 Julian Sun1,2†
Julian Sun1,2†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 26 June 2024
Sec. Adrenal Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1356870
Aims: Primary pigmented nodular adrenocortical disease (PPNAD), as a rare kind of Cushing’s syndrome, is frequently misdiagnosed. To get a better understanding of the disease, we analyzed the clinical characteristics and pathogenic variants of PPNAD.
Methods: Databases were searched, and the pathogenic variants and clinical manifestations of patients were summarized from the relevant articles.
Results: A total of 210 patients in 86 articles were enrolled with a median age of 22 and a female-to-male ratio of 2:1. Sixty-six (31.43%) patients were combined with Carney complex (CNC) and 94.29% were combined with osteoporosis/osteopenia. Among 151 patients who underwent genetic testing, 87.42% (132/151) had pathogenic variants. Six gene mutations (PRKAR1A, PDE11A, PRKACA, CTNNB1, PDE8B, and ARMC5) were detected in the patients. The most common mutation was PKAR1A, accounting for 79.47% (120/151). There was a significant correlation between PRKAR1A pathogenic variant and spotty skin pigmentation in CNC concurrent with PPNAD (p < 0.05). Among pregnant patients with PPNAD, those without surgical treatment and with bilateral adrenalectomy suffered from a high-risk perinatal period. However, patients with unilateral adrenalectomy presented a safe perinatal period.
Conclusions: For young patients with Cushing’s syndrome, especially female patients with spotty skin pigmentation and osteoporosis/osteopenia, PPNAD should be considered. Unilateral adrenal resection may be considered as an option for women with fertility needs. In view of the difficulty of PPNAD diagnosis, genetic testing before surgery might be a reasonable option. Patients with PPNAD with spotty skin pigmentation should consider the PRKAR1A pathogenic variant and pay attention to CNC.
Systematic review registration: https://www.crd.york.ac.uk/prospero, identifier CRD42023416988.
Primary pigmented nodular adrenocortical disease (PPNAD), as a subclass of bilateral micronodular adrenocortical disease, is a rare but an important cause of endogenous Cushing’s syndrome (CS) especially in children and young adults. Characterized by small, black and brown pigmented micronodules in the adrenal cortex, PPNAD can be combined with the “complex of myxomas, spotty skin pigmentation, and endocrine overactivity,” or Carney complex (CNC) (cPPNAD) (1). Meanwhile, 10%–20% of patients with PPNAD can also occur in patients without CNC [denoted as isolated PPNAD (i-PPNAD)] (2). Pathologically, the diagnosis of PPNAD is mainly based on histological findings, which displays multiple adrenocortical nodules with cytoplasmic pigmentation and inter-nodular cortical atrophy. However, radiology may underestimate the presence of bilateral adrenal gland involvement in patients with PPNAD due to the discrete nodular formations (sizes < 1 cm). These micronodules are often cortisol-producing and composed of lipid-poor cortical cells; therefore, hypercortisolism in PPNAD can be overt, subclinical, cyclic, or atypical (3, 4). All these make the diagnosis of PPNAD difficult. In addition, cPPNAD is causally related with inactivating mutations of the regulatory subunit type 1A of the cAMP-dependent protein kinase (PRKAR1A) gene and yet unknown defect(s) in other gene(s). PPNAD is a rare autosomal dominant disorder with variable penetrance and uncertain genotype/phenotype correlation. Delineation of a genotype–phenotype correlation for patients with PPNAD is essential for understanding potential gene functions and providing counseling and preventive care. In this review, we summarize the clinical features, pathogenic variants, and recent progress in investigation and therapy of PPNAD.
Five electronic databases (i.e., PubMed, Web of Science, Embase, the China National Knowledge Infrastructure, and Wanfang) were used to search for relevant studies with the following terms: “PPNAD” or “iPPNAD” or “familial isolated primary pigmented nodular adrenocortical disease” or “isolated primary pigmented nodular adrenocortical disease” or “primary pigmented nodular adrenocortical disease” or “micronodular adrenal disease”. Analyses of pathogenic variants and clinical features were conducted from inception to 12 September 2023. We selected studies in the English or Chinese language. Eligible studies met the following criteria: (1) PPNAD diagnosed by pathology after adrenalectomy according to the 2022 WHO classification of adrenal cortical tumors {PPNAD is composed of multiple beaded pigmented micronodules (<10 mm, often 2–5 mm). PPNAD is characterized by multiple subcentimeter micronodules composed of eosinophilic adrenocortical cells with different pigment deposits. Atrophy of the intertubercular cortex is commonly observed. Micronodules were positive for CYP11B1, confirming cortisol production (5)}. (2) The pathogenic variant sites were described. (3) The main clinical data of the patients were described. The flow diagram of the search process is provided in Figure 1.
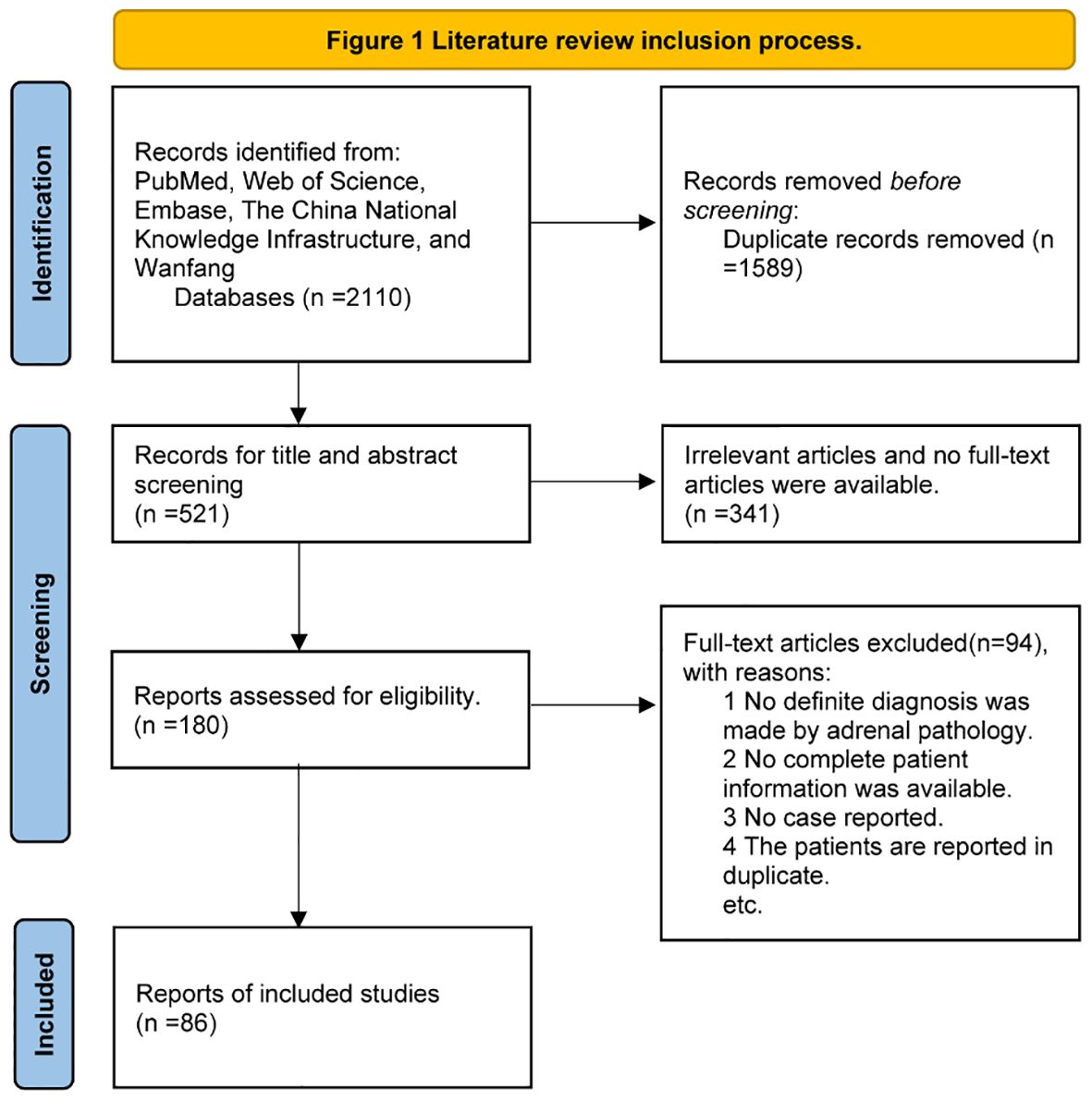
Figure 1 Literature review inclusion process.
The following data were extracted from the eligible studies: (1) country, (2) sex, (3) age at diagnosis of PPNAD, (4) pathogenic variant, (5) treatment, (6) clinical features, and (7) complication of CNC. Diagnostic criteria of CNC are shown in Table 1. The CNC diagnostic criteria were based on the criteria proposed by Stratakis et al. (1).

Table 1 Diagnostic criteria for CNC*.
Liddle GW was the first person using dexamethasone to assess CS in 1960. The original low-dose dexamethasone suppression test (LDDST) and high-dose dexamethasone suppression test (HDDST) were named Liddle tests, which were widely used in the evaluation of CS (6).
The epidemiological and clinical characteristics, and laboratory indexes of patients were described utilizing simple summary statistics. Fisher exact tests were used to test for association between qualitative variables. All tests were two-sided, and p-values < 0.05 were considered statistically significant. Statistical analysis was performed using the Statistical Package for the Social Sciences version 26 for Windows (SPSS). Since certain data in some patients were missing, the total number of patients was mentioned in each analysis.
Eighty-six articles including 210 patients were enrolled. The top three countries were France (65/210, 30.95%), China (43/210, 20.48%), and Canada (27/210, 12.86%). The patients were distributed in 23 countries on six continents. Europe has the highest number of cases (87/210, 41.43%), followed by Asia (64/210, 30.48%), North America (47/210, 22.38%), Africa and Oceania (8/210, 3.81%), and South America (4/210, 1.90%) (Table 2; Figure 2). A total of 26 patients were from 11 families, and the others were considered as sporadic.
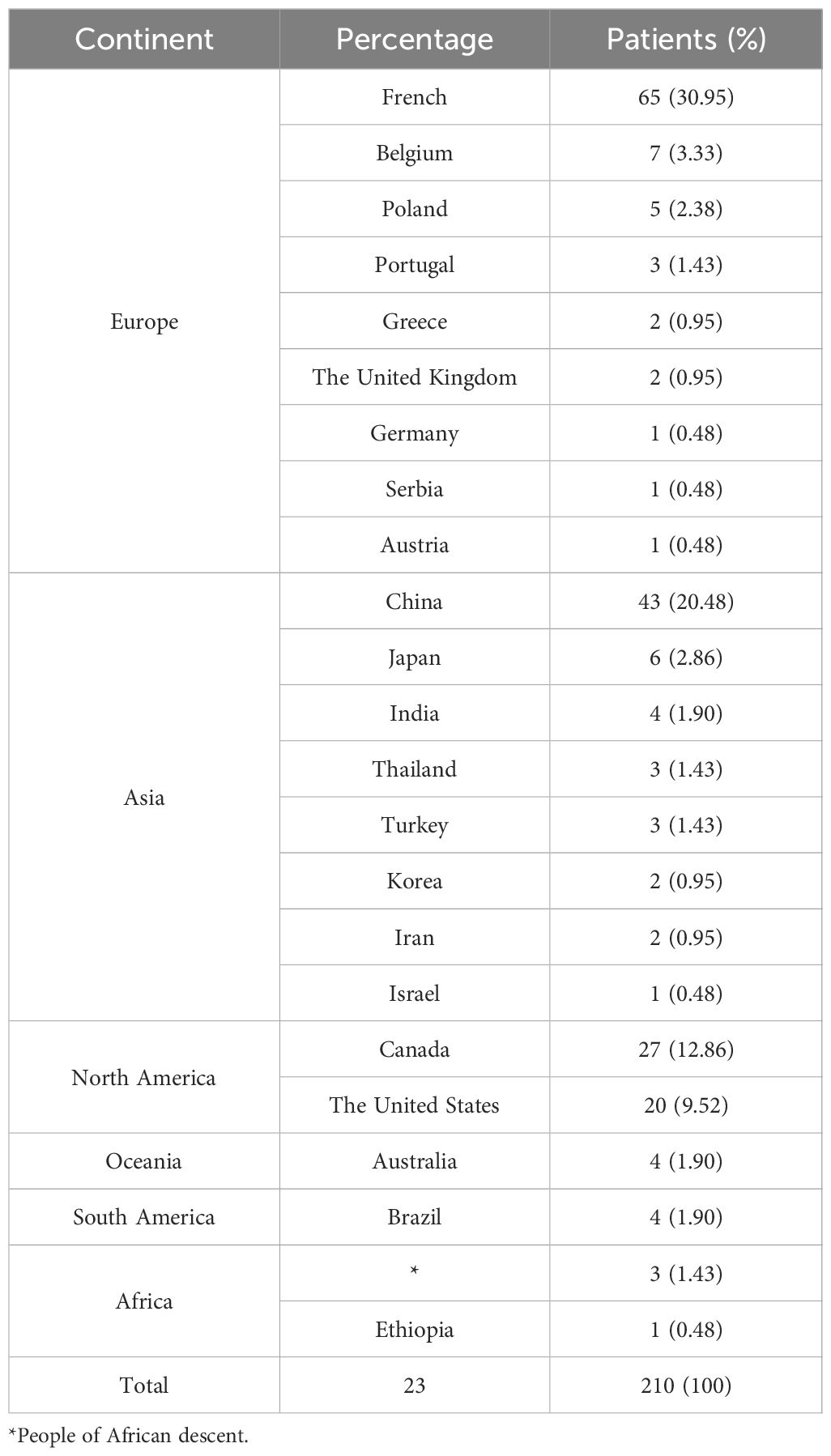
Table 2 The detailed information of patients with PPNAD.
The ages of patients ranged from 1 to 61 years old, with a median of 22 (quartiles 14–28), and 71.88% (151/210) of patients were 10–30 years old at diagnosis of PPNAD. Among them, female patients were 144 (144/210, 68.57%), with a female-to-male ratio of 2:1.
The clinical data are shown in Figure 2. In addition, BMI at diagnosis was only available for 56 patients, among which the prevalence of obesity (BMI ≥ 30 kg/m2) was 14.29% (8/56), overweight (BMI: 25–29.9 kg/m2) was 30.36% (17/56), and normal weight was 51.79% (29/56) with a median of 24.45 (quartiles 22.29–26.48) kg/m2. In addition, 28 patients had growth retardation, aged 1 to 19 years old with a median of 11 (quartiles 9–12). Clinical manifestations were not available in all patients, which included mainly osteoporosis or low bone mineral density (33/35, 94.29%), hypertension (81/120, 67.50%), and weight gain (71/120, 59.17%).
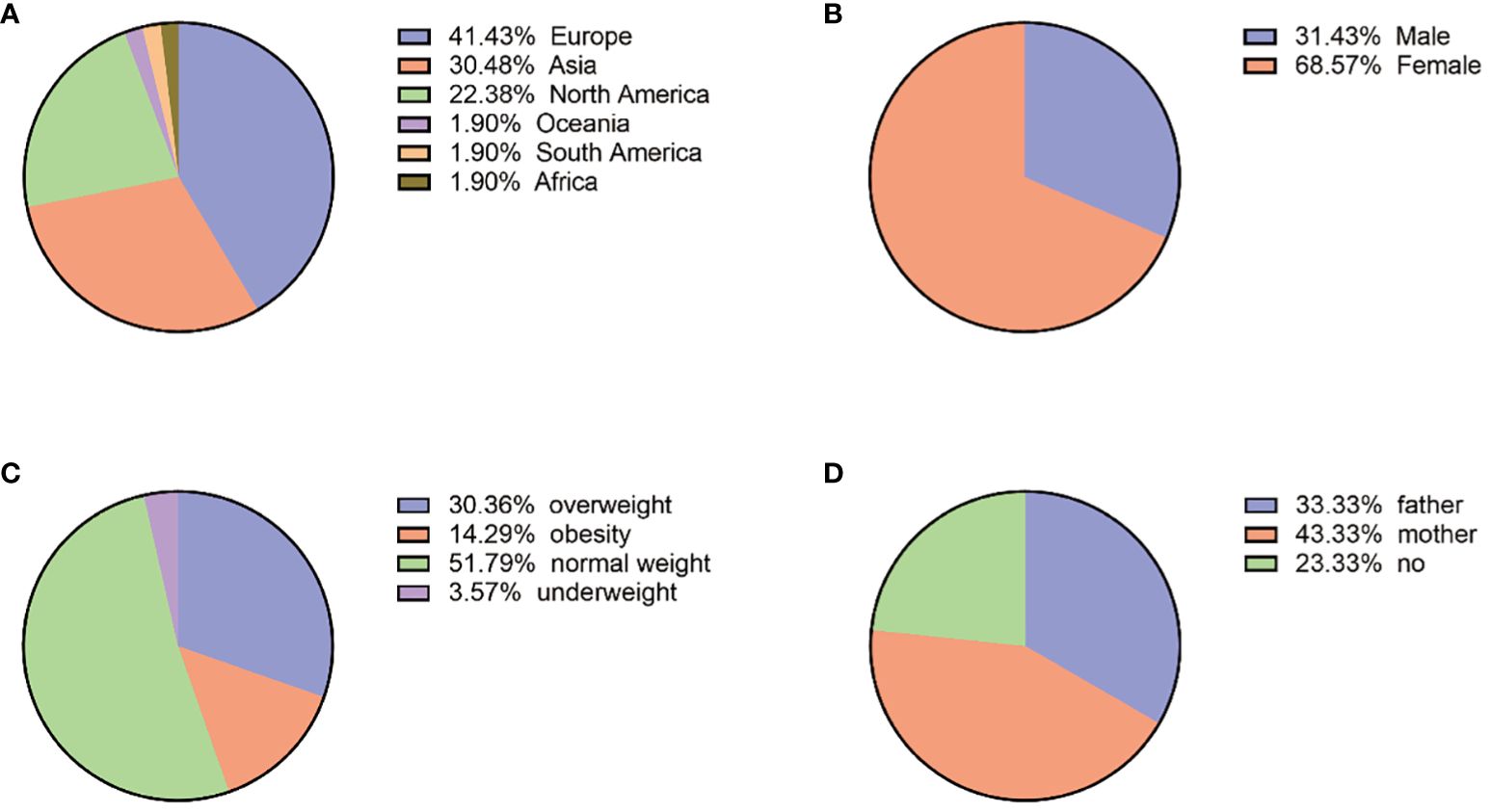
Figure 2 (A) Geographical country distribution. (B) Gender distribution. (C) BMI (N:56). (D) Mutated genes from parents (N:30).
A total of 66 (66/210, 31.43%) patients were cPPNAD and had details of the diagnosis of CNC, with 40 (40/66, 60.61%) being women, 47 (47/66, 71.21%) having spotty skin pigmentation with a typical distribution, 19 (19/66, 28.79%) having cutaneous or cardiac myxomas, and 7 (7/40, 17.50%) having multiple breast nodules or carcinoma. Particularly, one patient with PPNAD only showed spotty pigmentation of the skin, with suspected CNC (7).
ACTH data and normal criteria were available in 98 patients, 77 (77/98, 78.57%) of whom had low or undetectable ACTH, and 21 (21/98, 21.43%) were within the lower limit of the normal range of ACTH levels.
Seventy-one patients mentioned diurnal cortisol changes, among which 70 (70/71, 98.59%) lost their cortisol circadian rhythm. Moreover, regarding the dexamethasone suppression test, 31 patients were tested for urinary cortisol after high-dose dexamethasone test or Liddle test, and all (31/31, 100%) of them showed no significant suppression or even paradoxical increase. Plasma cortisol showed similar results and was not suppressed in all the patients with low-dose dexamethasone and high-dose dexamethasone inhibition tests.
Plasma dehydroepiandrosterone sulfate (DHEA-S) was available in 28 patients, normal in 11 (11/28, 39.29%) patients, decreased in 14 (14/28, 50.00%), and elevated in 3 (3/28, 10.71%) patients. Plasma growth hormone (GH) was provided in 13 patients, which was normal in 7 (7/13, 53.85%), decreased in 2 (2/13, 15.38%), and elevated in 4 (4/13, 30.77%) patients.
A total of 151 patients underwent genetic testing, and 132 (132/151, 87.42%) patients found pathogenic variants. Six different mutations, PRKAR1A, PDE11A, PRKACA, CTNNB1(β- catenin), PDE8B, and ARMC5, were identified. The most common mutation was PKAR1A, accounting for 79.47% (120/151) of cases; the second most common was PED11A mutation, found in 26.49% (40/151) of cases, while other mutations were relatively rare. A total of 33 patients had both PRKAR1A and PDE11A mutations (Tables 3, 4).
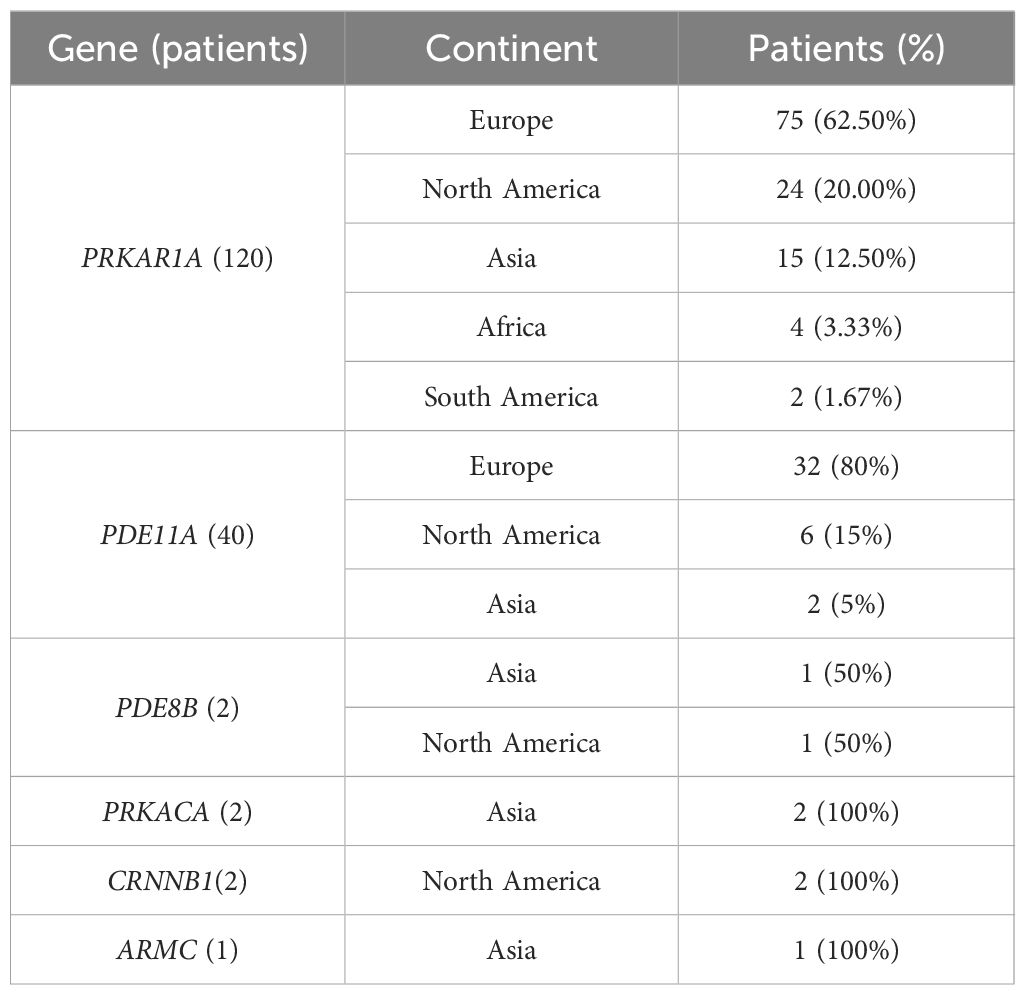
Table 3 Geographic distribution of genes.
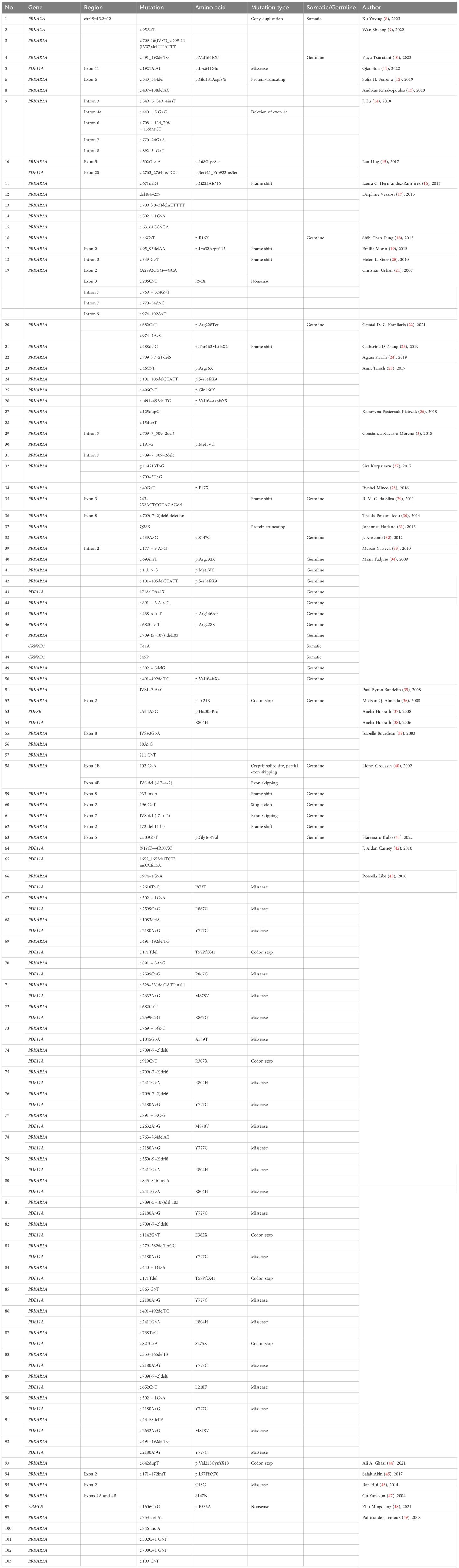
Table 4 Mutations in patients with PPNAD.
The family genetic history was recorded in 30 cases; the pathogenic variants of 10 cases (10/30, 33.33%) were from their fathers, those of 13 cases (13/30, 43.33%) were from their mothers, and those of the remaining 7 cases were not from their parents. Among the 21 cases with mutated PRKAR1A gene, 11 (11/21, 52.38%) were inherited from the mothers and 6 (6/21, 28.57%) were from the fathers. Among the six cases with mutated PDE11A gene, three (3/6, 50%) were inherited from the mothers and two (2/6, 33.33%) were inherited from the fathers. The geographic distribution of gene mutation is shown in Figure 2.
The proportion of spotty skin pigmentation with a typical distribution in patients with cPPNAD with or without PRKAR1A gene mutation is presented in Figure 3. Spotty skin pigmentation was presented in 33 patients (33/45, 73.33%) with PRKAR1A gene mutation. There was no patient who presented with spotty skin pigmentation without PRKAR1A gene mutation. There was significant correlation between PRKAR1A gene mutation and spotty skin pigmentation with a typical distribution in cPPNAD (p < 0.05).
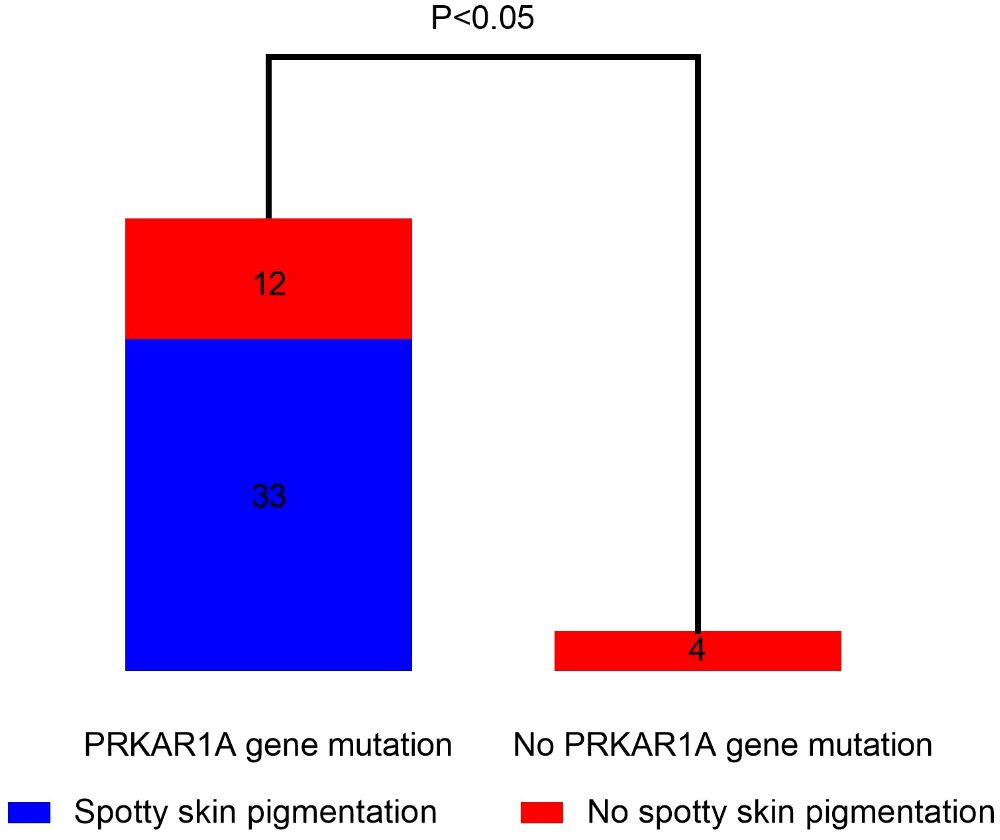
Figure 3 Correlation analysis of spotty skin pigmentation and PRKAR1A gene mutation in PPNAD patients with CNC.
The treatment regimens were recorded for 122 patients. A total of 62 patients (62/122, 50.82%) underwent bilateral adrenalectomy, 41 patients (41/122, 33.61%) underwent unilateral adrenalectomy, and 15 patients (15/122, 12.30%) underwent two-stage bilateral adrenalectomy (since unilateral adrenalectomy could not control the cortisol elevation, the other side of the adrenal gland was removed). Three patients (3/122, 2.46%) underwent unilateral total adrenalectomy and contralateral subtotal adrenalectomy, and one patient (1/122, 0.82%) underwent unilateral subtotal adrenalectomy.
Regarding the fertility status of four patients with PPNAD, notably, a 31-year-old woman with cPPNAD without adrenalectomy developed severe perinatal preeclampsia and gave birth to a baby girl with moderate respiratory distress syndrome (50). Moreover, a 28-year-old woman with CS secondary to cPPNAD had a successful pregnancy 3 years after bilateral adrenalectomy, but the perinatal period was not uneventful because of the need for hormone replacement (51). There were two women with PPNAD who had difficulty in pregnancy and gave birth to healthy babies after unilateral adrenalectomy with an uneventful perinatal period (31, 52).
Here are the following suggestions for choosing which side of the adrenal gland to remove. The side with more cortisol secretion with adrenal venous sampling (AVS) was selected (53, 54), the side with the larger volume with the use of 3D radiologic imaging volume analysis to compare the adrenal size was selected (55), the side with the highest uptake with 131-cholesterol removal radionuclide scan of adrenal gland was selected (56), and the side showing obvious unilateral adrenal nodules with CT or MRI scan was selected (56).
The present study is the first systemic review to investigate the different clinical features and pathogenic variants of PPNAD in 23 countries on six continents. Our study showed that most of the patients with PPNAD were between 10 and 30 years old (71.88%), with a female-to-male ratio of 2:1. Of the patients with PPNAD, 94.29% had osteoporosis or osteopenia. Moreover, almost all patients lost the cortisol circadian rhythm (98.59%), and both plasma and urinary cortisol cannot be suppressed in all patients tested with both low-dose or high-dose dexamethasone and Liddle test (100%). In terms of genetic mutations, six different gene mutations, PRKAR1A, PDE11A, PRKACA, CTNNB1(β-catenin), PDE8B, and ARMC5, were identified. In particular, there was significant correlation between PRKAR1A gene mutation and spotty skin pigmentation with a typical distribution in cPPNAD (p < 0.05). More than half of the patients underwent bilateral adrenalectomy, and 33.61% of the patients underwent unilateral adrenalectomy.
Abnormal bone metabolism and growth retardation are more common in PPNAD compared to other causes of CS (57, 58). In our study, the age of patients with growth retardation was 1 to 19 years old, which may be secondary to early onset and prolonged exposure to hypercortisolemia, impairing growth and delaying skeletal maturation. Moreover, PRKAR1A gene mutation might lead to abnormal osteoblast differentiation (59–61). The main reason for the unsatisfactory height after treatment is the lack of catch-up growth. To solve this problem, children with PPNAD should be tested for GH and IGF-1 in time after surgery. If GH is insufficient, the application of human GH is encouraged. Early treatment and a longer remaining growth period contribute to the achievement of ideal adult height (62). There are also four patients with acromegaly due to GH-producing pituitary adenoma in our study, which together form CNC. Thus, patients with PPNAD should be highly suspected of pituitary adenoma if there is an increase in GH.
In our study, we found a total of six different gene mutations, PRKAR1A, PRKACA, PDE11A, PDE8B, ARMC5, and CTNNB1 (β-catenin). Early studies had demonstrated that the development, proliferation, and function of adrenocortical cells are mainly regulated by the cyclic adenosine monophosphate protein kinase A (cAMP-PKA) pathway (63, 64). In particular, CTNNB1 (β-catenin) gene mutation only occurs in the greater adrenal tubercle, but as a somatic mutation, it may also be involved in secondary tumors based on primary hyperplasia (34, 40). Although PRKAR1A gene mutation is a supplemental criterion for CNC diagnosis, the diagnosis of CNC cannot be ruled out if PAKAR1A gene mutation is not detected. Genetic testing results of PRKAR1A, PRKACA, or PDE11A mutations are helpful for the diagnosis of CNC (11). I-PPNAD may be closely related to C. 709(-7–2) del6 or M1V mutation of PRKAR1A (65–67). As a genetic modifying factor for the development of testicular and adrenal tumors in patients with germline PRKAR1A mutation, PDE11A is probably a phenotype modifying gene but not a causative gene. Therefore, PDE11A mutation might indicate the occurrence of PPNAD and other types of adrenal tumors (42, 68). ARMC5 constitutional variant was reported “nonsense” in an Asian patient with PPNAD, but the P536A is a missense variant, and frequent especially in an Asian population (MAF = 0.002) (48). Some cases of corticotropin-independent macronodular adrenal hyperplasia often appear to be with inactivating mutations of ARMC5 (69, 70). Therefore, patients with PPNAD with ARMC5 gene mutation might combine with macronodular adrenal hyperplasia.
The study showed that the correlation between spotty skin pigmentation and PRKAR1A gene mutation was analyzed in patients with cPPNAD. The results showed that there was significant correlation between PRKAR1A gene mutation and spotty skin pigmentation in patients with cPPNAD. This is consistent with the findings of Jerome Bertherat et al. (71). PKA signaling promotes melanogenesis in melanocytes by phosphorylating CREB, which results in increased MITF transcription and subsequent increased expression of tyrosinase, TRP-1, and TRP-2. PRKAR1A deficiency produces large amounts of melanin and presents as darkly pigmented cutaneous papules or nodules (72). Therefore, the occurrence of spotty skin pigmentation in patients with CS should be highly suspicious of cPPNAD and PAKAR1A gene mutation.
Among all the patients included in this review, no mutated genes were reported in 19 cases. The reason is partly due to the limited gene sequence template selection, and the other is the somatic events. The tumor specimen might not obtain the corresponding tissue and was contaminated by the surrounding tissue. In case of somatic events, laser-captured micro-dissected cells can be used (36), and blood tissue can also be tested for genetic mutations to identify germline or somatic mutations.
Patients with PPNAD presented with an ACTH-independent form of CS, characterized by decreased or undetectable ACTH levels, elevated serum cortisol concentrations with loss of circadian rhythm, and paradoxical increase in UFC excretion after HDDST (Liddle test), with a maximum specificity of 100% (6, 39, 73, 74). Stratakis et al. found that all patients with PPNAD had a 100% or greater increase in urinary free cortisol excretion on day 6 of the Liddle test (6). The paradoxical rise in serum cortisol levels following Liddle’s test implied that glucocorticoids can locally regulate adrenocortical steroidogenesis in the majority of PPNAD. In at least some PPNAD tissues, aberrant coupling of the glucocorticoid receptor (GR) to the cAMP–PKA pathway instead of GR overexpression may be the culprit for the dexamethasone-induced rise in cortisol production (31). Different from the pathological features of isolated micronodular adrenocortical disease (i-MAD), PPNAD has small nodular staining and internodular atrophy, and the cortex was clearly segmented (75, 76). However, adrenal tissue acquisition was invasive, and a patient with a psychiatric disorder concealed a history of exogenous cortisol intake, while laboratory findings were consistent with PPNAD, leading physicians to misdiagnose and remove the normal adrenal gland (77). In the future, PPNAD gene mutation diagnosis due to blood might be the first choice for diagnosis and next-generation sequencing (NGS) should be preferred (22).
DHEA-S is a specific and stable marker of adrenal androgen secretion, which may also be a good predictor of risk for postoperative adrenal insufficiency, and ACTH plays an important role in its regulation (78, 79). Circulating DHEA-S levels are significantly reduced in patients with CS due to adrenocortical adenomas (78). However, in our study, patients with PPNAD can have a higher level of DHEA-S than normal, when combined with pituitary adenoma (21) or adrenal carcinoma (19). Therefore, if there is abnormal increase in DHEA-S, attention should be paid to whether it is complicated with other types of adrenal tumors and pituitary tumors.
Clinical management is also a complex issue to be discussed for patients with PPNAD. Xu et al. reported that 12 of 13 patients with PPNAD had clinical and laboratory remission of CS after unilateral adrenalectomy (56). Regarding the fertility status of four women with PPNAD, a woman without adrenalectomy developed severe perinatal preeclampsia and gave birth to a baby girl with moderate respiratory distress syndrome (50). Furthermore, a woman experienced a not uneventful perinatal period after bilateral adrenalectomy due to the need for hormone replacement (51). There were two women with PPNAD who gave birth to healthy babies after unilateral adrenalectomy with an uneventful perinatal period (31, 52). According to the included pregnant women, it is recommended that pre-pregnant women with PPNAD should undergo unilateral adrenalectomy to correct hypercortisolemia before pregnancy, and hydrocortisone can be given during delivery as a preventive measure (51, 52).
For patients who failed to effectively control elevated cortisol before or after adrenalectomy, ketoconazole, metyrapone, mitotane, and trilostane are effective in correcting hypercortisolemia through inhibition of steroidogenesis. Fluconazole has recently been proposed as a safer alternative to ketoconazole (80). After unilateral adrenalectomy, an individualized approach with close follow-up can lead to good clinical outcomes; dexamethasone stimulation test and adrenal MRI can be used for postoperative monitoring of PPNAD; if CS recurs during follow-up, contralateral adrenalectomy should be performed, followed by lifelong glucocorticoid therapy (56).
Among patients with cPPNAD, 28.79% were associated with cardiac or cutaneous myxoma, which is the most lethal manifestation of CNC and requires vigilant preoperative examination by a cardiologist and careful postoperative follow-up. Myxomas alter valve function through outflow obstruction and valve growth and pose an embolic threat to the brain and other organs (81). The incidence of embolism was 18% in the isolated atrial myxoma group and 40% in the recurrent myxoma group; CNC should be considered in all patients with cardiac myxoma; CNC is more common in patients with recurrent cardiac myxomas, and often involves two or more cavities. Testing for mutations in patients with isolated myxomas or multiple myxomas at atypical sites and screening for mutations in their immediate relatives may help establish an early diagnosis of the disease and implement appropriate clinical follow-up to detect recurrence in these patients (82).
PPNAD is the most common endocrine tumor in CNC, and in order to detect other complications of CNC in time, especially life-threatening cardiac myxoma, pediatric patients should have an annual follow-up examination, which should include echocardiography, thyroid ultrasound, and pituitary function examination, and twice a year if myxoma is found. Oral glucose tolerance, thyroid hormone release, and pituitary function tests can detect growth-hormone-producing pituitary adenomas early in children before clinical symptoms (such as acromegaly) appear. Adolescent patients should also be closely monitored for abnormal changes in growth rate and pubertal status caused by large cell calcifying Sertoli cell tumors (LCCSCT) (21, 83, 84). In addition, testicular ultrasound examination is recommended for men, and abdominal and pelvic ultrasound examination and breast imaging are recommended for female patients (1, 85). One patient had a cPPNAD with bilateral papillary thyroid carcinoma occurring 11 years apart. Thus, follow-up means decades (86).
Our study has several limitations. Firstly, because of the long incubation period of comorbidities in CNC, there may be bias in the diagnosis of CNC in some patients who do not yet have comorbidities. Secondly, the limited number of pregnant patients treated by unilateral adrenalectomy (n:2) is another limitation. Although the number of pregnant women included is small, this article included pregnant women with bilateral adrenalectomy, unilateral adrenalectomy, and no surgery, to comprehensively understand the conditions of the perinatal. Thirdly, when analyzing the characteristics of patients with PPNAD, there are cases with insufficient clinical information, which prevented us from analyzing the characteristics of some rare mutations. Furthermore, there may be a selection bias in this study because many patients with PPNAD did not undergo genetic testing. The mechanisms by which different mutations lead to different clinical features and whether the mutated gene detected is the pathogenic gene of PPNAD remain unclear. Further studies are needed to explain the molecular mechanism of PPNAD more precisely.
Our systematic review, despite the above limitations, is, to our knowledge, the most comprehensive review on patients with PPNAD published to date and provides clinicians with vital information on the common presentation features that may help with the diagnosis and highlight management options. In particular, our study analyzed special groups of pregnant women and children, summarized treatment options and prognosis from the included patients, and summarized treatment options for these groups. Moreover, laboratory tests were also analyzed in detail, and patient tests for PPNAD with other endocrine tumors were analyzed. This study contributes to a comprehensive understanding of PPNAD, including clinical manifestations, laboratory findings, treatment, prognosis, and follow-up.
In conclusion, our study is the first systematic review to investigate the different clinical features, pathogenic variants, and treatments of PPNAD. For the young patients with CS, especially female patients with growth retardation, spotty skin pigmentation, and osteoporosis/low bone mineral density, PPNAD should be considered. For treatment, unilateral adrenalectomy is recommended, especially in women who are preparing for pregnancy. In view of the trauma and difficulty of pathology in PPNAD diagnosis, genetic testing before surgery might be a reasonable option. Patients with PPNAD with spotty skin pigmentation should consider PRKAR1A gene mutation and pay attention to CNC. In the future, both short- and long-term evaluations of the complications of PPNAD need to be carried out.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
JS: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. LD: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. LH: Data curation, Writing – original draft. HF: Data curation, Writing – original draft. RL: Writing – original draft. JF: Writing – original draft. JD: Writing – review & editing. LL: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by National Natural Science Foundation of China Grants (No. 82170847).
We thank all participants who provided help in the process of conducting this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. (2001) 86:4041–6. doi: 10.1210/jcem.86.9.7903
2. Cazabat L, Ragazzon B, Groussin L, Bertherat J. PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary. (2006) 9(3):211–9. doi: 10.1007/s11102-006-0266-1
3. Navarro Moreno C, Delestienne A, Marbaix E, Aydin S, Hörtnagel K, Lechner S, et al. Familial forms of Cushing syndrome in primary pigmented nodular adrenocortical disease presenting with short stature and insidious symptoms: A clinical series. Horm Res Paediatr. (2018) 89(6):423–33. doi: 10.1159/000488761
4. Jarial KDS, Walia R, Nahar U, Bhansali A. Primary bilateral adrenal nodular disease with Cushing's syndrome: varying aetiology. BMJ Case Rep. (2017) 2017:bcr-2017-220154. doi: 10.1136/bcr-2017-220154
5. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol. (2022) 33(1):155–96. doi: 10.1007/s12022-022-09710-8
6. Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. (1999) 131(8):585–91. doi: 10.7326/0003-4819-131-8-199910190-00006
7. George J, Vimal MV, Bandgar T, Menon PS, Shah NS. An unusual variant of Cushing syndrome. Endocr Pract. (2008) 14:717–20. doi: 10.4158/EP.14.6.717
8. Xu YY, Li YY, Chen QL, Ma HM, Zhang J, Guo S. [A case of primary pigmented nodular adrenocortical disease caused by somatic variation of the PRKACA gene]. Zhonghua Er Ke Za Zhi. (2023) 61(1):76–8. doi: 10.3760/cma.j.cn112140-20220626-00589
9. Wan S, Zhang TT, Chen T, Zhang D, Mo D, Xu J, et al. Primary pigmented nodular adrenal disease: a report of three cases. Zhonghua nei ke za zhi. (2022) 61(8):944–7. doi: 10.3760/cma.j.cn112138-20211031-00760
10. Tsurutani Y, Kiriyama K, Kondo M, Hasebe M, Sata A, Mizuno Y, et al. Carney Complex Complicated with Primary Pigmented Nodular Adrenocortical Disease without Cushing's Syndrome Recurrence for Five Years after Unilateral Adrenalectomy. Intern Med. (2022) 61(2):205–11. doi: 10.2169/internalmedicine.7418-21
11. Sun Q, Song J, Feng W, Wang C, Yang X, Zhang M, et al. Carney complex presenting as subclinical Cushing syndrome in a child due to a novel Phosphodiesterase 11A mutation. Heliyon. (2022) 8(12):e12077. doi: 10.1016/j.heliyon.2022.e12077
12. Ferreira SH, Costa MM, Rios E, Santos Silva R, Costa C, Castro-Correia C, et al. Carney complex due to a novel pathogenic variant in the PRKAR1A gene - a case report. J Pediatr Endocrinol Metab. (2019) 32(2):197–202. doi: 10.1515/jpem-2018-0199
13. Kiriakopoulos A, Linos D. Carney syndrome presented as a pathological spine fracture in a 35-year-old male. Am J Case Rep. (2018) 19(8):1366–9. doi: 10.12659/AJCR.911962
14. Fu J, Lai F, Chen Y, Wan X, Wei G, Li Y, et al. A novel splice site mutation of the PRKAR1A gene, C.440+5 G>C, in a Chinese family with Carney complex. J Endocrinol Invest. (2018) 41(8):909–17. doi: 10.1007/s40618-017-0817-5
15. Lan L, Zhang G, Deng W, Wang H, Ye L, Ning G. To report and analyze the gene mutation in a family with primary pigmented nodular adrenocortical disease. Shandong Med J. (2017) 57(26):86–8. doi: 10.3969/j.issn.1002-266X.2017.26.029.
16. Hernández-Ramírez LC, Tatsi C, Lodish MB, Faucz FR, Pankratz N, Chittiboina P, et al. Corticotropinoma as a component of carney complex. J Endocr Soc. (2017) 1(7):918–25. doi: 10.1210/js.2017-00231
17. Vezzosi D, Tenenbaum F, Cazabat L, Tissier F, Bienvenu M, Carrasco CA, et al. Hormonal, radiological, NP-59 scintigraphy, and pathological correlations in patients with Cushing's syndrome due to primary pigmented nodular adrenocortical disease (PPNAD). J Clin Endocrinol Metab. (2015) 100(11):4332–8. doi: 10.1210/jc.2015-2174
18. Tung SC, Hwang DY, Yang JW, Chen WJ, Lee CT. An unusual presentation of Carney complex with diffuse primary pigmented nodular adrenocortical disease on one adrenal gland and a nonpigmented adrenocortical adenoma and focal primary pigmented nodular adrenocortical disease on the other. Endocr J. (2012) 59(9):823–30. doi: 10.1507/endocrj.EJ12-0040
19. Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab. (2012) 97(2):E202–6. doi: 10.1210/jc.2011-2321
20. Storr HL, Metherell LA, Dias R, Savage MO, Rasmussen AK, Clark AJ, et al. Familial isolated primary pigmented nodular adrenocortical disease associated with a novel low penetrance PRKAR1A gene splice site mutation. Horm Res Paediatr. (2010) 73(2):115–9. doi: 10.1159/000277629
21. Urban C, Weinhäusel A, Fritsch P, Sovinz P, Weinhandl G, Lackner H, et al. Primary pigmented nodular adrenocortical disease (PPNAD) and pituitary adenoma in a boy with sporadic carney complex due to a novel, de novo paternal PRKAR1A mutation (R96X). J Pediatr Endocrinol Metab. (2007) 20(2):247–52. doi: 10.1515/JPEM.2007.20.2.247
22. Kamilaris CDC, Faucz FR, Andriessen VC, Nilubol N, Lee CR, Ahlman MA, et al. First somatic PRKAR1A defect associated with mosaicism for another PRKAR1A mutation in a patient with Cushing syndrome. J Endocr Soc. (2021) 5(4):bvab007. doi: 10.1210/jendso/bvab007
23. Zhang CD, Pichurin PN, Bobr A, Lyden ML, Young WF, Bancos I. Cushing syndrome: uncovering Carney complex due to novel PRKAR1A mutation. Endocrinol Diabetes Metab Case Rep. (2019) 2019:18–0150. doi: 10.1530/EDM-18-0150
24. Kyrilli A, Lytrivi M, Bouquegneau MS, Demetter P, Lucidi V, Garcia C, et al. Unilateral adrenalectomy could be a valid option for primary nodular adrenal disease: evidence from twins. J Endocr Soc. (2018) 3(1):129–34. doi: 10.1210/js.2018-00261
25. Tirosh A, Lodish MB, Lyssikatos C, Belyavskaya E, Papadakis GZ, Stratakis CA. Circadian plasma cortisol measurements reflect severity of hypercortisolemia in children with different etiologies of endogenous Cushing syndrome. Horm Res Paediatr. (2017) 87(5):295–300. doi: 10.1159/000464463
26. Pasternak-Pietrzak K, Stratakis CA, Moszczyńska E, Lecka-Ambroziak A, Staniszewski M, Watrobińska U, et al. Detection of new potentially pathogenic mutations in two patients with primary pigmented nodular adrenocortical disease (PPNAD) - case reports with literature review. Endokrynol Pol. (2018) 69(6):675–81. doi: 10.5603/EP.a2018.0063
27. Korpaisarn S, Trachoo O, Panthan B, Aroonroch R, Suvikapakornkul R, Sriphrapradang C. A novel PRKAR1A mutation identified in a patient with isolated primary pigmented nodular adrenocortical disease. Case Rep Oncol. (2017) 10(2):769–76. doi: 10.1159/000479585
28. Mineo R, Tamba S, Yamada Y, Okita T, Kawachi Y, Mori R, et al. A Novel Mutation in the type Iα Regulatory Subunit of Protein Kinase A (PRKAR1A) in a Cushing's Syndrome Patient with Primary Pigmented Nodular Adrenocortical Disease. Intern Med. (2016) 55(17):2433–8. doi: 10.2169/internalmedicine.55.6605
29. da Silva RM, Pinto E, Goldman SM, Andreoni C, Vieira TC, Abucham J. Children with Cushing's syndrome: Primary Pigmented Nodular Adrenocortical Disease should always be suspected. Pituitary. (2011) 14(1):61–7. doi: 10.1007/s11102-010-0260-5
30. Poukoulidou T, Maiter D, Bertherat J, Beauloye V. A rare case of familial Cushing's syndrome with a common presentation of weight gain due to a mutation of the PRKAR1A gene causing isolated primary pigmented nodular adrenocortical disease. J Pediatr Endocrinol Metab. (2014) 27(9-10):1005–9. doi: 10.1515/jpem-2014-0018
31. Hofland J, de Herder WW, Derks L, Hofland LJ, van Koetsveld PM, de Krijger RR, et al. Regulation of steroidogenesis in a primary pigmented nodular adrenocortical disease-associated adenoma leading to virilization and subclinical Cushing's syndrome. Eur J Endocrinol. (2012) 168(1):67–74. doi: 10.1530/EJE-12-0594
32. Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, et al. A large family with carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab. (2012) 97(2):351–9. doi: 10.1210/jc.2011-2244
33. Peck MC, Visser BC, Norton JA, Pasche L, Katznelson L. A novel PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease and the Carney complex. Endocr Pract. (2010) 16(2):198–204. doi: 10.4158/EP09245
34. Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf). (2008) 69(3):367–73. doi: 10.1111/j.1365-2265.2008.03273.x
35. Bandelin PB, Moreno AJ, Lemar HJ, Stratakis CA, Oliver TG. The use of positron emission tomography-computed tomography scan in the evaluation of a patient with carney complex. J Clin Endocrinol Metab. (2008) 93(8):2946–7. doi: 10.1210/jc.2008-0313
36. Almeida MQ, Brito LP, Domenice S, Costa MH, Pinto EM, Osório CA, et al. Absence of PRKAR1A loss of heterozygosity in laser-captured microdissected pigmented nodular adrenocortical tissue from a patient with carney complex caused by the novel nonsense mutation p.Y21X. Arq Bras Endocrinol Metabol. (2008) 52(8):1257–63. doi: 10.1590/S0004-27302008000800009
37. Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med. (2008) 358:750–2. doi: 10.1056/NEJMc0706182
38. Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, et al. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res. (2006) 66(24):11571–5. doi: 10.1158/0008-5472.CAN-06-2914
39. Bourdeau I, Lacroix A, Schürch W, Caron P, Antakly T, Stratakis CA. Primary Pigmented nodular adrenocortical disease: Paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. (2003) 88(8):3931–7. doi: 10.1210/jc.2002-022001
40. Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, et al. Mutations of the PRKAR1A gene in Cushing's syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab. (2002) 87(9):4324–9. doi: 10.1210/jc.2002-020592
41. Kubo H, Tsurutani Y, Sugisawa C, Sunouchi T, Hirose R, Saito J. Phenotypic variability in a family with carney complex accompanied by a novel mutation involving PRKAR1A. Tohoku J Exp Med. (2022) 257(4):337–45. doi: 10.1620/tjem.2022.J051
42. Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol. (2010) 34(4):547–55. doi: 10.1097/PAS.0b013e3181d31f49
43. Libé R, Horvath A, Fratticci A, Vezzosi D, Coste J, Guillaud-Bataille M, et al. ESE Young Investigator Award Frequent phosphodiesterase 11a (pde11a4) gene mutations in patients with carney complex (cnc) due to prkar1a mutations and adrenal (ppnad) and Sertoli cell tumors (lccsct): A digenic disorder? Endocrine Abstracts. (2010) 22:H1.3. doi: 10.1210/jc.2010-1704
44. Ghazi AA, Mandegar MH, Abazari M, Behzadnia N, Sadeghian T, Torbaghan SS, et al. A novel mutation in PRKAR1A gene in a patient with Carney complex presenting with pituitary macroadenoma, acromegaly, Cushing's syndrome and recurrent atrial myxoma. Arch Endocrinol Metab. (2021) 65(3):376–80. doi: 10.20945/2359-3997000000369
45. Akin S, Noyan S, Dagdelen S, Pasaoglu I, Kaynaroglu V, Askun MM, et al. Unusual presentations of Carney Complex in patient with a novel PRKAR1A mutation. Neuroendocrinol Lett. (2017) 38(4):248–54.
46. Ran H, Ma X, Wang Q, Xie Z, Ding Y, Qin G. A pedigree study of a patient with primary pigmented nodular adrenocortical disease and familial gene mutation. Zhonghua nei ke za zhi. (2014) 53(5):398–402. doi: 10.3760/cma.j.issn.0578-1426.2014.05.015
47. Gu Y, Chen Y, Song H, Li X, Luo T, Qiao J, et al. Clinical and molecular research in a case of familial Carney complex. Zhonghua nei ke za zhi. (2004) 43:764–8.
48. Zhu M, Dong G, Huang K, Chen X, Zhang L, Dai Y, et al. Central precocious puberty with primary pigmented nodular adrenocortical disease:One case report. Chin J Endocrinol Metab. (2021) 37:240–4. doi: 10.3760/cma.j.cn311282-20200212-00057
49. de Cremoux P, Rosenberg D, Goussard J, Brémont-Weil C, Tissier F, Tran-Perennou C, et al. Expression of progesterone and estradiol receptors in normal adrenal cortex, adrenocortical tumors, and primary pigmented nodular adrenocortical disease. Endocrine-Related Cancer. (2008) 15(2):465–74. doi: 10.1677/ERC-07-0081
50. Schulz S, Redlich A, Köppe I, Reschke K, Weise W. Carney complex - An unexpected finding during puerperium. Gynecol Obstet Invest. (2001) 51(3):211–3. doi: 10.1159/000052927
51. Kalelioglu I, Mert M, Has R, Kale T, Iyibozkurt C, Aral F. Carney's complex: A successful pregnancy after bilateral adrenalectomy. Arch Med Sci. (2012) 8(1):175–7. doi: 10.5114/aoms.2012.27300
52. Cohen O, Bogat S, Dolitzki M, Karasik A. Successful pregnancy after unilateral adrenalectomy in a case of primary pigmented adrenocortical disease. J Matern Fetal Neonatal Med. (2005) 17(2):161–3. doi: 10.1080/14767050500043210
53. Peng X, Yu Y, Ding Y, Yang F, Chen X, Chang C. Adrenal venous sampling as used in a patient with primary pigmented nodular adrenocortical disease. Transl Cancer Res. (2017) 6(6):1117–22. doi: 10.21037/tcr.2017.12.03
54. Hackman KL, Davis AL, Curnow PA, Serpell JW, McLean CA, Topliss DJ. Cushing syndrome in a young woman due to primary pigmented nodular adrenal disease. Endocr Pract. (2010) 16(1):84–8. doi: 10.4158/EP09177.CR
55. Chrysostomou PP, Lodish MB, Turkbey EB, Papadakis GZ, Stratakis CA. Use of 3-dimensional volumetric modeling of adrenal gland size in patients with primary pigmented nodular adrenocortical disease. Horm Metab Res. (2016) 48(4):242–6. doi: 10.1055/s-0042-103686
56. Xu Y, Rui W, Qi Y, Zhang C, Zhao J, Wang X, et al. The role of unilateral adrenalectomy in corticotropin-independent bilateral adrenocortical hyperplasias. World J Surg. (2013) 37(7):1626–32. doi: 10.1007/s00268-013-2059-9
57. Chen S, Li R, Lu L, Duan L, Zhang X, Tong A, et al. Efficacy of dexamethasone suppression test during the diagnosis of primary pigmented nodular adrenocortical disease in Chinese adrenocorticotropic hormone-independent Cushing syndrome. Endocrine. (2018) 59(1):183–90. doi: 10.1007/s12020-017-1436-9
58. Zhou J, Zhang M, Bai X, Cui S, Pang C, Lu L, et al. Demographic characteristics, etiology, and comorbidities of patients with Cushing's syndrome: A 10-year retrospective study at a large general hospital in China. Int J Endocrinol. (2019) 2019:7159696. doi: 10.1155/2019/7159696
59. Savage MO, Scommegna S, Carroll PV, Ho JT, Monson JP, Besser GM, et al. Growth in disorders of adrenal hyperfunction. Horm Res. (2002) 58 Suppl 1:39–43. doi: 10.1159/000064767
60. Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous Cushing's syndrome. J Clin Endocrinol Metab. (1994) 79:1082–5. doi: 10.1210/jcem.79.4.7962277
61. Maillet M, Bourdeau I, Lacroix A. Update on primary micronodular bilateral adrenocortical diseases. Curr Opin Endocrinol Diabetes Obes. (2020) 27:132–9. doi: 10.1097/MED.0000000000000538
62. Lebrethon MC, Grossman AB, Afshar F, Plowman PN, Besser GM, Savage MO. Linear growth and final height after treatment for Cushing's disease in childhood. J Clin Endocrinol Metab. (2000) 85(9):3262–5. doi: 10.1210/jcem.85.9.6817
63. Lodish M, Stratakis CA. A genetic and molecular update on adrenocortical causes of Cushing syndrome. Nat Rev Endocrinol. (2016) 12:255–62. doi: 10.1038/nrendo.2016.24
64. Mazzuco TL, Durand J, Chapman A, Crespigio J, Bourdeau I. Genetic aspects of adrenocortical tumours and hyperplasias. Clin Endocrinol (Oxf). (2012) 77(1):1–10. doi: 10.1111/j.1365-2265.2012.04403.x
65. Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat. (2010) 31(4):369–79. doi: 10.1002/humu.21178
66. Pereira AM, Hes FJ, Horvath A, Woortman S, Greene E, Bimpaki E, et al. Association of the M1V PRKAR1A mutation with primary pigmented nodular adrenocortical disease in two large families. J Clin Endocrinol Metab. (2010) 95(1):338–42. doi: 10.1210/jc.2009-0993
67. Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab. (2006) 91(5):1943–9. doi: 10.1210/jc.2005-2708
68. Libé R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab. (2011) 96(1):E208–14. doi: 10.1210/jc.2010-1704
69. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med. (2013) 369(22):2105–14. doi: 10.1056/NEJMoa1304603
70. Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab. (2014) 99(6):E1113–9. doi: 10.1210/jc.2013-4280
71. Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. (2009) 94(6):2085–91. doi: 10.1210/jc.2008-2333
72. Cohen JN, Joseph NM, North JP, Onodera C, Zembowicz A, LeBoit PE. Genomic analysis of pigmented epithelioid melanocytomas reveals recurrent alterations in PRKAR1A, and PRKCA genes. Am J Surg Pathol. (2017) 41(10):1333–46. doi: 10.1097/PAS.0000000000000902
73. Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé R, Cabrol S, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab. (2009) 94(7):2406–13. doi: 10.1210/jc.2009-0031
74. Tizianel I, Barbot M, Ceccato F. Subtyping of Cushing's syndrome: a step ahead. Exp Clin Endocrinol Diabetes. (2024). doi: 10.1055/a-2299-5065
75. Shenoy BV, Carpenter PC, Carney JA. Bilateral primary pigmented nodular adrenocortical disease. Rare cause of the Cushing syndrome. Am J Surg Pathol. (1984) 8:335–44. doi: 10.1097/00000478-198405000-00002
76. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing's syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. (2007) 3:748–57. doi: 10.1038/ncpendmet0648
77. Pineyro MM, Redes L, De Mattos S, Sanchez L, Brignardello E, Bianchi V, et al. Factitious Cushing's syndrome: A diagnosis to consider when evaluating hypercortisolism. Front Endocrinol (Lausanne). (2019) 10:129. doi: 10.3389/fendo.2019.00129
78. Yamaji T, Ishibashi M, Sekihara H, Itabashi A, Yanaihara T. Serum dehydroepiandrosterone sulfate in Cushing's syndrome. J Clin Endocrinol Metab. (1984) 59(6):1164–8. doi: 10.1210/jcem-59-6-1164
79. Morio H, Terano T, Yamamoto K, Tomizuka T, Oeda T, Saito Y, et al. Serum levels of dehydroepiandrosterone sulfate in patients with asymptomatic cortisol producing adrenal adenoma: comparison with adrenal Cushing's syndrome and non-functional adrenal tumor. Endocr J. (1996) 43(4):387–96. doi: 10.1507/endocrj.43.387
80. Riedl M, Maier C, Zettinig G, Nowotny P, Schima W, Luger A. Long term control of hypercortisolism with fluconazole: case report and in vitro studies. Eur J Endocrinol. (2006) 154(4):519–24. doi: 10.1530/eje.1.02120
81. Rothschild JA, Kreso M, Slodzinski M. Sudden death in a patient with carney's complex. Anesth Pain Med. (2013) 2:182–5. doi: 10.5812/aapm
82. Vargas-Barrón J, Vargas-Alarcón G, Roldán F, Vázquez-Antona C, Vásquez Ortiz Z, Erdmenger-Orellana J, et al. Cardiac Myxomas and the Carney Complex. Revista Española de Cardiolog a (English Edition) (2008) 61(11):1205–9. doi: 10.1157/13127852
83. Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. (2006) 1:21. doi: 10.1186/1750-1172-1-21
84. Espiard S, Vantyghem MC, Assié G, Cardot-Bauters C, Raverot G, Brucker-Davis F, et al. Frequency and incidence of carney complex manifestations: A prospective multicenter study with a three-year follow-up. J Clin Endocrinol Metab. (2020) 105(3):dgaa002. doi: 10.1210/clinem/dgaa002
85. Stratakis CA, Ball DW. A concise genetic and clinical guide to multiple endocrine neoplasias and related syndromes. J Pediatr Endocrinol Metab. (2000) 13:457–65. doi: 10.1515/JPEM.2000.13.5.457
Keywords: PRKAR1A, Cushing’s syndrome, clinical characteristics, pathogenic variants, primary pigmented nodular adrenocortical disease
Citation: Sun J, Ding L, He L, Fu H, Li R, Feng J, Dong J and Liao L (2024) The clinical characteristics and pathogenic variants of primary pigmented nodular adrenocortical disease in 210 patients: a systematic review. Front. Endocrinol. 15:1356870. doi: 10.3389/fendo.2024.1356870
Received: 16 December 2023; Accepted: 31 May 2024;
Published: 26 June 2024.
Edited by:
Mirko Parasiliti-Caprino, University of Turin, ItalyReviewed by:
Valentina Morelli, Istituto Auxologico Italiano, ItalyCopyright © 2024 Sun, Ding, He, Fu, Li, Feng, Dong and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianjun Dong, dongjianjun@sdu.edu.cn; Lin Liao, liaolin@sdfmu.edu.cn
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.