 Zehra Yavas Abalı
Zehra Yavas Abalı Tulay Guran
Tulay Guran- Department of Pediatric Endocrinology and Diabetes, School of Medicine, Marmara University, Istanbul, Türkiye
Prenatal-onset androgen excess leads to abnormal sexual development in 46,XX individuals. This androgen excess can be caused endogenously by the adrenals or gonads or by exposure to exogenous androgens. The most common cause of 46,XX disorders/differences in sex development (DSD) is congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, comprising >90% of 46,XX DSD cases. Deficiencies of 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, and P450-oxidoreductase (POR) are rare types of CAH, resulting in 46,XX DSD. In all CAH forms, patients have normal ovarian development. The molecular genetic causes of 46,XX DSD, besides CAH, are uncommon. These etiologies include primary glucocorticoid resistance (PGCR) and aromatase deficiency with normal ovarian development. Additionally, 46,XX gonads can differentiate into testes, causing 46,XX testicular (T) DSD or a coexistence of ovarian and testicular tissue, defined as 46,XX ovotesticular (OT)-DSD. PGCR is caused by inactivating variants in NR3C1, resulting in glucocorticoid insensitivity and the signs of mineralocorticoid and androgen excess. Pathogenic variants in the CYP19A1 gene lead to aromatase deficiency, causing androgen excess. Many genes are involved in the mechanisms of gonadal development, and genes associated with 46,XX T/OT-DSD include translocations of the SRY; copy number variants in NR2F2, NR0B1, SOX3, SOX9, SOX10, and FGF9, and sequence variants in NR5A1, NR2F2, RSPO1, SOX9, WNT2B, WNT4, and WT1. Progress in cytogenetic and molecular genetic techniques has significantly improved our understanding of the etiology of non-CAH 46,XX DSD. Nonetheless, uncertainties about gonadal function and gender outcomes may make the management of these conditions challenging. This review explores the intricate landscape of diagnosing and managing these conditions, shedding light on the unique aspects that distinguish them from other types of DSD.
Introduction
Disorders/differences in sex development (DSD) refer to conditions due to the discrepant development of chromosomal, gonadal, and phenotypic sex (1). Androgen overproduction caused by abnormalities in the adrenal cortex and gonads or exposure to androgens from an ectopic/exogenous source may affect the normal sexual development of the individual (2). Fetal exposure to androgens causes DSD in an individual with 46,XX chromosomes and may be diagnosed with ambiguous genitalia in the newborn (3).
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is the most common etiology of 46,XX DSD, and other rare forms of CAH causing 46,XX DSD include 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, and P450-oxidoreductase (POR) deficiency (1).
Non-CAH 46,XX DSD are categorized as follows: (1) disorders with excessive amounts of endogenous androgens, such as primary glucocorticoid resistance (PGCR) and aromatase deficiency; (2) increased exogenous androgen exposure, such as gestational hyperandrogenism; (3) disorders/differences of gonadal differentiation [testicular (T)/ovotesticular (OT)-DSD, ovarian dysgenesis (OD)]; (4) others, classified as Mayer–Rokitansky–Küster–Hauser syndrome (MRKHS) (types I and II) and variants—complex syndromic disorders like cloacal exstrophy (4) (Table 1).
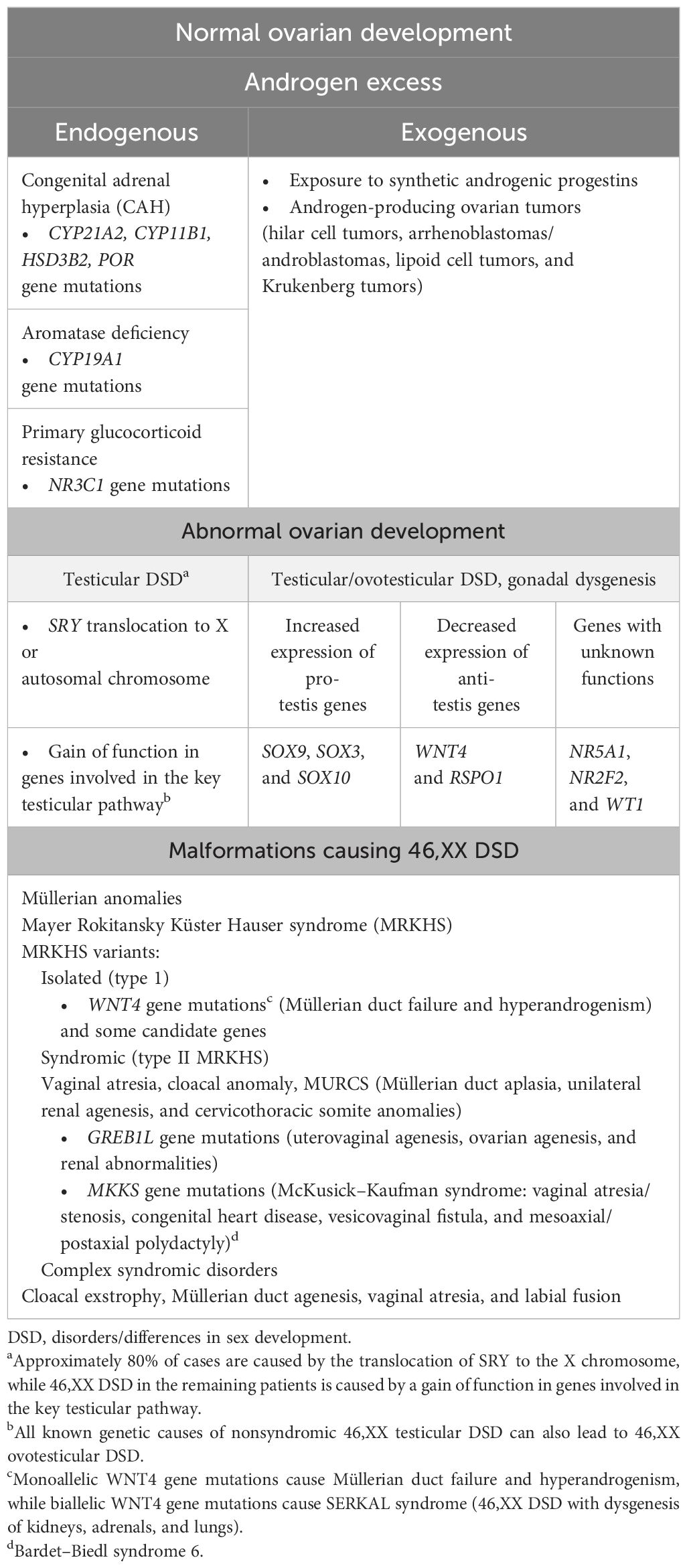
Table 1 Etiologies of 46,XX DSD.
Differential diagnosis of these conditions and identification of the underlying etiology may affect the patient’s management and long-term prognosis (Figure 1).
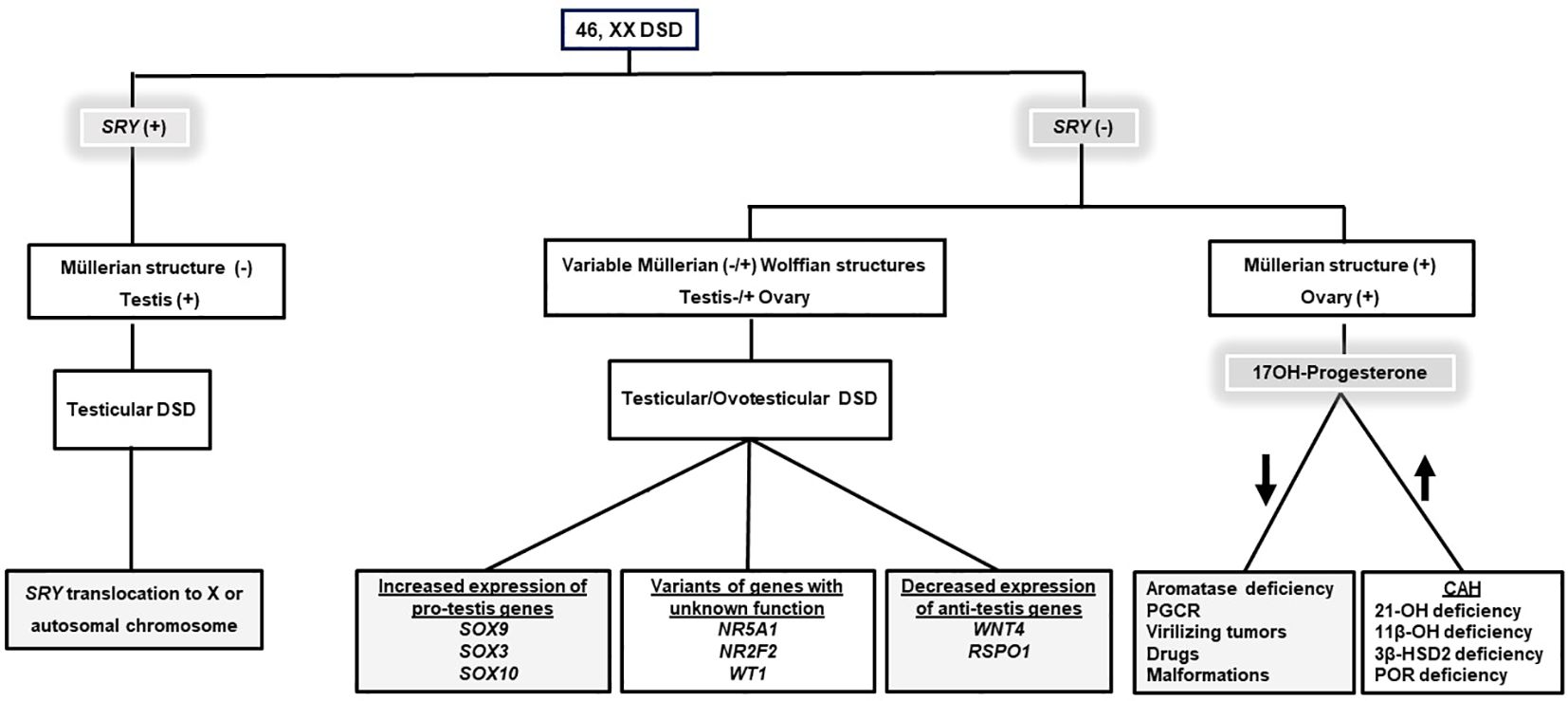
Figure 1 An algorithm for the differential diagnosis of 46, XX DSD. Gray boxes indicate the etiologies causing non-CAH 46, XX DSD. Information on SRY gene expression and 170H-Progesterone concentration is essential in the differential diagnosis of 46, XX DSD. DSD, Disorders/Differences in sex development; CAH, Congenital adrenal hyperplasia; 21-OH deficiency, 21α-Hydroxylase deficiency; 11β-OH deficiency, 11β-Hydroxylase deficiency; 3β-HSD2 deficiency, 3β-Hydroxysteroid dehydrogenase type 2 deficiency; POR deficiency P450 oxidoreductase deficiency, PGCR; Primary glucocorticoid resistance.
In this chapter, we aim to emphasize the distinctive features of non-CAH 46,XX DSD and the challenges in the management of these conditions.
Androgen excess in 46,XX with normal ovarian development
Primary glucocorticoid resistance
Glucocorticoids (GCs) are synthesized in the zona fasciculata of the adrenal gland and function as the end products of the stress-responsive hypothalamic–pituitary–adrenal axis. They play an important role in both basal physiology and stress response (5). The effects of GCs are controlled by the glucocorticoid receptor (GR) (nuclear receptor subfamily 3, group C, member 1, NR3C1, MIM* 138040). GR is an intracellular receptor, a member of the steroid/sterol/thyroid/retinoid/orphan receptor superfamily, and it is widely distributed in various tissues (6). After binding to its ligand, this receptor communicates from the cytoplasm to the nucleus, and it controls the transcription rate of GC-responsive genes here (Figure 2A). Upon ligand-dependent activation of GR, these genes are influenced directly or indirectly (7).
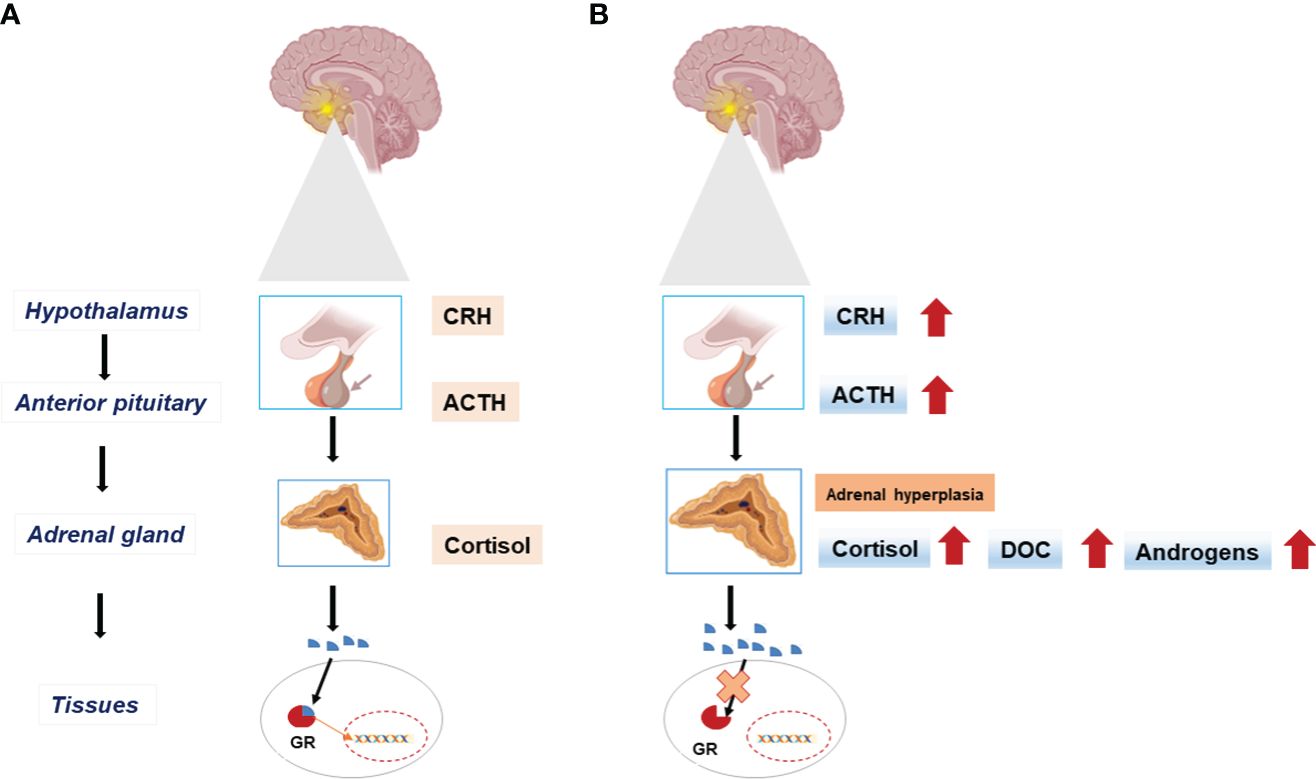
Figure 2 The hypothalamic-pituitary-adrenal (HPA) axis in physiologic state (A) and in Primary Glucocorticoid Resistance (B). End-organ insensitivity to glucocorticoids (Cortisol) and impaired feedback mechanisms result in excess adrenocorticotrophic hormone (ACTH) secretion with increased circulating cortisol concentration. Excess ACTH leads to adrenal cortex hyperplasia and activates the synthesis of mineralocorticoids (DOC), and androgens. CRH, Corticotropin-releasing hormone; GR, Glucocorticoid receptor; DOC, Deoxycorticosterone.
GCs also have an important role in the treatment of inflammatory, autoimmune, lymphoproliferative, and allergic diseases. The pathologic or therapeutic effects of the GR, including genetic alterations in the human GR (hGR) gene, the development of GR ligands with selective GR actions, and disease-associated GR regulatory molecules, are extremely important (8).
Primary glucocorticoid resistance (PGCR) occurs due to inactivating mutations on the hGR gene NR3C1 and causes systemic, partial GC insensitivity. Partial end-organ insensitivity to GCs and altered feedback mechanisms result in excess adrenocorticotrophic hormone (ACTH) production and thus increased cortisol concentrations (8, 9). Excess ACTH leads to adrenal cortex hypertrophy and increases mineralocorticoid (MC) and androgen synthesis (Figure 2B).
PGCR, also defined as Chrousos syndrome, is an extremely rare disease with a wide spectrum from asymptomatic to severe hyperandrogenism and MC excess (hypertension, hypokalemic alkalosis, and fatigue) (9). Acne, hirsutism, male pattern baldness, atypical genitalia, premature pubarche, precocious puberty, and subfertility may be observed due to hyperandrogenemia. PGCR may also cause irregular menstrual cycles and amenorrhea. The clinical findings of GC deficiency are subtle, like fatigue in childhood; however, growth retardation and hypoglycemia have also been reported (10, 11).
Plasma ACTH and serum cortisol concentrations are elevated in the majority of cases of PGCR. Hypertension and hypokalemia occur due to excess deoxycorticosterone (DOC) and increased cortisol availability at the MC receptor, causing a high ratio of cortisol concentration that overcomes the activity of 11β-hydroxysteroid dehydrogenase type 2 (HSD11B2). Most affected individuals present with hypertension, hypokalemia, and suppressed renin concentrations in childhood. The initial tests include the measurement of cortisol, androstenedione, testosterone, DHEAS, 11-deoxycortisol, and DOC concentrations (5, 12).
Although the circadian rhythm is maintained, it is primed to a high concentration in PGCR. The 24-h urinary cortisol (UFC) excretion is increased, and the serum concentrations of adrenal androgens (androstenedione, DHEA, and DHEA-S-) and MCs (DOC and corticosterone) are also increased due to compensatory ACTH elevation (8, 13).
A detailed medical history should be taken, and the signs of MC and/or androgen excess should be examined. Serum cortisol measurement and 24-h UFC excretion are required (two or three consecutive days). The HPA axis is resistant to dexamethasone suppression in PGCR; however, this may vary depending on the severity. Patients diagnosed with PGCR may respond to high doses of dexamethasone (14).
Differential diagnoses of PGCR include Cushing’s disease, pseudo-Cushing (depression and generalized anxiety disorder), conditions with increased cortisol-binding globulin (CBG) (pregnancy and estrogen treatment), hyperaldosteronism, essential hypertension, hyperandrogenism [polycystic ovary syndrome (PCOS), idiopathic hirsutism, and CAH] (12). Bone mineral density measurement is useful to differentiate PGCR from Cushing’s syndrome, and it is maintained in patients with GCR and increased in women due to androgen excess (15). In conditions with a clinical suspicion of PGCR, resistance can also be observed by the typical response of serum thyroid-stimulating hormone (TSH) to thyrotropin-releasing hormone (TRH) administration and/or the growth hormone response to insulin-induced hypoglycemia. These responses are compromised in Cushing’s disease (8).
The diagnosis of PGCR is confirmed by sequencing analysis of the NR3C1 gene (8). To date, 56 variants of NR3C1 have been described. Among these loss-of-function variants, 37 of them were missense, accompanied by frameshift and nonsense variants (16). Both biallelic and monoallelic variants that cause PGCR have been reported. A dominant negative effect on the wild-type GR is responsible for the effects of monoallelic variants causing GCR (8).
An estimate of the prevalence of NR3C1 variants in a cohort of patients with adrenal hyperplasia, hypertension, and/or increased cortisol despite the absence of Cushing’s syndrome was first provided by Vitellius et al. (17). A total of 5% of these patients had monoallelic NR3C1 variants. The variants [p.(R469*) and p.(R477S)] were identified in the DNA-binding domain, and the others [p.(R491*), p.(Q501H), and p.(L672P)] in the ligand-binding domain of hGR (17, 18). This prevalence rate indicates that many cases with NR3C1 variants may be present but not diagnosed. Therefore, clinicians should advise NR3C1 sequencing in patients with symptoms and signs indicative of PGCR (8).
A 3-year-old girl who was evaluated for hypertension, hypoglycemic seizures, and hypokalemia was reported by Tatsi et al. (19). The patient had findings of PGCR, and she was treated with antihypertensives, dexamethasone, and potassium. The heterozygous NR3C1 variant p.(R714Q) was detected in this case, and it was first reported by Nader et al. (11). At the age of nearly 12 years, the patient was using high-dose dexamethasone, and she had radiological findings of hypertensive encephalopathy. In the exome sequencing (ES) analysis, Tatsi et al. identified two heterozygous variants: the one previously published by Nader et al. and a novel p.(E198*) variant. This was the first published case of biallelic NR3C1 variants (compound heterozygous in the trans position) causing PGCR (11, 19).
In a Brazilian girl with clitoromegaly, urogenital sinus, and posterior labioscrotal fusion, a homozygous p.(V571A) variant was reported. This variant resulted in a marked reduction in receptor function. Although the phenotype might have been variable, severe GRα variants may manifest with mild 46,XX DSD (20). Next-generation sequencing (NGS) technologies have significantly contributed to the detection of pathogenic/likely pathogenic NR3C1 variants. Considerable progress has been made in understanding the mechanism of a specific genetic defect in NR3C1 that leads to a conformational change in hGRα (8). To date, no precise correlations between genotype and phenotype have been observed in PGCR (21).
Treatment of PGCR aims to reduce excess ACTH, suppress MCs, and/or regulate adrenal androgens. Supraphysiological doses of dexamethasone are used to elicit a physiological response in the presence of a poorly functioning receptor. Typically, the treatment starts with 0.25–0.5 mg/day and is adjusted gradually to suppress the ACTH, consequently decreasing androgens and controlling blood pressure. Depending on individual requirements, the dose of dexamethasone may be reduced. Hypertension may necessitate treatment with mineralocorticoid receptor antagonists (22). Thiazides and loop diuretics should be avoided due to the risk of hypokalemia. MC receptor antagonists can be used to manage hypertension, offering potential benefits owing to their anti-androgenic and potassium-sparing effects (8, 23).
Aromatase deficiency
Aromatase is a microsomal cytochrome P450 enzyme that catalyzes the conversion of C19 steroids (androgens) to C18 steroids (estrogens) (24). The androgenic precursors, androstenedione, testosterone, and 16-α-hydroxy dehydroepiandrosterone sulfate, were converted by aromatase to estrone (E1), estradiol (E2), and estriol (E3), respectively (25) (Figure 3).
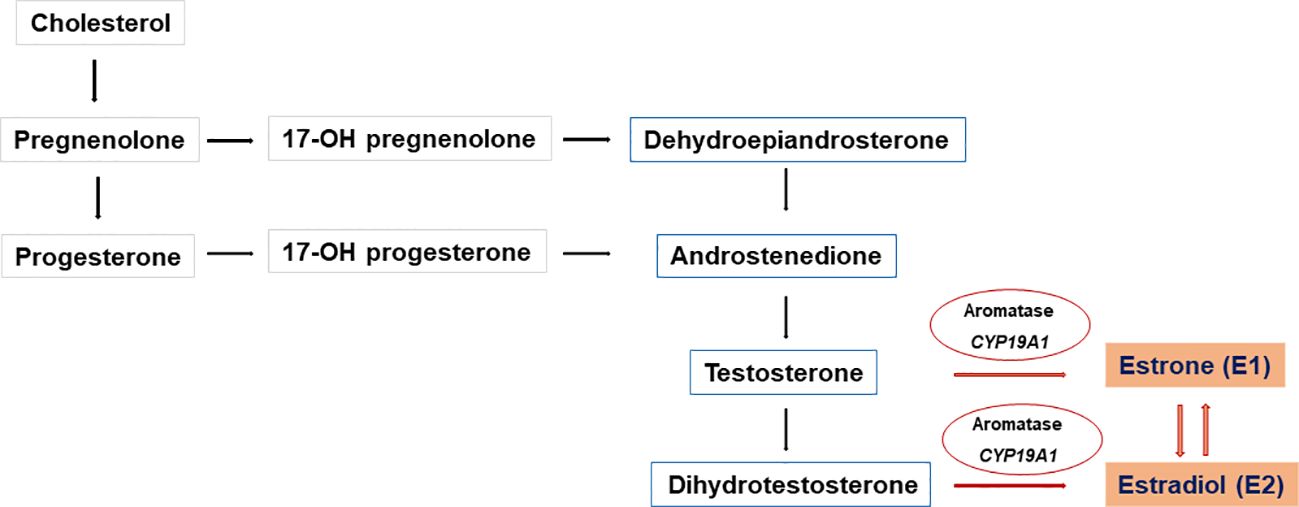
Figure 3 Biosynthesis of C18 steroids (estrogens). Aromatase (CYP19A1) converts C19 steroids (androgens) to C18 steroids (estrogens) Androstenedione, testosterone, and 16-α-hydroxy dehydroepiandrosterone sulfate are converted to estrone (E1), estradiol (E2), and estriol (E3), respectively E1 is converted to the biologically active E2 in target tissues by enzymatic processes with 17β-HSD activity.
Aromatase enzyme activity affects androgen metabolism and also influences the androgen–estrogen ratio in tissues (26). The aromatase enzyme is expressed in multiple tissues (ovary, placenta, adipose tissue, breast, brain, and bone). It is controlled by several tissue-specific promoters and has a role in the peripheral aromatization of androstenedione in adipocytes and skin fibroblasts (27). E1 has a weak estrogenic effect, and it is converted to estrone sulfate, which serves as a reserve for E1 in other tissues. E1 is converted to the active E2 in target tissues by enzymatic processes with reductive 17β-HSD activity. Androstenedione is the major substrate for aromatase activity in the target tissues. Aromatase has an important role in local estrogen production and estrogen synthesis from the ovary at the time of puberty (28).
Aromatase deficiency is caused by biallelic pathogenic/likely pathogenic variants in the CYP19A1 gene (MIM*107910). Loss of function of this gene results in excess testosterone production by an otherwise normal ovary. CYP19A1 is localized on chromosome 15q21.2 and consists of 10 exons (exon 1 has a role in tissue-specific gene expression) (26).
Shozu et al. were the first to describe aromatase deficiency, and subsequent studies determined that when the CYP19A1 gene was knocked out in female mice, a male body habitus, small/polycystic ovaries, a small uterus, and infertility were observed (29). To date, many cases with variable clinical presentations have been reported, and 95 variants have been reported in CYP19A1 (33 missense/nonsense) (30, 31).
Placental aromatase expression protects the mother from the virilizing effects of fetal adrenal androgens in physiologic pregnancy conditions. Disorders including POR deficiency, maternal androgen-producing tumors, and aromatase deficiency may cause virilization in the fetus and mother (32). In cases of aromatase deficiency, the placenta is unable to synthesize estrogen and large amounts of testosterone and androstenedione are transferred into the fetal and maternal circulation, causing virilization of the 46,XX fetus, and mother. This is also described as placental aromatase deficiency (29). Maternal virilization, such as increased hair growth, acne, and voice changes, becomes apparent after the second trimester of pregnancy, and generally, these symptoms resolve after the birth of the baby (32). However, it is worth noting that maternal virilization does not consistently manifest in every patient with aromatase deficiency (33, 34).
46,XX cases with aromatase deficiency may exhibit clitoromegaly, posterior fusion, scrotalization of the labioscrotal folds, and, in some infants, a urogenital sinus (26). Affected female individuals (46,XX) have Müllerian structures. In infancy, the histology of the ovaries is normal; however, due to FSH stimulation in aromatase deficiency, multiple, enlarged follicular cysts may be observed. At puberty, affected 46,XX patients have hypergonadotropic hypogonadism, they fail to develop female secondary sex characteristics, and exhibit progressive virilization. Plasma androstenedione and testosterone concentrations are elevated, with low or not measurable E1 and E2 concentrations. The ovaries enlarge and develop multiple cysts at puberty. Hypergonadotropic hypogonadism and the large multicystic ovaries respond to estrogen therapy, but treatment with an anti-androgen is required in some cases (32, 35).
Hypoplastic ovaries may also be observed in patients with aromatase deficiency. Therefore, hypergonadotropic hypogonadism in aromatase deficiency may develop secondary to impaired estrogen biosynthesis or hypoplastic ovaries (36–38). The clinical and molecular characteristics of aromatase deficiency in 46,XX patients are summarized in Table 2.
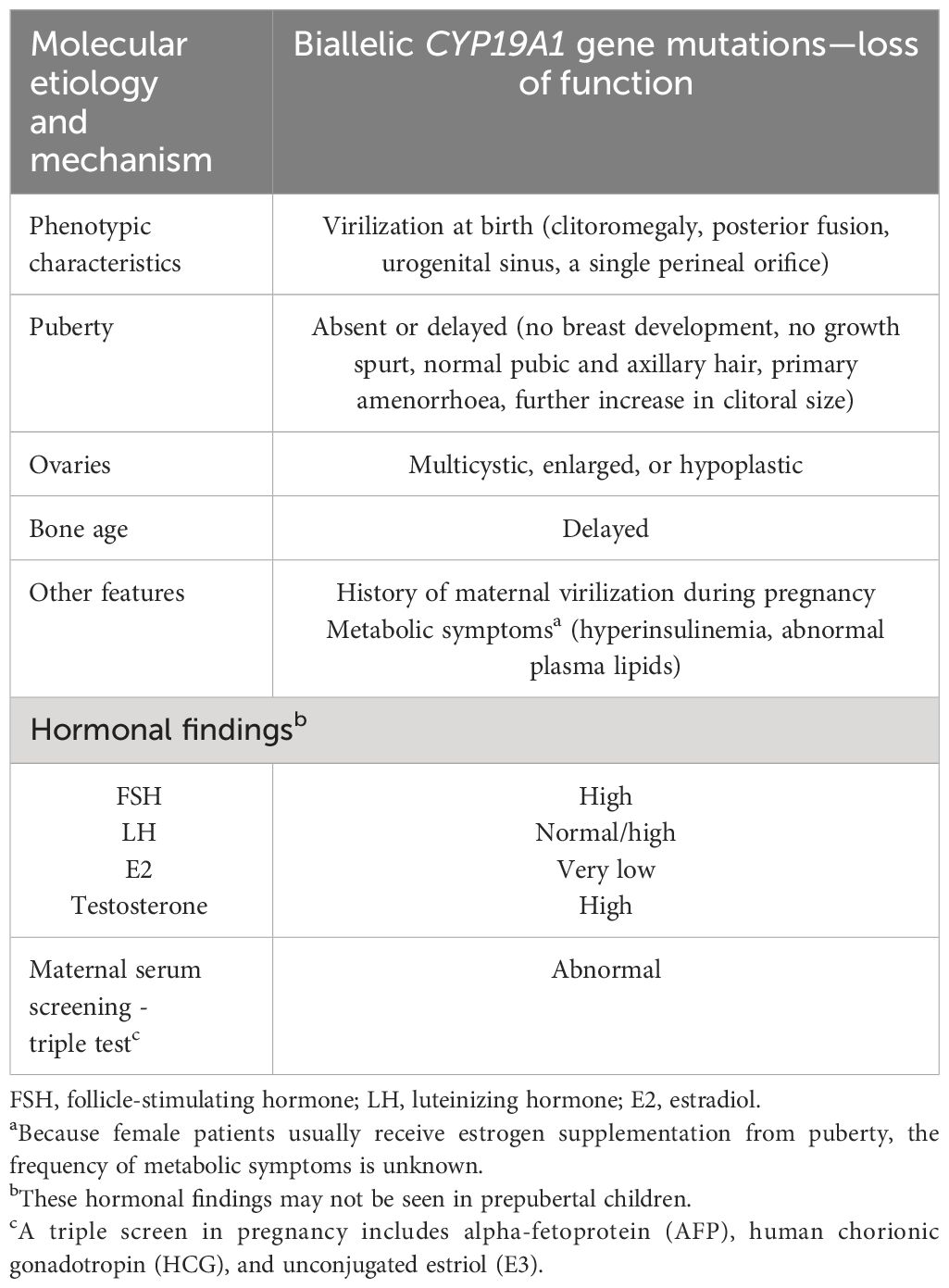
Table 2 Clinical and molecular characteristics of aromatase deficiency in 46,XX patients.
Estrogen is involved in the development of secondary sexual characteristics and the regulation of gonadotropin secretion in women and causes epiphysial closure, bone mass maintenance, regulation of lipoprotein synthesis, and carbohydrate metabolism (39, 40). Bone development, metabolism, and immune function are affected by aromatase deficiency, as described in the follow-up of a small number of 46,XX cases with aromatase deficiency and in studies of aromatase knockout mice. Dyslipidemia and hyperinsulinemia have also been reported, which may reflect estrogen insufficiency but may also reflect specific actions of aromatase itself (41). The demonstration of normal psychosexual development in adolescent or adult patients with aromatase deficiency suggests that estrogen does not have a critical role in the sex differentiation of the human brain. The long-term prognosis of female patients with aromatase deficiency is poorly understood, and it is still not clear whether these patients are fertile (32).
Functional studies for the aromatase activity have shown severe loss of enzyme activity in all cases, other than approximately 1% activity for the p.(R435C) variant detected in a compound heterozygote with the p.(C437Y) variant (42).
Cases of partial aromatase deficiency have also been described. A homozygous p.(R435C) variant has been described in a 46,XX girl who presented with atypical genitalia in the neonatal period but had breast development in adolescence. A single deletion of phenylalanine (Phe234del) causing partial loss of aromatase activity has been described in a virilized 46,XX female patient with Tanner stage 4 breast development at puberty (37).
Lin L et al. discovered that, in aromatase deficiency cases, even minimal aromatase activity can lead to breast development and estrogen production, particularly with elevated androgens. While alternative pathways for estrogen synthesis may exist, it is uncertain if they occur in humans. Individual variability in enzyme regulation and degradation of compounds could influence estrogen and androgen responses. Patients with complete aromatase deficiency have not shown estrogenization at puberty. Functional studies indicate a correlation between aromatase activity and the extent of estrogenization, with 0.7%–1.5% activity achieving Tanner breast stage 2 and 16%–19% allowing progression to breast stage 4 and full uterine growth (37).
Increased exogenous androgen exposure
Gestational hyperandrogenism
In a 46,XX case with normal ovarian and adrenal function, virilization may occur due to exposure to maternal androgens or synthetic androgenic progestins. The aromatase enzyme produced by the placenta converts androgens to estrogens; hence, high concentrations of maternal androgens may exceed placental aromatase activity and cause fetal virilization. Maternal luteomas or theca lutein cysts may result in maternal virilization during pregnancy (43). Androgen-producing ovarian tumors including hilar cell tumors, arrhenoblastomas/androblastomas, lipoid cell tumors, and Krukenberg tumors, may also cause virilization. Although extremely rare, androgen-secreting tumors of the adrenals can also occur during pregnancy (44).
Progestogens (or progestins), which include both bioidentical progesterone from plants and synthetic progestogens, are drugs with progesterone-like effects. They are commonly prescribed to reproductive-aged women for contraception, preventing threatened miscarriage and preterm birth. Adverse effects on reproductive development, like virilization in female infants, have been reported in case studies after exposure to progestogens in the first trimester. Similar effects have been observed in animal studies following in utero exposure to certain synthetic progestogens. More research is required to better understand the potential association of prenatal exposure to progestogens and adverse pregnancy outcomes, congenital malformation incidence, and longer-term health outcomes in prenatally exposed offspring (45).
Disorders/differences in gonadal differentiation (abnormal ovarian development)
46,XX testicular (T) DSD and 46,XX ovotesticular (OT) DSD
The chromosomal sex determines the differentiation of the primitive gonad into an ovary or testis. However, this process relies on a complex network of genes in addition to the presence or absence of the SRY (sex-determining region on the Y chromosome), with either activation of the testicular pathway and repression of the ovarian pathway or vice versa (46). The fetal testis and ovary are indifferent (bipotential gonad) until the 6th gestational week (gw). Morphological changes that promote ovarian development occur at the same developmental time point when a 46,XY bipotential gonad commences the organization of the testis structure. Without SRY, ovarian differentiation begins on the 7th gw in female individuals. Despite the absence of visible morphological changes, gene expression patterns within the 46,XX somatic cells of the bipotential gonad have been described that drive the differentiation of granulosa cells and steroid-producing theca cells. The initiation signals for the differentiation of granulosa cells are not clearly understood. The specific genetic mechanisms controlling ovarian development are being clarified, and some of the regulators have been identified. The signaling factors WNT4 and RSPO1 increase and stabilize the expression of CTNNB1 (β-catenin). CTNNB1 also maintains WNT4 expression, represses male-specific SOX9 expression, and promotes germ cell proliferation. In the 46,XX gonad, WNT, RSPO1, CTNNB1, FOXL2, and FST have a role in ovarian development and suppress testicular development. RSPO1 enhances β-catenin signaling through WNT4 in both humans and mice (47, 48). Figure 4 illustrates ovarian development and maintenance.
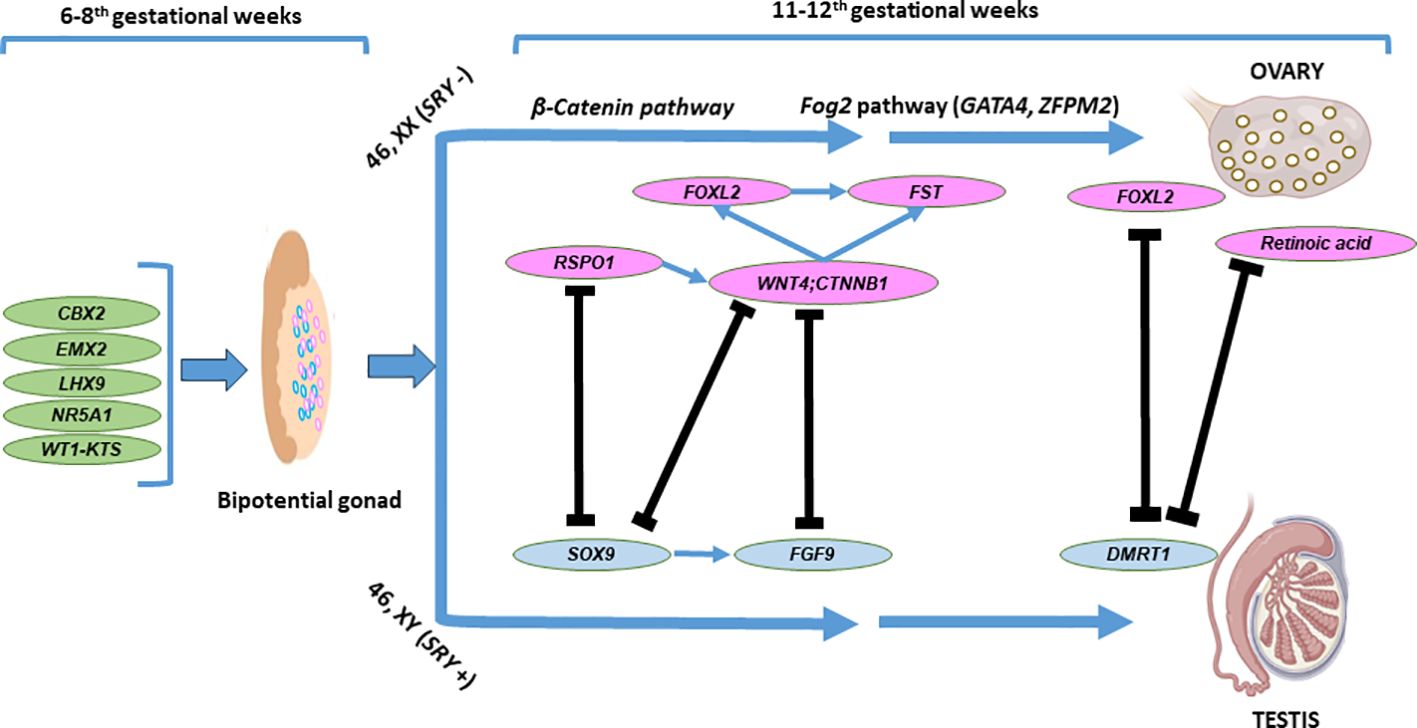
Figure 4 Basic genes and molecular pathways involved in ovarian development. The bipotential gonadal differentiates into ovary as a result of complex interactions between the testicular and ovarian developmental pathways Two components of the Wnt signalling pathway, WNT4 and RSP01 act synergistically to stabilise β-catenin encoded by CTNNB1, which promotes the expression of key ovarian genes, such as WNT4 and FST. Fog2 pathway (GATA4, ZFPM2) is also involved in the ovarian development. The ovary specific genes FOXL2, RSPO1. WNT4 and CTNNB1 (β-catenin) counteract the testicular development through inhibition of SOX9 and FGF9 expression. FOXL2 and ovarian retinoic acid is required in the adult ovary to suppress DMRT1 expression and in the maintenance of granulosa and theca cell populations of the adult ovary.
In 46,XX T-DSD, gonadal development progresses in the direction of the testicular pathway, and the degree of testosterone and anti-Müllerian hormone (AMH) production determines the external and internal genitalia (49). The gonad may be either a normal testis or a dysgenetic testis. External genitalia may change from male to ambiguous genitalia. Azoospermia, absence of Müllerian structures, and absence of congenital anomalies are characteristic of the nonsyndromic form of 46,XX T-DSD. Approximately 15% of individuals with 46,XX T-DSD present with atypical genitalia at birth, while the remaining are present after puberty (2). Diagnosis is based on clinical, endocrinological, and genetic testing (cytogenetic and/or molecular genetic). Hypergonadotropic hypogonadism due to testicular failure is observed. The cytogenetic analysis reveals a 46,XX chromosome structure (50). Translocation of SRY to the X chromosome causes approximately 80% of XX T-DSD cases, particularly in patients with male external genitalia. 46,XX T-DSD in most other patients is caused by a gain of function in genes involved in the key testicular pathway. Molecular genetic diagnosis is rarely determined in 46,XX T-DSD, and OT-DSD with atypical genitalia (51).
In 46,XX OT-DSD, ovarian and testicular tissues are present, either as asymmetrically developed gonads or as ovotestes in one individual. Diagnosis is generally made by histology, but hormonal evaluation and imaging studies may be suggestive. Although the chromosomal structure is generally 46,XX, 46,XX/46,XY (chimerism) or 46,XX/47,XXY may be detected in rare cases. This is a phenotypic spectrum and is characterized by variable penetrance. Several families have been reported in which some 46,XX individuals have OT-DSD, some have T-DSD, and others are asymptomatic carriers (52). The estimated incidence of OT-DSD is <1/20,000 people and accounts for 3%–10% of all DSD phenotypes (53–56). 46,XX OT-DSD cases are often SRY negative, with few reported cases having a Yp;Xp translocation, including SRY. Although the molecular genetic etiology has not been established for the majority of cases, gain-of-function of pro-testicular genes and their regulatory regions or decreased expression of pro-ovarian genes have been detected in OT-DSD. Copy number variations (CNV) of SOX genes or regulatory regions of SOX (SOX3, SOX9, and SOX10) have also been described (53, 57).
An ovotestis is present in approximately two-thirds of the affected individuals with OT-DSD. The gonad may also appear as a streak gonad (non-functional dysgenetic tissue with fibrosis), and the characteristics of gonadal tissue may be detected on biopsy. However, the probability of bias in the sampling of a gonadal biopsy may miss the ovarian portion of the gonads (58). OT-DSD cases may have a uterus or hemiuterus; however, Müllerian structures are absent in 46,XX T-DSD. The development of reproductive organs is closely associated with the gonads, and due to the AMH effect, there is no fallopian tube or uterus on the side of the testes, whereas, on the side of the ovary, the fallopian tube, unicornuate uterus, and vagina may be seen (59).
The differences between 46,XX T-DSD, and OT-DSD are shown in Table 3.
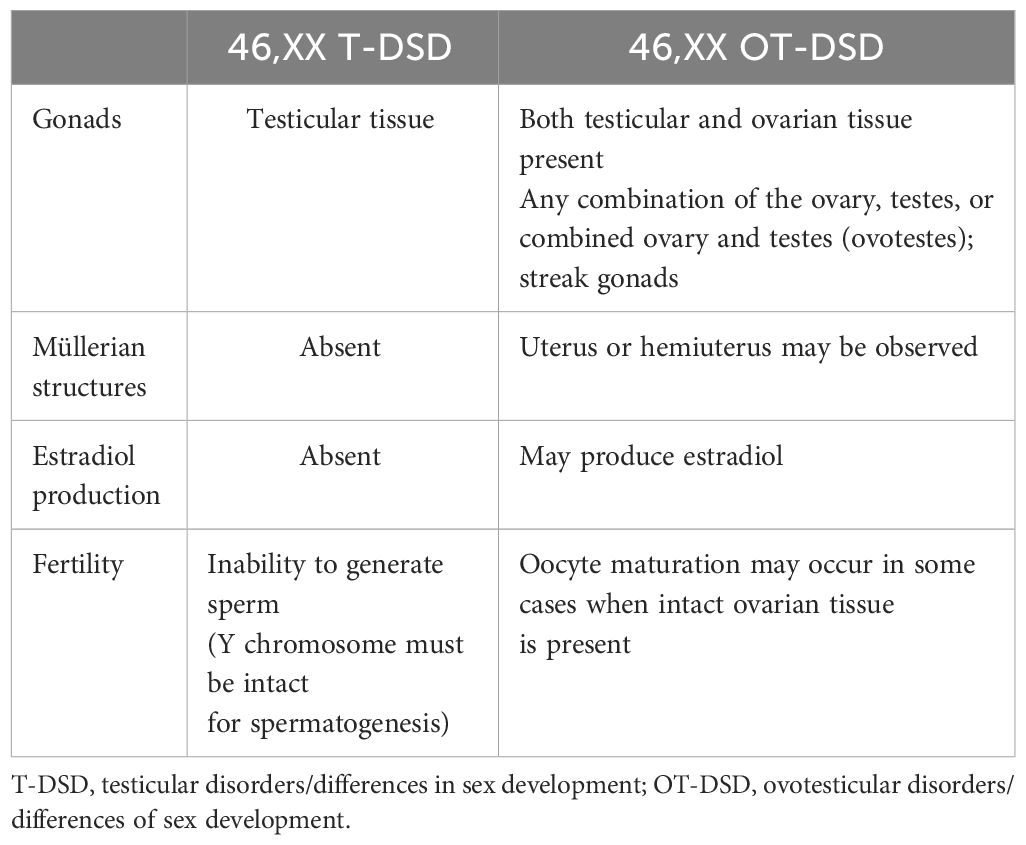
Table 3 Differences between 46,XX T-DSD and OT-DSD.
Recent evidence suggests that XX T-DSD and OT-DSD are the phenotypic spectrum of the same underlying defect in gonadal development (60, 61). All genetic etiologies causing 46,XX T-DSD can also lead to 46,XX OT-DSD (50, 53). SRY translocations, CNVs of FGF9, NR0B1, NR2F2, SOX3, SOX9, SOX10, and SPRY2, and sequence variants of NR5A1, NR2F2, RSPO1, SOX9, WNT4, WNT2B, and WT1 are responsible for the genetic mechanisms associated with 46,XX T/OT-DSD (62).
Molecular etiologies of 46,XX T-DSD and OT-DSD
Presence of SRY
SRY (MIM*480000) encodes a transcription factor that is a member of the high mobility group (HMG)-box family of DNA-binding proteins. In the developing gonad, the presence of SRY promotes the activation of testicular pathways; hence, the translocation of SRY to the X chromosome or an autosome causes XX T-DSD. Approximately 20% of all 46,XX T/OT-DSD cases (~80% of T-DSD) are caused by the translocation of SRY to the tip of the X chromosome detected by FISH or chromosomal microarray (CMA). This is caused by an inappropriate recombination between the X and Y chromosomes during paternal meiosis (50, 61). SRY-positive XX T-DSD is generally caused by de novo translocation of the Y and the X chromosomes. In conditions with SRY translocation to the autosome or when fertility is preserved, sex-limited autosomal dominant inheritance is observed rarely (50, 54).
Y; autosome translocation is extremely rare. X;Y translocations leading to 46,XX T-DSD are frequently associated with nonallelic homologous recombination, whereas the underlying mechanism of Y; autosome translocations remains to be clarified. Unbalanced Y; autosome translocations can occur between two low-similarity sequences. Nonhomologous end joining may play a significant role in the development of Y-chromosomal translocations (63).
NR5A1 variants
NR5A1 [(MIM*184757), steroidogenic factor 1 (SF1)] has a fundamental role in gonadal development and testicular differentiation, and it is an important etiology of 46,XY DSD. In addition to testis development, it also has a role in the activation of early ovary-determining genes, which occurs by upregulation of NR0B1 (nuclear receptor subfamily 0 group B member 1, a repressor of SOX9) and β-catenin (Figure 4) (64). To date, 324 variants in NR5A1 have been reported (31). In approximately 10%–20% of 46,XX T-DSD or OT-DSD cases, heterozygous variants in NR5A1 have been detected (52, 64, 65). Almost all of these variants affect a single amino acid residue, suggesting gain-of-function variants that cause inappropriate activation of testicular pathways in a 46,XX gonad. A specific variant in the NR5A1 gene [c.274C>T; p.(Arg92Trp); p.(R92W)] results in XX T/OT-DSD in some family members, while others are asymptomatic carriers (53). This variant represses the female-specific WNT signaling pathways (66). The exact mechanism is still not clear, but the hypothesis includes that p.(R92W) interferes with the NR5A1-mediated activation of ovarian development by an impaired interaction with β-catenin and by a loss of NR0B1-mediated suppression of SOX9 (52). Different studies demonstrated that no loss of SOX9 repression occurred and that NR5A1 p.(R92W) and novel p.(A260V) variants did not decrease the NR0B1 promoter activity (66). The variants disrupt the β-catenin-mediated activation of this promoter and result in increased repression of WNT signaling, resulting in reduced NR0B1 activity. WNT signaling is controlled by the NR5A1/β-catenin complex in a dose-dependent manner. In addition, a p.(R92Q) variant in the NR5A1 has been described in patients with and without OT-DSD (65). This variant did not show a reduced interaction with β-catenin (64). Pathogenic variants in NR5A1 associated with 46,XX T-DSD, or OT-DSD are inherited as an autosomal dominant trait with variable expressivity and incomplete penetrance. A heterozygous parent (if fertile) will transmit the variant to 50% of the offspring—offspring who is 46,XX, and at risk for T/OT-DSD (50).
Increased SOX3 expression
SOX3 (MIM* 313430, single exon) is localized in a highly conserved region of the X chromosome (Xq27.1) and is expressed in the brain, pituitary, and gonads, encoding a transcription factor very similar to SRY (44). Duplications or translocations in the regulatory regions of SOX3 may cause an ectopic expression of SOX3 in the 46,XX developing gonad, causing activation of testicular pathways (44). The first reports of 46,XX DSD due to SOX3 duplications suggested that SOX3 can act as SRY through increased expression. SOX3 can act synergistically with SF1 to upregulate SOX9 expression and activate testicular differentiation (67). Rare cases with SOX3 duplication in 46,XX OT-DSD, or T-DSD have been reported (16).
46,XX OT-DSD has been reported in a patient with a 774-kilobase (kb) insertion that is translocated from chromosome 1 to a region 82 kb distal to SOX3 (upregulation of SOX3 expression) (68). In this case, the gonads were testis in ultrasound, but one of them was ovarian tissue on biopsy, causing an OT-DSD phenotype. The translocation, including SOX3, was inherited from a fertile mother (68). The different phenotypes of the mother and proband may be ascribed to differential X inactivation in the developing gonad. T-DSD has been suggested in the other five individuals with SOX3-associated 46,XX DSD, but histologically demonstrated in only one case (67).
To date, in all known individuals with CNVs in or around the SOX3 gene with parental segregation analysis, it has been demonstrated that the disorder was caused by a de novo variant, and the risk of transmission to sibs is low (50). However, de Oliveira FM et al. described de novo SOX3 duplication in two siblings with atypical genitalia, suggesting germline mosaicism as the etiology (69).
SOX9 duplication
The transcription factor encoded by SOX9 (MIM*608160) functions downstream of SRY and is required for testicular development. Duplications of the SOX9 locus and its upstream regulatory region have been reported in 46,XX T-DSD (57). DSD is caused by a gain-of-function CNV in the distal upstream regulatory region that duplicates one or more enhancer elements of the SOX9 gene. Duplications can be localized up to 650 kb upstream of SOX9 and induce testicular development by increasing the number of enhancers, which finally results in SOX9 upregulation (70, 71). These duplications cause 46,XX DSD to varying degrees, and incomplete penetrance has also been documented (72). SOX9 duplications do not cause skeletal abnormalities. Hence, the gain- or loss-of-function variants in the SOX9 gene appear to demonstrate a sex-limited manifestation/inheritance (71).
CMA detects large CNVs (including the regulatory regions around SOX3 and SOX9) that cannot be detected by sequencing and small chromosomal rearrangements that may not be detected by karyotype. Small duplications or triplications in the regulatory regions of SOX9 have been documented, which affect SOX9 enhancers located up to two megabases (Mb) upstream of SOX9. A balanced translocation involving the 17q24.3 region has also been reported and should be observed in chromosome analysis (70). CNVs in or around SOX9 have been inherited as autosomal dominant; however, only those individuals with a 46,XX chromosome are affected (50).
CNVs in the SOX9 enhancer named RevSex cause XX sex reversal. The first case of 46,XX OT-DSD due to RevSex duplication demonstrated a male phenotype in affected XX family members. The father carrying the same variant was unaffected, which can be explained in the model where XY male individuals express high SOX9 levels during gonadogenesis, and further amplification of SOX9 expression does not deter from a pattern of typical male sexual development. Over the last decade, several cases of 46,XX OT-DSD caused by RevSex CNVs have been reported, exhibiting a range of genital presentations (typical to atypical genitalia). This phenomenon suggests that other genetic or non-genetic factors may contribute to the spatiotemporal expression of SOX9 during development, leading to variable clinical outcomes (70, 73).
SOX9 duplications more frequently result in 46,XX OT-DSD compared to 46,XX T-DSD. The two phenotypes have not been seen in the same family to date. To evaluate the risk of recurrence, the carrier status of the father should be analyzed. If parents are not carriers (de novo), the risk is no higher than the empirical risk in the general population. If the father is heterozygous for a CNV in or around SOX9, all reported 46,XY carriers have been fertile, anatomically male. One such 46,XY father (the father of a proband with OT-DSD) was reported to have inherited the duplication from his 46,XX mother. The risk of inheriting the CNV from the sibs is 50%. 46,XX individuals who carry the CNV are at risk of having 46,XX T, or OT-DSD. Individuals with 46,XY chromosomes will be fertile males (70).
WT1 variants
A DNA-binding protein containing four zinc fingers encoded by WT1 (MIM* 607102) is essential for normal urogenital development (51). Pathogenic variants of WT1 cause abnormal testis development, resulting in 46,XY DSD (74). The first report describing a WT1 variant causing 46,XX T-DSD was published in 2017. In a male with microcephaly, dysgenetic testis, small uterus, and male external genitalia, the p.(Arg495Gly) variant was detected in WT1 (75). Several cases of 46,XX T/OT DSD caused by pathogenic variants affecting the fourth zinc finger of WT1 have been reported to date. This led to the identification of many male sex-determining genes and the suppression of FOXL2 in a dominant-negative manner. Mutant WT1 showed an interaction with β-catenin and upregulation of SOX9 (75, 76). T-DSD is observed more frequently compared to OT-DSD in 46,XX cases, with frameshift and missense variants affecting the ZF4 domain of WT1. Of the nine individuals reported to date, four have histologically determined T-DSD, and two have OT-DSD. Of six reported individuals with WT1-related 46,XX T-DSD, only one had palpable gonads and typical male genitalia (75–77).
To date, all known individuals with a pathogenic WT1 variant that causes 46,XX T-DSD whose parents have undergone molecular genetic testing have the disorder as a result of a de novo pathogenic variant, and the risk of transmission to the sibs is low (50).
NR2F2 variants
NR2F2 (MIM*107773) is localized on chromosome 15q26.2, which encodes chicken ovalbumin upstream promoter transcription factor 2 (COUP-TF2). COUP-TFs are members of the steroid/thyroid hormone receptor superfamily. COUP-TF homologs, which have been cloned from humans, suggest that their protein sequences are highly homologous across species, defining functional conservation (62, 78). NR2F2 is primarily expressed in mesenchymal cells (78). Compatible with the expression pattern of NR2F2, congenital heart defects are the well-known phenotypes associated with the variants of this gene (78). To date, 35 different variants have been described in NR2F2, and the majority of these are related to congenital heart defects (16).
In developing testicular tissue, NR2F2 expression is observed in Leydig cells from 7 to 10 gw but is downregulated at 15 gw and repressed throughout fetal life. Previous studies concluded that NR2F2 repression is important for fetal Leydig cell differentiation (62). According to rodent studies, the target genes of NR2F2 in Leydig cells are AMHR2, INSL3, and genes encoding steroidogenic enzymes (62).
To date, four 46,XX T/OT-DSD cases with loss-of-function variants of NR2F2 have been reported, suggesting that NR2F2 is an anti-testicular gene. The first three cases with the frameshift variants were reported by Bashamboo et al., while one case with a 3-Mb deletion encompassing NR2F2 was reported by Carvalheira et al. (79, 80). All four patients were 46,XX with testicular development. The mechanism underlying testicular development associated with NR2F2 variants is not clear (62). Bashamboo et al. described that the two patients had the same 7-bp deletion, while one individual had a nearly identical 7-bp deletion. The variants were de novo in two patients, but the segregation of parents in the third case was not performed (80).
In a young man with XX OT-DSD, blepharophimosis–ptosis–epicantus syndrome (BPES), and coarctation of the aorta, a de novo 3-Mb deletion resulting in partial 15q monosomy of an evolutionarily conserved region was described. The deletion included the NR2F2 and SPATA8 genes, as well as three noncoding genes, three pseudogenes, and regulatory regions (79).
Copy number variants around NR0B1
An 80-kb deletion involving the NR0B1 and putative MAGEB regulatory regions in a patient with 46,XX OT-DSD was reported by Dangle et al. It has been hypothesized that the combination of a loss of one copy of anti-testicular NR0B1 and overexpression of pro-testicular MAGEB resulted in testicular development in their patient (62, 81).
Loss of function variants in genes repressing testicular pathways
The regulatory pathways controlling ovarian development have remained elusive in humans in contrast to testis formation. The pro-ovarian WNT4/RSPO1 and beta-catenin pathways are activated in the absence of SRY (Figure 4) (82). Likely pathogenic/pathogenic variants in these genes may cause 46,XX T/OT-DSD and are often associated with abnormalities of other systems.
WNT4 variants
WNT4 (wingless-type MMTV integration site family member 4, MIM* 603490) controls the development of the female reproductive structure in the absence of SRY in 46,XX (83). WNT4 has five coding exons and is localized on chromosome 1p36.12. The action of this gene occurs through the RSPO1-assisted canonical beta-catenin pathways. The mice study reported that excessive Wnt4 supports feminization in male individuals, and insufficient Wnt4 produces virilization in female individuals. WNT4 and its frizzled cell surface receptors have a role in the induction of female sex differentiation, and when both alleles of WNT4 are inactive, SERKAL syndrome (sex reversal, kidneys, adrenal, and lung dysgenesis; MIM# 611812) occurs (84).
RSPO1 variants
R-spondin proteins are agonists of the canonical WNT/β-catenin signaling pathway (85). Biallelic RSPO1 (MIM*609595) variants cause a 46,XX T/OT DSD, palmoplantar keratoderma, and a predisposition to squamous cell carcinoma (86, 87). To date, nine variants in RSPO1 have been reported, and only six of these are related to XX sex reversal (16).
Management of 46,XX T-DSD and OT-DSD
The management of OT-DSD is complicated by numerous uncertainties about the etiology, gonadal function, and ultimate sex outcomes; therefore, a combination of medical, surgical, and psychological interventions must be implemented.
Inguinal or labioscrotal gonads, a hemiuterus, and a normal adrenal steroid profile in a virilized 46,XX infant should suggest OT-DSD. The AMH concentration, which reflects the amount of testicular tissue, is typically in the intermediate range in children with 46,XX OT-DSD. Since AMH tends to decrease in the initial weeks of postnatal life, serial AMH measurements after 1 to 2 months increase diagnostic certainty. AMH is invaluable for evaluating the presence of testicular tissue, its clinical utility decreases during the prepubertal period due to declining serum AMH with increasing intratesticular testosterone concentrations (53).
Normal serum gonadotropin concentrations in a XX OT-DSD suggest the existence of functional gonadal tissue. Unstimulated testosterone concentration is reliable during mini-puberty and from puberty onward. To adequately measure Leydig cell function in the first 2 to 3 weeks of life and childhood, an hCG test is required (88). However, a reliable functional test demonstrating the presence of ovarian tissue has not been described. In the study by Mendez et al., a human menopausal gonadotropin (hMG) stimulation test was performed on children with atypical genitalia. In patients with subsequent histologic OT-DSD, E2 levels increased above 80 pg/mL after the hMG challenge. All responders had ovarian or ovotestis, whereas none of the non-responders had histologic evidence of ovarian tissue. Nonetheless, further studies in larger numbers of patients are needed to confirm the sensitivity and specificity of this test (89).
Laparoscopic evaluation of genital structures and gonadal biopsies may be required for diagnosis. Nonetheless, in some situations, gonadal biopsies often do not reflect the characteristics of gonadal tissue and should not be regarded as a test for definitive diagnosis and decision making. Chromosomal analysis (exclusion of Y chromosome material by FISH or PCR) and NGS techniques (targeted gene panel, ES) focusing on the pathology of genes causing T/OT DSD and CMA for genomic imbalances are exclusively important for diagnosis (53).
Sex assignment is complicated in newborns with OT-DSD. Patient management must be highly individualized, and decisions should be made by a specialized multidisciplinary team and the parents. The severity of virilization, the approximate amount of gonadal tissue (testis or ovary), and the presence of the uterus may affect the final decisions. Gender outcome is unpredictable; hence, early genital surgery, like (partial) gonadectomy must be avoided in OT-DSD (53, 90). When gender identity is uncertain, GnRH analogs may be an alternative for use in childhood (91). The data about the fertility outcomes of OT-DSD are scarce. XX male individuals are infertile due to the absence of the Y chromosome. The characteristics of chimeric OT-DSD are unknown, but few reports indicate that XX males with testicular tissue had normal prepubertal progression, while testosterone concentrations decreased gradually. Hormone replacement may be required for these patients. In female individuals with ovarian tissue in place, menstruation may be observed, and some pregnancies have also been reported (92).
A gonadectomy may be required after puberty, provided that the gender identity is stable. In patients with a demarcated gonadal structure, gonad-sparing partial gonadectomy may be considered in the ovotestis (93). Monitoring of AMH and E2 concentrations is essential to ensuring that the relevant tissues have been completely removed. Nonetheless, it will be inevitable to completely extract the ovotestis.
Due to the possibility of persistence of functional gonadal tissue after partial gonadectomy in adolescents with OT-DSD, these patients should be followed up regularly. The residual tissue may cause clitoromegaly in female individuals (53).
The risk factors predisposing to malignancy are dysgenetic gonads, an intraabdominal gonad, and the presence of a Y chromosome. Knowledge about the risk of gonadal germ cell cancer (GCC) occurrence in OT-DSD is limited. Due to the absence of the Y chromosome, a low risk of GCC has been suggested, specifically in the gonadal tissues (94). The risk of GCC in OT-DSD is low compared to other DSDs. However, gonadoblastoma, seminoma, dysgerminoma, and yolk sac carcinomas have been described. This low-risk ratio likely reflects the fact that the majority of OT-DSD individuals are SRY-negative 46,XX cases (95). The ovarian reserve must be monitored in pubertal girls with OT-DSD to evaluate the possibility of oocyte cryopreservation (96).
Despite the identification of various genetic causes leading to defects in sex determination/differentiation, genetic factors alone cannot account for the diverse range of health or psychological issues that an individual with a DSD might encounter. Nonetheless, the detection of multiple developmental genes has increased our knowledge about the pathophysiology of DSD, which may help to follow the long-term effects of these etiologies and treatment outcomes in these individuals (58).
It is important to note that the management of these patients must be individualized, and decisions should be made with a team (endocrinologists, urologists, geneticists, and mental health professionals). Gonadal biopsy and early genital surgery, including gonadectomy, should be avoided in OT-DSD because of the unpredictability of rearing sex and adult outcome. The aim must be to provide comprehensive care that addresses both the physical and psychosocial aspects of the condition.
46,XX gonadal (ovarian) dysgenesis
46,XX gonadal (ovarian) dysgenesis (OD) is heterogeneous condition and may present with primary amenorrhea and infertility.
Multiple genes have been implicated in the etiology of OD, including BMP15, PSMC3IP, MCMDC1, SOHLH1, NUP107, MRPS22, ESR2, SPIDR, FIGNL1, and ZSWIM7 (97, 98). Advancements in NGS techniques are progressively enhancing our understanding of 46,XX OD.
BPES is inherited in both dominant and recessive forms, and abnormal eyelids can be observed with OD (type I). Monoallelic variants of FOXL2 (MIM* 605597) have been detected in 90% of BPES cases. Genomic rearrangements causing total or partial deletion of FOXL2 account for approximately 12% of cases, while the remaining cases are caused by intragenic variants. Ovarian phenotypes are highly variable in female individuals with type I BPES, ranging from primary amenorrhea to irregular menstruation (99).
Perrault syndrome is a recessively inherited condition characterized by 46,XX OD, sensorineural hearing loss (in both sexes), and neurological findings in some cases. Mild intellectual disability with cerebellar and peripheral nervous system involvement may be observed. Perrault syndrome is clinically heterogeneous and is classified into type I (without neurological disease) and type II (progressive neurological disease) (100). Pathogenic/likely pathogenic variants in HARS2, CLPP, LARS2, TWNK, ERAL1, and PROPR may cause Perrault syndrome-related OD.
Malformations causing 46,XX DSD
Isolated malformations of the internal genital tracts (vagina, uterus, and fallopian tubes) may be observed in some females, and these malformations may be related to the incomplete development of Müllerian structures or the presence of abnormal structures. A family history of malformations, which suggests a genetic etiology, may be detected in some patients, although the etiology is rarely elucidated. Aplasia or hypoplasia of the uterus and the fallopian tubes and a bicornuate/bipartite uterus that may be associated with malformations in other systems or tissues may occur. MRKHS (MIM%277000) is defined as the absence of a uterus and vagina in a phenotypically female 46,XX case. The two subtypes of MRKHS are an isolated type I and a type II with extragenital malformations. The majority of cases are sporadic. MURCS syndrome (MIM% 601076) includes Mullerian aplasia, renal aplasia, and cervical–thoracic somite abnormalities (101).
Molecular cytogenetic studies (CMA and MLPA) identified CNVs in different chromosomal regions, including the TAR susceptibility locus (1q21.1), chromosome 16p11.2, and 17q12 and 22q11.21 microduplication and deletion regions. The sequencing analysis of LHX1, TBX6, and RBM8A revealed other MRKH-associated genes. An analysis of WNT9B determined some causative variants in MRKHS (4).
In a group of patients with hyperandrogenemia, variants of WNT4 are causative. Monoallelic WNT4 variants cause Mullerian duct failure and hyperandrogenism, while biallelic WNT4 variants cause SERKAL syndrome (46,XX DSD, dysgenetic kidneys, adrenals, and lungs) (102).
Conclusion
The etiology of non-CAH 46,XX DSD includes a heterogeneous group of disorders. Progress in cytogenetic and molecular genetic techniques has significantly increased our knowledge about the etiology of these disorders. Nonetheless, uncertainties about the gonadal function and gender outcomes may still make the clinical management of these conditions complicated, and these patients must be monitored by an experienced multidisciplinary team.
Author contributions
ZYA: Investigation, Writing – original draft, Writing – review & editing. TG: Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McElreavey K, Bashamboo A. Monogenic forms of DSD: an update. Horm Res Paediatr. (2023) 96:144–68. doi: 10.1159/000521381
2. Alkhzouz C, Bucerzan S, Miclaus M, Mirea AM, Miclea D. 46,XX DSD: developmental, clinical and genetic aspects. Diagnostics (Basel). (2021) 11:1379. doi: 10.3390/diagnostics11081379
3. Hughes IA, Nihoul-Fékété C, Thomas B, Cohen-Kettenis PT. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab. (2007) 21:351–65. doi: 10.1016/j.beem.2007.06.003
4. Ledig S, Wieacker P. Clinical and genetic aspects of Mayer-Rokitansky-Küster-Hauser syndrome. Med Genet. (2018) 30:3–11. doi: 10.1007/s11825-018-0173-7
5. Nicolaides NC, Charmandari E. Primary generalized glucocorticoid resistance and hypersensitivity syndromes: A 2021 update. Int J Mol Sci. (2021) 22:10839. doi: 10.3390/ijms221910839
6. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. (1995) 83:835–9. doi: 10.1016/0092-8674(95)90199-X
7. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. (2002) 16:61–71. doi: 10.1096/fj.01-0245com
8. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. (2008) 93:1563–72. doi: 10.1210/jc.2008-0040
9. Ali SR, Bryce J, Haghpanahan H, Lewsey JD, Tan LE, Atapattu N, et al. Real-world estimates of adrenal insufficiency-related adverse events in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2021) 106:e192–203. doi: 10.1210/clinem/dgaa694
10. McMahon SK, Pretorius CJ, Ungerer JP, Salmon NJ, Conwell LS, Pearen MA, et al. Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in Helix 12 of the ligand binding domain of the glucocorticoid receptor gene (NR3C1). J Clin Endocrinol Metab. (2010) 95:297–302. doi: 10.1210/jc.2009-1003
11. Nader N, Bachrach BE, Hurt DE, Gajula S, Pittman A, Lescher R, et al. A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab. (2010) 95:2281–5. doi: 10.1210/jc.2009-2463
12. Huang H, Wang W. Molecular mechanisms of glucocorticoid resistance. Eur J Clin Invest. (2023) 53:e13901. doi: 10.1111/eci.13901
13. Nicolaides NC, Charmandari E. Chrousos syndrome: from molecular pathogenesis to therapeutic management. Eur J Clin Invest. (2015) 45:504–14. doi: 10.1111/eci.12426
14. Lamberts SW, Koper JW, Biemond P, den Holder FH, de Jong FH. Cortisol receptor resistance: the variability of its clinical presentation and response to treatment. J Clin Endocrinol Metab. (1992) 74:313–21. doi: 10.1210/jcem.74.2.1309833
15. Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab. (2004) 89:1939–49. doi: 10.1210/jc.2003-030450
16. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human gene mutation database (HGMD®): 2003 update. Hum Mutation. (2003) 21:577–81. doi: 10.1002/humu.10212
17. Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, et al. Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: results of the French MUTA-GR Study. Eur J Endocrinology. (2018) 178:411–23. doi: 10.1530/EJE-17-1071
18. Nicolaides NC, Chrousos GP. Bilateral adrenal hyperplasia and NR3C1 mutations causing glucocorticoid resistance: Is there an association? Eur J Endocrinol. (2018) 179:C1–c4. doi: 10.1530/EJE-18-0471
19. Tatsi C, Xekouki P, Nioti O, Bachrach B, Belyavskaya E, Lyssikatos C, et al. A novel mutation in the glucocorticoid receptor gene as a cause of severe glucocorticoid resistance complicated by hypertensive encephalopathy. J Hypertens. (2019) 37:1475–81. doi: 10.1097/HJH.0000000000002048
20. Mendonca BB, Leite MV, de Castro M, Kino T, Elias LL, Bachega TA, et al. Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab. (2002) 87:1805–9. doi: 10.1210/jcem.87.4.8379
21. Mauri S, Nieto-Moragas J, Obón M, Oriola J. The glucocorticoid resistance syndrome. Two cases of a novel pathogenic variant in the glucocorticoid receptor gene. JCEM Case Rep. (2024). 2:luad153. doi: 10.1210/jcemcr/luad153
22. van Rossum EFC, van den Akker ELT. Glucocorticoid resistance. Endocr Dev. (2011) 20:127–36. doi: 10.1159/000321234
23. Nicolaides NC, Kino T, Chrousos G, Charmandari E. Primary Generalized Glucocorticoid Resistance or Chrousos Syndrome. Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. South Dartmouth (MA): MDText.com, Inc. Copyright (2000).
24. Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev. (2009) 30:343–75. doi: 10.1210/er.2008-0016
25. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. (2011) 32:81–151. doi: 10.1210/er.2010-0013
26. Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res. (2009) 72:321–30. doi: 10.1159/000249159
27. Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev. (2005) 57:359–83. doi: 10.1124/pr.57.3.6
28. Peltoketo H, Luu-The V, Simard J, Adamski J. 17beta-hydroxysteroid dehydrogenase (HSD)/17-ketosteroid reductase (KSR) family; nomenclature and main characteristics of the 17HSD/KSR enzymes. J Mol Endocrinol. (1999) 23:1–11. doi: 10.1677/jme.0.0230001
29. Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. (1991) 72:560–6. doi: 10.1210/jcem-72-3-560
30. Acar S, Erbaş İM, Paketçi A, Onay H, Çankaya T, Gürsoy S, et al. A novel compound heterozygous variant in CYP19A1 resulting in aromatase deficiency with normal ovarian tissue. Turk J Pediatr. (2020) 62:826–30. doi: 10.24953/turkjped.2020.05.015
31. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. (2003) 21:577–81. doi: 10.1002/humu.10212
32. Fukami M, Ogata T. Congenital disorders of estrogen biosynthesis and action. Best Pract Res Clin Endocrinol Metab. (2022) 36:101580. doi: 10.1016/j.beem.2021.101580
33. Bouchoucha N, Samara-Boustani D, Pandey AV, Bony-Trifunovic H, Hofer G, Aigrain Y, et al. Characterization of a novel CYP19A1 (aromatase) R192H mutation causing virilization of a 46,XX newborn, undervirilization of the 46,XY brother, but no virilization of the mother during pregnancies. Mol Cell Endocrinol. (2014) 390:8–17. doi: 10.1016/j.mce.2014.03.008
34. Dursun F, Ceylaner S. A novel homozygous CYP19A1 gene mutation: aromatase deficiency mimicking congenital adrenal hyperplasia in an infant without obvious maternal virilisation. J Clin Res Pediatr Endocrinol. (2019) 11:196–201. doi: 10.4274/jcrpe
35. Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. (1997) 82:1739–45. doi: 10.1210/jc.82.6.1739
36. Akçurin S, Türkkahraman D, Kim WY, Durmaz E, Shin JG, Lee SJ. A novel null mutation in P450 aromatase gene (CYP19A1) associated with development of hypoplastic ovaries in humans. J Clin Res Pediatr Endocrinol. (2016) 8:205–10. doi: 10.4274/jcrpe
37. Lin L, Ercan O, Raza J, Burren CP, Creighton SM, Auchus RJ, et al. Variable phenotypes associated with aromatase (CYP19) insufficiency in humans. J Clin Endocrinol Metab. (2007) 92:982–90. doi: 10.1210/jc.2006-1181
38. Gagliardi L, Scott HS, Feng J, Torpy DJ. A case of Aromatase deficiency due to a novel CYP19A1 mutation. BMC Endocr Disord. (2014) 14:16. doi: 10.1186/1472-6823-14-16
39. Bulun SE. Aromatase and estrogen receptor α deficiency. Fertil Steril. (2014) 101:323–9. doi: 10.1016/j.fertnstert.2013.12.022
40. Herrmann BL, Janssen OE, Hahn S, Broecker-Preuss M, Mann K. Effects of estrogen replacement therapy on bone and glucose metabolism in a male with congenital aromatase deficiency. Horm Metab Res. (2005) 37:178–83. doi: 10.1055/s-2005-861292
41. Verma N, Jain V, Birla S, Jain R, Sharma A. Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J Pediatr Endocrinol Metab. (2012) 25:1185–90. doi: 10.1515/jpem-2012-0152
42. Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, et al. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. (1997) 337:91–5. doi: 10.1056/NEJM199707103370204
43. Hougen HY, Seideman CA, Adam MP, Amies Oelschlager AM, Fechner PY, Ramsell L, et al. Congenital virilization of female infants recognized after pregnancies with retained levonorgestrel intrauterine devices. J Pediatr Urol. (2020) 16:241–3. doi: 10.1016/j.jpurol.2020.03.008
44. Flück CE, Güran T. Ambiguous Genitalia in the Newborn. Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. South Dartmouth (MA): MDText.com, Inc.Copyright © (2000).
45. NTP research report on the scoping review of prenatal exposure to progestogens and adverse health outcomes: research report 17. (2020). National Toxicology P. NTP Research ReportsResearch Triangle Park (NC): National Toxicology Program;.
46. Rey R, Josso N, Racine C. Sexual Differentiation. Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. South Dartmouth (MA): MDText.com, Inc. Copyright (2000).
47. Biason-Lauber A. WNT4, RSPO1, and FOXL2 in sex development. Semin Reprod Med. (2012) 30:387–95. doi: 10.1055/s-00000072
48. Nef S, Schaad O, Stallings NR, Cederroth CR, Pitetti JL, Schaer G, et al. Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol. (2005) 287:361–77. doi: 10.1016/j.ydbio.2005.09.008
49. Flanagan SE, Xie W, Caswell R, Damhuis A, Vianey-Saban C, Akcay T, et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. Am J Hum Genet. (2013) 92:131–6. doi: 10.1016/j.ajhg.2012.11.017
50. Délot EC, Vilain EJ. Nonsyndromic 46,XX Testicular Disorders/Differences of Sex Development. Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al, editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © (1993).
51. Grinspon RP, Rey RA. Disorders of sex development with testicular differentiation in SRY-negative 46,XX individuals: clinical and genetic aspects. Sex Dev. (2016) 10:1–11. doi: 10.1159/000445088
52. Baetens D, Stoop H, Peelman F, Todeschini AL, Rosseel T, Coppieters F, et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med. (2017) 19:367–76. doi: 10.1038/gim.2016.118
53. Syryn H, Van De Vijver K, Cools M. Ovotesticular difference of sex development: genetic background, histological features, and clinical management. Horm Res Paediatr. (2023) 96:180–9. doi: 10.1159/000519323
54. Baetens D, Verdin H, De Baere E, Cools M. Update on the genetics of differences of sex development (DSD). Best Pract Res Clin Endocrinol Metab. (2019) 33:101271. doi: 10.1016/j.beem.2019.04.005
55. Mekkawy MK, Kamel AK, Dessouky N, Elgharbawy M, Mazen I. Cytogenetic spectrum of ovotesticular difference of sex development (OT DSD) among a large cohort of DSD patients and literature review. Sex Dev. (2019) 13:221–7. doi: 10.1159/000508153
56. Bashamboo A, McElreavey K. Human sex-determination and disorders of sex-development (DSD). Semin Cell Dev Biol. (2015) 45:77–83. doi: 10.1016/j.semcdb.2015.10.030
57. Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. (2015) 23:1025–32. doi: 10.1038/ejhg.2014.237
58. Arboleda VA, Sandberg DE, Vilain E. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol. (2014) 10:603–15. doi: 10.1038/nrendo.2014.130
59. Deng S, Sun A, Chen R, Yu Q, Tian Q. Gonadal dominance and internal genitalia phenotypes of patients with ovotesticular disorders of sex development: report of 22 cases and literature review. Sex Dev. (2019) 13:187–94. doi: 10.1159/000507036
60. Maciel-Guerra AT, de Mello MP, Coeli FB, Ribeiro ML, Miranda ML, Marques-de-Faria AP, et al. XX Maleness and XX true hermaphroditism in SRY-negative monozygotic twins: additional evidence for a common origin. J Clin Endocrinol Metab. (2008) 93:339–43. doi: 10.1210/jc.2007-1115
61. Lambert S, Peycelon M, Samara-Boustani D, Hyon C, Dumeige L, Peuchmaur M, et al. SRY-negative 46,XX testicular/ovotesticular DSD: Long-term outcomes and early blockade of gonadotropic axis. Clin Endocrinol (Oxf). (2021) 94:667–76. doi: 10.1111/cen.14389
62. Hattori A, Fukami M. Nuclear receptor gene variants underlying disorders/differences of sex development through abnormal testicular development. Biomolecules. (2023) 13:691. doi: 10.3390/biom13040691
63. Uehara E, Hattori A, Shima H, Ishiguro A, Abe Y, Ogata T, et al. Unbalanced Y;7 translocation between two low-similarity sequences leading to SRY-positive 45,X testicular disorders of sex development. Cytogenet Genome Res. (2019) 158:115–20. doi: 10.1159/000501378
64. Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. (2016) 25:3446–53. doi: 10.1093/hmg/ddw186
65. Swartz JM, Ciarlo R, Guo MH, Abrha A, Weaver B, Diamond DA, et al. A 46,XX ovotesticular disorder of sex development likely caused by a steroidogenic factor-1 (NR5A1) variant. Horm Res Paediatr. (2017) 87:191–5. doi: 10.1159/000452888
66. Knarston IM, Robevska G, van den Bergen JA, Eggers S, Croft B, Yates J, et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum Mutat. (2019) 40:207–16. doi: 10.1002/humu.23672
67. Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. (2011) 121:328–41. doi: 10.1172/JCI42580
68. Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab. (2015) 100:E815–20. doi: 10.1210/jc.2014-4383
69. de Oliveira FM, Barros BA, Dos Santos AP, Campos NLV, Mazzola TN, Filho PL, et al. SOX3 duplication in a boy with 46,XX ovotesticular disorder of sex development and his 46,XX sister with atypical genitalia: Probable germline mosaicism. Am J Med Genet A. (2023) 191:592–8. doi: 10.1002/ajmg.a.63051
70. Croft B, Ohnesorg T, Hewitt J, Bowles J, Quinn A, Tan J, et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat Commun. (2018) 9:5319. doi: 10.1038/s41467-018-07784-9
71. Sajan SA, Brown CM, Davis-Keppen L, Burns K, Royer E, Coleman JAC, et al. The smallest likely pathogenic duplication of a SOX9 enhancer identified to date in a family with 46,XX testicular differences of sex development. Am J Med Genet A. (2023) 191:2831–6. doi: 10.1002/ajmg.a.63367
72. Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. (2011) 48:825–30. doi: 10.1136/jmedgenet-2011-100255
73. Qian Z, Grand K, Freedman A, Nieto MC, Behlmann A, Schweiger BM, et al. Whole genome sequencing identifies a cryptic SOX9 regulatory element duplication underlying a case of 46,XX ovotesticular difference of sexual development. Am J Med Genet A. (2021) 185:2782–8. doi: 10.1002/ajmg.a.62373
74. Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. (1997) 17:467–70. doi: 10.1038/ng1297-467
75. Eozenou C, Gonen N, Touzon MS, Jorgensen A, Yatsenko SA, Fusee L, et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms' tumor 1 (WT1) gene. Proc Natl Acad Sci U S A. (2020) 117:13680–8. doi: 10.1073/pnas.1921676117
76. Gomes NL, de Paula LCP, Silva JM, Silva TE, Lerário AM, Nishi MY, et al. A 46,XX testicular disorder of sex development caused by a Wilms' tumour Factor-1 (WT1) pathogenic variant. Clin Genet. (2019) 95:172–6. doi: 10.1111/cge.13459
77. Sirokha D, Gorodna O, Vitrenko Y, Zelinska N, Ploski R, Nef S, et al. A novel WT1 mutation identified in a 46,XX testicular/ovotesticular DSD patient results in the retention of intron 9. Biol (Basel). (2021) 10:1248. doi: 10.3390/biology10121248
78. Polvani S, Pepe S, Milani S, Galli A. COUP-TFII in health and disease. Cells. (2019) 9:101. doi: 10.3390/cells9010101
79. Carvalheira G, Malinverni AM, Moysés-Oliveira M, Ueta R, Cardili L, Monteagudo P, et al. The natural history of a man with ovotesticular 46,XX DSD caused by a novel 3-mb 15q26.2 deletion containing NR2F2 gene. J Endocr Soc. (2019) 3:2107–13. doi: 10.1210/js.2019-00241
80. Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, et al. Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46,XX children. Am J Hum Genet. (2018) 102:487–93. doi: 10.1016/j.ajhg.2018.01.021
81. Dangle P, Touzon MS, Reyes-Múgica M, Witchel SF, Rajkovic A, Schneck FX, et al. Female-to-male sex reversal associated with unique Xp21.2 deletion disrupting genomic regulatory architecture of the dosage-sensitive sex reversal region. J Med Genet. (2017) 54:705–9. doi: 10.1136/jmedgenet-2016-104128
82. Lamothe S, Bernard V, Christin-Maitre S. Gonad differentiation toward ovary. Ann Endocrinol (Paris). (2020) 81:83–8. doi: 10.1016/j.ando.2020.04.004
83. Philibert P, Biason-Lauber A, Gueorguieva I, Stuckens C, Pienkowski C, Lebon-Labich B, et al. Molecular analysis of WNT4 gene in four adolescent girls with mullerian duct abnormality and hyperandrogenism (atypical Mayer-Rokitansky-Küster-Hauser syndrome). Fertil Steril. (2011) 95:2683–6. doi: 10.1016/j.fertnstert.2011.01.152
84. Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. (2008) 82:39–47. doi: 10.1016/j.ajhg.2007.08.005
85. Naasse Y, Bakhchane A, Charoute H, Jennane F, Bignon-Topalovic J, Malki A, et al. A novel homozygous missense mutation in the FU-CRD2 domain of the R-spondin1 gene associated with familial 46,XX DSD. Sex Dev. (2017) 11:269–74. doi: 10.1159/000485393
86. Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. (2008) 29:220–6. doi: 10.1002/humu.v29:2
87. Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, et al. R-spondin1 is essential in sex determination, skin differentiation and Malignancy. Nat Genet. (2006) 38:1304–9. doi: 10.1038/ng1907
88. Ahmed SF, Keir L, McNeilly J, Galloway P, O'Toole S, Wallace AM. The concordance between serum anti-Mullerian hormone and testosterone concentrations depends on duration of hCG stimulation in boys undergoing investigation of gonadal function. Clin Endocrinol (Oxf). (2010) 72:814–9. doi: 10.1111/j.1365-2265.2009.03724.x
89. Mendez JP, Schiavon R, Diaz-Cueto L, Ruiz AI, Canto P, Söderlund D, et al. A reliable endocrine test with human menopausal gonadotropins for diagnosis of true hermaphroditism in early infancy. J Clin Endocrinol Metab. (1998) 83:3523–6. doi: 10.1210/jc.83.10.3523
90. Flück C, Nordenström A, Ahmed SF, Ali SR, Berra M, Hall J, et al. Standardised data collection for clinical follow-up and assessment of outcomes in differences of sex development (DSD): recommendations from the COST action DSDnet. Eur J Endocrinol. (2019) 181:545–64. doi: 10.1530/EJE-19-0363
91. Hembree WC, Cohen-Kettenis PT, Gooren L, Hannema SE, Meyer WJ, Murad MH, et al. Endocrine treatment of gender-dysphoric/gender-incongruent persons: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2017) 102:3869–903. doi: 10.1210/jc.2017-01658
92. Verkauskas G, Jaubert F, Lortat-Jacob S, Malan V, Thibaud E, Nihoul-Fékété C. The long-term followup of 33 cases of true hermaphroditism: a 40-year experience with conservative gonadal surgery. J Urol. (2007) 177:726–31. doi: 10.1016/j.juro.2006.10.003
93. Delforge X, Brachet C, Damry N, Segers V, Luyckx S, Heinrichs C, et al. A novel approach in the intraoperative management of ovotesticular DSD. J Pediatr Urol. (2020) 16:768–70. doi: 10.1016/j.jpurol.2020.10.007
94. Costanzo M, Touzon MS, Marino R, Guercio G, Ramirez P, Mattone MC, et al. Gonadal tumor development in 46,XX disorders of gonadal development. Eur J Endocrinol. (2022) 187:451–62. doi: 10.1530/EJE-22-0283
95. Kilberg MJ, McLoughlin M, Pyle LC, Vogiatzi MG. Endocrine management of ovotesticular DSD, an index case and review of the literature. Pediatr Endocrinol Rev. (2019) 17:110–6. doi: 10.17458/per.vol17.2019.kmv.endocrineovotesticulardsd
96. Gomes NL, Chetty T, Jorgensen A, Mitchell RT. Disorders of sex development-novel regulators, impacts on fertility, and options for fertility preservation. Int J Mol Sci. (2020) 21:2292. doi: 10.3390/ijms21072282
97. Florsheim N, Naugolni L, Zahdeh F, Lobel O, Terespolsky B, Michaelson-Cohen R, et al. Loss of function of FIGNL1, a DNA damage response gene, causes human ovarian dysgenesis. Eur J Endocrinol. (2023) 189:K7–k14. doi: 10.1093/ejendo/lvad127
98. Demain LAM, Metcalfe K, Boetje E, Clayton P, Martindale EA, Busby G, et al. Homozygous missense variants in BMPR15 can result in primary ovarian insufficiency. Reprod BioMed Online. (2022) 45:727–9. doi: 10.1016/j.rbmo.2022.05.003
99. Elzaiat M, Todeschini AL, Caburet S, Veitia RA. The genetic make-up of ovarian development and function: the focus on the transcription factor FOXL2. Clin Genet. (2017) 91:173–82. doi: 10.1111/cge.12862
100. Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet. (2010) 87:282–8. doi: 10.1016/j.ajhg.2010.07.007
101. Granada ML, Audí L. The laboratory in the multidisciplinary diagnosis of differences or disorders of sex development (DSD): I) Physiology, classification, approach, and methodologyII) Biochemical and genetic markers in 46,XX DSD. Adv Lab Med. (2021) 2:468–93. doi: 10.1515/almed-2021-0042
Keywords: non-CAH 46, XX DSD, disorders/differences in sex development, primary glucocorticoid resistance, aromatase deficiency, testicular/ovotesticular disorders/differences in sex development, DSD, gonadal dysgenesis
Citation: Yavas Abalı Z and Guran T (2024) Diagnosis and management of non-CAH 46,XX disorders/differences in sex development. Front. Endocrinol. 15:1354759. doi: 10.3389/fendo.2024.1354759
Received: 12 December 2023; Accepted: 01 April 2024;
Published: 15 May 2024.
Edited by:
Yukihiro Hasegawa, Tokyo Metropolitan Children’s Medical Center, JapanReviewed by:
Tomohiro Ishii, Keio University, JapanMagnus R. Dias da Silva, Federal University of São Paulo, Brazil
Copyright © 2024 Yavas Abalı and Guran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tulay Guran, dHVsYXlndXJhbkBtYXJtYXJhLmVkdS50cg==