 Mark Yorek1,2,3*
Mark Yorek1,2,3*- 1Department of Internal Medicine, University of Iowa, Iowa City, IA, United States
- 2Department of Veterans Affairs Iowa City Health Care System, Iowa City, IA, United States
- 3Fraternal Order of Eagles Diabetes Research Center, University of Iowa, Iowa City, IA, United States
In 2022, the Center for Disease Control and Prevention reported that 11.3% of the United States population, 37.3 million people, had diabetes and 38% of the population had prediabetes. A large American study conducted in 2021 and supported by many other studies, concluded that about 47% of diabetes patients have peripheral neuropathy and that diabetic neuropathy was present in 7.5% of patients at the time of diabetes diagnosis. In subjects deemed to be pre-diabetes and impaired glucose tolerance there was a wide range of prevalence estimates (interquartile range (IQR): 6%-34%), but most studies (72%) reported a prevalence of peripheral neuropathy ≥10%. There is no recognized treatment for diabetic peripheral neuropathy (DPN) other than good blood glucose control. Good glycemic control slows progression of DPN in patients with type 1 diabetes but for patients with type 2 diabetes it is less effective. With obesity and type 2 diabetes at epidemic levels the need of a treatment for DPN could not be more important. In this article I will first present background information on the “primary” mechanisms shown from pre-clinical studies to contribute to DPN and then discuss mono- and combination therapies that have demonstrated efficacy in animal studies and may have success when translated to human subjects. I like to compare the challenge of finding an effective treatment for DPN to the ongoing work being done to treat hypertension. Combination therapy is the recognized approach used to normalize blood pressure often requiring two, three or more drugs in addition to lifestyle modification to achieve the desired outcome. Hypertension, like DPN, is a progressive disease caused by multiple mechanisms. Therefore, it seems likely as well as logical that combination therapy combined with lifestyle adjustments will be required to successfully treat DPN.
Diabetic peripheral neuropathy
About 50% of the diabetes population is affected by DPN. After many years of pre-clinical and clinical research a life changing treatment remains elusive. Animal studies have shown that the cause of DPN is complex with numerous mechanisms being involved at different cellular levels including vascular, nerve and Schwann cells. This complex etiology is believed to be the primary factors contributing to sensory and motor nerve impairment (1–5). Elevated and dysregulated blood glucose levels were thought to be the sole cause of peripheral neuropathy in patients with type 1 diabetes. In patients with type 1 diabetes good regulation of blood glucose levels and hemoglobin A1C values was shown to slow the progression of DPN. However, good glycemic control does not prevent, nor is it sufficient to significantly alter the course of DPN in patients with type 2 diabetes (1, 2, 5). To further complicate this issue research in human subjects has shown that about 30% of those with impaired glucose tolerance and insulin resistance develop forms of sensory neuropathy (6, 7). This observation has been duplicated in studies with diet-induced obese rats that develop sensory neuropathy related deficits (8).
Etiology of DPN
In 2004 Dr. Brownlee presented the Banting lecture that focused on a unifying hypothesis for diabetes complications (9). In this presentation four pathways were highlighted, and these are briefly reviewed below. Unfortunately, the etiology of diabetes complications and DPN is not that straightforward. There are many other factors that contribute to these complications although ultimately increased oxidative stress and/or inflammation is a contributing factor and maybe the common element linking many of these other pathways to the pathology of DPN. Below I also briefly review the pathology of oxidative and inflammatory stress in DPN and why combination therapies may be the best approach to ultimately finding a treatment that will work.
Aldose reductase pathway
One of the early mechanisms to receive a great deal of attention was the aldose reductase pathway or polyol pathway. This pathway consists of two enzymes: aldose reductase and sorbitol dehydrogenase (10). In the initial step aldose reductase reduces glucose to sorbitol in a reaction requiring NADPH. This is followed by sorbitol reacting with NAD-dependent sorbitol dehydrogenase to form fructose (Figure 1). The increase in cellular sorbitol induces osmotic stress that is compensated at the cellular level by decreasing the levels of taurine and myo-inositol. Besides reduction of intracellular levels of myo-inositol and taurine other potential negative pathological consequences of this pathway are the generation of excess fructose, a potent glycating agent, and a decrease in nerve Na+/K+ ATPase activity.
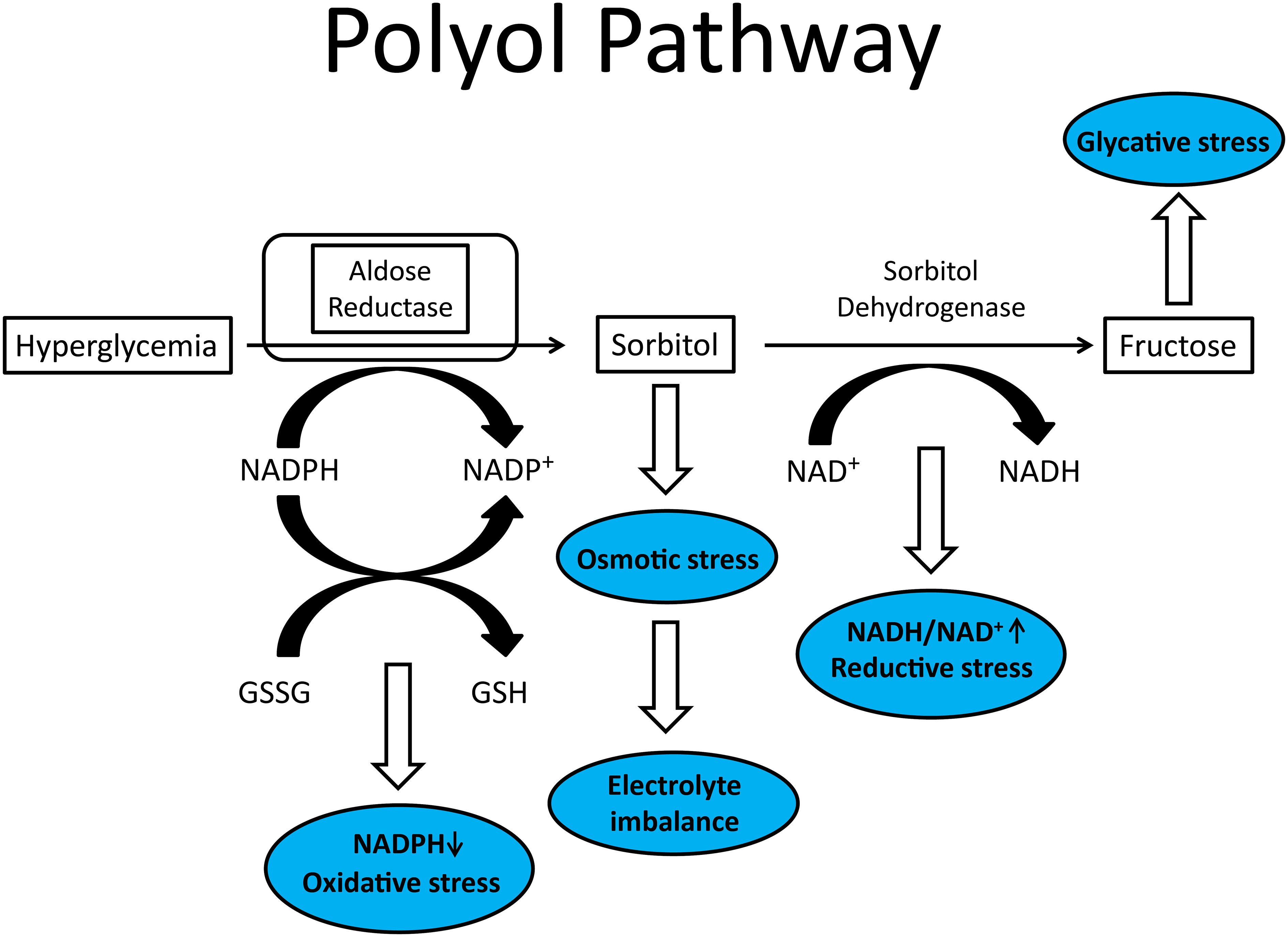
Figure 1 Illustration of polyol pathway with blue ovals representing conditions that contributes to diabetic peripheral neuropathy.
Preclinical studies treating primarily diabetic rodents with aldose reductase inhibitors demonstrated improvement in multiple endpoints associated with DPN (11–13). This led investigators to propose what became known as the compatible osmolyte hypothesis: myo-inositol depletion and abnormal signaling by phosphatidylinositol causes a decrease in peripheral nerve Na+/K+ ATPase activity (14). Na+/K+ ATPase is important for maintaining the sodium and potassium transmembrane potential that is required for nerve impulse conduction as well as the sodium gradient used for uptake of substrates that are Na+-dependent (15). Treatment with aldose reductase inhibitors not only corrected the depletion of intracellular myo-inositol and taurine in tissues but sciatic nerve Na+-ATPase activity was also corrected as was DPN as determined by measurement of nerve conduction velocity. Supplementing diets with myo-inositol or taurine was also shown to partially reverse peripheral neuropathy in diabetic rodents (16–18). My laboratory has demonstrated that treating streptozotocin-induced type 1 diabetic rats with an aldose reductase inhibitor or myo-inositol dietary replenishment improved endoneurial blood flow and dilatory function of epineurial arterioles that provide blood flow to the sciatic nerve (19). This study was important, because we had previously demonstrated that decreased vascular relaxation to acetylcholine by epineurial arterioles preceded the slowing of nerve conduction velocity suggesting that vascular dysregulation is a contributing factor to DPN (20).
The effect of aldose reductase inhibitors has been studied in human subjects with DPN. As expected, the design of these studies varied including the number of subjects enrolled, duration of treatment, dose, and endpoints examined. Wide variation in study design often leads to mixed results. Some of these human studies with aldose reductase inhibitors reported significant improvement in nerve conduction velocity and axonal atrophy (21–23) while others reported non-significant results (24–26). The results from these trials were summarized nicely by Pfeifer et al. (27) stating that “future trials should be designed with adequate statistical power, with consideration of the variability of the endpoint measurements for long enough duration, and with rigorous quality control to definitively confirm the utility of aldose reductase inhibitors in the treatment of diabetic distal symmetrical polyneuropathy and autonomic neuropathy”. This review was written in 1997 and the same problems are still a challenge for adequate clinical trials for DPN in 2023. Presently, in Japan, Epalrestat is the only aldose reductase inhibitor being used clinically for treatment of DPN (28, 29). Despite these mixed results, aldose reductase is still considered to be a potential therapeutic target for neurodegeneration (30).
Non-enzymatic glycation
Along with the polyol pathway, increased non-enzymatic glycation was the other pathway that was recognized early to contribute to DPN. When present in over abundant levels reducing sugars like glucose or fructose react non-enzymatically with free amino groups of proteins, lipids, or nucleic acids to initially form Schiff bases or Amadori adducts (Figure 2) (31–33). Left unchallenged these products will continue to undergo reactions that lead to the formation of advanced glycation endproducts (32). In diabetes, this accumulation can cause structural and functional damage to tissues and organs including components of peripheral nerves (34, 35). Besides their effect on nerves, advanced glycation endproducts can alter vascular structure and function, reducing blood flow, and leading to ischemia. Pathology attributed to advanced glycation endproducts can also occur through binding of these compounds to the receptor for advanced glycation endproducts (RAGE) that is expressed in Schwann and endothelial cells (36, 37). Activation of RAGE has been demonstrated to increase expression of markers associated with increased oxidative stress (38, 39). In regard to DPN, treatment with inhibitors of advanced glycation end-product formation has been shown to improve endoneurial blood flow as well as other DPN related endpoints; thereby further linking vascular and neural complications to DPN (19, 40). In mice lacking RAGE the induction of type 1 diabetes by streptozotocin did not reduce nerve conduction velocity as compared to type 1 diabetic wild type mice (41, 42). Treating diabetic wild type mice with soluble RAGE (sRAGE), a treatment intended to sequester RAGE ligands prior to their binding to RAGE also improved DPN (42). An early inhibitor of formation and activity of advanced glycation endproducts was aminoguanidine (43). It has been widely shown that treating diabetic rats with aminoguanidine improved DPN (19, 43–48).
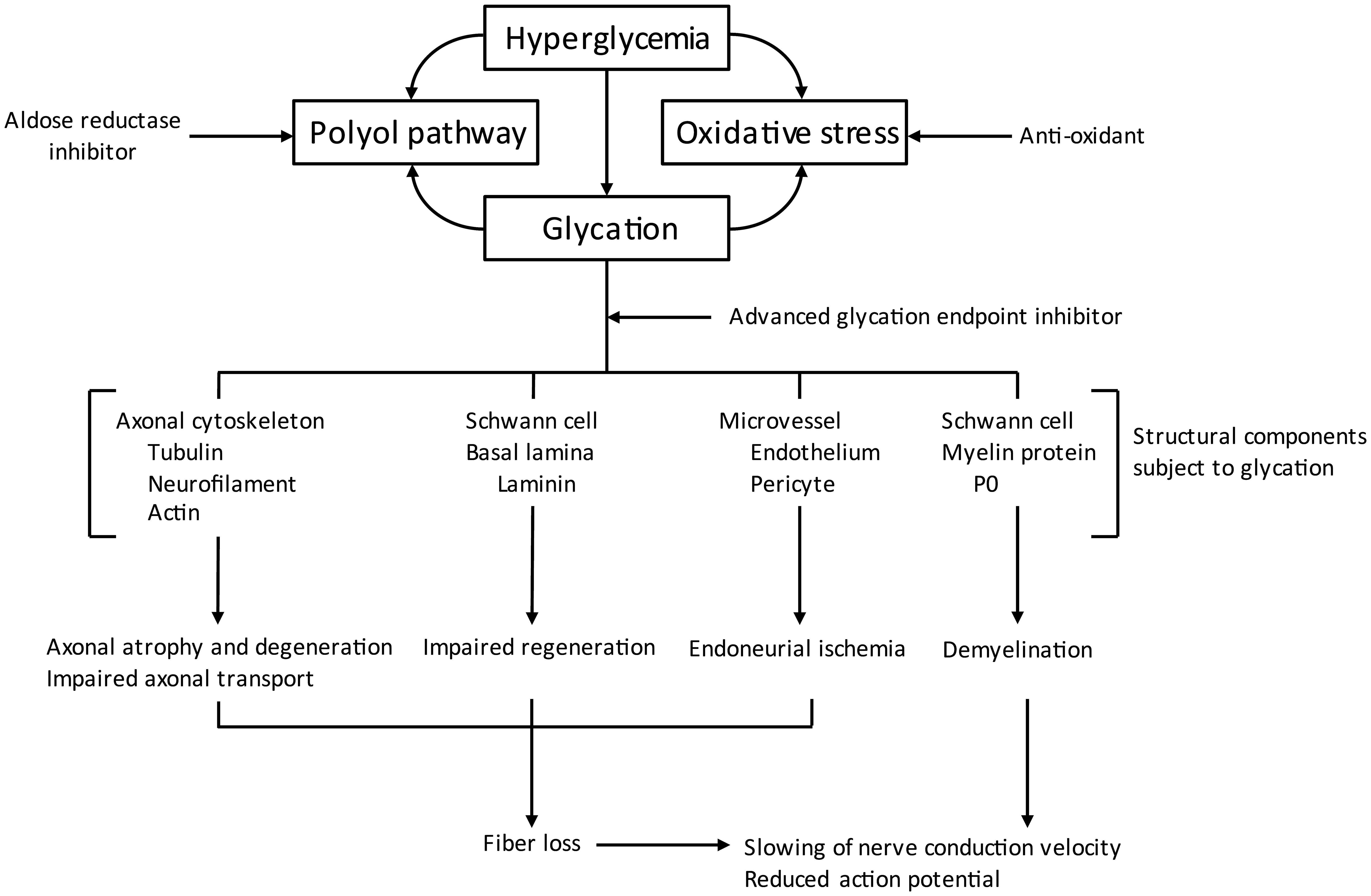
Figure 2 Pathogenic mechanisms associated with non-enzymatic glycation (modified from (31).
Protein kinase C pathway
Diabetes induced elevation in circulating glucose levels leads to the accumulation of glucose in cells independent of insulin for glucose transport. Once transported into these cells, there is an increase in glycolysis and concentrations of diacylglycerol are elevated, which can activate PKC (49). Activity of PKC in neural tissue including the vasculature in diabetes is complex. When PKC is activated by intracellular hyperglycemia the expression of genes that target a variety of cellular systems is altered. For example, in vascular tissue diabetes increases the activity of PKCβ and endothelial nitric oxide synthase is decreased thereby reducing production of nitric oxide a vasodilator and the production of the vasoconstrictor entothelin-1 is increased (50). It is likely that PKCβ activation induces DPN through reduction of vascular blood flow and impairment of microvascular tissue rather than directly injuring neurons (50). Several laboratories have reported that inhibition of PKCβ improves vascular and neural function in diabetic rodents (51–54). In addition, dysregulation of Schwann cells in part through altered PKC activity can contribute to DPN (50, 55–58). In streptozotocin-induced diabetic rats’ activity of PKCα was decreased in neurons and Schwann cells that was corrected by treatment with methylcobalamin (59). In contrast, up regulation of PKCϵ in DPN has been associated with endoplasmic reticulum stress, autophagic formation and pain (60).
Hexosamine pathway
Cellular hyperglycemia is also critical to the pathology of this pathway as is in the polyol pathway and excess production of diacylglycerol for activation of PKC. When excess glucose is being metabolized by the glycolytic pathway an excess amount of the fructose-6-phosphate that is formed gets diverted into the hexosamine pathway leading to the over production of uridine diphosphate N-acetyl glucosamine. This excess glucosamine gets incorporated onto the serine and threonine residues of transcription factors such as Sp1 resulting in pathologic changes in gene expression of fibrinogen activator inhibitor-1 and transforming growth factor-β (61). Treatment of rats with benfotiamine, a synthetic analogue of thiamine (vitamin B1) and an inhibitor of hexosamine pathway and formation of advanced glycation end products, has been shown to improve DPN (62). In human subjects with DPN it has also been shown to be beneficial, however; it is not commonly used and less effective than α-lipoic acid (63–65).
Oxidative stress
In the Banting lecture Brownlee referred to overproduction of superoxide (O2-) by the mitochondrial electron transport chain as the one element linking the different pathogenic pathways to diabetic complications (9).
Oxidative stress occurs is a result of an inability of cellular antioxidant mechanisms to neutralize increased generation of reactive oxygen species (ROS) (66). The most common forms of ROS are O2-, hydroxyl radical (OH-), hydrogen peroxide (H2O2), and peroxynitrite (ONOO-) (67). These compounds can be generated throughout the body under both normal and pathological conditions (67). O2-, the most biologically active form of ROS, is created via multiple sources including the electron transport chain, and various enzymes: NADH oxidase, NAD(P)H oxidase, lipoxygenase, cyclooxygenase, xanthine oxidase, cytochrome P-450, and, during periods of tetrahydrobiopterin deficiency, by nitric oxide synthase (67). O2- can react with superoxide dismutase (SOD) acquiring an electron it is converted to H2O2. SOD consists of a family of enzymes: mitochondrial derived Mn-SOD and two isoforms of Cu,Zn-SOD, which are located either in the cytosol or extracellularly (67). H2O2 is converted to water by the action of catalase or glutathione peroxidase in the presence of reduced glutathione (67). In the presence of iron, H2O2 can form OH- via a process known as the Fenton reaction (67). ONOO- formation is the result of a reaction between O2- and nitric oxide (NO). Referred to as nitrosative stress it causes damage primarily to vascular tissue and has been demonstrated to be increased with diabetes (67–69). ONOO- biologically has a short half-life but has the ability to diffuse across cell membranes and depending on the cell environment can cause a nitrosylation of proteins and peroxidation of lipids (69).
As previously discussed, endothelial dysfunction occurs early in diabetes and is a primary factor in the development of diabetic vascular disease, which contributes to the development of DPN. Studies from numerous laboratories including my own have demonstrated that reducing oxidative stress in a diabetes setting improves vascular function and blood flow. Combined, these studies have demonstrated that the generation of ROS in diabetes such as O2- and ONOO- contributes to oxidative stress and vascular dysfunction including decreased endothelium-dependent vascular relaxation of epineurial arterioles of the sciatic nerve (19, 20, 70, 71).
The most often studied antioxidant in a diabetes setting and DPN has been α-lipoic acid (72, 73). Using diabetic rats, we have observed that α-lipoic acid provides good protection against oxidative stress (70). We observed improvement in diabetes-induced decrease in endoneurial blood flow, endothelium-dependent vascular relaxation in arterioles that provide circulation to the sciatic nerve to acetylcholine, and nerve conduction velocity. α-Lipoic acid also reduced the production of O2- by the aorta and O2- and ONOO- by epineurial arterioles. α-Lipoic acid is a good metal chelator and is capable of scavenging hydroxyl radicals, hypochlorous acid and singlet oxygen, but not O2- or peroxyl radicals (73–76). However, in its reduced form, as dihydrolipoic acid, it is a good scavenger of O2- and prevents initiation of lipid peroxidation (73–76). In vivo, α-lipoic acid can be converted into dihydrolipoic acid (73, 74) (Figure 3). This reaction requires NADPH; however, levels of this co-factor is reduced in diabetes due to the increased flux of glucose through the polyol pathway and the formation of sorbitol as discussed above (66, 78). To overcome this pathological condition a combination therapy using an aldose reductase inhibitor to protect the availability of NADPH with α-lipoic acid should provide improved treatment for DPN compared to monotherapy. In theory this combination will promote the formation of dihydrolipoic acid, thereby enhancing the antioxidant potential of α-lipoic acid and possibly providing a synergistic effect. To test this hypothesis my laboratory examined the effect of the combination of α-lipoic acid and fidarestat, an aldose reductase inhibitor, on vascular and neural complications in a type 1 diabetic rat model (79). We demonstrated that this combination was more efficacious in preventing diabetes-induced vascular and neural dysfunction than the use of α-lipoic acid and fidarestat alone. We attributed this outcome to the verified increased conversion of α-lipoic acid to the more efficient antioxidant dihydrolipoic acid (Figure 3) (77, 79). This approach has also been applied clinically. Two different teams in China compared the efficacy of epalrestat and α-lipoic acid vs. monotherapy Both teams reported that the combination of α-lipoic acid and epalrestat is better than monotherapy clinically for DPN and improvement of motor and sensory nerve conduction velocity (80–82).
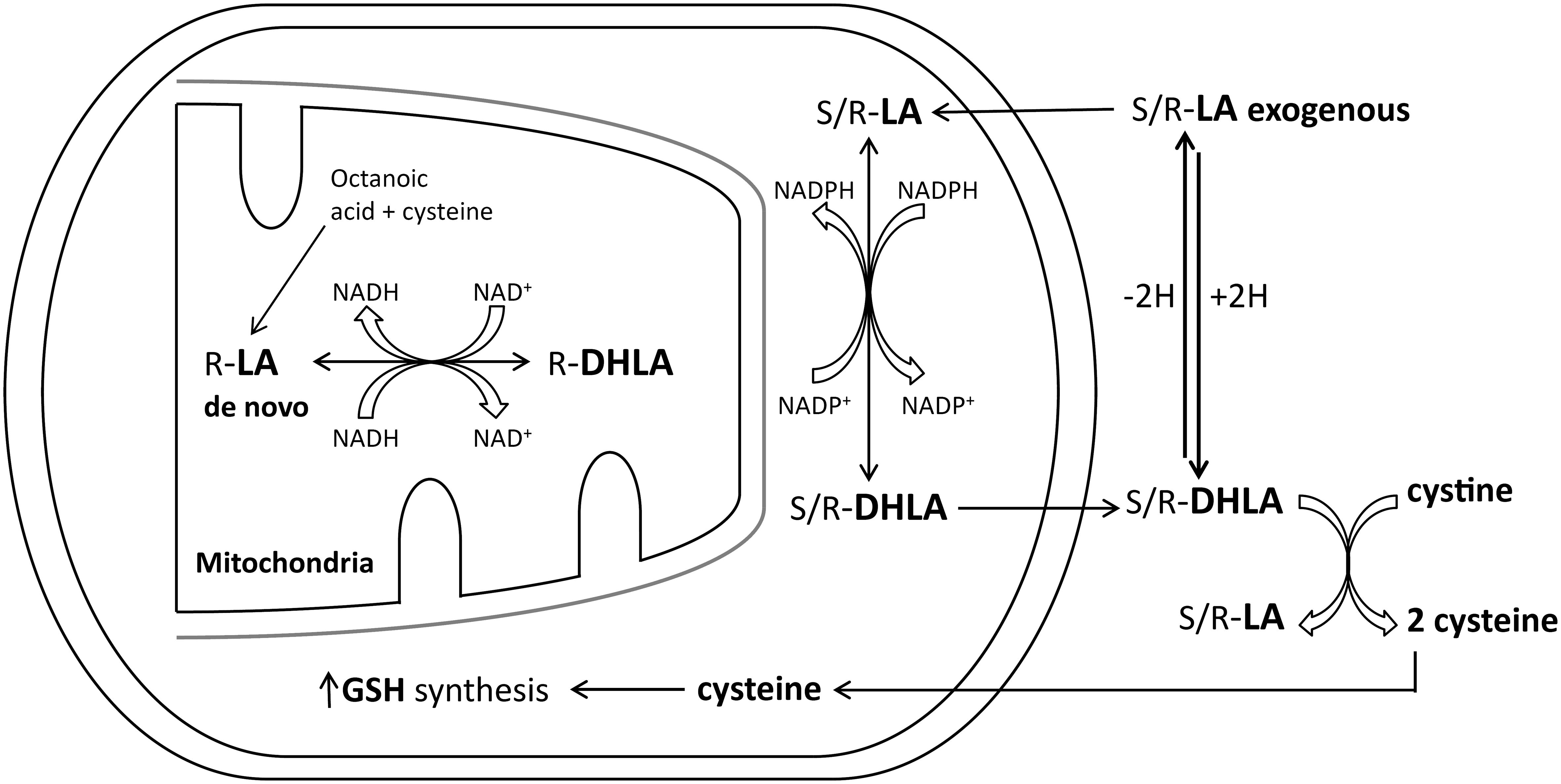
Figure 3 Lipoic acid (LA) and dihydrolipoic acid (DHLA) metabolism (modified from (77).
There have been other combination therapies tested using α-lipoic acid. In a pre-clinical study using type 1 diabetic rats the combination of coenzyme Q10 with α-lipoic acid was found to reduce oxidative stress and prevent apoptosis and degeneration of dorsal root ganglion neurons (83). In a study using the combination of prostaglandin E1 and α-lipoic acid the investigators found that the combination therapy was superior to monotherapy in improving neuropathic symptoms and nerve conduction velocity in patients with DPN (84). Another team studying type 1 diabetic rats used the combination of a prostaglandin E1 analogue and the aldose reductase inhibitor, statil, improved nerve conduction velocity better than monotherapy (85). This team concluded that a multiple-drug therapy with different mechanisms of action has greater effects on DPN than a single-drug therapy and is worthy of clinical consideration (85).
Inflammatory stress
A condition that is almost inseparable from oxidative stress is inflammatory stress. Inflammatory stress is defined as an overactivation of the immune system, causing an imbalance between accumulation of inflammatory molecules and ability anti-inflammation systems to neutralize their abundance. Chronic inflammation is an essential component of all chronic diseases including diabetes. Hyperglycemia coupled with loss of insulin signaling (type 1 diabetes) and/or insulin resistance (type 2 diabetes) including dysregulation of lipid metabolism and dyslipidemia, lead to systemic inflammation and vicious cycles of oxidative/nitrosative stress, endoplasmic and mitochondrial stress, and accumulating cellular damage (86). Besides nerve bodies, Schwann cells and microvasculature are also impaired by oxidative and inflammatory stress. Thus, an effective treatment for diabetes complications including DPN should include some form of an anti-inflammatory component.
Mechanism related therapies
Pre-clinical studies have provided a wide array of potential mechanisms and effective treatments for DPN (50, 61, 87). However, when tested in human subjects there was a general failure although α-lipoic acid and epalrestat are used clinically for DPN in Germany and Japan, respectively. These treatments are not recommended universally by any guidelines (88). Why success in pre-clinical studies for DPN primarily in rodents did not translate to human subjects has been a highly debated subject. Obvious responses include that DPN in rodents does replicate human DPN well enough for translation. This is likely true for some DPN endpoints used for studies with diabetic rodents such as nerve conduction velocities. Slowing of nerve conduction velocity develops within weeks in diabetic rats while in humans it takes years to manifest clinically (20, 89). However, pre-clinical endpoints examining decrease in sensory nerve density in the skin and cornea of diabetic rodents does seem to correspond well to the human condition (8, 90). Other reasons for the failure of translating pre-clinical finding to human disease are the endpoints used in clinical studies for DPN are not sensitive or require greater period for reversal then is allowed for in most studies. The gold standard for clinical trials of DPN has been nerve conduction velocity or subjective determination of nerve sensitivity. There is a need for objective endpoints encompassing earlier detection. Determination of sensory nerve density in the skin or cornea have been proposed to meet these criteria but clinical trials have not been performed to decide of their value (91). Another possibility that has not been given enough consideration is need for combination therapy. The etiology of DPN is complex and is impacted by dysfunction of numerous known and unknown pathways of metabolic and molecular disorders as well as structural damage. It is irrational to believe that monotherapy will be successful. For instance, hypertension is commonly treated with multiple drugs each directed at improving a causative event. The same approach needs to be considered for DPN.
If combination therapy is the best approach what evidence is there to suggest a specific combination of agents? We have extensively studied the effect of the combination of several different treatments in diabetic rodents focusing on neural and vascular endpoints. An important issue is that individually each of these agents have been safely used in humans and in combination would be predicted to have a good safety profile. Another consideration of the benefit of combination therapy is that precise dosing of each agent would not necessarily be needed to achieve the desired effect, thereby reducing the potential of unwanted side effects.
Our early studies demonstrated that vascular dysfunction of epineural arterioles that provide circulation to the sciatic nerve preceded slowing of nerve conduction velocity in streptozotocin-induced diabetes (20). This indicated a major role for vascular impairment in DPN. Vascular reactivity of these epineurial arterioles is mediated by different vasoactive compounds including acetylcholine, endothelial derived hyperpolarizing factor, calcitonin gene-related peptide (CGRP), bradykinin, insulin, and type C natriuretic peptide (CNP) (92, 93). Three of these peptides CGRP, bradykinin, and CNP are degraded by neutral endopeptidase (NEP) (94, 95). NEP activity increases with obesity and diabetes (96, 97). This stimulated our interest in the role NEP may have in regulating vascular function and DPN.
Studies in both types 1 and 2 diabetic rats have demonstrated that impaired vascular relaxation of the small blood vessels that provide circulation to the sciatic nerve occurs prior to the development of nerve dysfunction such as slowing of motor nerve conduction velocity (19, 20). In addition, our studies have indicated that increased oxidative and inflammatory stress are factors leading to impaired vascular function of these resistance vessels in diabetes (70, 98). These data have led us to focus our pre-clinical studies on interventions that reduce oxidative and inflammatory stress to improve vascular and nerve function.
AVE7688 & LCZ7696
After a series of studies our interest turned to a combination therapy consisting of inhibition of angiotensin converting enzyme (ACE) and NEP or neprilysin. We and other had shown in animal models and human subjects that inhibition of ACE improved diabetic peripheral neuropathy (99, 100). In a clinical retrospective cohort study of patients with type 2 diabetes it was concluded that pharmacological inhibition of the angiotensin system is beneficial to prevent DPN (101). Our previous studies had also demonstrated that vascular relaxation by epineurial arterioles that provide blood flow to the sciatic nerve is mediated by CNP and CGRP (95, 102). CNP is expressed in the outer membrane of endothelial cells of epineurial arterioles, whereas CGRP is expressed in sensory nerves that innervate these blood vessels (95). Interestingly, NEP degrades natriuretic peptides such as CNP, CGRP, adrenomedullin, bradykinin, and endothelin (103). NEP expression is widespread and found in many tissues including vascular tissue, and in regard to diabetes, its activity is increased by fatty acids and glucose in human microvascular cells (104–108). We have discussed that diabetes leads to an increase in protein kinase C and in vascular tissue protein kinase C stimulates NEP activity (109, 110). We have shown that this increase of NEP expression and activity occurs in epineurial arterioles of the sciatic nerve of streptozotocin-treated rats (95). The first inhibitor combination of ACE and NEP we studied was AVE7688, a vasopeptidase inhibitor, later to be called Ilepatril. In those studies, we used both type 1 or type 2 diabetic rats and an intervention protocol to determine the efficacy of AVE7688 in DPN. AVE7688 reversed the diabetes-induced decrease in endoneurial blood flow, improved acetylcholine-and CGRP-mediated vascular relaxation by epineurial arterioles and reduced superoxide and nitrotyrosine levels in these resistance vessels, prevented the development of hypoalgesia in the hindpaw and significantly improved motor and sensory nerve conduction velocity (95, 111). These studies suggest that this class of drug may be an effective approach for the treatment of diabetic vascular and DPN. However, in studies with human subjects Ilepatril was found to increase the risk for angioedema and clinical development of this drug was stopped (112). As one door closes another open, and this led to the development of a new drug combination of angiotensin receptor blocker and neprilysin inhibitor. This new combination drug that highlights the use of angiotensin II receptor blocker rather than ACE inhibitor reduced the risk of angioedema (112). LCZ696 (ENTRESTO®) was the first representative of this class of drug and combines the dual action of sacubitril and valsartan (112). Sacubitril is a neprilysin/NEP inhibitor and valsartan is a well-known angiotensin II receptor antagonist. In a clinical trial sacubitril/valsartan was shown to be an effective treatment for heart failure with reduced ejection fraction and side effects comparable to the ACE inhibitor enalapril (113). Sacubitril/valsartan has obtained Food and Drug Administration (FDA) approval for treatment of heart related conditions but had not been tested for DPN. Because vasopeptidase inhibitors like AVE7688 and sacubitril/valsartan have similar targets we were interested in examining the efficacy of sacubitril/valsartan on diabetic vascular and DPN. A comprehensive study was designed using early and late intervention protocols. Treatment was initiated 4- or 12-weeks after the induction of hyperglycemia in a late-stage type 2 diabetic rat model. Treatment was given daily by gavage for 12 weeks followed by an extensive evaluation of vascular and neural endpoints (114). The highlights of the results were efficacy of sacubitril/valsartan treatment were superior in improving vascular and neural function than valsartan alone. In the early intervention protocol sacubitril/valsartan treatment slowed the progression of DPN impacting all relative endpoints, and intervention 12-weeks after induction of hyperglycemia stimulated restoration of vascular reactivity, motor and sensory nerve conduction velocities and sensitivity/regeneration of sensory nerves of the skin and cornea. We attributed the beneficial effects observed with sacubitril/valsartan treatment to reduction in oxidative stress and protection of neuro- and vaso-active peptides. These pre-clinical studies provide support for this class of drugs as a potential new treatment for DPN. Even though LCZ696 has FDA approval for another important clinical problem and has a good safety profile no further research has been done with this compound for DPN.
Enalapril, α-lipoic acid & menhaden oil
Based upon individual results as mono-therapies we tested the combination of enalapril, α-lipoic acid and menhaden (fish) oil on peripheral neuropathy (115, 116). In a study with type 2 diabetic rats treatment consisting of enalapril, menhaden oil or α-lipoic acid as monotherapies or their combination were initiated 16 weeks after induction of hyperglycemia and treatment lasted 12 weeks (116). Prior to and after treatments analyses of an array of neural and vascular endpoints including cornea sensitivity, corneal nerve density, vascular reactivity of epineurial arterioles, motor and sensory nerve conduction velocity, intraepidermal nerve fiber density and thermal nociception were performed. Untreated diabetes caused an impairment of 20-40% in all endpoints. Vascular relaxation, nerve conduction velocity and thermal nociception worsened in untreated diabetic rats over the period of the study. The monotherapy treatment of diabetic rats was effective in improving neural and vascular deficits with menhaden oil being the most efficacious. Glucose clearance, a marker for insulin resistance, was impaired in untreated diabetic rats and significantly improved only with combination therapy. Diabetes caused steatosis, elevated serum lipid levels, slowed motor and sensory nerve conduction velocity, caused thermal hypoalgesia and reduction in intraepidermal nerve fiber density, decreased cornea sub-basal nerve fiber length and corneal sensitivity and caused impairment in vascular relaxation to acetylcholine and CGRP in epineurial arterioles of the sciatic nerve. Treating diabetic rats with the combination of enalapril, menhaden oil or α-lipoic acid served as a form of rehabilitation therapy, with nearly all the vascular and nerve deficits to revert to near control values with the lone exception of motor nerve conduction velocity, which was also significantly improved compared to diabetic rats but remained significantly decreased compared to control rats. As mono therapies each of these compounds/drugs have an excellent safety profile but have never been tested in humans as a combination. Each alone have been shown in humans to provide benefit to peripheral nerve dysfunction and other problems such as cardiovascular disease (100, 117, 118). It would be predicted that a combination of these compounds would be safe and have the potential to improve DPN but has not been tested in human subjects.
Menhaden oil & salsalate
Increasing consumption of omega-3 (n-3) polyunsaturated fatty acids, commonly found in marine mammals, (eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) through the diet or by supplements has been shown to improve cardiovascular disease (117, 119). Increased inflammatory stress is associated with a poor n-6 to n-3 ratio of polyunsaturated fatty acids in circulation (120, 121). Metabolites of EPA, E series resolvins and DHA, neuroprotectin D1, D series resolvins and maresins have been shown to have anti-inflammatory and neuroprotective properties (122–124). Salicylsalicylic acid (salsalate), a non-acetylated salicylate, is a nonsteroidal anti-inflammatory agent that inhibits the synthesis of prostaglandins by inactivating cyclooxygenase enzymes (Figure 4) (125). Salsalate, like aspirin, in combination with fish oil increases the production of resolvins from EPA and DHA (Figure 5) (126). Salsalate is insoluble in the stomach and moves to the small intestine where it is hydrolyzed into two salicylic acid molecules. Treatment of human subjects with type 2 diabetes with salsalate has been shown to improve insulin resistance (127, 128). This led us to examine the combination of omega-3 polyunsaturated fatty acids (fish oil) and salsalate on DPN. We hypothesized that the addition of salsalate to omega-3 polyunsaturated fatty acids would increase the formation of omega-3 polyunsaturated fatty acid metabolites including E and D series resolvins that we have previously shown to be as effective as fish oil alone in improving DPN in type 2 diabetic mice (126). In a study with type 2 diabetic rats, we verified our hypothesis by showing that adding salsalate to high fat diets enriched with 10% or 25% kcal of menhaden (fish) oil prevented the diabetes-induced decrease in motor and sensory nerve conduction velocity, intraepidermal nerve fiber density thermal nociception, cornea nerve density and sensitivity as well as vascular relaxation to acetylcholine and CGRP, to a greater extent than 10% or 25% menhaden oil alone (129). Our study also showed that the combination of salsalate with menhaden oil increased the circulating levels of resolvin D1 significantly compared to menhaden oil alone. When using a higher dose of menhaden oil alone (45% kcal) we found that the vascular and neural functions were maximally protected and adding salsalate did not provide any additional benefit. In addition, the use of salsalate alone in the high fat diet of type 2 diabetic rats provided only minimal protection/improvement of vascular and neural dysfunction. The primary message from these studies was that the effects of dietary salsalate in combination with lower amounts of menhaden oil are at minimum additive and perhaps synergistic toward diabetes-induced vascular and peripheral neuropathy endpoints.
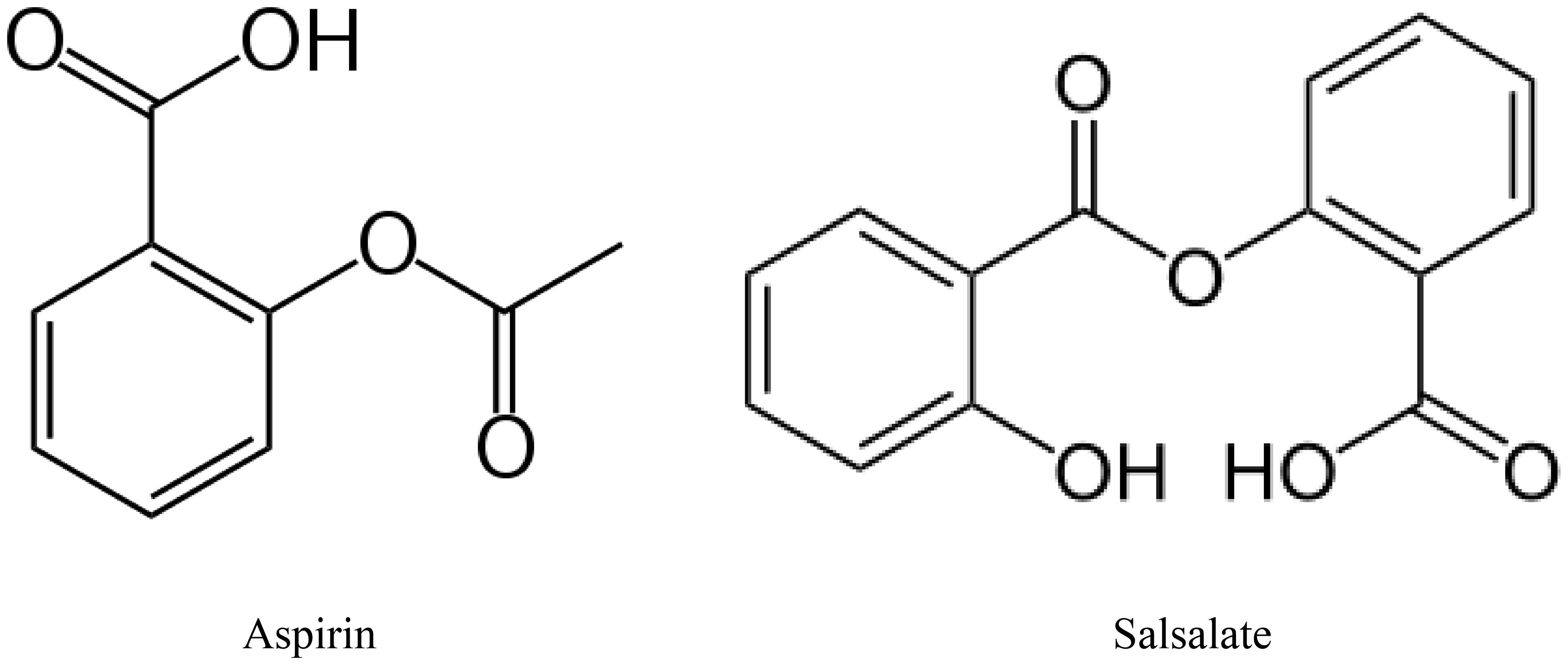
Figure 4 Metabolites of EPA and DHA.
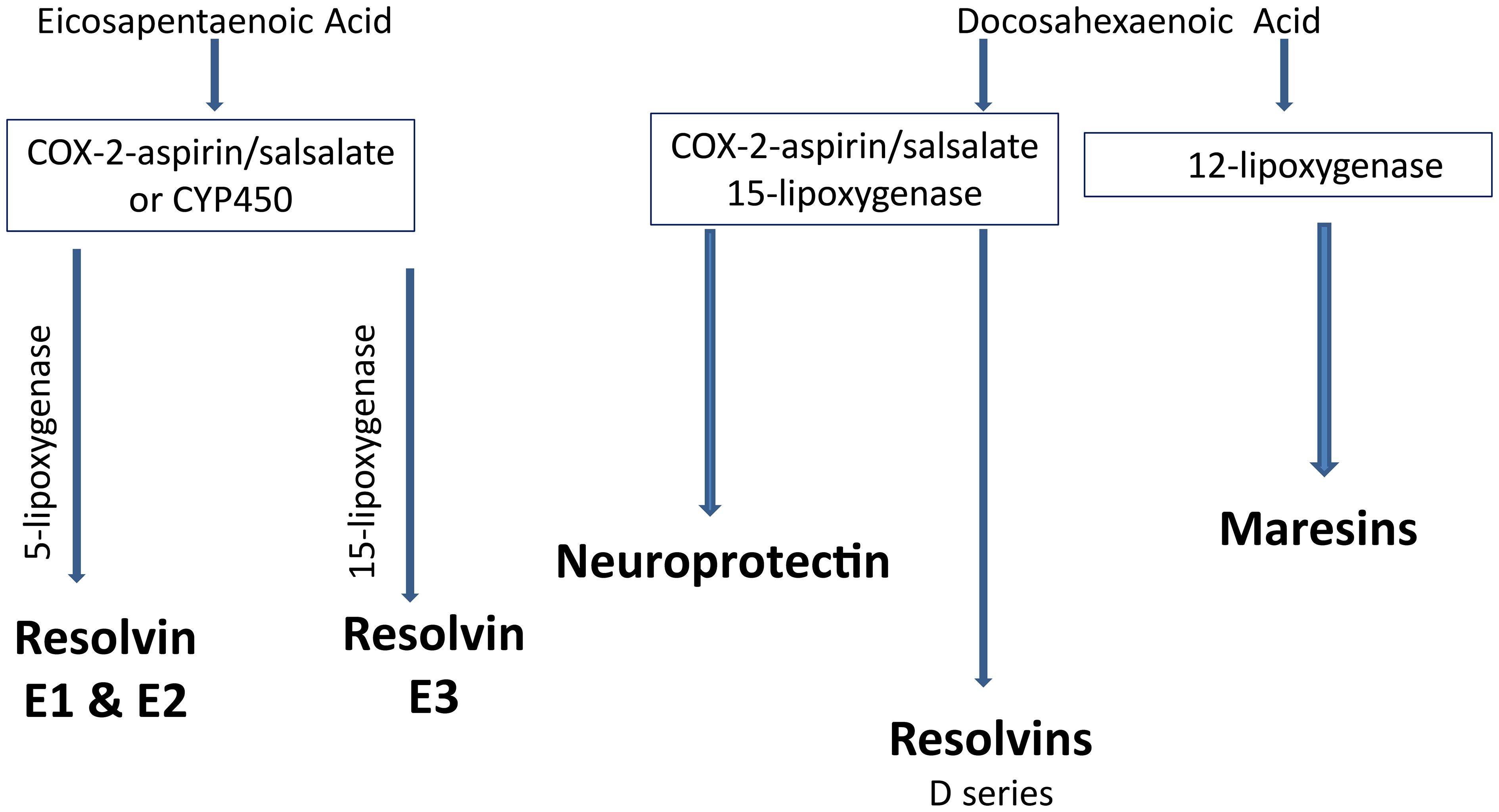
Figure 5 Metabolites of EPA and DHA.
Summary
DPN is a disease with multiple etiologies thereby providing many potential targets for treatment. Even though a mono-therapeutic approach has provided success in rodent studies successful translation to humans has not occurred. There is a movement toward combination therapy to alleviate pain for painful diabetic neuropathy (130, 131). Therefore, it is difficult to understand the reluctance to take the same approach to provide a disease modifying therapy for DPN. Above I have summarized several combinations that my laboratory has used to treat DPN in rodent models. The literature provides more examples that were not reviewed here. The encouraging fact that for each drug or compound in these combinations have been shown to be safe for humans. Future clinical trials should consider using updated primary endpoints that do not take years to develop such as such as nerve conduction deficits and a combination of drugs/compounds that are safe for human use combined with proper lifestyle modifications in diet and exercise (132).
If asked what a successful combination treatment for DPN would look like I would base my answer on the pre-clinical studies we have performed. I would consider a combination of angiotensin converting enzyme inhibitor or angiotensin receptor blocker, omega-3 polyunsaturated fatty acid supplement of 2 – 3 g per day, aldose reductase inhibitor and α-lipoic acid. Each of these compounds have been used by human subjects with a high degree of safety, thereby reducing the likelihood of unknown side effects. However, when combining drugs there is always a risk of unknown or additional side effects and this would need to be monitored with treatment. In addition, to these treatments improving diet and exercise should be recommended using the guidelines provided by the American Diabetes Association. When discussing weight loss consideration of treatment with a glucagon-like peptide-1 receptor agonist needs to be acknowledged. This class of drugs are not new but have gained attention recently as an effective treatment for obesity and type 2 diabetes (133–135). There is also evidence that these drugs are effective for DPN (136, 137). There are few pre-clinical studies available about the interaction of glucagon-like peptide-1 receptor agonists with the other drugs discussed above and if included in combination with other drugs monitoring of unknown side effects would be needed.
In conclusion, to find a successful treatment for DPN there is a need for standardized diagnostic criteria for early detection, reliable biomarkers, and innovative combination treatment approaches as discussed above (138).
Author contributions
MY: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding for this project was provided by NIH DK126837 and by Veterans Affairs RX000889 and RX003826.
Acknowledgments
Lawrence Coppey, Eric Davidson, Hanna Shevalye, and Alexander Obrosov each contributed to the studies from my laboratory mentioned in this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat Rev Dis Primers. (2019) 5:41. doi: 10.1038/s41572-019-0097-9
2. Callaghan BC, Hur J, Feldman EL. Diabetic neuropathy: one disease or two? Curr Opin Neurol. (2012) 25:536–41. doi: 10.1097/WCO.0b013e328357a797
3. Jolivalt CG, Frizzi KE, Guernsey L, Marquez A, Ochoa J, Rodriguez M, et al. Peripheral neuropathy in mouse models of diabetes. Curr Protoc Mouse Biol. (2016) 6:223–55. doi: 10.1002/cpmo.11
4. Yorek MA. Alternatives to the streptozotocin-diabetic rodent. Int Rev Neurobiol. (2016) 127:89–112. doi: 10.1016/bs.irn.2016.03.002
5. Pop-Busui R, Boulton AJ, Feldman EL, Bril V, Freeman R, Malik RA, et al. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care. (2017) 40:136–54. doi: 10.2337/dc16-2042
6. Cortez M, Singleton JR, Smith AG. Glucose intolerance, metabolic syndrome, and neuropathy. Handb Clin Neurol. (2014) 126:109–22. doi: 10.1016/B978-0-444-53480-4.00009-6
7. Papanas N, Ziegler D. Polyneuropathy in impaired glucose tolerance: is postprandial hyperglycemia the main culprit? A mini-review. Gerontology. (2013) 59:193–8. doi: 10.1159/000343988
8. Davidson EP, Coppey LJ, Kardon RH, Yorek MA. Differences and similarities in development of corneal nerve damage and peripheral neuropathy and in diet-induced obesity and type 2 diabetic rats. Invest Ophthalmol Vis Sci. (2014) 55:1222–30. doi: 10.1167/iovs.13-13794
9. Brownlee M. The pathobiology of diabetic complications. Diabetes. (2005) 54:1615–25. doi: 10.2337/diabetes.54.6.1615
10. Gabbay KH. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Curr Diabetes Rep. (2004) 4:405–8. doi: 10.1007/s11892-004-0047-z
11. Oates PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Curr Drug Targets. (2008) 9:14–36. doi: 10.2174/138945008783431781
12. Kuzumoto Y, Kusunoki S, Kato N, Kihara M, Low PA. Effect of the aldose reductase inhibitor fidarestat on experimental diabetic neuropathy in the rat. Diabetologia. (2006) 49:3085–93. doi: 10.1007/s00125-006-0400-7
13. Greene DA, Sima AA, Stevens MJ, Feldman EL, Killen PD, Henry DN, et al. Aldose reductase inhibitors: an approach to the treatment of diabetic nerve damage. Diabetes Metab Rev. (1993) 9:189–217. doi: 10.1002/dmr.5610090304
14. Greene DA, Lattimer SA, Sima AA. Are disturbances of sorbitol, phosphoinositide, and Na+-K+-ATPase regulation involved in pathogenesis of diabetic neuropathy? Diabetes. (1988) 37:688–93. doi: 10.2337/diab.37.6.688
15. Greene DA, Lattimer SA. Recent advances in the therapy of diabetic peripheral neuropathy by means of a aldose reductase inhibitor. Am J Med. (1985) 79:13–7. doi: 10.1016/0002-9343(85)90505-4
16. Gillon KR, Hawthorne JN, Tomlinson DR. Myo-inositol and sorbitol metabolism in relation to peripheral nerve function in experimental diabetes in the rat: effect of aldose reductase inhibition. Diabetologia. (1983) 25:365–71. doi: 10.1007/BF00253203
17. Pop-Busui R, Sullivan KA, Van Huysen CV, Bayer L, Cao X, Towns R, et al. Depletion of taurine in experimental diabetic neuropathy: implications for nerve metabolic, vascular, and functional deficits. Exp Neurol. (2001) 168:259–72. doi: 10.1006/exnr.2000.7591
18. Stevens MJ, Lattimer SA, Kamijo M, Van Huysen C, Sima AA, Greene DA. Osmotically-induced nerve taurine depletion and the compatible osmolyte hypothesis in experimental diabetic neuropathy in the rat. Diabetologia. (1993) 36:608–14. doi: 10.1007/BF00404069
19. Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Yorek MA. Effect of treating streptozotocin-induced diabetic rats with sorbinil, myo-inositol or aminoguanidine on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Int J Diabetes Res. (2002) 3:21–36. doi: 10.1080/15604280212525
20. Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. Int J Exp Diabetes Res. (2000) 1:131–43. doi: 10.1155/edr.2000.131
21. Sima AA, Bril V, Nathaniel V, McEwen TA, Brown MB, Lattimer SA, et al. Regeneration and repair of myelinated fibers in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with sorbinil. N Engl J Med. (1988) 319:548–55. doi: 10.1056/NEJM198809013190905
22. Sima AA, Prashar A, Nathaniel V, Bril V, Werb MR, Greene DA. Overt diabetic neuropathy: repair of axo-glial dysjunction and axonal atrophy by aldose reductase inhibition and its correlation to improvement in nerve conduction velocity. Diabetes Med. (1993) 10:115–21. doi: 10.1111/j.1464-5491.1993.tb00027.x
23. Gieron MA, Malone JI, Lowitt S, Korthals JK. Improvement in peripheral nerve function after one year of Sorbinil. Neuroreport. (1991) 2:348–50. doi: 10.1097/00001756-199106000-00012
24. Lewin IG, O’Brien IA, Morgan MH, Corrall RJ. Clinical and neurophysiological studies with the aldose reductase inhibitor, sorbinil, in symptomatic diabetic neuropathy. Diabetologia. (1984) 26:445–8. doi: 10.1007/BF00262218
25. O’Hare JP, Morgan MH, Alden P, Chissel S, O’Brien IA, Corrall RJ. Aldose reductase inhibition in diabetic neuropathy: clinical and neurophysiological studies of one year’s treatment with sorbinil. Diabetes Med. (1988) 5:537–42. doi: 10.1111/j.1464-5491.1988.tb01047.x
26. Fagius J, Brattberg A, Jameson S, Berne C. Limited benefit of treatment of diabetic polyneuropathy with an aldose reductase inhibitor: a 24-week controlled trial. Diabetologia. (1985) 28:323–9. doi: 10.1007/BF00283137
27. Pfeifer MA, Schumer MP, Gelber DA. Aldose reductase inhibitors: the end of an era or the need for different trial designs? Diabetes. (1997) 46 Suppl 2:S82–9. doi: 10.2337/diab.46.2.s82
28. Hotta N, Kawamori R, Fukuda M, Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabetes Med. (2012) 29:1529–33. doi: 10.1111/j.1464-5491.2012.03684.x
29. Ramirez MA, Borja NL. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. (2008) 28:646–55. doi: 10.1592/phco.28.5.646
30. Chang K-C, Rao M. Aldose reductase is a potential therapeutic target for neurodegeneration. Chem Biol Interact. (2024) 5:110856. doi: 10.1016/j.cbi.2024.110856
31. Sima AA, Sugimoto K. Experimental diabetic neuropathy: an update. Diabetologia. (1999) 42:773–88. doi: 10.1007/s001250051227
32. Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. (1988) 318:1315–21. doi: 10.1056/NEJM198805193182007
33. Zochodne D. Mechanisms of diabetic neuron damage: molecular pathways. Handb Clin Neurol. (2014) 126:379–99. doi: 10.1016/B978-0-444-53480-4.00028-X
34. Ryle C, Leow CK, Donaghy M. Nonenzymatic glycation of peripheral and central nervous system proteins in experimental diabetes mellitus. Muscle Nerve. (1997) 20:577–84. doi: 10.1002/(sici)1097-4598(199705)20:5<577:aid-mus6>3.0.co;2-6
35. Cullum NA, Mahon J, Stringer K, McLean WG. Glycation of rat sciatic nerve tubulin in experimental diabetic mellitus. Diabetologia. (1991) 34:387–9. doi: 10.1007/BF00403175
36. Forbes J, Cooper M. Mechanisms of diabetic complications. Physiol Rev. (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
37. Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann N Y Acad Sci. (2005) 1043:598–604. doi: 10.1196/annals.1338.067
38. Yamagishi S, Nakamura K, Matsui T, Noda Y, Imaizumi T. Receptor for advanced glycation end products (RAGE): a novel therapeutic target for diabetic vascular complication. Curr Pharm Des. (2008) 14:487–95. doi: 10.2174/138161208783597416
39. Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end product (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. (2008) 17:983–96. doi: 10.1517/13543784.17.7.983
40. Cameron NE, Gibson TM, Nangle MR, Cotter MA. Inhibitors of advanced glycation end product formation and neurovascular dysfunction in experimental diabetes. Ann N Y Acad Sci. (2005) 1043:784–92. doi: 10.1196/annals.1333.091
41. Juranek JK, Geddis MS, Song F, Zhang J, Garcia J, Rosario R, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. (2013) 62:931–43. doi: 10.2337/db12-0632
42. Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. (200) 83:876–86. doi: 10.1007/s00109-005-0688-7
43. Yagihashi S, Kamijo M, Baba M, Yagihashi N, Nagai K. Effect of aminoguanidine on functional and structural abnormalities in peripheral nerve of STZ-induced diabetic rats. Diabetes. (1992) 41:47–52. doi: 10.2337/diab.41.1.47
44. Cameron NE, Cotter MA, Dines KC, Love A. Effects of aminoguanidine on peripheral nerve function and polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. (1992) 35:946–50. doi: 10.1007/BF00401423
45. Miyauchi Y, Shikama H, Takasu T, Okamiya M, Umeda E, Hirasaki I, et al. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozotocin-induced diabetic rats. Eur J Endocrinol. (1996) 134:467–73. doi: 10.1530/eje.0.1340467
46. Sugimoto K, Yagihashi S. Effects of aminoguanidine on structural alterations of microvessels in peripheral nerve of streptozotocin diabetic rats. Microvasc Res. (1997) 53:105–12. doi: 10.1006/mvre.1996.2002
47. Kihara M, Schmelzer JD, Poduslo JF, Curran GL, Nickander KK, Low PA. Aminoguanidine effects on nerve blood flow, vascular permeability, electrophysiology, and oxygen free radicals. Proc Natl Acad Sci USA. (1991) 88:6107–11. doi: 10.1073/pnas.88.14.6107
48. Cameron NE, Cotter MA. Rapid reversal by aminoguanidine of the neurovascular effects of diabetes in rats: modulation by nitric oxide synthase inhibition. Metabolism. (1996) 45:1147–52. doi: 10.1016/s0026-0495(96)90015-3
49. Ighodaro OM. Molecular pathways associated with oxidative stress in diabetes mellitus. BioMed Pharmacother. (2018) 108:656–62. doi: 10.1016/j.biopha.2018.09.058
50. Singh R, Kishore L, Kaur N. Diabetic peripheral neuropathy: Current perspective and future directions. Pharmacol Res. (2014) 80:21–35. doi: 10.1016/j.phrs.2013.12.005
51. Ishii H, Koya D, King GL. Protein kinase C activation and its role in the development of vascular complications in diabetes mellitus. J Mol Med. (1998) 76:21–31. doi: 10.1007/s001090050187
52. Cameron NE, Cotter MA. Effects of protein kinase C β inhibition on neurovascular dysfunction in diabetic rats: interaction with oxidative stress and essential fatty acid dysmetabolism. Diabetes Metab Res Rev. (2002) 18:315–23. doi: 10.1002/dmrr.307
53. Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, et al. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. (1999) 48:2090–5. doi: 10.2337/diabetes.48.2090
54. Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci. (2002) 103:311–21. doi: 10.1042/cs1030311
55. Li J, Guan R, Pan L. Mechanism of Schwann cells in diabetic peripheral neuropathy: a review. Medicine. (2023) 102:e32653. doi: 10.1097/MD.0000000000032653
56. Vincent AM, Callaghan BC, Smith AL, Feldman EL. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol. (2011) 7:573–83. doi: 10.1038/nrneurol.2011.137
57. Naruse K. Schwann cells as crucial players in diabetic neuropathy. Adv Exp Med Biol. (2019) 1190:345–56. doi: 10.1007/978-981-32-9636-7_22
58. Hu X, Buhl CS, Sjogaard MB, Schousboe K, Mizrak HI, Kufaishi H, et al. Structural changes in Schwann cells and nerve fibers in type 1 diabetes: relationship with diabetic polyneuropathy. Diabetologia. (2023) 66:2332–45. doi: 10.1007/s00125-023-06009-z
59. Mizukami H, Ogasawara S, Yamagishi S-I, Takahashi K, Yagihashi S. Methylcobalamin effects on diabetic neuropathy and nerve protein kinase C in rats. Eur J Clin Invest. (2011) 41:442–50. doi: 10.1111/eci.2011.41.issue-4
60. Kan Y-Y, Chang Y-S, Liao W-C, Chao T-N, Hsieh Y-L. Roles of neuronal protein kinase Cϵ on endoplasmic reticulum stress and autophagic formation in diabetic neuropathy. Mol Neurobiol. (2023). doi: 10.1007/s12035-023-03716-x
61. Zhu J, Hu Z, Luo Y, Liu Y, Luo W, Du X, et al. Diabetic peripheral neuropathy: pathogenetic mechanisms and treatment. Front Endocrinol. (2024) 14:1265372. doi: 10.1186/s13098-021-00718-4
62. Sánchez-Ramírez GM, Caram-Salas NL, Rocha-González HI, Vidal-Cantú GC, Medina-Santillán R, Reyes-García G, et al. Benfotiamine relieves inflammatory and neuropathic pain in rats. Eur J Pharmacol. (2006) 530:48–53. doi: 10.1016/j.ejphar.2005.11.016
63. Winkler G, Nagybeganyi BPE, Ory I, Porochnavec M, Kempler P. Effectiveness of different benfotiamine dosage regimens in the treatment of painful diabetic neuropathy. Arzneimittelforschung. (1999) 49:220–4. doi: 10.1055/s-0031-1300405
64. Haupt E, Ledermann H, Kopcke W. Benfotiamine in the treatment of diabetic peripheral neuropathy-a three -week randomized, controlled pilot study (BEDIP study). Int J Clin Pharmacol Ther. (2005) 43:71–7. doi: 10.5414/cpp43071
65. Ang CD, Alviar MJM, Dans AL, Bautista-Velez GGP, Villaruz-Sulit MVC, tan JJ, et al. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst Rev. (2008) 16:CD004573. doi: 10.1002/14651858.CD004573.pub3
66. van Dam PS. Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes/Metabolism Res Rev. (2002) 18:176–84. doi: 10.1002/dmrr.287
67. Schnackenberg CG. Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am J Physiol Regul Integr Comp Physiol. (2002) 282:R335–42. doi: 10.1152/ajpregu.00605.2001
68. Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radical Biol Med. (2001) 30:463–88. doi: 10.1016/s0891-5849(00)00373-7
69. Fredstrom S. Nitric oxide, oxidative stress, and dietary antioxidants. Nutrition. (2002) 18:537–9. doi: 10.1016/S0899-9007(02)00758-X
70. Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. (2001) 50:1927–37. doi: 10.2337/diabetes.50.8.1927
71. Nassar T, Kadery B, Lotan C, Da’as N, Kleinman Y, Haj-Yehia A. Effects of the superoxide dismutase-mimetic compound tempol on endothelial dysfunction in streptozotocin-induced diabetic rats. Eur J Pharmacol. (2002) 436:111–8. doi: 10.1016/s0014-2999(01)01566-7
72. Kocak G, Aktan F, Canbolat O, Ozoğul C, Elbeğ S, Yildizoglu-Ari N, et al. Alpha-lipoic acid treatment ameliorates metabolic parameters, blood pressure, vascular reactivity and morphology of vessels already damaged by streptozotocin-diabetes. Diabetes Nutr Metab. (2000) 13:308–18.
73. Coleman MD, Eason RC, Bailey CJ. The therapeutic use of lipoic acid in diabetes: a current perspective. Environ Toxicol Pharmacol. (2001) 10:167–72. doi: 10.1016/s1382-6689(01)00080-1
74. Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. (2001) 17:888–95. doi: 10.1016/S0899-9007(01)00658-X
75. Dincer Y, Telci A, Kayah R, Yilmaz IA, Cakatay U, Akcay T. Effect of α-lipoic acid on lipid peroxidation and anti-oxidant enzyme activities in diabetic rats. Clin Exp Pharmacol Physiol. (2002) 29:281–4. doi: 10.1046/j.1440-1681.2002.03642.x
76. Jones W, Li X, Zhi-Chao Q, Perriott L, Whitesell RR, May JM. Uptake, recycling , and antioxidant actions of α-lipoic acid in endothelial cells. Free Rad Biol Med. (2002) 33:83–93. doi: 10.1016/s0891-5849(02)00862-6
77. Rochette L, Ghibu S, Muresan A, Vergely C. Alpha-lipoic acid: molecular mechanisms and therapeutic potential in diabetes. Can J Physiol Pharmacol. (2015) 93:1021–7. doi: 10.1139/cjpp-2014-0353
78. Bravenboer B, Kappelle AC, Hamers FPR, van Buren T, Erkelens DW, Gispen WH. Potential use of glutathione for the prevention and treatment of diabetic neuropathy in the streptozotocin-induced diabetic rat. Diabetologia. (1992) 35:813–7. doi: 10.1007/BF00399926
79. Yorek MA, Coppey LJ, Gellett JS, Davidson EP, Lund DD. Effect of fidarestat and alpha-lipoic acid on diabetes-induced epineurial arteriole vascular dysfunction. Exp Diabesity Res. (2004) 5:123–35. doi: 10.1080/15438600490277824
80. Wang X-T, Lin H-X, Xu S-A, Lu Y-K. Lipoic acid combined with epalrestat versus lipoic acid in treating diabetic peripheral neuropathy: a meta-analysis. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2017) 39:656–64. doi: 10.3881/j.issn.1000-503X.2017.05.010
81. Wang X, Lin H, Xu S, Jin Y, Zhang R. Alpha lipoic acid combined with epalrestat: a therapeutic option for patients with diabetic peripheral neuropathy. Drug Des Devel Ther. (2018) 12:2827–40. doi: 10.2147/DDDT.S168878
82. Zhao M, Chen J-Y, Chu Y-D, Zhu Y-B, Luo L, Bu S-Z. Efficacy of epalrestat plus α-lipoic acid combination therapy versus monotherapy in patients with diabetic peripheral neuropathy: a meta-analysis of 20 randomized controlled trials. Neural Regener Res. (2018) 13:1087–95. doi: 10.4103/1673-5374.233453
83. Galeshkalami NS, Abdollahi M, Najafi R, Baeeri M, Jamshidzade A, Falak R, et al. Alpha-lipoic acid and coenzyme Q10 combination ameliorates experimental diabetic neuropathy by modulating oxidative stress and apoptosis. Life Sci. (2019) 216:101–10. doi: 10.1016/j.lfs.2018.10.055
84. Jiang D-E, Li M-X, Ma Y-J, Wang Y, Wang Y. Efficacy and safety of prostaglandin EI plus lipoic acid combination therapy versus monotherapy for patients with diabetic peripheral neuropathy. J Clin Neurosci. (2016) 27:8–16. doi: 10.1016/j.jocn.2015.07.028
85. Yasuda H, Sonobe M, Hisanaga T, Kawabata T, Maeda K, Kikkawa R, et al. A combination of the aldose reductase inhibitor, statil, and the prostaglandin E1 analogue, OP1206.alpha CD, completely improves sciatic motor nerve conduction velocity in streptozotocin-induced chronically diabetic rats. Metabolism. (1992) 41:778–82. doi: 10.1016/0026-0495(92)90320-A
86. Liu Y-Z, Wang Y-X, Jiang C-L. Inflammation: The common pathway of stress-related diseases. Front Hum Neurosci. (2017) 11:316. doi: 10.3389/fnhum.2017.00316
87. Eid SA, Rumora AE, Beirowski B, Bennett DL, Hur J, Savelieff MG, et al. New perspectives in diabetic neuropathy. Neuron. (2023) 111:2623–41. doi: 10.1016/j.neuron.2023.05.003
88. Rafiullah M, Siddiqui K. Pharmacological treatment of diabetic peripheral neuropathy: an update. Drug Targets. (2022) 21:884–900. doi: 10.2174/1871527320666210303111939
89. Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD, Yorek MA. Progression of vascular and neural dysfunction in sciatic nerves of Zucker diabetic fatty and Zucker rats. Am J Physiol Endocrinol Metab. (2005) 289:E113–22. doi: 10.1152/ajpendo.00594.2004
90. Davidson EP, Coppey LJ, Holmes A, Yorek MA. Changes in corneal innervation and sensitivity and acetylcholine-mediated vascular relaxation of the posterior ciliary artery in a type 2 diabetic rat. Invest Ophthalmol Vis Sci. (2012) 53:1182–7. doi: 10.1167/iovs.11-8806
91. Petropoulos IN, Ponirakis G, Ferdousi M, Azmi S, Kalteniece A, Khan A, et al. Corneal confocal microscopy: A biomarker for diabetic peripheral neuropathy. Clin Ther. (2021) 43:1457–75. doi: 10.1016/j.clinthera.2021.04.003
92. Terata K, Coppey LJ, Davidson EP, Dunlap JA, Gutterman DD, Yorek MA. Acetylcholine-induced arteriolar dilation is reduced in streptozotocin-induced diabetic rats with motor nerve dysfunction. Br J Pharmacol. (1999) 128:837–43. doi: 10.1038/sj.bjp.0702856
93. Coppey LJ, Gellett JS, Yorek MA. Mediation of vascular relaxation in epineurial arterioles of the sciatic nerve: effect of diabetes in type 1 and type 2 diabetic rat models. Endothelium. (2003) 10:89–94. doi: 10.1080/10623320303366
94. Davidson EP, Coppey LJ, Holmes A, Yorek MA. Effect of inhibition of angiotensin converting enzyme and/or neutral endopeptidase on vascular and neural complications in high fat fed/low dose streptozotocin-diabetic rats. Eur J Pharmacol. (2012) 677:180–7. doi: 10.1016/j.ejphar.2011.12.003
95. Davidson EP, Kleinschmidt TL, Oltman CL, Lund DD, Yorek MA. Treatment of streptozotocin-induced diabetic rats with AVE7688, a vasopeptidase inhibitor: effect on vascular and neural disease. Diabetes. (2007) 56:355–62. doi: 10.2337/db06-1180
96. Standeven KF, Hess K, Carter AM, Rice GI, Cordell PA, Balmforth AJ, et al. Neprilysin, obesity and the metabolic syndrome. Int J Obes (Lond). (2011) 35:1031–40. doi: 10.1038/ijo.2010.227
97. Nalivaeva NN, Zhuravin IA, Turner AJ. Neprilysin expression and functions in development, ageing and disease. Mech Ageing Dev. (2020) 192:111363. doi: 10.1016/j.mad.2020.111363
98. Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Salvemini D, et al. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Br J Pharmacol. (2001) 134:21–9. doi: 10.1038/sj.bjp.0704216
99. Coppey LJ, Davidson EP, Rinehart TW, Gellett JS, Oltman CL, Lund DD, et al. ACE inhibitor or angiotensin II receptor antagonist attenuates diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetes. (2006) 55:341–8. doi: 10.2337/diabetes.55.02.06.db05-0885
100. Malik RA, Williamson S, Abbott C, Carrington AL, Iqbal J, SChady W, et al. Effect of angiotensin-converting-enzyme (ACE) inhibitor trandolapril on human diabetic neuropathy: randomised double-blind controlled trial. Lancet. (1998) 352:1978–81. doi: 10.1016/S0140-6736(98)02478-7
101. Iwane S, Nemoto W, Miyamoto T, Hayashi T, Tanaka M, Uchitani K, et al. Clinical and preclinical evidence that angiotensin-converting enzyme inhibitors and angiotensin receptor blockers prevent diabetic peripheral neuropathy. Sci Rep. (2024) 14:1039. doi: 10.1038/s41598-024-51572-z
102. Yorek MA, Coppey LJ, Gellett JS, Davidson EP. Sensory nerve innervation of epineurial arterioles of the sciatic nerve containing calcitonin gene-related peptide: effect of streptozotocin-induced diabetes. Exp Diabesity Res. (2004) 5:187–93. doi: 10.1080/15438600490486732
103. Pu Q, Schiffrin EL. Effect of ACE/NEP inhibition on cardiac and vascular collagen in stroke-prone spontaneously hypertensive rats. Am J Hypertens. (2001) 14:1067–72. doi: 10.1016/s0895-7061(01)02157-4
104. Gonzalez W, Soleilhac JM, Fournie-Zaluski MC, Roques BP, Michel JB. Characterization of neutral endopeptidase in vascular cells, modulation of vasoactive peptide levels. Eur J Pharmacol. (1998) 345:323–31. doi: 10.1016/s0014-2999(98)00038-7
105. Vatter H, Schilling L, Schmiedek P, Ehrenreich H. Evidence for functional endothelin-converting enzyme activity in isolated rat basilar artery: effect of inhibitors. J Cardiovasc Pharmacol. (1998) 31:S64–7. doi: 10.1097/00005344-199800001-00021
106. Edwards RM, Pullen M, Nambi P. Distribution of neutral endopeptidase activity along the rat and rabbit nephron. Pharmacology. (1999) 59:45–50. doi: 10.1159/000028304
107. Ebihara F, Di Marco GS, Juliano MA, Casarini DE. Neutral endopeptidase expression in mesangial cells. J Renin Angiotensin Aldosterone Syst. (2003) 4:228–33. doi: 10.3317/jraas.2003.037
108. Muangman P, Spenny ML, Tamura RN, Gibran NS. Fatty acids and glucose increase neutral endopeptidase activity in human microvascular endothelial cells. Shock. (2003) 19:508–12. doi: 10.1097/01.shk.0000055815.40894.16
109. Suzki T, Ino K, Kikkawa F, Uehara C, Kajiyama H, Shibata K, et al. Neutral endopeptidase/CD10 expression during phorbol ester-induced differentiation of choriocarcinoma cells through the protein kinase C- and extracellular signal-regulated kinase-dependent signaling pathway. Placenta. (2002) 23:475–82. doi: 10.1053/plac.2002.0820
110. Kikkawa F, Shibata K, Suzuki T, Kajiyama H, Ino K, Nomura S, et al. Signal pathway involved in increased expression of neutral endopeptidase by gonadotropin releasing hormone in choriocarcinoma cells. Placenta. (2004) 25:176–83. doi: 10.1016/j.placenta.2003.09.002
111. Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Yorek MA. Treatment of Zucker diabetic fatty rats with AVE7688 improves vascular and neural dysfunction. Diabetes Obes Metab. (2009) 11:223–33. doi: 10.1111/j.1463-1326.2008.00924.x
112. Pickering TG. The rise and fall of omapatrilat. J Clin Hypertens. (2002) 4:371–3. doi: 10.1111/j.1524-6175.2002.01506.x
113. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. (2014) 371:993–1004. doi: 10.1056/NEJMoa1409077
114. Davidson EP, Coppey LJ, Shevalye H, Obrosov A, Yorek MA. Vascular and neural complications in type 2 diabetic rats: improvement by sacubitril/valsartan greater than valsartan alone. Diabetes. (2018) 67:1616–26. doi: 10.2337/db18-0062
115. Yorek MS, Obrosov A, Shevalye H, Coppey LJ, Kardon RH, Yorek MA. Early vs. late intervention of high fat/low dose streptozotocin treated C57Bl/6J mice with enalapril, α-lipoic acid, menhaden oil or their combination: Effect on diabetic neuropathy related endpoints. Neuropharmacol. (2017) 116:122–31. doi: 10.1016/j.neuropharm.2016.12.022
116. Davidson EP, Holmes A, Coppey LJ, Yorek MA. Effect of combination therapy consisting of enalapril, α-lipoic acid, and menhaden oil on diabetic neuropathy in a high fat/low dose streptozotocin treated rat. Eur J Pharmacol. (2015) 765:258–67. doi: 10.1016/j.ejphar.2015.08.015
117. Siscovick DS, Barringer TA, Fretts AM, Wu JH, Lichtenstein AH, Costello RB, et al. Omega-3 polyunsaturated fatty acid (Fish oil) supplementation and the prevention of clinical cardiovascular disease: A science advisory from the american heart association. Circulation. (2017) 135:e867–84. doi: 10.1161/CIR.0000000000000482
118. Papanas N, Ziegler D. Efficacy of α-lipoic acid in diabetic neuropathy. Expert Opin Pharmacother. (2014) 15:2721–31. doi: 10.1517/14656566.2014.972935
119. Yanai H, Masui Y, Katsuyama H, Adachi H, Kawaguchi A, Hakoshima M, et al. An improvement of cardiovascular risk factors by omega-3 polyunsaturated fatty acids. J Clin Med Res. (2018) 10:281–9. doi: 10.14740/jocmr3362w
120. Zárate R, El Jaber-Vazdekis N, Tejera N, Pérez JA, Rodríguez C. Significance of long chain polyunsaturated fatty acids in human health. Clin Transl Med. (2017) 6:25. doi: 10.1186/s40169-017-0153-6
121. Yang LG, Sing ZX, Yin H, Wang YY, Shu GF, Lu HX, et al. Low n-6/n-3 PUFA ratio improves lipid metabolism, inflammation, oxidative stress and endothelial function in rats using plant oils as n-3 fatty acid source. Lipids. (2016) 51:49–59. doi: 10.1007/s11745-015-4091-z
122. Shevalye H, Yorek MS, Coppey LJ, Holmes A, Harper MM, Kardon RH, et al. Effect of enriching the diet with menhaden oil or daily treatment with resolvin D1 on neuropathy in a mouse model of type 2 diabetes. J Neurophysiol. (2015) 114:199–208. doi: 10.1152/jn.00224.2015
123. Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. (2011) 111:5922–43. doi: 10.1021/cr100396c
124. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
125. Higgs GA, Salmon JA, Henderson B, Vane JR. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc Natl Acad Sci USA. (1987) 84:1417–20. doi: 10.1073/pnas.84.5.1417
126. Yorek MS, Coppey LJ, Shevalye H, Obrosov A, Kardon RH, Yorek MA. Effect of treatment with salsalate, menhaden oil, combination of salsalate and menhaden oil, or resolvin D1 of C57Bl/6J type 1 diabetic mouse on neuropathic endpoints. J Nutr Metab. (2016) 2016:5905891. doi: 10.1155/2016/5905891
127. Desouza CV. An overview of salsalate as a potential antidiabetic therapy. Drugs Today (Barc). (2010) 46:847–53. doi: 10.1358/dot.2010.46.11.1534820
128. Hasan I, Rainsford KD, Ross JS. Salsalate: a pleotrophic anti-inflammatory drug in the treatment of diabetes, obesity, and metabolic diseases. Inflammopharmacology. (2023) 31:2781–97. doi: 10.1007/s10787-023-01242-9
129. Davidson EP, Coppey LJ, Shevalye H, Obrosov A, Yorek MA. Effect of dietary content of menhaden oil with or without salsalate on neuropathic endpoints in high-fat-fed/low-dose streptozotocin-treated sprague dawley rats. J Diabetes Res. (2018) 2018:2967127. doi: 10.1155/2018/2967127
130. Dantas JM, Oliveira MdeJ, Silva LAO, Batista S, Dagostin CS, Schachter DC. Monotherapy versus combination therapy in the treatment of painful diabetic neuropathy: a systematic review and meta-analysis. Clin Drug Investig. (2023) 43:905–14. doi: 10.1007/s40261-023-01318-y
131. Saul H, Deeney B, Imison C, Tesfaye S. Combination therapy for painful diabetic neuropathy is safe and effective. BHJ. (2023) 21:381. doi: 10.1136/bmj.p866
132. Guldiken YC, Malik A, Petropoulos IN, Gad H, Elgassim E, Salivon I, et al. Where Art Thou O treatment for diabetic neuropathy: the sequel. Expert Rev Neurother. (2023) 23:845–51. doi: 10.1080/14737175.2023.2247163
133. Wang JY, Wang QW, Yang XY, Yang W, Li DR, Jin JY, et al. GLP-1 receptor agonists for the treatment of obesity: Role as a promising approach. Front Endocrinol. (2023) 14:1085799. doi: 10.3389/fendo.2023.1085799
134. Scheen AJ. Dual GIP/GLP-1 receptor agonists: New advances for treating type-2 diabetes. Ann Endocrinol. (2023) 84:316–21. doi: 10.1016/j.ando.2022.12.423
135. Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes – state-of-the-art. Mol Metab. (2021) 46:101102. doi: 10.1016/j.molmet.2020.101102
136. Garcia-Casares N, Gonzalez-Gonzalez G, de la Cruz-Cosme C, Garzon-Maldonado FJ, de Rojas-Leal C, Ariza MJ. Effects of GLP-1 receptor agonists on neurological complications of diabetes. Rev Endocr Metab Disord. (2023) 24:655–72. doi: 10.1007/s11154-023-09807-3
137. Mehta K, Behl T, Kumar A, Uddin MS, Zengin G, Arora S. Deciphering the neuroprotective role of glucagon-like peptide-1 agonists in diabetic neuropathy: Current perspective and future directions. (2021). Curr Protein Pept Sci. (2021) 22:4–18. doi: 10.2174/1389203721999201208195901
Keywords: diabetes, peripheral neuropathy, animal models, oxidative stress, inflammatory stress
Citation: Yorek M (2024) Combination therapy is it in the future for successfully treating peripheral diabetic neuropathy? Front. Endocrinol. 15:1357859. doi: 10.3389/fendo.2024.1357859
Received: 18 December 2023; Accepted: 16 April 2024;
Published: 15 May 2024.
Edited by:
Simone Baltrusch, University Hospital Rostock, GermanyReviewed by:
Zoltan Kender, Heidelberg University, GermanyCorinne Genevieve Jolivalt, University of California, San Diego, United States
Copyright © 2024 Yorek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Yorek, bWFyay15b3Jla0B1aW93YS5lZHU=