
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 26 March 2024
Sec. Pediatric Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1364234
 Silvia Ventresca1,2
Silvia Ventresca1,2 Francesca Romana Lepri3Sabrina Criscuolo2,4Giorgia Bottaro2
Francesca Romana Lepri3Sabrina Criscuolo2,4Giorgia Bottaro2 Antonio Novelli5
Antonio Novelli5 Sandro Loche6
Sandro Loche6 Marco Cappa6*
Marco Cappa6*Silver-Russell syndrome (SRS, OMIM, 180860) is a rare genetic disorder with a wide spectrum of symptoms. The most common features are intrauterine growth retardation (IUGR), poor postnatal development, macrocephaly, triangular face, prominent forehead, body asymmetry, and feeding problems. The diagnosis of SRS is based on a combination of clinical features. Up to 60% of SRS patients have chromosome 7 or 11 abnormalities, and <1% show abnormalities in IGF2 signaling pathway genes (IGF2, HMGA2, PLAG1 and CDKN1C). The underlying genetic cause remains unknown in about 40% of cases (idiopathic SRS). We report a novel IGF2 variant c.[-6-2A>G] (NM_000612) in a child with severe IUGR and clinical features of SRS and confirm the utility of targeted exome sequencing in patients with negative results to common genetic analyses. In addition, we report that long-term growth hormone treatment improves height SDS in this patient.
Insulin-like growth factor 2 (IGF2) plays a pivotal role in intrauterine growth and development, and is encoded by a paternally expressed gene on chromosome 11p15.5 (1, 2). Recently, paternally inherited IGF2 loss-of-function variants have been discovered in clusters of families with a SRS-like phenotype (3–5).
SRS (SRS, OMIM, 180860) is a rare genetic disorder with a wide spectrum of symptoms. The most common features are IUGR, poor postnatal development, macrocephaly, triangular face, prominent forehead, body asymmetry, and feeding problems. The diagnosis of SRS is based on a combination of clinical features according to the Netchine-Harbison Clinical Score System (NH-CSS) (6).
Up to 60% of SRS patients have chromosome 7 or 11 abnormalities, and <1% show abnormalities in IGF2 signalling pathway genes (IGF2, HMGA2, PLAG1 and CDKN1C). Rare familial cases of SRS have been reported with mutations in a single gene (7–9). The underlying genetic cause remains unknown in about 40% of cases (idiopathic SRS). In these cases, a comprehensive search for IGF2 variants is recommended by the, 2017 SRS consensus (6, 8, 10–13). The use of whole exome sequencing has been reportedly shown as an effective strategy to improve the diagnostic yield in individuals with growth retardation due to genetic heterogeneity (9, 10, 14, 15).
Growth hormone (GH) is an approved growth-promoting therapy for short children born small for gestational age (SGA), including children with SRS (16). Smeets et al. reported the results of long-term GH therapy in SRS (17) in a study comparing the growth response to GH treatment in 62 SRS patients and 227 short, non-syndromic individuals born SGA. They found that mean total height gain was comparable in the two groups. While SRS subjects did not reach the same adult height due to their significant height disadvantage at the beginning of GH, the effectiveness of GH treatment was comparable in SRS and non-SRS SGA subjects. All subtypes of SRS seem to benefit from GH treatment, with a tendency towards maternal uniparental disomy for chromosome 7 and idiopathic SRS having the greatest height increase.
In this study, we report a novel paternally inherited IGF2 variant identified by a next-generation sequencing panel in an Italian boy with a clinical diagnosis of SRS. Furthermore, we report the long term response to GH treatment in our patient. Our findings expand the spectrum of disease-causing IGF2 variants and confirm the efficacy of GH therapy in these patients.
The proband was referred to our institute for suspected SRS at the age of 2 years. His parents were phenotypically normal, unrelated, and clinically healthy. His father and mother’s heights were 179 and 167 cm, respectively. He was born by caesarean section performed at 37 weeks gestation for severe IUGR. He was small for gestational age (SGA) (weight -2.9 SD, length -4 SD) and showed a SRS phenotype characterized by pre- and postnatal growth failure, pronounced frontal bossing, feeding difficulties, and low BMI (NH-CSS=4). Recombinant GH was started at the age of 3 years (height -3 SDS) at a dose of 0.33 mg/kg/week.
This study was carried out at the Bambino Gesù Children’s Hospital in Rome, Italy.
Written informed consent was obtained from the parents of the patient for publication of this article. The study protocol conforms to the ethical guidelines of the, 1975 Declaration of Helsinki and has been approved by the Institution’s Human Research Committee.
Genomic DNA was extracted from blood leukocytes. SNP array and Methylation-specific MLPA were performed, ruling out abnormalities of chromosome 7 and 11. Using the Illumina NextSeq550 platform, mutational analysis was carried out by the TruSight One Sequencing Panel (Illumina, San Diego, California). The BWA Enrichment application of BaseSpace (Illumina, San Diego, CA, USA) was used to call variants, and the Geneyx Analysis program (Geneyx Genomex) was used to annotate variants and prioritize potential genes based on phenotypes.
We found a heterozygous splice variant c.[-6-2A>G] (NM_000612) in the IGF2 gene. The American College of Medical Genetics (ACMG) rated the variation as possibly pathogenic. Using established procedures, Sanger sequencing was used for segregation analysis.
The detected genetic variation segregates on the paternal side. The patient’s father inherited the mutation from his mother (Figure 1) Due to the maternal imprinting of the IGF2 gene, the patient’s father is an asymptomatic carrier.
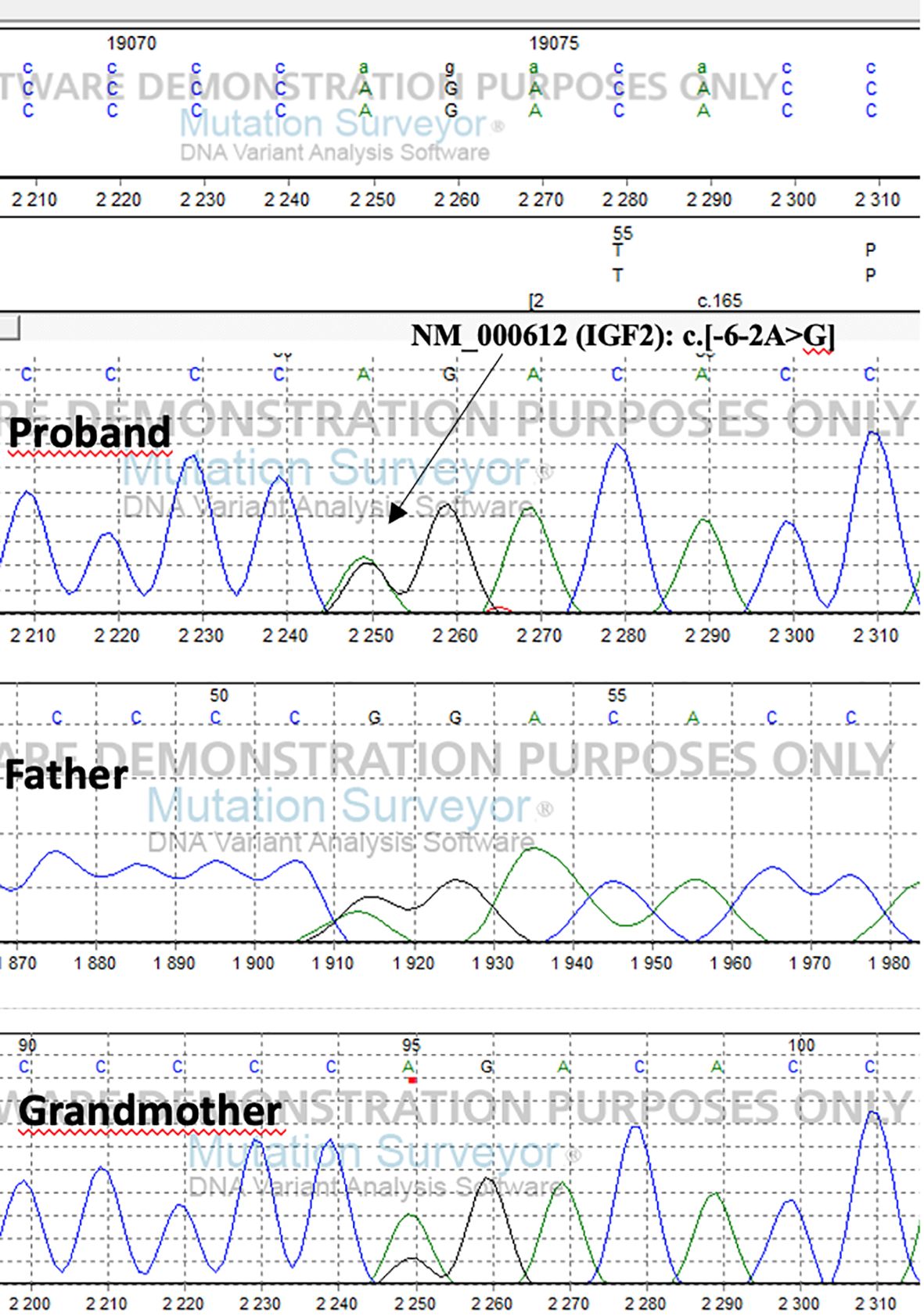
Figure 1 Results of sequence analysis of the proband, his father and grandmother.
GH therapy produced a positive growth response. His growth rate increased from 5 cm/y at baseline to 8 cm/y during the first year of treatment. At the age of 8 his growth rate was 7 cm/year with a bone age of 7 years. (Figure 2). His height SDS increased from -3.50 SDS at baseline to -1.29 SDS at the age of 8. No side effects and no serious illnesses were observed during treatment. Psychomotor development was normal.
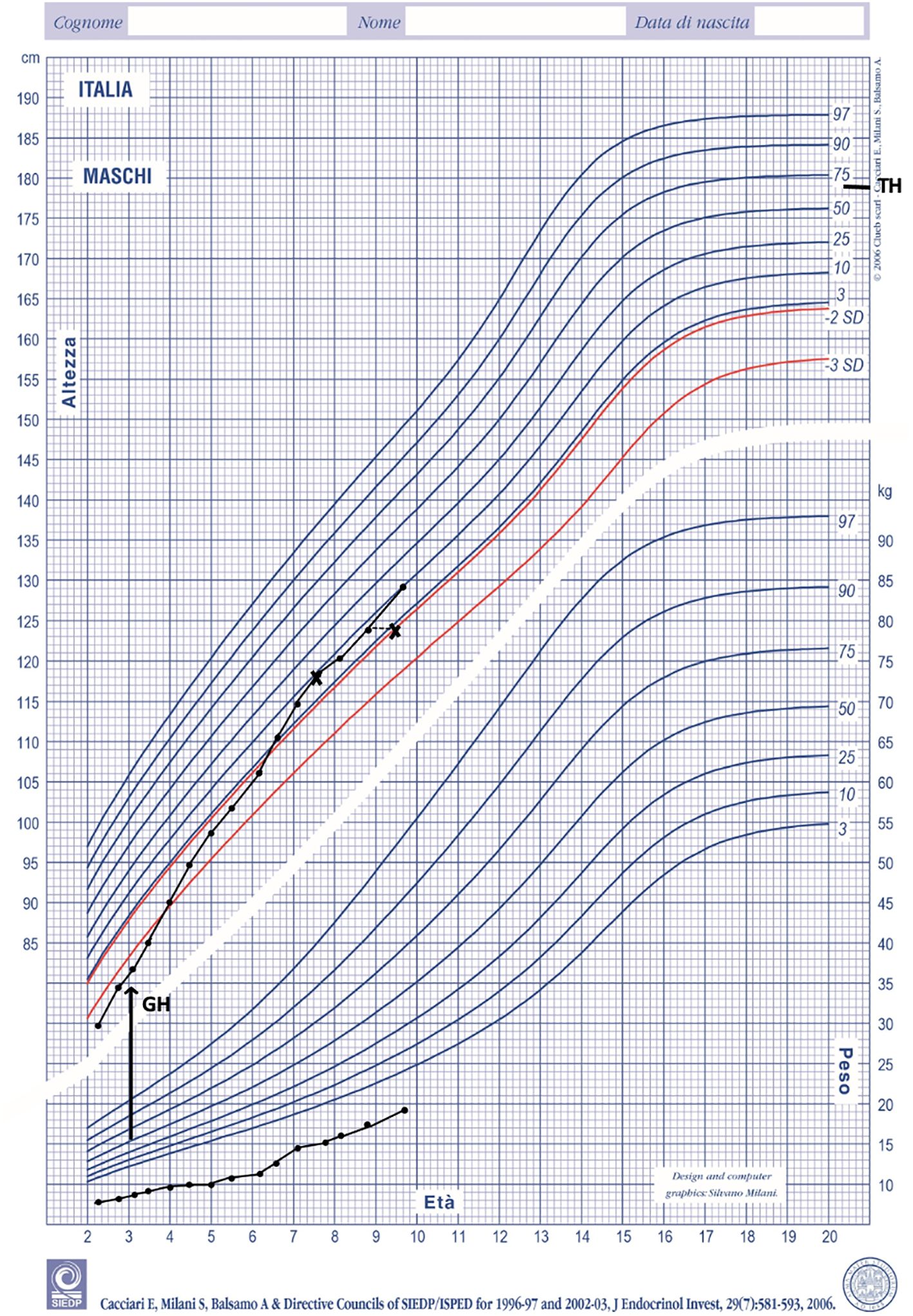
Figure 2 Height and weight during GH treatment plotted on Italian growth chart for boys (18). TH, target height.
Serum IGF1 levels were normal at baseline and increased but remained in the normal range during the first 1,5 years. Then increased above 2 SDS and remained consistently elevated throughout the treatment period (last measurement +3.17 SDS) (Table 1).
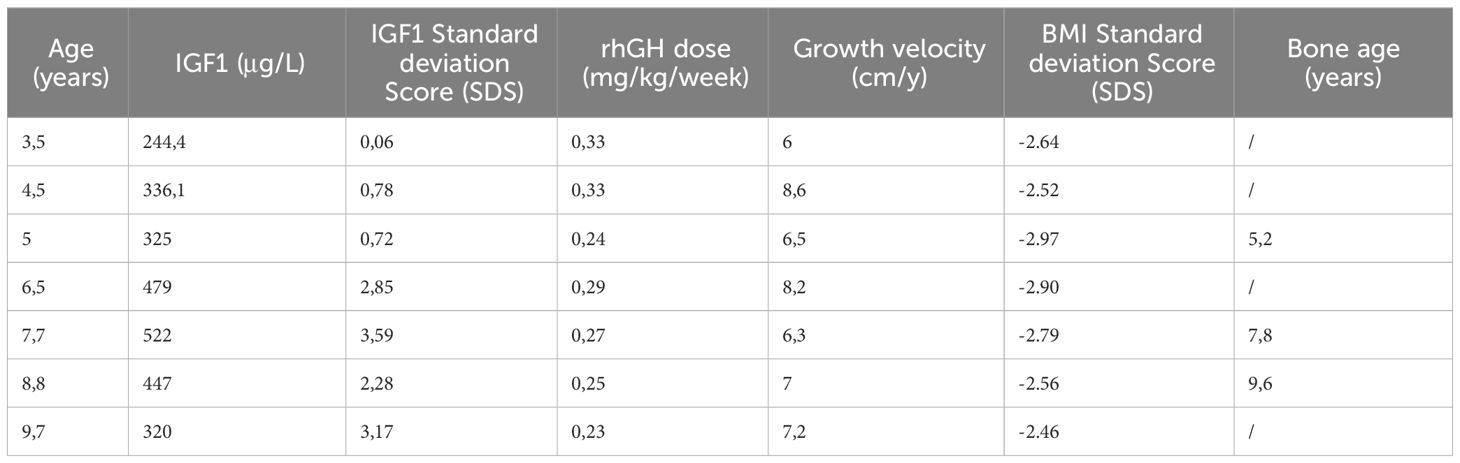
Table 1 IGF1 concentration, GH dose, growth velocity, BMI and bone age during treatment.
We identified a novel genetic abnormality (variation c.[-6-2A>G] NM_000612) in the IGF2 gene in a young child whit pre- and postnatal growth retardation and clinical features of SRS. Exome capture was performed on the patient and parental DNA samples after exclusion of molecular abnormalities on chromosomes 7 and 11 (maternal uniparental disomy, ICR1 hypomethylation, submicroscopic imbalances, and CDKN1C variations) by SNP array and methylation-specific MLPA. This variant has not been observed in gnomAD, 1,000 Genomes, or Sequencing Initiative Suomi database (SISu), it has not been reported in dbSNP, ClinVar, or HGMD and is classified as possibly pathogenic by the American College of Medical Genetics. The presence of the IGF2 variant was confirmed by Sanger sequencing, and segregates on the paternal side. Thus, it was inherited from the father, who had inherited it from his mother. There were no similar cases in the family. These findings are consistent with maternal imprinting of the IGF2 gene.
Our results confirm that specific molecular analyses are useful to elucidate the diverse molecular spectrum of SRS and clinically related disorders. Confirming previous observations (5), whole genome sequencing, long-read sequencing (third generation sequencing) and transcriptomics allow the identification of most molecular abnormalities in a single genetic study.
Meyer et al. (10) performed genetic analysis in 75 SRS phenotypes, and identified a disease-causing variant in 21/75 patients, including variants in known SRS genes (IGF2, PLAG1, HMGA2). Several patients carried variants in genes not previously linked to the diagnosis of SRS. A recent clinical study in Japan (4) found five patients with clinical features of SRS and novel IGF2 polymorphisms in the paternal allele. IGF2 epimutations are clinically associated with SRS with various phenotypic abnormalities. This is most likely due to the different IGF2 expression patterns in the target organs (4). The study by Yamoto et al. (5) of a family with a paternally inherited nonsense mutation in IGF2 confirmed that IGF2 variations could account for the combined phenotypes of the four SRS individuals (extrinsic finger, hypo-masculinization, developmental delay and placental hypoplasia). In addition, they observed that serum IGF1 and IGFBP3 levels were significantly higher than normal in these patients (19). In the study of Smeets et al. (17) baseline IGF1 concentrations were in the normal range in patients with SRS. However, after one year of GH treatment median IGF1concentration was above 2 SDS in the subgroup of patients with 11p15 aberrations. Consistent with these findings, our patient also had normal baseline IGF1 concentrations which increased above 2 SDS on GH treatment. IGF1 remained above 2 SDS even after reduction of the GH dose in our patient. The reasons for the elevated IGF1 concentrations are not clear. Previous studies have suggested reduced IGF1 sensitivity in SRS patients with 11p15 epimutation (20). Reduced IGF1 sensitivity may thus be speculated also for our patient.
Short children born with SGA are a recognized indication for GH therapy. Treated subjects reach a mean height gain of 1.5 SDS compared with 0.25 SDS in untreated subjects (21). The dose of GH recommended for SGA (0.22 mg/kg/week) is higher than that commonly used in children with GHD (21). Recently, beneficial effects of GH treatment on growth and body composition have also been reported in SGA children with SRS (17). Total height gain during GH treatment was similar in SGA children and in SGA children with SRS at a dose of 0.22 mg/kg/week. In addition, the growth response was not significantly different between children with different genetic subtypes. Interestingly, a correlation between age at start of therapy and long-term outcome has been recently reported in short children born SGA (22). Confirming these findings, our patient exhibited a positive response to GH treatment. Our patient’s primary diagnosis was SRS, and given the pronounced height deficit already present at the age of 3 years we asked permission to the ad hoc local Committee to start treatment with GH. We started GH treatment using the maximum allowed dose of 0,33 mg/Kg/week and did not adjust the dose according to IGF1 concentration, but rather to growth velocity. We have reduced the dose gradually as growth velocity reached a plateau. After 5 years of treatment, his height SDS increased from -3.50 at baseline to -1.29 with a total height gain of 1,89 SDS. Weight did not increase over height during treatment and as a result BMI was consistently low. To our knowledge there are no studies on the effect of long-term GH treatment on BMI in children with SRS. A recent report, however, speculated that lower BMI could result from differences in body composition including reduced fat mass in GH‐treated individuals (23).
We have confirmed that targeted exome sequencing to search for IGF2 gene mutations is useful in children with IUGR and SRS-like phenotype. All IGF2 variants associated with an SRS-like phenotype reported so far (Table 2), including this novel one, presented with pre- and postnatal growth restriction and feeding difficulties, and 80% had also developmental delay and/or intellectual disability. Our patient had severe prenatal growth restriction, feeding difficulties and severe growth retardation, which improved with GH treatment.
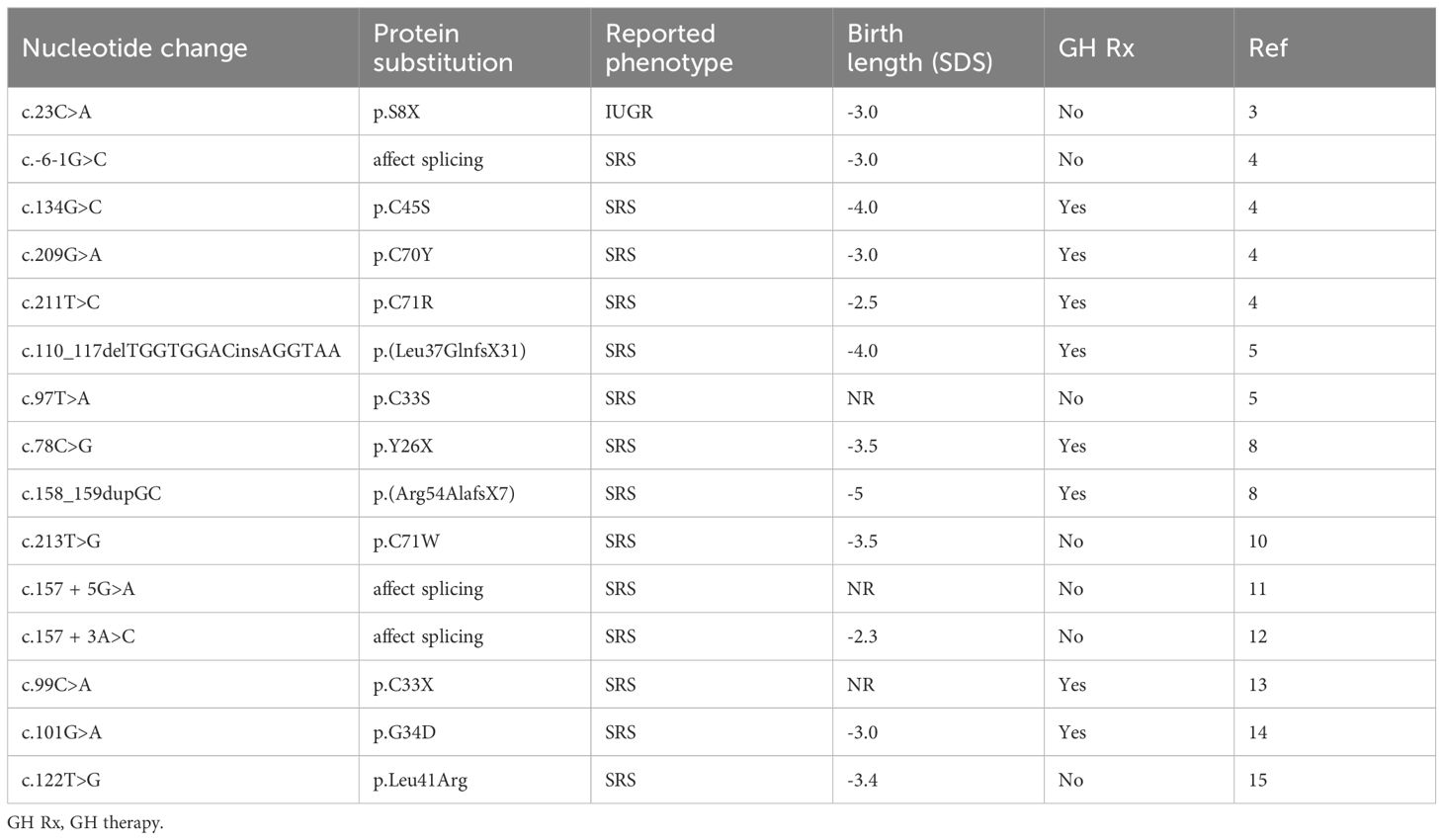
Table 2 Summary of the IGF2 variants reported in patients with SRS phenotype.
In conclusion, the use of a next-generation sequencing protocol that analyses multiple genes simultaneously allows early detection of pathogenetic mutations in various genetic conditions with high standards of coverage and quality. This can lead to early diagnosis, especially in patients with mild or atypical features, and more appropriate genetic counselling and clinical management, including the possibility of early initiation of GH therapy.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
The studies involving humans were approved by the Ethics Committee of Bambino Gesù Children Hospital of Rome. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
SV: Writing – original draft, Writing – review & editing. FL: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. GB: Writing – original draft, Writing – review & editing. AN: Writing – original draft, Writing – review & editing. SL: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gicquel C, Le Bouc Y. Hormonal regulation of fetal growth. Horm Res. (2006) 65:28–33. doi: 10.1159/000091503
2. Argente J. Genetic causes of growth disorders. Curr Opin Endocr Metab Res. (2020) 14:7–14. doi: 10.1016/j.coemr.2020.03.007
3. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel H, et al. Paternally Inherited IGF2 mutation and growth restriction. N Engl J Med. (2006) 373:349–56. doi: 10.1056/NEJMoa1415227
4. Masunaga Y, Inoue T, Yamoto K, Fujisawa Y, Sato Y, Kawashima-Sonoyama Y, et al. IGF2 mutations: Report of five cases, review of the literature, and comparison with H19/IGF2:IG-DMR epimutations. J Clin Endocrinol Metab. (2020) 105:dgz034. doi: 10.1210/clinem/dgz034
5. Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Muut. (2017) 38:953–58. doi: 10.1002/humu.23253
6. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. (2017) 13:105–24. doi: 10.1038/nrendo.2016.138
7. Brereton RE, Nickerson SL, Woodward KJ, Edwards T, Sivamoorthy S, Ramos Vasques Walters F, et al. Further heterogeneity in Silver-Russell syndrome: PLAG1 deletion in association with a complex chromosomal rearrangement. Amer J Med Genet. (2021) 185:3136–45. doi: 10.1002/ajmg.a.62391
8. Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. (2018) 20:250–58. doi: 10.1038/gim.2017.105
9. Inoue T, Nakamura A, Iwahashi-Odano M, Tanase-Nakao K, Matsubara K, Nishioka J, et al. Contribution of gene mutations to Silver-Russell syndrome phenotype: multigene sequencing analysis in 92 etiology-unknown patients. Clin Epigenet. (2020) . 12:86. doi: 10.1186/s13148-020-00865-x
10. Meyer R, Begemann M, Hübner CT, Dey D, Kuechler A, Elgizouli M, et al. One test for all: whole exome sequencing significantly improves the diagnostic yield in growth retarded patients referred for molecular testing for Silver-Russell syndrome. Orphanet J Rare Dis. (2021) 16:42. doi: 10.1186/s13023-021-01683-x
11. Xia CL, Lyu Y, Li C, Li H, Zhang ZT, Yin SW, et al. Rare de novo IGF2 variant on the paternal allele in a patient with Silver-Russell Syndrome. Front Genet. (2019) 10:1161. doi: 10.3389/fgene.2019.01161
12. Poulton C, Azmanov D, Atkinson V, Beilby J, Ewans L, Gration D, et al. Silver Russel syndrome in an aboriginal patient from Australia. Amer J Med Genet. (2018) 176:2561–63. doi: 10.1002/ajmg.a.40502
13. James KN, Clark MM, Camp B, Kint C, Schols P, Batalov S, et al. Partially automated whole-genome sequencing reanalysis of previously undiagnosed pediatric patients can efficiently yield new diagnoses. NPJ Genom Med. (2020) 11:5–33. doi: 10.1038/s41525-020-00140-1
14. Liu D, Wang Y, Yang XA, Liu D. De novo mutation of paternal IGF2 gene causing Silver-Russell syndrome in a sporadic patient. Front Genet. (2017) 8:105. doi: 10.3389/fgene.2017.00105
15. Loid P, Lipsanen-Nyman M, Ala-Mello S, Hannula-Jouppi K, Kere J, Mäkitie O, et al. Case report: A novel de novo IGF2 missense variant in a Finnish patient with Silver-Russell syndrome. Front Pediatr. (2022) 4:969881. doi: 10.3389/fped.2022.969881
16. Van Pareren Y, Mulder P, Houdijk M, Jansen M, Reeser M, Hokken-Koelega ACS. Adult height after long-term, continuous growth hormone (GH) treatment in short children born small for gestational age: Results of a randomized, double-blind, dose-response GH trial. J Clin Endocrinol Metab. (2003) 88:3584–90. doi: 10.1210/jc.2002-021172
17. Smeets CC, Zandwijken GR, Renes JS, Hokken-Koelega ACS. Long-term results of GH treatment in Silver-Russell Syndrome (SRS): do they benefit the same as non-SRS short-SGA? J Clin Endocrinol Metab. (2016) 101:2105–12. doi: 10.1210/jc.2015-4273
18. Cacciari E, Milani S, Balsamo A, Spada E, Bona G, Cavallo L, et al. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol Invest. (2006) 29:581–93. doi: 10.1007/BF03344156
19. Cooke DW, Divall SA, Radovick S. Normal and aberrant growth in children. Williams Textbook of Endocrinology. 13th Ed. Elsevier (2016) p. 964–1073, ISBN: 9780323297387. doi: 10.1016/B978-0-323-29738-7.00024-1
20. Binder G, Seidel AK, Martin DD, Schweizer R, Schwarze CP, Wollmann HA, et al. The endocrine phenotype in Silver-Russell syndrome is defined by the underlying epigenetic alteration. J Clin Endocrinol Metab. (2008) 93:1402–07. doi: 10.1210/jc.2007-1897
21. Hokken-Koelega ACS, van der Steen M, Boguszewski MCS, Cianfarani S, Dahlgren J. Horikawa R et alInternational consensus guideline on small for gestational age: Etiology and management from infancy to early adulthood. Endocr Rev. (2023) 44:539–65. doi: 10.1210/endrev/bnad002
22. Juul A, Backeljauw P, Cappa M, Pietropoli A, Kelepouris N, Linglart A, et al. Early growth hormone initiation leads to favorable long-term growth outcomes in children born small for gestational age. J Clin Endocrinl Metab. (2023) 108:1043–52. doi: 10.1210/clinem/dgac694
Keywords: IGF2 variant, Silver-Russel syndrome, children, growth retardation, GH therapy
Citation: Ventresca S, Lepri FR, Criscuolo S, Bottaro G, Novelli A, Loche S and Cappa M (2024) Case report: Long term response to growth hormone in a child with Silver-Russell syndrome-like phenotype due to a novel paternally inherited IGF2 variant. Front. Endocrinol. 15:1364234. doi: 10.3389/fendo.2024.1364234
Received: 01 January 2024; Accepted: 12 March 2024;
Published: 26 March 2024.
Edited by:
Mariacarolina Salerno, University of Naples Federico II, ItalyReviewed by:
Tommaso Aversa, University of Messina, ItalyCopyright © 2024 Ventresca, Lepri, Criscuolo, Bottaro, Novelli, Loche and Cappa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Cappa, bWFyY28uY2FwcGFAb3BiZy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.