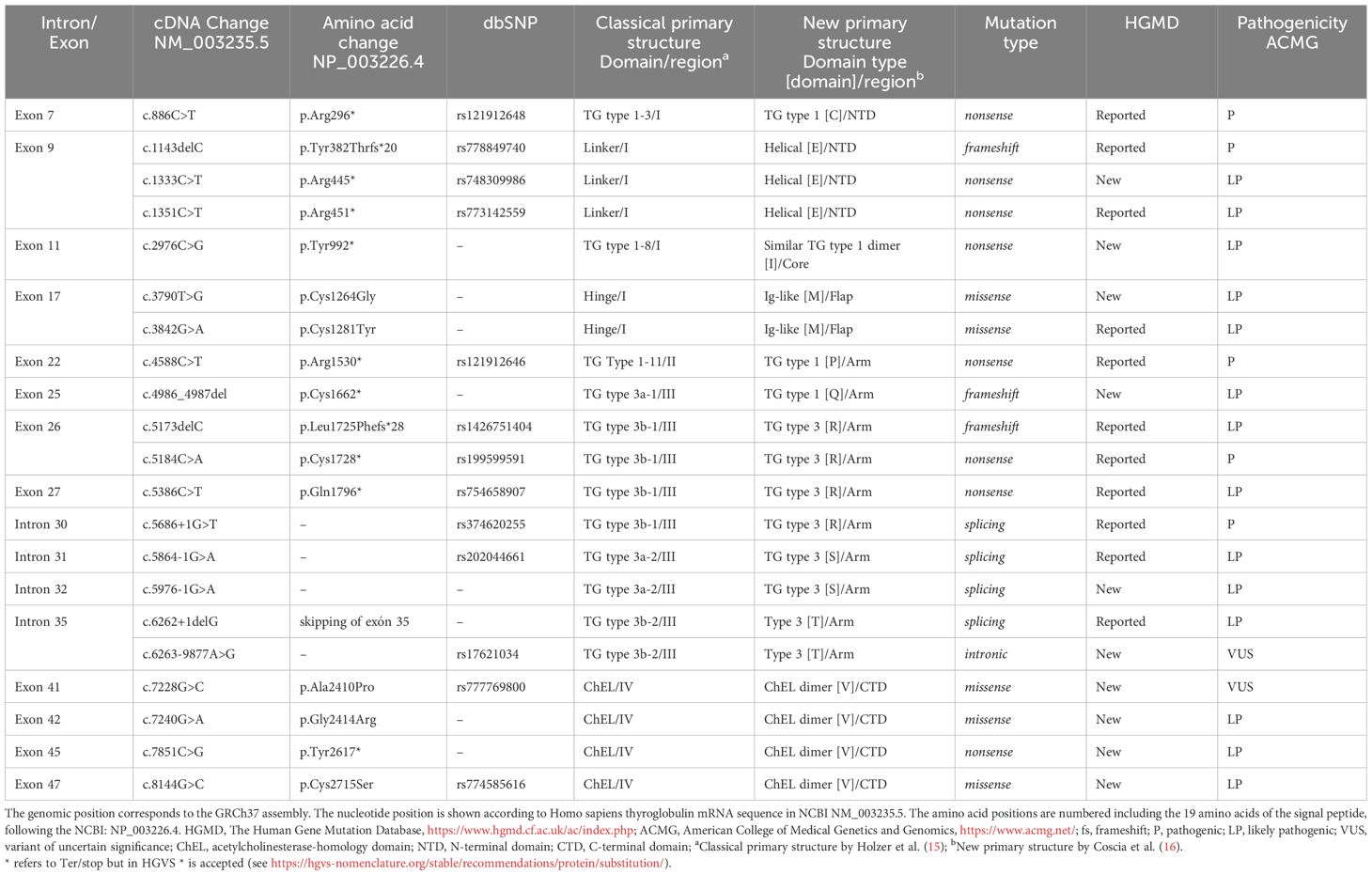
Table 2 Thyroglobulin variants detected in patients with THD.
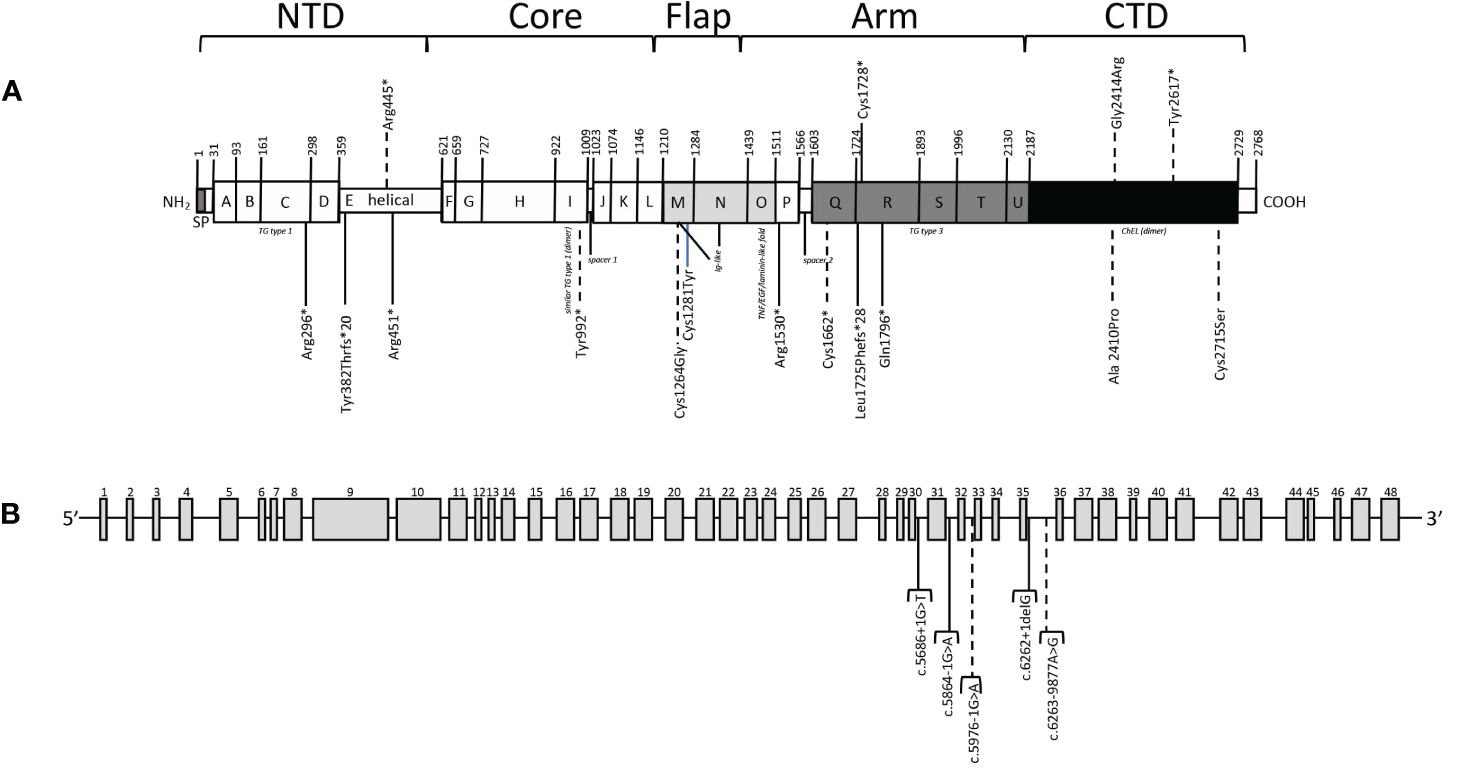
Figure 1 Variants detected in our patients. (A) Location of variants in primary TG structure. Thyroglobulin signal peptide (SP) and TG type 1 (the A, B, C, D, F, G, H, J, K, L and P domains), helical domain (E), TG type 3 (the Q, R, S, T and U domains), similar TG type 1 (dimer) (the I domain), Ig-like (the M and N domains), TNF/EGF/laminin-like fold (the O domain) and acetylcholinesterase-homology (ChEL (dimer), the V domain) domain types, are represented by boxes. The thyroglobulin monomer is organized in five structural regions: N-Terminal Domain (NTD), Core, Flap, Arm and C-Terminal Domain (CTD). The amino acid positions are numbered including the 19 amino acids of the signal peptide, following the NCBI: NP_003226.4 and Uniprot: P01266 numbering. (B) Location of intronic variants. Dashed lines indicate non reported variants. * refers to Ter/stop but in HGVS * is accepted (see https://hgvs-nomenclature.org/stable/recommendations/protein/substitution/).
In this study, we also identified another 16 patients from the Spanish cohort who presented TG variants, which in themselves do not explain their THD classic phenotype. These cases were categorized into 3 groups: a) unconfirmed TG diagnosis: patients with heterozygous variants in TG gene (n=8); b) oligogenic group: patients with heterozygous variants in different related genes (n=5); and c) other genes: patients with monoallelic TG variant and homozygous or compound heterozygous variants in another gene that would explain their THD phenotype (n=3) (Supplementary Table S2).
All patients were diagnosed with primary CH at birth by the Neonatal Screening Program in Catalonia followed by a venous-blood TSH assay for confirmation. Gestational age was 39.6 ± 2.3 weeks (mean ± SD), with a weight of 3299.9 ± 528.7 g and length of 49.5 ± 2.6 cm at birth. Only patient P20 was premature with a gestational age of 32 weeks, 1680 g and 41 cm at birth (Supplementary Table S1).
The median age at THD diagnosis was 14 (range 7-42) days after birth, with median and range TSH of 375 (22–835) mIU/L, fT4 of 0.49 (0.2-1.6) ng/dL and TG of 0.2 (0.1-73.5) ng/mL (Table 1).
Ten patients had normal thyroid uptake at THD diagnosis, eight had high iodide uptake, and goiter was evident at diagnosis in P20, who developed goiter probably because the diagnosis of THD was made late (42 days after birth) due to her prematurity and her late TSH peak. Of the 17 index cases, four had a family history of CH and two had an affected sibling (P7 and P8). A PDT was performed in five patients, all of whom had a negative result.
Eight out of 19 patients (42.1%) were reevaluated at approximately 3 years of age after one month of discontinuation of LT4 treatment. In addition, seven patients were not reevaluated because their LT4 requirements and genetic results were suggestive of permanent hypothyroidism, and in the remaining four data from reevaluation had not been obtained because they were less than 3 years old. All reevaluated patients had severe permanent hypothyroidism.
CH was more severe biochemically (p<0.05) in patients with confirmed molecular diagnosis (homozygous or compound heterozygous pathogenic variants in the TG gene), comparing TSH, fT4 and TG levels at diagnosis between patient groups (confirmed TG diagnosis group vs unconfirmed TG diagnosis and oligogenic groups) (Table 1). All patients with unconfirmed TG diagnosis were reevaluated, except P32; one had severe permanent hypothyroidism, three had mild permanent hypothyroidism and three had transient hypothyroidism. All patients of the oligogenic group, except P9, were reevaluated: two had mild permanent hypothyroidism and two transient hypothyroidism (Supplementary Table S2). Most of the patients in these groups had a high serum TG protein level and, at follow up, presented with a transient or mild phenotype, which is consistent with no TG pathogenic variant being clearly involved (only one affected allele or/and a VUS variant).
High-throughput sequencing analysis identified 21 TG gene variants in 19 patients (17 index cases) with MAF <0.01 compared to public databases, and in silico prediction as pathogenic, likely pathogenic or VUS (Table 2).
Variants in homozygous state were identified in 2/19 patients (10.5%) and compound heterozygous variants in 17/19 (89.5%). These 19 patients showed a genotype-phenotype correlation and were considered diagnosed. These patients presented pathogenic [p.(Arg296*), p.(Tyr382Thrfs*20), p.(Arg1530*), p.(Cys1728*), c.5686 + 1G>T]; likely pathogenic [p.(Arg445*), p.(Arg451*), p.(Tyr992*), p.(Cys1662*), p.(Leu1725Phefs*28), p.(Gln1796*), c.5864-1G>A, c.5976-1G>A, c.6262 + 1delG, p.(Gly2414Arg), p.)Tyr2617*)]; or initially VUS [p.(Cys1264Gly), p.(Cys1281Tyr), p.(Cys2715Ser), c.6263-9877A>G, p.(Ala2410Pro)] TG variants. The presence of these TG variants could plausibly have contributed to the THD phenotype and cosegregation studies allowed the reclassification of p.(Cys1264Gly), p.(Cys1281Tyr) and p.(Cys2715Ser) missense variants as likely pathogenic. Family cosegregation studies have been carried out for 17 of the patients and, in all cases, the parents were heterozygous carriers of the detected variants (Table 1).
Variants detected in TG gene included missense variants (n=5), nonsense variants (n=8), frameshift deletions (n=3) and intronic variants (n=5), 4 of them in splicing nucleotides. Among the missense variants, four were initially VUS but only one remains classified as VUS, as segregation has been not possible. Furthermore, none of the four were previously described. Three of eight nonsense variants were not previously described and all of them were classified as pathogenic (n=3) or likely pathogenic (n=5) (Table 2).
Two of the frameshift variants, p.(Tyr382Thrfs*20) and p.(Leu1725Phefs*28), were previously described (29, 30), yet frameshift c.4986_4987del;p.(Cys1662*) variant was new (Table 2).
Three of the intronic variants affecting the canonical intronic splice site region, c.5686 + 1G>T, c.5864-1G>A and c.6262 + 1delG, were previously described (30, 31) and classified as pathogenic or likely pathogenic. The other intronic variant affecting the canonical intronic splice site, c.5976-1G>A, was novel and classified as likely pathogenic. In the other groups of patients, three of the four intronic variants observed were not previously described and classified as VUS (Supplementary Table S3), whereas the fourth is pathogenic and affects to the canonical site c.5975 + 5G>C.
The most frequent variant in the TG gene in our THD patient cohort was the pathogenic nonsense variant c.886C>T;p.(Arg296*) and is present in 12 of the 19 patients (63.2%).
Twenty-seven relatives (14 mothers, 13 fathers) of 17 different families were analyzed. De novo variants have not been detected in our cohort. Patient P20 is a result of an in vitro fecundation from sperm and oocyte donors, and the cosegregation study of P33 was not performed because the patient moved to another city. In both cases, we consider that TG variants are responsible for their THD phenotype, assuming that the two alleles are affected, since their clinical characteristics are consistent with pathogenic TG.
Eight patients had a non-confirmed diagnosis of THD due to TG defects (unconfirmed TG diagnosis group, Supplementary Table S2). Two of them (P5, P14) were compound heterozygous for TG variants, but with a maternal allele with a likely benign TG variant, following ACMG criteria. Moreover, according to the clinical and biochemical characteristics of these two patients, TG variants would not explain the phenotype. The other six patients of these group (P15, P16, P18, P22, P27, P32) were heterozygous for a VUS, likely pathogenic or pathogenic TG variant.
Three patients (P24, P28, P29; other gene group, Supplementary Table S2) were solved by compound heterozygous pathogenic DUOX2 variants or the homozygous pathogenic variant in the TPO gene. Their phenotype and clinical characteristics correlated with variants in DUOX2 and TPO, respectively.
Among the previously undescribed missense variants detected in our patients, a multiple sequence alignment of the human TG with sequences of other species revealed that wild-type cysteine residues at positions 1264 and 2715, wild-type alanine residue at position 2410, and wild-type glycine residue at position 2414 are strictly conserved in all TG species analyzed (Supplementary Figure S1), except residue 2410 that in G. gallus is a methionine. These four variants were detected in patients P17, P4, P20 and P8 siblings, respectively, in combination with pathogenic or likely pathogenic variants in the other allele.
In this study, we used a high-throughput sequencing panel with CH-related genes to perform a comprehensive genetic screening of patients with THD. All patients were diagnosed with primary CH at birth (serum TSH≥10 mUI/L) by the Neonatal Screening Program in Catalonia.
Dyshormonogenesis due to TG gene variants is a cause of CH and is inherited as an autosomal recessive trait. Only homozygous or compound heterozygous pathogenic variants in the TG gene were thought to be the cause of THD in our patients (“confirmed TG diagnosis” group). TSH, fT4 and TG values of this group at diagnosis are consistent with the molecular analysis. This finding confirms that very low TG levels are a good indication for TG gene variant screening [0.2 ng/mL (0.1-73.5) vs 219 ng/mL (10-1230) or 341 ng/mL (41.1-2031) in “unconfirmed TG diagnosis” or “oligogenic” groups, respectively]. The results of our study are in line with Targovnik et al. (32). In addition, genetically solved cases had a more severe phenotype than ambiguous cases at the reevaluation stage. However, the patients unsolved by pathogenic TG variants (unconfirmed TG diagnosis and oligogenic groups) included one case of severe permanent CH (P27) and five of mild permanent CH (P3, P14, P15, P16, P26) (Supplementary Table S2). In these cases, we assumed that these variants could coexist with an additional undetected copy number variation (CNV), intronic, or regulatory mutation in the TG gene or in another related gene that could explain the THD phenotype.
Our high-throughput sequencing analysis identified 21 different TG gene variants, ten (47.6%) of them not previously described. Of these, eight were classified as likely pathogenic and two as VUS. The pathogenicity of each variant was carefully evaluated based on clinical data including correlations with clinical phenotypes, hormonal results, previously published information, in silico prediction tool results, familiar cosegregation and location of variants in regions of interest of the TG protein.
The most frequent TG variant in our THD patient cohort (63.2%) was the pathogenic variant c.886C>T;p.(Arg296*). This variant was reported as the most frequent variant detected in Caucasian populations [originally published as p.(Arg277*)], taking homozygous, compound heterozygous or monoallelic variants (8, 10, 33–37). The functional consequence of p.(Arg296*) truncated protein is the complete loss of the central and carboxy-terminal hormonogenic domains, and consequently, limited ability to generate TH. However, p.(Arg296*) TG peptide retains its ability to synthesize T4 because it still harbors both the acceptor tyrosine24 and the donor tyrosine149. Affected cases usually have a CH phenotype (10, 38).
The presence of TG variants (pathogenic, likely pathogenic or VUS) in patients could plausibly be contributing to their THD (Table 2). Homozygous variants were identified in patients P11 and P31 [p.(Gln1796*) and p.(Arg296*), respectively], while compound heterozygous variants were identified in the rest of the patients. Twenty-one different TG variants were identified in this group of patients, ten of which were previously undescribed. Of these, likely pathogenic variant c.5976-1G>A affected the canonical intronic splice site, probably leading to aberrant splicing. Exon skipping in the TG gene can be caused by nucleotide substitutions in acceptor or donor splice sites involving -1 intronic position. Therefore, mRNA studies of patient P20 may help to clarify this. In addition, four undescribed variants were nonsense: p.(Arg445*), p.(Tyr992*), p.(Cys1662*) and p.(Tyr2617*). The deleterious effect of these nonsense variants is assumed, following the ACMG criteria (25), as variant loss of function (LOF) is a known mechanism of disease in the TG gene (pLoF 0.89) and they are not reported in population database (gnomAD v4.0.0). Moreover, they are predicted to undergo non-sense-mediated decay (NMD) because they are not located in the last exon or the last 50bp of the preliminary exon. Truncated proteins as a consequence of nonsense variants could represent adequate targets for the nonsense mediated mRNA decay (NMD) pathway, a known RNA surveillance mechanism that detects and then selectively and rapidly degrades mRNAs that contain premature terminated codons (39).
Three novel missense variants detected in these patients [p.(Cys1264Gly), p.(Cys2715Ser), p.(Ala2410Pro)] were initially classified as VUS, but the two first were reclassified as likely pathogenic after segregation. However, c.7240G>A p.(Gly2414Arg) is a variant which also affects the first nucleotide of the 42 exon and predicts a loosing of the acceptor site that may result in the skipping of the whole exon, which was why it was classified as likely pathogenic following ACMG guidelines. Multiple sequence alignment of human TG with sequences from other homologous species revealed that cysteine at positions 1264 and 2715, alanine at position 2410 and glycine residue at position 2414 are highly conserved (Supplementary Figure S1). Although familial segregation studies have helped in the interpretation of these missense novel genetic variants, functional in vitro studies or in vivo evidence of impaired TSH-stimulated mutant TG production will help to definitively confirm their pathogenicity (13). Such analyses have not been undertaken, but the novel identified variants were rare, conserved between species and segregated with phenotype and hormonal results, suggesting that in combination with another pathogenic TG allele it resulted in the THD phenotype.
Sequencing analysis of the TG gene revealed missense mutations, which were described involving cysteine residues, indicating that the loss of cysteine residues may eliminate disulfide bonds and alter the normal conformational structure of the TG (10, 32). The novel p.(Cys1264Gly) in patient P17 and p.(Cys2715Ser) in patient P4 could have remove the disulfide bond, altering the normal structure of TG, and in combination with nonsense variants p.(Arg296*) and p.(Tyr2617*), respectively, in the other alleles this may have led to CH.
Several missense mutations associated with CH were reported in the ChEL domain (32, 40), which is essential for the intracellular transport of TG to the site of its hormonogenesis via the secretory pathway (41–43). This domain is also required for protein dimerization and consequently plays a critical structural and functional role in the TG protein. It is well documented that pathogenic missense mutations in the ChEL domain can result in the intracellular retention of TG in the endoplasmic reticulum (ER) and premature degradation. This could be the explanation for the low TG levels in P8 siblings and P20, who were carriers of p.(Gly2414Arg) and p.(Ala2410Pro), respectively, in combination with a nonsense p.(Arg296*) and a splicing c.5976-1G>A variants in the other allele.
The previously described c.6262 + 1delG causes an inframe loss of exon 35 in the TG gene, generating a polypeptide that had lost 21 amino acids compared with the wild-type TG (31). The resulting protein is synthesized but cannot be released into the follicle lumen. Instead, this protein is retained and processed intracellularly. This variant was also present in a reported Galician family with several members who have congenital goitrous hypothyroidism caused by the same two variants detected in patient P12.
Three patients (P2, P13 and P33) carried frameshift variants, which were previously described (29, 30). Both P3 and P13 had the nonsense variant p.(Arg296*), therefore in these patients neither of the two alleles can synthesize a complete TG protein. The variant in patient P33 [p.(Cys662*)] was not previously described. Specifically, this variant changed two nucleotides of the codon for Cys, at the position of the deletion, for a stop codon.
Eight patients had a non-confirmed diagnosis of THD due to TG defects (unconfirmed TG diagnosis group, Supplementary Table S2). Two of them (P5, P14) were compound heterozygous for TG variants, but with a maternal allele with a likely benign TG variant, following ACMG criteria. Moreover, according to the clinical and biochemical characteristics of these two patients, TG variants would not explain the phenotype. The other six patients of these group (P15, P16, P18, P22, P27, P32) were heterozygous for a VUS, likely pathogenic or pathogenic TG variant. Previous reports of CH due to TG pathogenic variants most commonly involved biallelic mutations (4). Therefore, it is unclear whether the mild hypothyroidism of these patients after reevaluation was attributable to the monoallelic variants or whether they harbored another non-detected variant. With our panel gene we only detected exonic and flanking intronic regions, but not deep-intronic or regulatory variants. In addition, four of the variants presented in these unsolved patients [p.(Arg296*), p.(Arg1250Leu), p.(Gln1796*), p.(Arg2585Trp)] have been previously described as associated with THD but in homozygosis or compound heterozygosis (4). Indeed, the TSH, fT4 and TG values of patients of these unsolved groups at diagnosis were not consistent with the presence of a mutated TG protein. We assumed that these variants could coexist with an additional undetected CNV, intronic, or regulatory mutation in the TG gene or in another related gene that could explain this THD mild phenotype.
Interestingly, three patients (P24, P28, P29; other gene group, Supplementary Table S2) were solved by compound heterozygous pathogenic DUOX2 variants or the homozygous pathogenic variant in the TPO gene. Their phenotype and clinical characteristics correlated with variants in DUOX2 and TPO, respectively. In these 3 patients, the VUS TG variants may partially modify or not influence the phenotype of the patients.
Most cases with CH due to TG gene mutations show decreased serum TG levels before TH replacement and this is a key factor for the diagnosis of TG defects (34, 44). Genetic variants in the TG gene may result in structurally defective proteins severely impairing the functional ability of TG to serve as a matrix for T3 and T4 generation. Misfolded TGs may cause TG retention in the ER and premature degradation by the ER-associated protein degradation pathways, causing ER distention, and ER storage disease, although, it is likely that hydrolysis of limited amounts of mutated TG molecules may escape from the ER and migrate to the colloid, allowing synthesis of small amounts of the thyroid hormone (44). This is a possible mechanism providing a minimum of TH to the body, allowing detection of circulating levels of TG in patients P7a (35.6 ng/mL), P10 (22.6 ng/mL), P13 (13.6 ng/mL) and P17 (73.5 ng/mL). Other cases of TG mutations manifesting high TG serum levels have been reported (40, 45–47). These four patients were compound heterozygous carriers of a nonsense pathogenic variant in one allele, whereas in the other allele they harbored p.(Cys1281Tyr), c.6263-9877A>G, p.(Leu1725Phefs*28) and p.(Cys1264Gly) variants, respectively. We hypothesize that the mutated TG molecules produced by these variants could escape from ER, reach the follicular lumen and produce circulating TG levels in these patients.
With the widespread use of newborn screening programs and the application of improved molecular techniques, some THD cases have been genetically diagnosed (8, 9, 48–50), although the genetic heterogeneity of CH makes its diagnosis complex. To date, all reported genes with digenic variants are involved in the same metabolic pathway: thyroid hormone biosynthesis, and therefore oligogenicity, has often been proposed to underlie the intrafamilial variability seen in known genetic causes of CH, especially in association with DUOX2 mutations (51). In our study, all oligogenic cases (n=5) (Supplementary Table S2) carried heterozygous TG variants in combination with heterozygous variants in three different genes: IYD (n=1), DUOX2 (n=3) and TPO (n=1). This may indicate that iodide organification defects may be more common in CH patients, even though the interpretation of the dominant effect of some variants is controversial. For example, there have been monoallelic variants reported in DUOX2 (52), and later this turned out to be associated with transient hypothyroidism (53, 54). In our six patients with oligogenicity, two presented mild permanent hypothyroidism, two presented a transient THD and two are awaiting reevaluation.
High-throughput sequencing has improved the detection of variants in candidate genes, therefore, their application after newborn screening and CH confirmation may be a valid strategy for rapidly obtaining an accurate diagnosis of CH due to THD. However, certain limitations were observed in our study. Firstly, 2/21 (9.5%) of TG variants detected in our patients were VUS, thus, in vitro functional studies would be necessary to confirm their pathogenicity. Secondly, our genetic analysis approach only detects single nucleotide substitution and short deletion/insertion of the coding region and 10-bp exon-intron boundaries of the nine CH causal genes analyzed. Therefore, we were unable to detect large CNVs, variants in other candidate genes, regulatory or deep intronic variants. To detect such variants, multiplex ligation-dependent probe amplification (MLPA) analysis, whole exome and/or whole genome sequencing should be performed, with the additional goal of identifying new candidate genes. Accordingly, we cannot discard the possibility that some of our monogenic “solved” cases carried additional variants in genes which were not screened in our study but could contribute to their CH phenotype.
In conclusion, we have identified 10 novel and 11 previously reported variants in the TG gene, in 19 Spanish patients from non-consanguineous families with CH due to THD. Although not all the variants found can explain the THD phenotype of patients, it is important to monitor them since some had mild to severe permanent hypothyroidism at reevaluation. This study contributes to the understanding of the molecular mechanisms involved in the development of dyshormonogenesis, and expands the clinical knowledge and evolution of patients with defective TG.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by Clinical Research Ethics Committee of the VHUH [PR (AMI) 390/2016). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MF-C: Writing – original draft, Writing – review & editing. MA: Writing – review & editing. MC: Writing – review & editing. AC-M: Writing – review & editing. EM: Writing – review & editing. NB-R: Writing – review & editing. JL-C: Writing – review & editing. GC-A: Writing – review & editing. EG-A: Writing – review & editing. LS-C: Writing – review & editing. NG-L: Writing – review & editing. NC-T: Writing – review & editing. DY: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Instituto de Salud Carlos III (www.isciii.es/; Madrid, Spain) and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER, http://www.ciberer.es; U-712).
We acknowledge the patients and their families for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1367808/full#supplementary-material
Supplementary Figure 1 | Homology of TG from different animal species using NCBI database. Partial protein alignment are shown with new amino acid changes detected in our patients. The amino acids are indicated by the single-letter code. The amino acid sequences are based on GenBank protein database (Homo sapiens TG, NP_003226.4).
Supplementary Table 1 | Clinical characteristics and anthropometric neonatal parameters of patients with confirmed thyroid dyshormonogenesis diagnosis due to variants in the TG gene.
Supplementary Table 2 | Clinical and genetic characteristics of “unconfirmed TG diagnosis”, “oligogenic” and “other gene” groups of patients with TG variants.
Supplementary Table 3 | Thyroglobulin variants of “unconfirmed TG diagnosis”, “oligogenic” and “other gene” groups of patients.
1. Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. (2010) 5:17. doi: 10.1186/1750-1172-5-17
2. Léger J, Olivieri A, Donaldson M, Torresani T, Krude H, van Vliet G, et al. European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J Clin Endocrinol Metab. (2014) 99:363–84. doi: 10.1210/jc.2013-1891
3. Kwak MJ. Clinical genetics of defects in thyroid hormone synthesis. Ann Pediatr Endocrinol Metab. (2018) 23:169–75. doi: 10.6065/apem.2018.23.4.169
4. van Trotsenburg P, Stoupa A, Léger J, Rohrer T, Peters C, Fugazzola L, et al. Congenital hypothyroidism: A 2020-2021 consensus guidelines update-an ENDO-european reference network initiative endorsed by the european society for pediatric endocrinology and the european society for endocrinology. Thyroid. (2021) 31:387–419. doi: 10.1089/thy.2020.0333
5. Rose SR, Wassner AJ, Wintergerst KA, Yayah-Jones NH, Hopkin RJ, Chuang J, et al. Congenital hypothyroidism: screening and management. Pediatrics. (2023) 151:e2022060419. doi: 10.1542/peds.2022-060419
6. Stoupa A, Kariyawasam D, Muzza M, de Filippis T, Fugazzola L, Polak M, et al. New genetics in congenital hypothyroidism. Endocrine. (2021) 71:696–705. doi: 10.1007/s12020-021-02646-9
7. Stoupa A, Kariyawasam D, Nguyen Quoc A, Polak M, Carré A. Approach to the patient with congenital hypothyroidism. J Clin Endocrinol Metab. (2022) 107:3418–27. doi: 10.1210/clinem/dgac534
8. Nicholas AK, Serra EG, Cangul H, Alyaarubi S, Ullah I, Schoenmakers E, et al. Comprehensive screening of eight known causative genes in congenital hypothyroidism with gland-in-situ. J Clin Endocrinol Metab. (2016) 101:4521–31. doi: 10.1210/jc.2016-1879
9. Yamaguchi T, Nakamura A, Nakayama K, Hishimura N, Morikawa S, Ishizu K, et al. Targeted next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J Clin Endocrinol Metab. (2020) 105:dgaa308. doi: 10.1210/clinem/dgaa308
10. Citterio CE, Rivolta CM, Targovnik HM. Structure and genetic variants of thyroglobulin: Pathophysiological implications. Mol Cell Endocrinol. (2021) 528:111227. doi: 10.1016/j.mce.2021.111227
11. Hishinuma A, Fukata S, Nishiyama S, Nishi Y, Oh-Ishi M, Murata Y, et al. Haplotype analysis reveals founder effects of thyroglobulin gene mutations C1058R and C1977S in Japan. J Clin Endocrinol Metab. (2006) 91:3100–4. doi: 10.1210/jc.2005-2702
12. Targovnik HM, Esperante SA, Rivolta CM. Genetics and phenomics of hypothyroidism and goiter due to thyroglobulin mutations. Mol Cell Endocrinol. (2010) 322:44–55. doi: 10.1016/j.mce.2010.01.009
13. Targovnik HM, Citterio CE, Rivolta CM. Thyroglobulin gene mutations in congenital hypothyroidism. Horm Res Paediatr. (2011) 75:311–21. doi: 10.1159/000324882
14. Citterio CE, Machiavelli GA, Miras MB, Gruñeiro-Papendieck L, Lachlan K, Sobrero G, et al. New insights into thyroglobulin gene: molecular analysis of seven novel mutations associated with goiter and hypothyroidism. Mol Cell Endocrinol. (2013) 365:277–91. doi: 10.1016/j.mce.2012.11.002
15. Di Jeso B, Arvan P. Thyroglobulin from molecular and cellular biology to clinical endocrinology. Endocr Rev. (2016) 37:2–36. doi: 10.1210/er.2015-1090
16. Mercken L, Simons MJ, De Martynoff G, Swillens S, Vassart G. Presence of hormonogenic and repetitive domains in the first 930 amino acids of bovine thyroglobulin as deduced from the cDNA sequence. Eur J Biochem. (1985) 147:59–64. doi: 10.1111/j.1432-1033.1985.tb08718.x
17. Malthiéry Y, Lissitzky S. Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur J Biochem. (1987) 165:491–8. doi: 10.1111/j.1432-1033.1987.tb11466.x
18. van de Graaf SA, Ris-Stalpers C, Pauws E, Mendive FM, Targovnik HM, de Vijlder JJ. Up to date with human thyroglobulin. J Endocrinol. (2001) 170:307–21. doi: 10.1677/joe.0.1700307
19. Mendive FM, Rivolta CM, Moya CM, Vassart G, Targovnik HM. Genomic organization of the human thyroglobulin gene: the complete intron-exon structure. Eur J Endocrinol. (2001) 145:485–96. doi: 10.1530/eje.0.1450485
20. Camats N, Baz-Redón N, Fernández-Cancio M, Clemente M, Campos-Martorell A, Jaimes N, et al. Phenotypic variability of patients with PAX8 variants presenting with congenital hypothyroidism and eutopic thyroid. J Clin Endocrinol Metab. (202) 106:e152–70. doi: 10.1210/clinem/dgaa711
21. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
22. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
23. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
24. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
25. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
26. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. (2016) 37:564–9. doi: 10.1002/humu.22981
27. Holzer G, Morishita Y, Fini JB, Lorin T, Gillet B, Hughes S, et al. Thyroglobulin represents a novel molecular architecture of vertebrates. J Biol Chem. (2016) 291:16553–66. doi: 10.1074/jbc.M116.719047
28. Coscia F, Taler-Verčič A, Chang VT, Sinn L, O'Reilly FJ, Izoré T, et al. The structure of human thyroglobulin. Nature. (2020) 578:627–30. doi: 10.1038/s41586-020-1995-4
29. Caron P, Moya CM, Malet D, Gutnisky VJ, Chabardes B, Rivolta CM, et al. Compound heterozygous mutations in the thyroglobulin gene (1143delC and 6725G–>A [R2223H]) resulting in fetal goitrous hypothyroidism. J Clin Endocrinol Metab. (2003) 88:3546–53. doi: 10.1210/jc.2002-021744
30. Abulí A, Boada M, Rodríguez-Santiago B, Coroleu B, Veiga A, Armengol L, et al. NGS-based assay for the identification of individuals carrying recessive genetic mutations in reproductive medicine. Hum Mutat. (2016) 37:516–23. doi: 10.1002/humu.22989
31. Peteiro-Gonzalez D, Lee J, Rodriguez-Fontan J, Castro-Piedras I, Cameselle-Teijeiro J, Beiras A, et al. New insights into thyroglobulin pathophysiology revealed by the study of a family with congenital goiter. J Clin Endocrinol Metab. (2010) 95:3522–6. doi: 10.1210/jc.2009-2109
32. Targovnik HM, Scheps KG, Rivolta CM. Defects in protein folding in congenital hypothyroidism. Mol Cell Endocrinol. (2020) 501:110638. doi: 10.1016/j.mce.2019.110638
33. van de Graaf SA, Ris-Stalpers C, Veenboer GJ, Cammenga M, Santos C, Targovnik HM, et al. A premature stopcodon in thyroglobulin messenger RNA results in familial goiter and moderate hypothyroidism. J Clin Endocrinol Metab. (1999) 84:2537–42. doi: 10.1210/jcem.84.7.5862
34. Siffo S, Adrover E, Citterio CE, Miras MB, Balbi VA, Chiesa A, et al. Molecular analysis of thyroglobulin mutations found in patients with goiter and hypothyroidism. Mol Cell Endocrinol. (2018) 473:1–16. doi: 10.1016/j.mce.2017.12.009
35. Zou M, Alzahrani AS, Al-Odaib A, Alqahtani MA, Babiker O, Al-Rijjal RA, et al. Molecular analysis of congenital hypothyroidism in Saudi Arabia: SLC26A7 mutation is a novel defect in thyroid dyshormonogenesis. J Clin Endocrinol Metab. (2018) 103:1889–98. doi: 10.1210/jc.2017-02202
36. Santos-Silva R, Rosário M, Grangeia A, Costa C, Castro-Correia C, Alonso I, et al. Genetic analyses in a cohort of Portuguese pediatric patients with congenital hypothyroidism. J Pediatr Endocrinol Metab. (2019) 32:1265–73. doi: 10.1515/jpem-2019-0047
37. Pio MG, Molina MF, Siffo S, Chiesa A, Rivolta CM, Targovnik HM. A novel mutation in intron 11 donor splice site, responsible of a rare genotype in thyroglobulin gene by altering the pre-mRNA splincing process. Cell expression and bioinformatic analysis. Mol Cell Endocrinol. (2021) 522:111124. doi: 10.1016/j.mce.2020.111124
38. Rivolta CM, Moya CM, Gutnisky VJ, Varela V, Miralles-García JM, González-Sarmiento R, et al. A new case of congenital goiter with hypothyroidism caused by a homozygous p.R277X mutation in the exon 7 of the thyroglobulin gene: a mutational hot spot could explain the recurrence of this mutation. J Clin Endocrinol Metab. (2005) 90:3766–70. doi: 10.1210/jc.2005-0278
39. Karousis ED, Nasif S, Mühlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA. (2016) 7:661–82. doi: 10.1002/wrna.1357
40. Pardo V, Vono-Toniolo J, Rubio IG, Knobel M, Possato RF, Targovnik HM, et al. The p.A2215D thyroglobulin gene mutation leads to deficient synthesis and secretion of the mutated protein and congenital hypothyroidism with wide phenotype variation. J Clin Endocrinol Metab. (2009) 94:2938–44. doi: 10.1210/jc.2009-0150
41. Park YN, Arvan P. The acetylcholinesterase homology region is essential for normal conformational maturation and secretion of thyroglobulin. J Biol Chem. (2004) 279:17085–9. doi: 10.1074/jbc.M314042200
42. Lee J, Di Jeso B, Arvan P. The cholinesterase-like domain of thyroglobulin functions as an intramolecular chaperone. J Clin Invest. (2008) 118:2950–8. doi: 10.1172/JCI35164
43. Lee J, Arvan P. Repeat motif-containing regions within thyroglobulin. J Biol Chem. (2011) 286:26327–33. doi: 10.1074/jbc.M111.242099
44. Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev. (1998) 19:173–202. doi: 10.1210/edrv.19.2.0327
45. Hishinuma A, Takamatsu J, Ohyama Y, Yokozawa T, Kanno Y, Kuma K, et al. Two novel cysteine substitutions (C1263R and C1995S) of thyroglobulin cause a defect in intracellular transport of thyroglobulin in patients with congenital goiter and the variant type of adenomatous goiter. J Clin Endocrinol Metab. (1999) 84:1438–44. doi: 10.1210/jcem.84.4.5633
46. Vono-Toniolo J, Kopp P. Thyroglobulin gene mutations and other genetic defects associated with congenital hypothyroidism. Arq Bras Endocrinol Metabol. (2004) 48:70–82. doi: 10.1590/S0004-27302004000100009
47. Watanabe Y, Sharwood E, Goodwin B, Creech MK, Hassan HY, Netea MG, et al. A novel mutation in the TG gene (G2322S) causing congenital hypothyroidism in a Sudanese family: a case report. BMC Med Genet. (2018) 19:69. doi: 10.1186/s12881-018-0588-7
48. Satoh M, Aso K, Ogikubo S, Yoshizawa-Ogasawara A, Saji T. Hypothyroidism caused by the combination of two heterozygous mutations: one in the TSH receptor gene the other in the DUOX2 gene. J Pediatr Endocrinol Metab. (2015) 28:657–61. doi: 10.1515/jpem-2014-0078
49. Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, et al. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol. (2018) 178:623–33. doi: 10.1530/EJE-17-1017
50. Yang R, Lu Y, Yang C, Wu X, Feng J, Zhu L, et al. Case report: expanding the digenic variants involved in thyroid hormone synthesis-10 new cases of congenital hypothyroidism and a literature review. Front Genet. (2021) 12:694683. doi: 10.3389/fgene.2021.694683
51. Muzza M, Rabbiosi S, Vigone MC, Zamproni I, Cirello V, Maffini MA, et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab. (2014) 99:E544–553. doi: 10.1210/jc.2013-3618
52. Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med. (2002) 347:95–102. doi: 10.1056/NEJMoa012752
53. Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang L, et al. Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clin Endocrinol (Oxf). (2014) 81:452–7. doi: 10.1111/cen.12469
Keywords: congenital hypothyroidism, thyroid dyshormonogenesis, thyroglobulin, TG, gene variant
Citation: Fernández-Cancio M, Antolín M, Clemente M, Campos-Martorell A, Mogas E, Baz-Redón N, Leno-Colorado J, Comas-Armangué G, García-Arumí E, Soler-Colomer L, González-Llorens N, Camats-Tarruella N and Yeste D (2024) Clinical and molecular study of patients with thyroid dyshormogenesis and variants in the thyroglobulin gene. Front. Endocrinol. 15:1367808. doi: 10.3389/fendo.2024.1367808
Received: 09 January 2024; Accepted: 27 May 2024;
Published: 08 July 2024.
Edited by:
Klaus Brusgaard, Odense University Hospital, DenmarkReviewed by:
Cintia E. Citterio, Chapman University, United StatesCopyright © 2024 Fernández-Cancio, Antolín, Clemente, Campos-Martorell, Mogas, Baz-Redón, Leno-Colorado, Comas-Armangué, García-Arumí, Soler-Colomer, González-Llorens, Camats-Tarruella and Yeste. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mónica Fernández-Cancio, bW9uaWNhLmZlcm5hbmRlei5jYW5jaW9Admhpci5vcmc=
†These authors share first authorship
‡ORCID: Mónica Fernández-Cancio, orcid.org/0000-0003-1872-3488
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.