 Zhaoqing Li
Zhaoqing Li Jia Liu
Jia Liu- Department of Thyroid Surgery, General Surgery Center, The First Hospital of Jilin University, Changchun, China
Recently, research into the link between thyroid dysfunction and Alzheimer’s disease (AD) remains a current topic of interest. Previous research has primarily concentrated on examining the impact of thyroid dysfunction on the risk of developing AD, or solely explored the mechanisms of interaction between hypothyroidism and AD, a comprehensive analysis of the mechanisms linking thyroid dysfunction, including hyperthyroidism and hypothyroidism, to Alzheimer’s disease (AD) still require further elucidation. Therefore, the aim of this review is to offer a thorough and comprehensive explanation of the potential mechanisms underlying the causal relationship between thyroid dysfunction and AD, highlighting the existence of a vicious circle. The effect of thyroid dysfunction on AD includes neuron death, impaired synaptic plasticity and memory, misfolded protein deposition, oxidative stress, and diffuse and global neurochemical disturbances. Conversely, AD can also contribute to thyroid dysfunction by affecting the stress repair response and disrupting pathways involved in thyroid hormone (TH) production, transport, and activation. Furthermore, this review briefly discusses the role and significance of utilizing the thyroid as a therapeutic target for cognitive recovery in AD. By exploring potential mechanisms and therapeutic avenues, this research contributes to our understanding and management of this devastating neurodegenerative disease.
1 Introduction
The dysfunction of the endocrine system is increasingly associated with the pathological processes of Alzheimer’s disease and other forms of dementia (1, 2). The thyroid, which is a crucial endocrine gland, significantly determines the development of the central nervous system (CNS). Regulated by the hypothalamic-pituitary-thyroid axis, the primary function of the thyroid gland is to produce, store, and secrete thyroxine (T4) and a lesser amount of triiodothyronine (T3). Thyroid hormone (TH), functioning as a crucial neuromodulator, is critical to the regulation of neuronal differentiation, synaptic development, and myelination (3). Normal thyroid function is essential for growth, behavior, as well as mental and neurological development. Hyperthyroidism is characterized by an excessive production and release of TH. Hyperthyroidism affects multiple organ systems and typically manifests with symptoms like increased appetite with weight loss, heat-related symptoms, tremors, anxiety, fatigue, disrupted sleep, and palpitations (4). Hypothyroidism is a disease characterized by hypometabolism due to insufficient synthesis and secretion or physiological effects of thyroxine. In children, hypothyroidism is characterized by delayed growth and development, reduced physiological function, and cognitive impairment (5). Signs of hypothyroidism in adults include weight gain, intolerance to cold, fatigue, hoarseness of voice, dry skin, hormonal imbalances, and memory impairment (6). However, the clinical presentation of thyroid dysfunction can vary significantly based on factors such as age, gender, and the duration between onset and diagnosis.
Alzheimer’s disease (AD), often colloquially referred to as late-onset dementia, is a gradually advancing degenerative disorder that specifically targets the CNS. Tobore (2019) offers a quintuple framework encompassing all known risk factors, including thyroid dysfunction, vitamin D deficiency, sex hormones, mitochondrial dysfunction, and oxidative stress, each of them has a direct effect on the development and physiological processes of AD, both independently and in combination with each other, as well as through genetic interactions (7). Numerous studies have consistently demonstrated that the β-Amyloid peptide (βA) deposition of extracellular senile plaque and the phosphorylation tau of intracellular neurofibrillary tangles are significant factors contributing to the initiation and advancement of the disease, and in addition, activated glial cells and enlarged endosomes can be observed under the microscope (8). But more research still needs to be done. The biological processes underlying neurodegeneration in Alzheimer’s disease are still hugely controversial, including other protein components that co-aggregate with βA, loss of synaptic homeostasis, clearance of damaged proteins, and network connectivity in the cortex (8, 9). Moreover, early diagnosis of the disease is a major challenge. These include the ambiguity of early diagnostic criteria, the limitations of diagnostic tools, and the fact that other neurodegenerative or cerebrovascular diseases can also cause early mild cognitive impairment (8). And the slow pathological changes in AD also make our early diagnosis difficult (10). Furthermore, the neurotoxic properties of βA and p-tau, along with their prion-like transmission nature, enable them to spread between neurons, sowing cytotoxic seeds that propagate cytotoxicity and exacerbate the pathological changes that characterize AD (11).
An increasing body of evidence suggests a strong link between thyroid dysfunction and Alzheimer’s disease (2, 3). Therefore, by analyzing the role of thyroid dysfunction as a potential cause and consequence of AD, as well as using the thyroid as a therapeutic target for treating AD, our objective is to gain a thorough understanding of the causal connection between thyroid dysfunction and AD. Additionally, we propose a concept of a “vicious circle” to describe this relationship, highlighting the reciprocal influence between the two conditions. We anticipate that these findings will lay the foundation for the creation of individualized and innovative treatment approaches for individuals who are impacted by AD.
2 The relevance between thyroid dysfunction and AD
The association between hyperthyroidism and heightened susceptibility to AD has been a subject of debate in the academic community. A cohort study conducted in 2006 by de Jong, den et al. challenged the idea that thyroid function significantly influences the onset of AD (12). Similarly, a 3-year follow-up study involving individuals over 85 years old concluded that thyroid function status has no impact on cognition (13). Another cross-sectional study conducted on 295 Chinese elderly patients with cognitive impairment, as reported by Hu, Wang et al. in 2016, observed no significant link between hypothyroidism or subclinical hyperthyroidism and AD or mild cognitive impairment (MCI) (14). Annerbo highlighted that low TSH levels are strongly linked to AD patients, but suspected that homocysteine rather than TSH might be associated with the development of AD and homocysteine was negatively correlated with TSH (15). Furthermore, a bidirectional two-sample Mendelian randomization study (16) and a meta-analysis (17) both concluded that there is no causal association between hyperthyroidism and AD. However, a study in 2010 found that although there was no association between TT3 and cognition in the control group, patients with MCI exhibited a negative relationship between serum TT3 levels and cognitive function. This implies that higher TT3 levels, combined with increased AD pathological changes, might contribute to a deterioration in cognitive function (18).
Some findings have also indicated that elevated levels of T4 are linked to an elevated likelihood of developing dementia and AD (19, 20). According to a longitudinal study conducted by Yeap, higher circulating levels of fT4 are an independent predictor of dementia in elderly men (21). At the same time, other researches have shown a closer linkage between lower TSH concentrations and a higher risk of AD. In comparison with individuals having normal TSH levels, a decrease in TSH by 6 months is linked to a 16% higher risk of developing dementia (22). A meta-analysis of cohort studies involving 344 248 individuals revealed that only subclinical hyperthyroidism exhibited a connection with an increased risk of dementia, with TSH concentrations ranging from 1.55-1.60mU/L being associated with the highest risk (23). In contrast, subclinical hypothyroidism, clinical hyperthyroidism and clinical hypothyroidism had no effect on dementia (23). In 2000, Kalmijn et al. conducted a prospective study focused on subclinical hyperthyroidism in the elderly, specifically examining its association with dementia and AD. Interestingly, after accounting for potential confounding factors such as age, gender, and atrial fibrillation, the study discovered that this correlation persevered and held substantial weight (24). Additionally, it was observed that individuals with positive TPO-Abs (Antibodies to Thyroid Peroxidase) coexists with low TSH levels were particularly vulnerable to developing dementia, which reminds us of the importance of autoimmune thyroid disease in AD (24).
Research has consistently demonstrated a strong link between hypothyroidism and an elevated likelihood of developing AD. Significant hypothyroidism can impair various cognitive functions, including thinking, speech, attention, concentration, general intelligence, perceptual function, psychomotor function, and executive function (25). Specifically, memory experiences the greatest impact (25). A reanalysis of eight case-control studies exposed a significantly heightened risk of AD in patients with hypothyroidism (26). Similarly, Quinlan’s prospective study in 2014 discovered an inverse correlation between lower levels of serum FT3 and the risk of AD development, and comparatively individuals in the lowest quartile of FT3 levels exhibited more than twice the risk of subsequently developing AD in comparison to those in the highest quartile (27).
We present our perspective on the ongoing debate surrounding the relationship between thyroid dysfunction and the risk of developing Alzheimer’s disease. One of the key issues in the research is the use of inadequate sample sizes and inappropriate proportions (13, 17, 20). Insufficient total sample size and a small proportion of the population with thyroid dysfunction can result in less accurate study effect estimates. Another important factor to consider is the limitations of the study methodology. In cross-sectional studies, it is challenging to determine the chronological order of thyroid dysfunction and Alzheimer’s disease (14, 18). Moreover, most prospective studies only assess thyroid status once during the follow-up period, without tracking any changes in the participants’ thyroid function (12, 13, 24). This lack of information regarding spontaneous normalization of thyroid function or other interventions can impact the study’s effectiveness. Additionally, the absence of sufficient follow-up time and imprecise assessment of cognitive status during follow-up are areas that require improvement and further development (12, 24). Misclassification of thyroid function is also a potential concern, as thyroid hormone requirements vary based on age, gender, and race (15, 17, 19, 20). More critically, few measurements of CNS levels of TH are currently available. Another challenge lies in the unclear diagnosis of Alzheimer’s patients (20). Most studies solely rely on cognitive status assessment and clinical presentation without a clear pathological diagnosis, making it difficult to determine whether individuals with thyroid dysfunction have developed Alzheimer’s disease pathology without cognitive impairment after several years of follow-up. It is also unclear whether pathological changes associated with Alzheimer’s disease were already present before the onset of thyroid dysfunction, and cognitive function had not yet been affected. In conclusion, future studies should focus on appropriate sample sizes and proportions, accurate diagnosis of thyroid functional status, and precise prospective studies for the diagnosis of Alzheimer’s disease.
3 Thyroid dysfunction as the result of AD
Viewed from the thyroid status in the early stages of AD, it appears that there is an increased risk of hyperthyroidism. Ren et al. created an early Alzheimer’s disease (AD) rat model by injecting okadaic acid (OA) into the rats’ hippocampus, which activated GSK-3, increased higher oxidative stress, generated tau hyperphosphorylation, and increased βA neurotoxicity. The study’s findings demonstrated a notable increase in FT3, FT4, TSH, and TRH levels within the brain tissues of rats in the early stages of AD, and there was a notably higher expression of TH receptors in the hippocampal tissues compared to the control group, along with FT4, TSH, and TRH were up in blood, with no change in FT3 (28). As a result, it came up that the hyperthyroid condition observed in the brain and circulation during early AD could be an early local and systemic stress repair response. Furthermore, Quinlan et al. discovered that elevated serum T4 levels in relatively early AD were not accompanied by elevated T3 levels, and that these patients did not have thyroid dysfunction or use drugs that could alter TH levels, which is consistent with Ren’s findings (29). These findings imply that peripheral T4 to T3 conversion is decreased in early AD. It remains unclear whether this result is due to a self-protective mechanism of high serum T4 in early Alzheimer’s disease or a result of lower deiodinase activity during the primary pathological process in AD patients. Unfortunately, both Ren and Quinlan et al. failed to detect and analyze deiodinase, despite both acknowledged its importance in this process. Therefore, the levels of deiodinase in the circulation and brain tissue of patients with early Alzheimer’s disease need to be further investigated.
As the pathogenic process of AD worsens, there is a continuous accumulation of toxic βA. The development of a hypothyroid state is linked with the disease’s progression to the middle and late stages. According to our viewpoint, the loss of compensation for the stress repair response and the expanding lesion are two essential factors that contribute to this. In the first study exploring the hypothalamic-pituitary-thyroid axis in AD, the levels of TRH, TSH and TH were significantly lower in patients with AD (30). In the AD cases, there were no significant correlations observed between TRH and TSH, TT3, TT4, FT3, and FT4, as well as between TSH and TT3, TT4, FT3, FT4 (30). However, in the healthy controls, TRH showed significant correlations with TSH and FT4, while TSH exhibited significant correlations with TT4 and FT4 (30). This shows a severe deterioration of the biofeedback regulation of hypothalamic-pituitary-thyroid axis. Similarly, Chen et al. discovered that serum TSH levels in AD patients lacked a circadian rhythm and were significantly lower than health controls (31). Hence, we speculate that diminished TRH and TSH secretion as well as the absence of normal feedback control caused by degeneration and impaired function of the hypothalamus and pituitary gland, result in the development of hypothyroidism in patients with AD during the mid to late stages. Insufficiently, to our knowledge, as the studies mentioned above have mostly analyzed the function of the hypothalamic-pituitary-thyroid axis in patients with Alzheimer’s disease through changes in the levels of thyroid hormones and related hormones, and there is a lack of definitive pathological diagnosis of definite hypothalamic and pituitary degeneration. In the future, a clear pathological diagnosis of hypothalamic and pituitary degeneration during disease progression in AD is needed.
As Alzheimer’s disease progresses, TH transport mechanisms may be also disrupted. A huge calcium inflow is caused by increased ryanodine receptor expression in AD patients, activating the nNOS expression in neurons and leading to overproduction of NO, which directly accelerating the evolution of the AD pathology and suppresses monocarboxylate transporter 1 (MCT1) expression in both neurons and glial cells (32). We speculate that the same mechanism may also apply to the thyroid hormone transporters MCT8 and MCT10, but unfortunately no evidence has yet been found to confirm our speculation. TTR (transthyretin) is a homotetrameric protein, with four identical subunits, the liver secretes it into the bloodstream while the choroid plexus secretes it into the cerebrospinal fluid (33). When toxic pathological products of AD disrupt the structure and function of the choroid plexus, the concentration of TTR in the brain decreases. TTR is responsible for the transfer of T4 from the bloodstream to the cerebrospinal fluid, thus ensuring that T4 is not lost across the blood-brain barrier and maintaining an optimal concentration of T4 in the brain (33). However, it has also been suggested that TTR was not necessary for the upkeep of thyroid hormone homeostasis (34). TTR has been reported to function as a pro-angiogenic agent during early stage AD development by upregulating the expression of VEGF, Ang-2, IL-6 and IL-8 (33). Meanwhile, TTR can also regulate βA metabolism by inhibiting βA production, accelerating βA clearance and attenuating βA toxicity (35). Therefore, the stability of TTR function is essential for the whole pathological process of AD and plays a key neuroprotective role. However, it remains unsatisfactorily investigated whether TH transport is impaired in AD. First, the mechanism of MCT1 downregulation during AD pathology is uncertain as to whether it applies to MCT8 or MCT10. Second, the role of TTR in maintaining TH concentrations in the brain is controversial. Hence, we are looking forward to further studies on TH transport during AD pathology.
Deiodination is the initial stage in the activation or inactivation of thyroid hormone, and deiodinases are important in this process (36). The exocyclic deiodination of T4 and its conversion to the active T3 are the primary reactions catalyzed by iodothyronine deiodinase 1 (DIO1) and iodothyronine deiodinase 2 (DIO2), which promote the activation pathway. In the inactivation pathway, iodothyronine deiodinase 3 (DIO3) acts as an essential endocyclic deiodinase by catalyzing the conversion of active T4 and T3 to inactive reverse-T3 and T2 (36). DIO2 and DIO3 are deiodinases that function principally in the central nervous system and are subcellularly localized in endoplasmic reticulum membranes and cell membranes, respectively (36). Based on a whole transcriptome study, a significant transcriptional difference specific to late-onset AD was found, in which the expression of DIO2 was reduced (37). The high oxidative stress in Alzheimer’s patients also decreases DIO2 activity and increases DIO3 activity (36). Wajner et al, by modelling deiodinase dysfunction under IL-6-induced oxidative stress, found that after partial correction of the oxidative stress state by n-acetylcysteine supplementation, DIO3 activity was fully restored, whereas DIO2 or DIO1 function was not (38). Therefore, how oxidative stress destroys deiodinase function, and whether there are etiological factors other than oxidative stress, for deiodinase dysfunction that occurs during the pathological process of Alzheimer’s disease are directions that need to be explored and investigated more in the future.
In our conclusion but not a proven fact, as the pathophysiological process of AD progresses, the functional state of the thyroid gland undergoes changes. It can be divided into two stages: early hyperthyroidism due to stress repair response, and a mid to late phase hypothyroid state caused by abnormalities in TH production, transport, and activation (Figure 1).
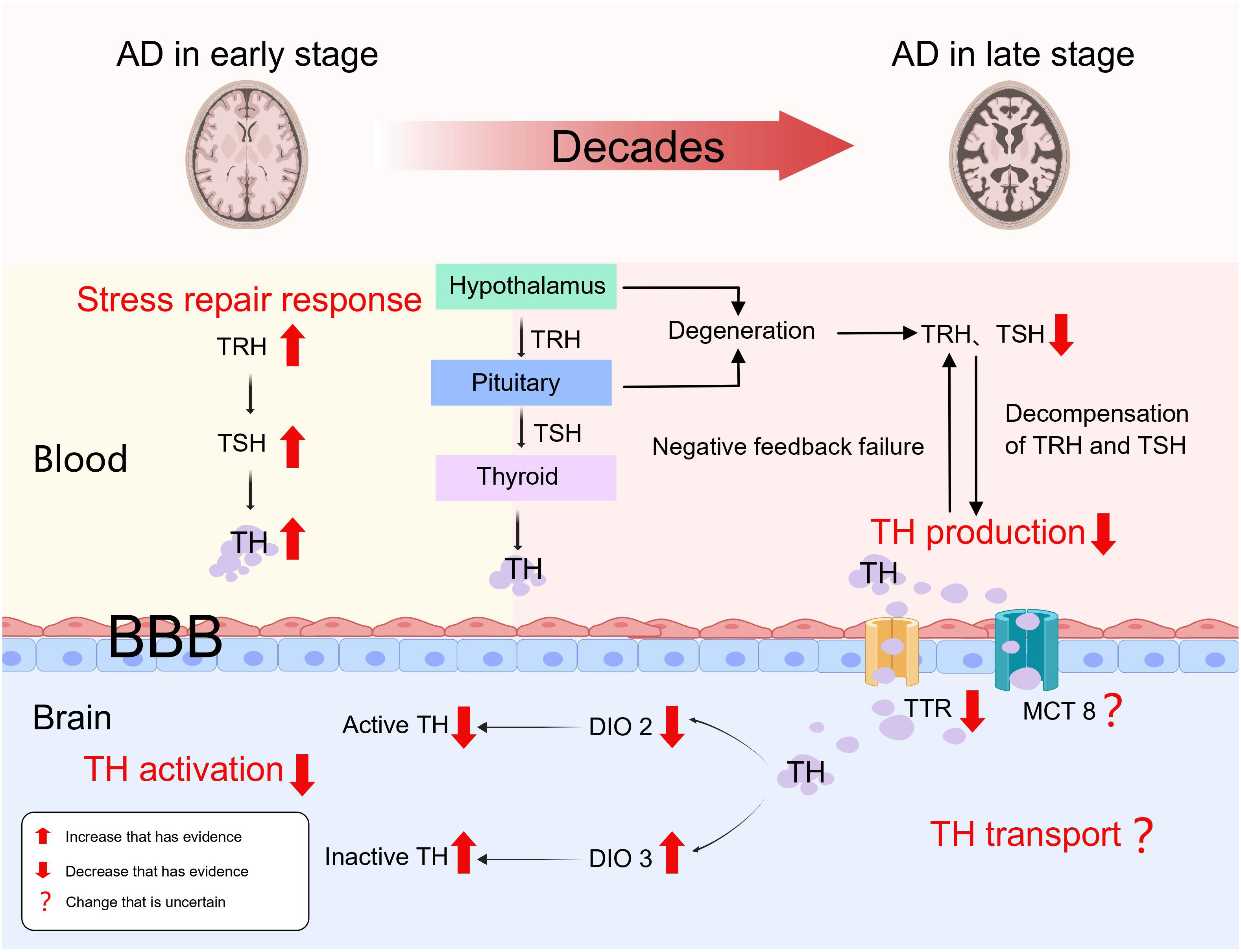
Figure 1 Thyroid dysfunction as a result of AD.
4 Thyroid dysfunction as the cause of AD
4.1 The effect of hyperthyroidism on AD
The underlying processes of the relationship between hyperthyroidism and Alzheimer’s disease must be thoroughly studied. According to an autopsy in 2009, more neurofibrillary tangles and neocortical plaques were found in patients having higher total thyroxine levels (39). For the first time, Li, Yang et al. discovered in 2020 that patients with hyperthyroidism exhibited notably elevated levels of total serum tau in comparison to those with normal thyroid function (40). These observations serve as a reminder that tau protein aggregation and phosphorylation are hallmarks of an AD pathological process that may be present in hyperthyroid patients. But unfortunately, in these studies, we were not able to find a causal relationship between high thyroid hormone levels and the development of AD. More importantly, plasma Tau levels only partially reflect the pathological process of AD and their diagnostic accuracy is poor compared to cerebrospinal fluid Tau levels (41). We look forward to finding more sensitive, specific and easily sampled biomarkers for AD in the future.
At the cellular level, Hyperthyroidism coexists with a lipid peroxidation state which can be inhibited by treatment of hyperthyroidism (42). In addition, liver mitochondria in the hyperthyroid state have a strong ability to scavenge H2O2, resulting in a substantial production of reactive oxygen species (ROS) and damaging redox equilibrium (43). However, these findings only demonstrate the high oxidative stress state of hyperthyroidism from peripheral tissues and do not prove whether the same mechanism applies to the central nervous system. Nonetheless, it is clear that correcting the hyperthyroid state improves the state of oxidative stress throughout the body (44). And the use of antioxidants can reduce the oxidative stress and neuronal cell death in the hippocampus of the brain caused by hyperthyroidism (45). Oxidative stress aggravates the pathogenic process of AD by promoting the accumulation of βA, hyperphosphorylation of tau, and the loss of synapses and neurons (46). A vicious loop is created when abnormal βA or phosphorylated tau protein builds up, further promoting redox imbalance (47). More crucially, when the amount of ROS produced in some neurodegenerative diseases—like Alzheimer’s disease—is too high, the delicate balance that fragile neurons maintain is more likely to be upset, which increases the likelihood of synapse loss and dendritic damage, ultimately resulting in neuronal death and axonal pathology (48). In addition, the excessive presence of thyroid hormone hampers the neuronal proliferation, regeneration and repair processes by impeding the expression of DNA synthesis and cell cycle-related antigens in the region of proliferating cells within the CNS (49). In a newly published study, hyperthyroidism exacerbates cognitive impairments and increases the accumulation of βA plaques in mice by the activation of neuroinflammation and the induction of brain tissue necroptosis via the RIPK3/MLKL pathway (50).
In addition, elevated thyroid hormone can cause the atrophy of hippocampal and temporal lobe structures (12). According to research findings, the administration of excessive thyroid hormone in young rats has been observed to lead to a notable reduction in long-term potentiation (LTP), deficits in cognitive functions, as well as significant neurochemical and morphological changes in the hippocampus (51). As reported, T4 has the ability to trigger long-term depression (LTD) and reduce LTP through a process known as non-genomic, membrane priming effect (52). But the molecular mechanisms of how TH mediates LTD and LTP await further anatomical and pharmacological analysis. It was also found that people diagnosed with hyperthyroidism have a reduced functional connectivity between the hippocampus and the cerebral cortex (53). In hyperthyroid mice, a reduction in the density of mature dendritic spines in the hippocampus has been observed, accompanied by impaired function of postsynaptic α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPAR) and N-methyl-D-aspartate receptors (NMDAR), leading to decreased excitatory synaptic transmission (54). Also, the expression of NMDAR subunit GRIN2B gene is downregulated in the hippocampus tissue, which affects spatial memory that depends on the hippocampus (55). Moreover, hyperthyroidism has been found to potentially result in a decline in brain-derived neurotrophic factor (BDNF) levels (56). As a result, patients with hyperthyroidism may face difficulties in learning tasks that heavily rely on the hippocampus and are prone to forgetting them at a faster rate. Additionally, as TRH levels drop, the MAPK pathway’s signal weakens, releasing its inhibition on GSK3-activity and causing tau to become hyperphosphorylated (57).
The brain metabolism of patients with hyperthyroidism is also significantly altered, with diffuse and global neurochemical disturbances. A seminal study conducted the first functional brain imaging investigation in individuals with thyrotoxicosis, revealing a noteworthy decrease in glucose metabolism within the limbic system, a brain region crucially involved in the consolidation of long-term memory (58). According to Danielsen et al., it was observed that there was a sustained disturbance in the glutamate-glutamine cycle within the cerebral white matter in cases of hyperthyroidism, and additionally, the levels of total choline and myo-inositol were significantly lower during the acute phase of hyperthyroidism compared to the control group (59). Also, the importance of TRH for the significant and long-lasting release of endogenous acetylcholine in the hippocampus and the increased acetylcholinesterase activity in hyperthyroidism remind us of the importance of hyperthyroidism on the metabolism of acetylcholine (60, 61). These studies suggest that there may be a loss of acetylcholine due to TRH depletion, the elevation of acetylcholinesterase activity or other reasons yet to be explored in the hyperthyroid state, leading to cognitive impairment. However, to our knowledge, no study has confirmed the reduction of acetylcholine in the hyperthyroid state. What’s more, the activation of the thyroid hormone response system has been observed to result in the upregulation of neuroserpin in the brain, which is the primary inhibitor of tissue plasminogen activator within the brain, and its upregulation leads to the deposition of βA in the brain (62). Besides, a nationwide study in Spain showed that hyperthyroidism increases the risk of cardiovascular disease (63). Meanwhile, the impact of cardiovascular disease on AD can be explained by the “two-hit hypothesis”, namely reduced cellular blood flow and blood-brain barrier dysfunction (64). These two events have a synergistic role in the accumulation of βA in the brain, which leads to neuronal loss and cognitive decline.
It is worth noting that these aforementioned influences do not exist in isolation, but rather interact and mutually affect each other. And we must emphasize the lack of convincing conclusions between hyperthyroidism and AD. By our speculation, hyperthyroidism could disturb the structure and function of neurons and the hippocampus, disturb the redox balance, alter the metabolic state of the brain, upregulate the expression of serine proteases, and even exposes CNS to a “two-hit” attack through the cardiovascular system. These concurrent mechanisms may all be involved in the pathogenesis of Alzheimer’s disease (Figure 2).
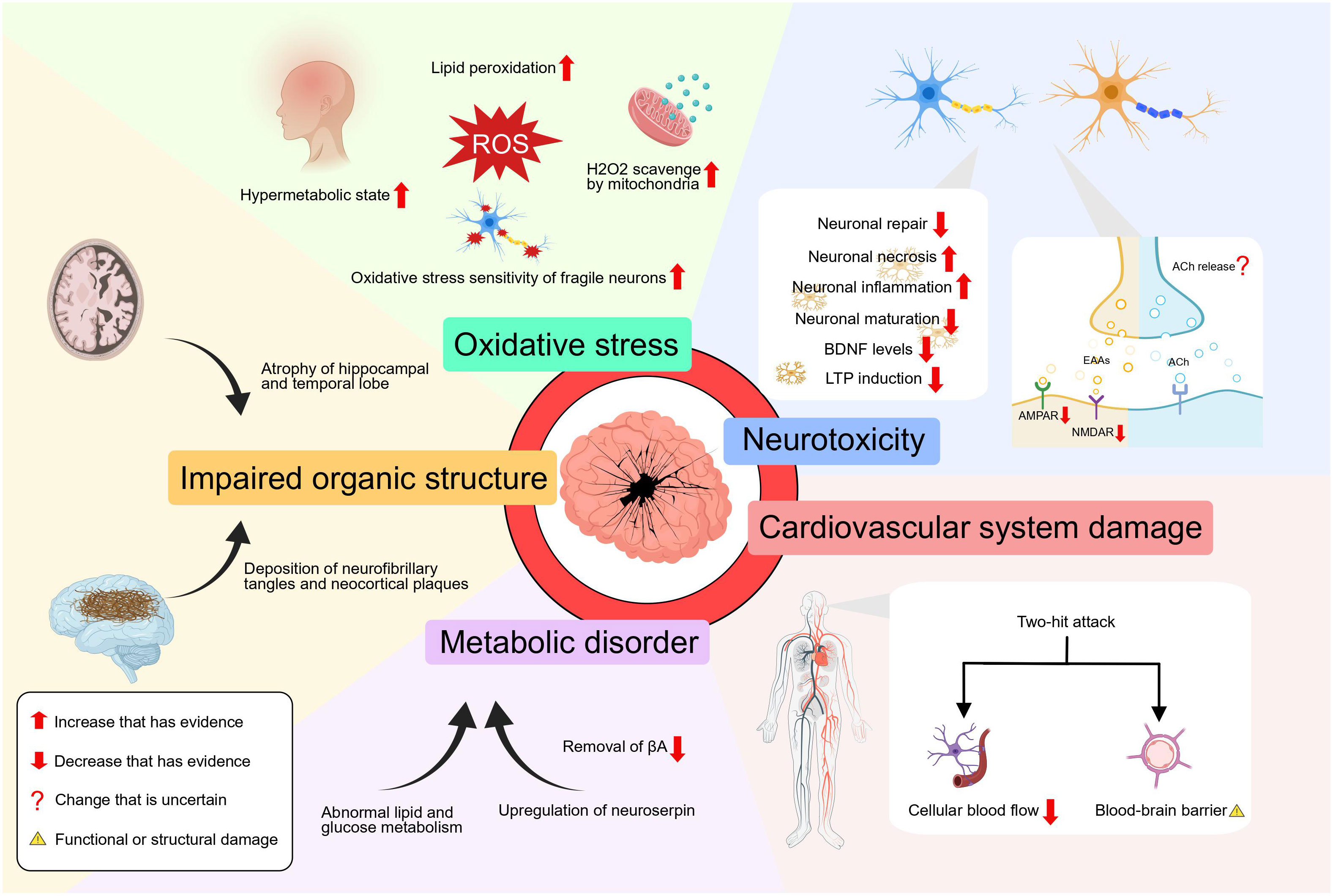
Figure 2 The effect of hyperthyroidism on AD.
4.2 The effect of hypothyroidism on AD
Thyroid hormone acts as a neuromodulator and crucially impacts mood regulation, neurotransmitter function and regulation, brain development and function, neuroprotection and brain metabolism. The hippocampus displays a high concentration of receptors for TH and dysregulation of TH results in significant impairment of both cognitive and memory functions (65). Thyroid hormone enhances the action of nerve growth factor and promotes the growth of neurites (66). Thyroid hormones can also affect adult hippocampal neurogenesis by stimulating the proliferation, survival and differentiation of dentate granule cell progenitors (67). Expression of significant components in the MAPK/ERK-CREB and Ca2+/Calmodulin pathways, which are crucial for synaptic plasticity, decreases in the hypothyroid state and restored after thyroid hormone supplementation (68). Meanwhile, antithyroid drug administration led to a reduction in serum BDNF levels and a developmental delay in primary hippocampal neurons in rats, and the addition of BDNF can rescue this variation (69). Input-output connections and LTP of the perforant pathway were greatly reduced in animals with hypothyroidism, and the ability of synapses between the perforant pathway and the dentate gyrus to undergo LTP was significantly impaired (70).
Hypothyroidism can lead to impaired glutamatergic synaptic function. On one hand, the release of the excitatory neurotransmitter glutamate is decreased (71) and there is also less expression of the postsynaptic neuronal NMDAR subunit NR1 mRNA in the hippocampus (72). This decrease causes a block and delay in synaptic transmission, it also inhibits the proliferation and differentiation of neural stem cells (73). On another, the reduction in thyroid hormone makes neurons more sensitive to the neurotoxic effects of glutamate, which may be attributed to the ability of thyroid hormone in regulating extracellular levels of glutamate by modulating transporter proteins on astrocytes (74). Hypothyroidism causes abnormalities in cholinergic neurons, with significant reductions in hippocampal acetylcholine content and choline acetyltransferase activity, and the cholinergic neurons exhibit abnormal electrophysiological properties including depolarization of resting membrane potentials and elevated thresholds (75–77). Also, evidence from many studies has shown the importance of thyroid hormone supplementation combined with the use of cholinesterase inhibitors in reversing synaptic damage and restoring cognitive function (75, 78).
Hypothyroidism facilitates AD via APP (Amyloid Precursor Protein) and TREM2 (Triggering Receptor Expressed on Myeloid Cells 2) gene expression. Preclinical research indicates that thyroid hormones block APP gene expression by reducing histone H3 acetylation and histone H3 lysine 4 methylation, and thus reduce βA formation (79). Regulation of TREM2 gene expression on microglia by thyroid hormones has also been demonstrated (80). If TREM2 dysfunction occurs, it may alter the response of microglia to βA and may be involved in the etiology of Alzheimer’s disease through immune and inflammatory pathways (81). Hypothyroidism also leads to an increase in pro-inflammatory factors such as IL1β, IL6 and TNF expression within the hippocampus (68). Activation of interleukin 1 (IL-1) prompts autophagy, which in turn, triggers the death of brain cells in the hippocampus resulting in impaired cognitive function in young rats (82). At the same time, autophagic vesicles possess the necessary components for βA synthesis, and the exaggerated autophagic pathway contributes to βA accumulation (83). Furthermore, research has also shown that hypothyroidism can upregulate autophagy and apoptosis in neurons of the cerebellum, and the autophagic process can be attenuated by thyroid hormone replacement (84). One study summarized the relevance of autophagy failure to ageing and AD, while suggesting the possibility of stimulating autophagy in AD therapy (85, 86). This suggests to us that the autophagy pathway may be overwhelmed by the accumulation of oxidative stress and cellular damage, resulting in relative autophagic insufficiency. How to effectively regulate autophagic activity may become a potential treatment for AD and other neurodegenerative diseases.
Hypothyroidism can induce damage to the hippocampus and trigger oxidative stress states by activating endoplasmic reticulum stress (87). Meanwhile, endoplasmic reticulum stress is a critical step in multiple neurodegenerative pathologies that are marked by the buildup of misfolded proteins and aggregates (88). Due to the important function of which thyroid hormones play in mitochondrial function and energy metabolism, mitochondrial dysfunction may also occur in thyroid hormone deficiency (89). Damage to mitochondria may increases the vulnerability of neurons to oxidative stress (90). And oxidative stress and mitochondrial dysfunction have key roles in the pathogenesis and progression of AD (91). In brain metabolism, thyroid hormone enhances insulin signaling in the hippocampus, decreases activation of glycogen synthase kinase 3 (GSK3) and tau protein levels of the hippocampus, and slows neurodegeneration (92). Age-related hypothyroidism increases transportation of APOE4-loaded exosomes from the liver to the brain, ultimately leading to disrupted lipid metabolism in the brain, accelerated accumulation of ROS and promotion of neuronal cell death (93). Meanwhile, new study in mouse model with subclinical hypothyroidism has demonstrated the possible participation of endoplasmic reticulum stress in lipid metabolism disorders (94). Cerebral blood flow to brain regions associated with memory is lower in patients with subclinical hypothyroidism and mild hypothyroidism (95, 96). Long-term inadequate blood flow to the brain specifically lowers neuronal activity in the limbic system, damages cognitive memory, and participates in the abnormal accumulation of βA and p-Tau that occur in AD (97).
As shown above, the impact of hypothyroidism on AD can be examined through various mechanisms, including diminished neuromodulation, alterations in gene expression, impaired autophagy, elevated oxidative stress, and cerebral metabolic dysfunctions (Figure 3). It should be noted that these elements do not operate independently, but instead collaborate synergistically to foster the onset and advancement of AD. And we must point out that these are only possible mechanisms for the link between hypothyroidism and Alzheimer’s disease. The exact mechanisms for the connections between the two are inconclusive and need to be determined in future studies.
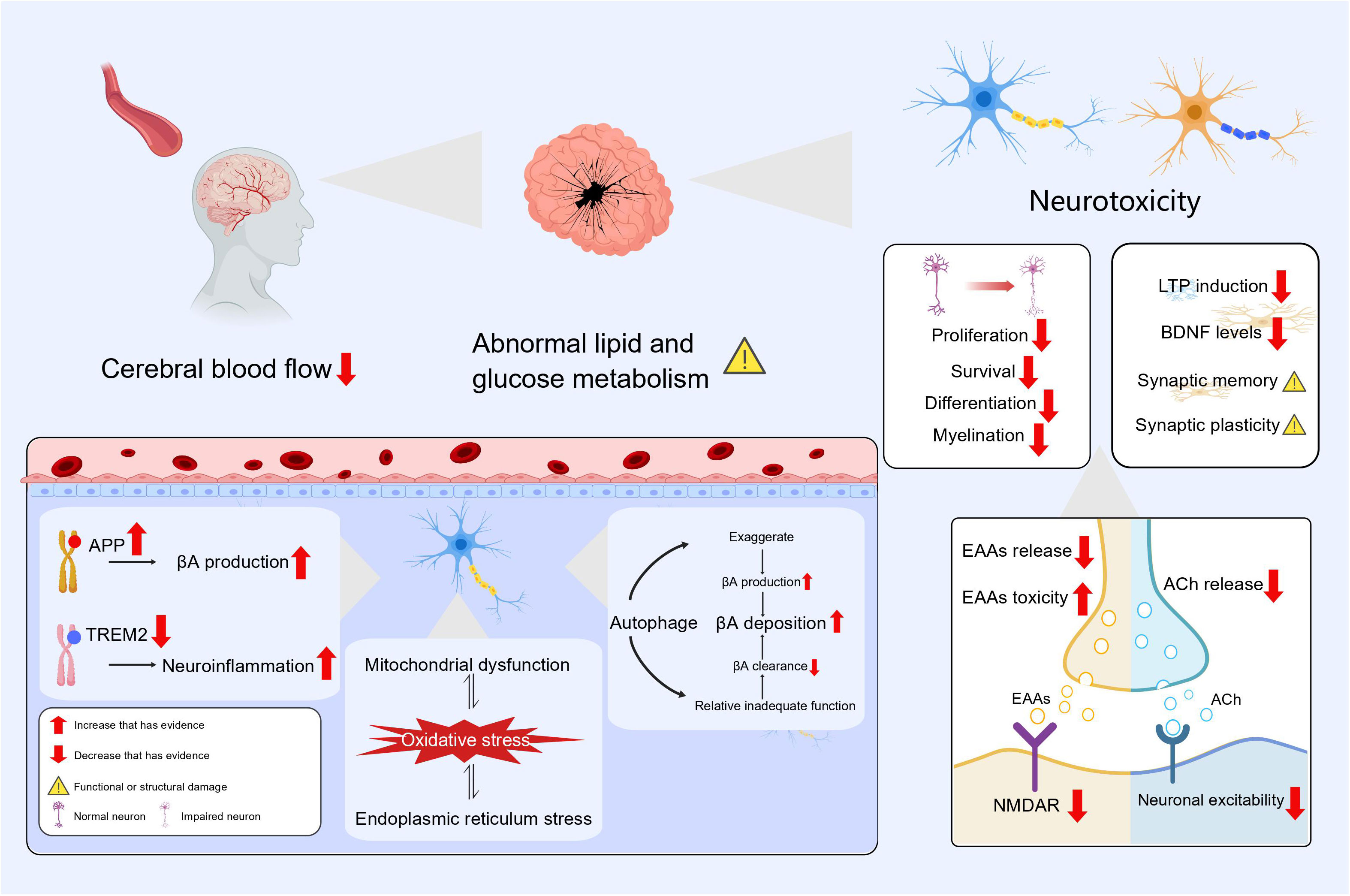
Figure 3 The effect of hypothyroidism on AD.
5 Thyroid as a target for AD therapy
In recent years, many studies have highlighted the significance of antithyroid therapy in enhancing cognitive function. A systematic review encompassing 31 high-quality studies conducted over the past two decades, focusing on the quality of life of patients with primary hyperthyroidism following thyroidectomy, strongly indicates that treatment of hyperthyroidism leads to a significant and enduring improvement in neurocognitive status (98). Antithyroid medication improves learning ability in individuals with hyperthyroid dementia, and parts of the frontoparietal lobe with uptake impairments gradually respond to the medicine (99). However, antithyroid therapy does not completely reverse the symptoms of dementia that occur in hyperthyroid patients. Kumar et al. discovered that, by examining changes in key metabolites in the posterior parietal cortex and the dorsolateral prefrontal cortex in hyperthyroid patients with an overactive thyroid before and after receiving medication to treat the condition, the proportion of neuro-metabolites in brain regions, while reversible, was not completely reversed even when normal thyroid function was achieved (100). Meanwhile, although functional brain connections and cognitive abilities were improved in hyperthyroid patients treated with antithyroid therapy, they were not totally recovered (101).
Similarly, proper care of hypothyroid patients is critical. Thyroid hormone supplementation therapy can reverse memory deficits and alleviate cognitive deficits by reducing βA production, decrease neuroinflammation, stimulating synaptic plasticity and memory enhancement, and restoring key indicators of cell signaling pathways in the hippocampus (68). Thyroid hormone replacement treatment is required for the restoration of hippocampus impairments caused by hypothyroidism (102). However, similar to the response to antithyroid therapy, the response to thyroid hormone replacement therapy is not complete or sufficient. Farag et al. found that thyroid hormone supplementation has the ability to repair and preserve synapses and dendrites within the hippocampus, but they also noted that this regeneration is insufficient and incomplete (103). Yet, that study only demonstrated histological hippocampus recovery and did not include additional functional and neurophysiological assessments (103). Meanwhile, a systematic review of thyroid hormone replacement therapy for cognitive dysfunction discovered that thyroid hormone benefits in the treatment of cognitive deficits resulting from hypothyroidism, but that this therapeutic effect is not fully and always effective, and may even result in side effects (104).
With the above, it can be seen that the management and treatment of patients with hyperthyroidism or hypothyroidism can partially ameliorate and alleviate symptoms, although it does not completely normalize cognitive function altogether. The incomplete recovery of cognitive function in patients after antithyroid therapy or thyroid hormone replacement may be attributed to several factors. Firstly, the multifaceted nature of the etiology of Alzheimer’s disease and the complexity of the pathology contribute to this issue (7, 8). Secondly, individual differences among patients, such as age, gender, genetics, lifestyle, the presence of underlying diseases, and even the duration of the abnormal thyroid status, also play a role. Thirdly, the limited action of thyroxine and the uncertainty of thyroxine levels in the brain., particularly the regionalized effects of thyroid hormones on brain metabolism and selective differential contributions to hippocampal circuitry and function, have been observed (79, 100). And in some studies, there was a slight improvement in serum thyroid hormone levels, but brain thyroid hormone levels were not re-established (103). Lastly, the limited sample size and insufficient follow-up period in previous studies suggests whether inadequate recovery is related to insufficient treatment duration (100, 101). Further research is needed with larger sample sizes and longer durations, including different time points for evaluation. In the future, normalization of thyroid status in combination with other interventions such as antioxidants, cholinesterase inhibitors, stabilization of neuronal calcium ions and modulation of autophagy have strong therapeutic potential for neurodegenerative diseases such as AD (45, 78, 83, 105).
Even in cognitively normal subjects with an abnormal brain βA burden, it usually takes more than 10 years before cognitive decline begins (10). More critically, maintaining homeostasis of thyroid hormone levels within the brain is critical to the proper functioning of the CNS, and even relatively minor deviations in brain hormone levels can cause severe behavioral and cognitive impairment (106). Therefore, it is vital to provide suitable therapy at the best time for each individual patient to prevent and treat cognitive deficits caused by thyroid dysfunction.
6 Conclusion
In this discussion, we have provided a detailed analysis of the interrelationship between thyroid dysfunction and Alzheimer’s disease (AD). However, we must admit that the exact mechanisms and the sequence of events between thyroid dysfunction and AD have not yet been convincingly concluded. We require longer-term, large-scale clinical trials and more in-depth basic research at various levels, including molecular, cellular, and animal models. Meanwhile, methods for measuring thyroid hormone levels in the central nervous system, more appropriate criteria for assessing thyroid function in specific populations, and more sensitive, specific and convenient diagnostic methods for Alzheimer’s disease need to be further explored. We look forward to more in-depth studies in the future to gradually improve the pathological mechanisms between thyroid dysfunction and Alzheimer’s disease, and to find a more comprehensive and convincing network of mechanisms. Such efforts will help develop more effective clinical treatment strategies. Moreover, it is crucial to prioritize early detection, diagnosis, and treatment by assessing thyroid function in individuals at a high risk of developing neurodegenerative diseases. This approach could significantly contribute to the prevention and deceleration of Alzheimer’s disease progression.
Author contributions
ZL: Writing – original draft. JL: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We are grateful to all the researchers who have worked on this topic. Incredibly grateful for Dr. Dan Li (North University of China, Taiyuan, China) for helping in this revision.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Palm R, Ayala-Fontanez N, Garcia Y, H-g L, MA S, Casadesus G. Neuroendocrinology-based therapy for alzheimer's disease. Biofactors (2012) 38:123–32. doi: 10.1002/biof.1011
2. Ge F, Zhu D, Tian M, Shi J. The role of thyroid function in alzheimer's disease. J Alzheimers Dis (2021) 83:1553–62. doi: 10.3233/JAD-210339
3. Bavarsad K, Hosseini M, Hadjzadeh MA, Sahebkar A. The effects of thyroid hormones on memory impairment and alzheimer's disease. J Cell Physiol (2019) 234:14633–40. doi: 10.1002/jcp.28198
4. Leo SD, Lee SY, Braverman LE. Hyperthyroidism. Lancet (2016) 388:906–18. doi: 10.1016/S0140-6736(16)00278-6
5. Salerno M, Capalbo D, Cerbone M, Luca FD. Subclinical hypothyroidism in childhood - current knowledge and open issues. Nat Rev Endocrinol (2016) 12:734–46. doi: 10.1038/nrendo.2016.100
6. Chaker L, Bianco AC, Jonklaas J, Peeters RP. Hypothyroidism. Lancet (2017) 390:1550–62. doi: 10.1016/S0140-6736(17)30703-1
7. Tobore TO. On the etiopathogenesis and pathophysiology of alzheimer's disease: A comprehensive theoretical review. J Alzheimers Dis (2019) 68:417–37. doi: 10.3233/JAD-181052
8. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers (2021) 7(1):33. doi: 10.1038/s41572-021-00269-y
9. Rahman MM, Lendel C. Extracellular protein components of amyloid plaques and their roles in alzheimer's disease pathology. Mol Neurodegener (2021) 16:59. doi: 10.1186/s13024-021-00465-0
10. Choi BW, Kim S, Kang S, Won KS, Yi H-A, Kim HW. Relationship between thyroid hormone levels and the pathology of alzheimer’s disease in euthyroid subjects. Thyroid (2020) 30:1547–55. doi: 10.1089/thy.2019.0727
11. Condello C, Merz GE, Aoyagi A, DeGrado WF, Prusiner. SB. Aβ and tau prions causing alzheimer's disease. Methods Mol Biol (2023) 2561:293–337. doi: 10.1007/978-1-0716-2655-9_16
12. de Jong FJ, den Heijer T, Visser TJ, de Rijke YB, Drexhage HA, Hofman A, et al. Thyroid hormones, dementia, and atrophy of the medial temporal lobe. J Clin Endocrinol Metab (2006) 91:2569–73. doi: 10.1210/jc.2006-0449
13. Formiga F, Ferrer A, Padros G, Contra A, Corbella X, Pujol R, et al. Thyroid Status and Functional and Cognitive Status at Baseline and Survival after 3 Years of Follow-Up: The Octabaix Study. Eur J Endocrinol (2014) 170:69–75. doi: 10.1530/eje-13-0722
14. Hu Y, Wang ZC, Guo QH, Cheng W, Chen YW. Is thyroid status associated with cognitive impairment in elderly patients in China? BMC Endocr Disord (2016) 16:11. doi: 10.1186/s12902-016-0092-z
15. Annerbo S, Kivipelto M, Lökk J. A prospective study on the development of alzheimer’s disease with regard to thyroid stimulating hormone and homocysteine. Dement Geriatr Cognit Disord (2009) 28:275–80. doi: 10.1159/000242439
16. Zou Y, Wang Q, Cheng X. Causal relationship between basal metabolic rate and alzheimer’s disease: A bidirectional two-sample mendelian randomization study. Neurol Ther (2023) 12:763–76. doi: 10.1007/s40120-023-00458-9
17. Salehipour A, Dolatshahi M, Haghshomar M, Amin J. The role of thyroid dysfunction in alzheimer’s disease: A systematic review and meta-analysis. J Prev Alzheimers Dis (2023) 10:276–86. doi: 10.14283/jpad.2023.20
18. Quinlan P, Nordlund A, Lind K, Gustafson D, Edman Å, Wallin A. Thyroid hormones are associated with poorer cognition in mild cognitive impairment. Dement Geriatr Cognit Disord (2010) 30:205–11. doi: 10.1159/000319746
19. de Jong FJ, Masaki K, Chen H, Remaley AT, Breteler MMB, Petrovitch H, et al. Thyroid function, the risk of dementia and neuropathologic changes: the honolulu–asia aging study. Neurobiol Aging (2009) 30:600–6. doi: 10.1016/j.neurobiolaging.2007.07.019
20. George KM, Lutsey PL, Selvin E, Palta P, Windham BG, Folsom AR. Association between thyroid dysfunction and incident dementia in the atherosclerosis risk in communities neurocognitive study. J Endocrinol Metab (2019) 9:82–9. doi: 10.14740/jem588
21. Yeap BB, Alfonso H, Chubb SAP, Puri G, Hankey GJ, Flicker L, et al. Higher free thyroxine levels predict increased incidence of dementia in older men: the health in men study. J Clin Endocrinol Metab (2012) 97:E2230–7. doi: 10.1210/jc.2012-2108
22. Folkestad L, Brandt F, Lillevang-Johansen M, Brix TH, Hegedüs L. Graves’ Disease and toxic nodular goiter, aggravated by duration of hyperthyroidism, are associated with alzheimer’s and vascular dementia: A registry-based long-term follow-up of two large cohorts. Thyroid (2020) 30:672–80. doi: 10.1089/thy.2019.0672
23. Tang X, Song Z-H, Wang D, Yang J, Cardoso MA, Zhou J-B, et al. Spectrum of thyroid dysfunction and dementia: A dose–response meta-analysis of 344,248 individuals from cohort studies. Endocr Connect (2021) 10:410–21. doi: 10.1530/EC-21-0047
24. Kalmijn S, Mehta KM, Pols HAP, Hofman A, Drexhage HA, Breteler MMB. Subclinical hyperthyroidism and the risk of dementia. The rotterdam study. Clin Endocrinol (Oxf) (2000) 53:733–7. doi: 10.1046/j.1365-2265.2000.01146.x
25. Samuels MH. Psychiatric and cognitive manifestations of hypothyroidism. Curr Opin Endocrinol Diabetes Obes (2014) 21:377–83. doi: 10.1097/MED.0000000000000089
26. Breteler MM, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Medical history and the risk of alzheimer's disease: A collaborative re-analysis of case-control studies. Int J Epidemiol (1991) 20 Suppl 2:S36–42. doi: 10.1093/ije/20.supplement_2.s36
27. Quinlan P, Horvath A, Wallin A, Svensson J. Low serum concentration of free triiodothyronine (Ft3) is associated with increased risk of alzheimer's disease. Psychoneuroendocrinology (2019) 99:112–9. doi: 10.1016/j.psyneuen.2018.09.002
28. Ren B, Ma J, Tao M, Jing G, Han S, Zhou C, et al. The disturbance of thyroid-associated hormone and its receptors in brain and blood circulation existed in the early stage of mouse model of alzheimer’s disease. Aging (Albany NY) (2023) 15(3):1591–602. doi: 10.18632/aging.204570
29. Quinlan P, Horvath A, Eckerstrom C, Wallin A, Svensson J. Altered thyroid hormone profile in patients with alzheimer's disease. Psychoneuroendocrinology (2020) 121:104844. doi: 10.1016/j.psyneuen.2020.104844
30. Yong-Hong L, Xiao-Dong P, Chang-Quan H, Bo Y, Qing-Xiu L. Hypothalamic-pituitary-thyroid axis in patients with alzheimer disease (Ad). J Investig Med (2013) 61:578–81. doi: 10.2310/JIM.0b013e318280aafb
31. Chen J-M, Huang C-Q, Ai M, Kuang L. Circadian rhythm of tsh levels in subjects with alzheimer’s disease (Ad). Aging Clin Exp Res (2013) 25:153–7. doi: 10.1007/s40520-013-0025-x
32. Tang X, Li Z, Zhang W, Yao Z. Nitric oxide might be an inducing factor in cognitive impairment in alzheimer's disease via downregulating the monocarboxylate transporter 1. Nitric Oxide (2019) 91:35–41. doi: 10.1016/j.niox.2019.07.006
33. Gião T, Saavedra J, Vieira JR, Pinto MT, Arsequell G, Cardoso I. Neuroprotection in early stages of alzheimer’s disease is promoted by transthyretin angiogenic properties. Alzheimers Res Ther (2021) 13:143. doi: 10.1186/s13195-021-00883-8
34. Palha JA, Episkopou V, Maeda S, Shimada K, Gottesman ME, Saraiva MJM. Thyroid hormone metabolism in a transthyretin-null mouse strai. J Biol Chem (1994) 269(52):33135–9.
35. Gião T, Saavedra J, Cotrina E, Quintana J, Llop J, Arsequell G, et al. Undiscovered roles for transthyretin: from a transporter protein to a new therapeutic target for alzheimer’s disease. Int J Mol Sci (2020) 21:2075. doi: 10.3390/ijms21062075
36. Sabatino L, Vassalle C, Seppia CD, Iervasi G. Deiodinases and the three types of thyroid hormone deiodination reactions. Endocrinol Metab (Seoul) (2021) 36:952–64. doi: 10.3803/EnM.2021.1198
37. Humphries CE, Kohli MA, Nathanson L, Whitehead P, Beecham G, Martin E, et al. Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset alzheimer's disease. J Alzheimers Dis (2015) 44:977–87. doi: 10.3233/JAD-141989
38. Wajner SM, Rohenkohl HC, Serrano T, Maia AL. Sodium selenite supplementation does not fully restore oxidative stress-induced deiodinase dysfunction: implications for the nonthyroidal illness syndrome. Redox Biol (2015) 6:436–45. doi: 10.1016/j.redox.2015.09.002
39. de Jong FJ, Masaki K, Chen H, Remaley AT, Breteler MMB, Petrovitch H, et al. Thyroid function, the risk of dementia and neuropathologic changes: the honolulu-asia aging study. Neurobiol Aging (2009) 30:600–6. doi: 10.1016/j.neurobiolaging.2007.07.019
40. Li L-X, Yang T, Guo L, Wang D-Y, Tang C-H, Li Q, et al. Serum tau levels are increased in patients with hyperthyroidism. Neurosci Lett (2020) 729:135003. doi: 10.1016/j.neulet.2020.135003
41. Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in alzheimer disease. Neurology (2016) 87:1827–35. doi: 10.1212/WNL.0000000000003246
42. Videla LA, Sir T, Wolff C. Increased lipid peroxidation in hyperthyroid patients: suppression by propylthiouracil treatment. Free Radic Res Commun (1988) 5:1–10. doi: 10.3109/10715768809068553
43. Venditti P, Napolitano G, Barone D, Coppola I, Meo SD. Effect of thyroid state on enzymatic and non-enzymatic processes in H2o2 removal by liver mitochondria of male rats. Mol Cell Endocrinol (2015) 403:57–63. doi: 10.1016/j.mce.2015.01.019
44. Larsen CB, Riis KR, Winther KH, Larsen EL, Ellervik C, Hegedüs L, et al. Treatment of hyperthyroidism reduces systemic oxidative stress, as measured by markers of RNA and DNA damage. J Clin Endocrinol Metab (2021) 106:e2512–e20. doi: 10.1210/clinem/dgab273
45. Rao G, Verma R, Mukherjee A, Haldar C, Agrawal NK. Melatonin alleviates hyperthyroidism induced oxidative stress and neuronal cell death in hippocampus of aged female golden hamster, mesocricetus auratus. Exp Gerontol (2016) 82:125–30. doi: 10.1016/j.exger.2016.06.014
46. Tobore TO. On the central role of mitochondria dysfunction and oxidative stress in alzheimer's disease. Neurol Sci (2019) 40:1527–40. doi: 10.1007/s10072-019-03863-x
47. Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sof MA, et al. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. BioMed Pharmacother (2015) 74:101–10. doi: 10.1016/j.biopha.2015.07.025
48. Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci (2010) 2:12. doi: 10.3389/fnagi.2010.00012
49. Fernandez M, Pirondi S, Manservigi M, Giardino L, Calza L. Thyroid hormone participates in the regulation of neural stem cells and oligodendrocyte precursor cells in the central nervous system of adult rat. Eur J Neurosci (2004) 20:2059–70. doi: 10.1111/j.1460-9568.2004.03664.x
50. Lou K, Liu S, Zhang F, Sun W, Su X, Bi W, et al. The effect of hyperthyroidism on cognitive function, neuroinflammation, and necroptosis in APP/PS1 mice. J Transl Med (2023) 21:657. doi: 10.1186/s12967-023-04511-x
51. Pavlides C, Westlind-Danielsson AI, Nyborg H, McEwen BS. Neonatal hyperthyroidism disrupts hippocampal LTP and spatial-learning. Exp Brain Res (1991) 85:559–64. doi: 10.1007/BF00231740
52. Bitiktas S, Tan B, Kavraal S, Yousef M, Bayar Y, Dursun N, et al. The effects of intra-hippocampal L-thyroxine infusion on long-term potentiation and long-term depression: A possible role for the Αvβ3 integrin receptor. J Neurosci Res (2017) 95:1621–32. doi: 10.1002/jnr.23985
53. Zhang W, Liu X, Zhang Y, Song L, Hou J, Chen B, et al. Disrupted functional connectivity of the hippocampus in patients with hyperthyroidism: evidence from resting-state fMRI. Eur J Radiol (2014) 83:1907–13. doi: 10.1016/j.ejrad.2014.07.003
54. Zhu W, Wu F, Li J, Meng L, Zhang W, Zhang H, et al. Impaired learning and memory generated by hyperthyroidism is rescued by restoration of ampa and nmda receptors function. Neurobiol Dis (2022) 171:105807. doi: 10.1016/j.nbd.2022.105807
55. Sahin L, Müsüroglu SK, Cevik OS, Cevik K, Temel GO. Hyperthyroidism leads learning and memory impairment possibly via GRIN2B expression alterations. Brain Res (2023) 1802:148209. doi: 10.1016/j.brainres.2022.148209
56. Abd-Elmawla MA, Essam RM, Ahmed KA, Abdelmonem M. Implication of Wnt/GSK-3beta/beta-catenin signaling in the pathogenesis of mood disturbances associated with hyperthyroidism in rats: potential therapeutic effect of naringin. ACS Chem Neurosci (2023) 14:2035–48. doi: 10.1021/acschemneuro.3c00013
57. Luo L, Yano N, Mao Q, Jackson IMD, Stopa EG. Thyrotropin releasing hormone (Trh) in the hippocampus of alzheimer patients. J Alzheimers Dis (2002) 4:97–103. doi: 10.3233/jad-2002-4204
58. Schreckenberger MF, Egle UT, Drecker S, Buchholz HG, Weber MM, Bartenstein P, et al. Positron emission tomography reveals correlations between brain metabolism and mood changes in hyperthyroidism. J Clin Endocrinol Metab (2006) 91:4786–91. doi: 10.1210/jc.2006-0573
59. Danielsen ER, Elberling TV, Rasmussen ÅK, Dock J, Hording M, Perrild H, et al. Reduced parietooccipital white matter glutamine measured by proton magnetic resonance spectroscopy in treated graves’ Disease patients. J Clin Endocrinol Metab (2008) 93:3192–8. doi: 10.1210/jc.2007-2161
60. Giovannini MG, Casamenti F, Nistri A, Paoli F, Pepeu G. Effect of thyrotropin releasing hormone (Trh) on acetylcholine release from different brain areas investigated by microdialysis. Br J Pharmacol (1991) 102:363–8. doi: 10.1111/j.1476-5381.1991.tb12179.x
61. Koohestani F, Brown CM, Meisami E. Differential effects of developmental hypo- and hyperthyroidism on acetylcholinesterase and butyrylcholinesterase activity in the spinal cord of developing postnatal rat pups. Int J Dev Neurosci (2012) 30:570–7. doi: 10.1016/j.ijdevneu.2012.07.006
62. Subhadra B, Schaller K, Seeds NW. Neuroserpin up-regulation in the alzheimer's disease brain is associated with elevated thyroid hormone receptor-veta1 and hud expression. Neurochem Int (2013) 63:476–81. doi: 10.1016/j.neuint.2013.08.010
63. Iglesias P, Díez JJ. Hyperthyroidism and cardiovascular risk factors: A nationwide study. Endocrinol Diabetes Nutr (Engl Ed) (2023) 70:255–61. doi: 10.1016/j.endien.2023.03.015
64. Leszek J, Mikhaylenko EV, Belousov DM, Koutsouraki E, Szczechowiak K, Kobusiak-Prokopowicz M, et al. The links between cardiovascular diseases and alzheimer's disease. Curr Neuropharmacol (2021) 19:152–69. doi: 10.2174/1570159x18666200729093724
65. Bauer M, Goetz T, Glenn T, Whybrow PC. The thyroid-brain interaction in thyroid disorders and mood disorders. J Neuroendocrinol (2008) 20:1101–14. doi: 10.1111/j.1365-2826.2008.01774.x
66. Ce O, Cj W, Ab W, S D, D L. Thyroid hormone enhances neurite outgrowth in neuroscreen 1 cells. Int J BioMed Investig (2018) 1:1–11. doi: 10.31531/2581-4745.1000104
67. Desouza LA, Ladiwala U, Daniel SM, Agashe S, Vaidya RA, Vaidya VA. Thyroid hormone regulates hippocampal neurogenesis in the adult rat brain. Mol Cell Neurosci (2005) 29:414–26. doi: 10.1016/j.mcn.2005.03.010
68. Chaalal A, Poirier R, Blum D, Laroche S, Enderlin V. Thyroid hormone supplementation restores spatial memory, hippocampal markers of neuroinflammation, plasticity-related signaling molecules, and Β-amyloid peptide load in hypothyroid rats. Mol Neurobiol (2019) 56:722–35. doi: 10.1007/s12035-018-1111-z
69. Yajima H, Amano I, Ishii S, Sadakata T, Miyazaki W, Takatsuru Y, et al. Absence of thyroid hormone induced delayed dendritic arborization in mouse primary hippocampal neurons through insufficient expression of brain-derived neurotrophic factor. Front Endocrinol (Lausanne) (2021) 12:629100. doi: 10.3389/fendo.2021.629100
70. Fernández-Lamo I, Montero-Pedrazuela A, Delgado-García JM, Guadaño-Ferraz A, Gruart A. Effects of thyroid hormone replacement on associative learning and hippocampal synaptic plasticity in adult hypothyroid rats. Eur J Neurosci (2009) 30:679–92. doi: 10.1111/j.1460-9568.2009.06862.x
71. Sánchez-Huerta KB, Montes S, Pérez-Severiano F, Alva-Sánchez C, Ríos C, Pacheco-Rosado J. Hypothyroidism reduces glutamate-synaptic release by ouabain depolarization in rat ca3-hippocampal region. J Neurosci Res (2012) 90:905–12. doi: 10.1002/jnr.22806
72. Lee PR, Brady D, Koenig JI. Thyroid hormone regulation of N-methyl-D-aspartic acid receptor subunit mRNA expression in adult brain. J Neuroendocrinol (2003) 15:87–92. doi: 10.1046/j.1365-2826.2003.00959.x
73. Chakraborty A, Murphy S, Coleman N. The role of nmda receptors in neural stem cell proliferation and differentiation. Stem Cells Dev (2017) 26:798–807. doi: 10.1089/scd.2016.0325
74. Mendes-de-Aguiar CBN, Alchini R, Decker H, Alvarez-Silva M, Tasca CI, Trentin AG. Thyroid hormone increases astrocytic glutamate uptake and protects astrocytes and neurons against glutamate toxicity. J Neurosci Res (2008) 86:3117–25. doi: 10.1002/jnr.21755
75. Wang F, Zeng X, Zhu Y, Ning D, Liu J, Liu C, et al. Effects of thyroxine and donepezil on hippocampal acetylcholine content, acetylcholinesterase activity, synaptotagmin-1 and snap-25 expression in hypothyroid adult rats. Mol Med Rep (2015) 11:775–82. doi: 10.3892/mmr.2014.2825
76. Patel AJ, Hayashi M, Hunt A. Selective persistent reduction in choline acetyltransferase activity in basal forebrain of the rat after thyroid deficiency during early life. Brain Res (1987) 422:182–5. doi: 10.1016/0006-8993(87)90556-7
77. Xu YX, Sun Y, Cheng J, Xia Q, Liu TT, Zhu DF, et al. Genetic difference of hypothyroidism-induced cognitive dysfunction in C57bl/6j and 129/Sv mice. Neurochem Res (2019) 44:1999–2006. doi: 10.1007/s11064-019-02836-y
78. Xu Y-X, Wang C, Li X-D, Guo W-L, Liu G-Y, Zhang H-B, et al. Activation of cholinergic basal forebrain neurons improved cognitive functions in adult-onset hypothyroid mice. BioMed Pharmacother (2022) 153:113495. doi: 10.1016/j.biopha.2022.113495
79. Belakavadia M, Dellb J, Grovera GJ, Joseph D. Fondella. Thyroid hormone suppression of Β-amyloid precursor protein gene expression in the brain involves multiple epigenetic regulatory events. Mol Cell Endocrinol (2011) 339:72–80. doi: 10.1016/j.mce.2011.03.016
80. Ferrara SJ, Chaudhary P, DeBell MJ, Marracci G, Miller H, Calkins E, et al. Trem2 is thyroid hormone regulated making the trem2 pathway druggable with ligands for thyroid hormone receptor. BioRxiv (2021) 29(2):239–48. doi: 10.1101/2021.01.25.428149
81. Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of trem2 in alzheimer's disease and other neurodegenerative disorders. Lancet Neurol (2018) 17:721–30. doi: 10.1016/S1474-4422(18)30232-1
82. Mishra J, Vishwakarma J, Malik R, Gupta K, Pandey R, Maurya SK, et al. Hypothyroidism induces interleukin-1-dependent autophagy mechanism as a key mediator of hippocampal neuronal apoptosis and cognitive decline in postnatal rats. Mol Neurobiol (2021) 58:1196–211. doi: 10.1007/s12035-020-02178-9
83. Tung Y-T, Wang B-J, Hu M-K, Hsu W-M, Lee H, Huang W-P, et al. Autophagy: A double-edged sword in alzheimer's disease. J Biosci (2012) 37:157–65. doi: 10.1007/s12038-011-9176-0
84. Vishwakarma J, Gupta K, Mishra J, Garg A, Malik R, Kashyap A, et al. Hypothyroidism induces motor deficit via altered cerebellar HB-EGF/EGFR and autophagy. J Endocrinol (2023) 257:e220338. doi: 10.1530/JOE-22-0338
85. Mary A, Eysert F, Checler F, Chami M. Mitophagy in alzheimer’s disease: molecular defects and therapeutic approaches. Mol Psychiatry (2022) 28:202–16. doi: 10.1038/s41380-022-01631-6
86. Tran M, Reddy PH. Defective autophagy and mitophagy in aging and alzheimer’s disease. Front Neurosci (2021) 14:612757. doi: 10.3389/fnins.2020.612757
87. Torres-Manzo AP, Franco-Colín M, Blas-Valdivia V, Pineda-Reynoso M, Cano-Europa E. Hypothyroidism causes endoplasmic reticulum stress in adult rat hippocampus: A mechanism associated with hippocampal damage. Oxid Med Cell Longev (2018) 2018:2089404. doi: 10.1155/2018/2089404
88. Ghemrawi R, Khair M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int J Mol Sci (2020) 21:6127. doi: 10.3390/ijms21176127
89. Cioffi F, Giacco A, Goglia F, Silvestri E. Bioenergetic aspects of mitochondrial actions of thyroid hormones. Cells-Basel (2022) 11:997. doi: 10.3390/cells11060997
90. Francelin C, Mitter SK, Qian Q, Barodia SK, Ip C, Qi X, et al. Bace1 inhibition increases susceptibility to oxidative stress by promoting mitochondrial damage. Antioxidants (Basel) (2021) 10:1539. doi: 10.3390/antiox10101539
91. Misrani A, Tabassum S, Yang L. Mitochondrial dysfunction and oxidative stress in alzheimer’s disease. Front Aging Neurosci (2021) 13:617588. doi: 10.3389/fnagi.2021.617588
92. Prieto-Almeida F, Panveloski-Costa AC, Crunfli F, da Silva Teixeira S, Nunes MT, Torrão ADS. Thyroid hormone improves insulin signaling and reduces the activation of neurodegenerative pathway in the hippocampus of diabetic adult male rats. Life Sci (2018) 192:253–8. doi: 10.1016/j.lfs.2017.11.013
93. Zhang M, Gong W, Zhang D, Ji M, Chen B, Chen B, et al. Ageing related thyroid deficiency increases brain-targeted transport of liver-derived apoe4-laden exosomes leading to cognitive impairment. Cell Death Dis (2022) 13:406. doi: 10.1038/s41419-022-04858-x
94. Zhou L, Ding S, Li Y, Wang L, Chen W, Bo T, et al. Endoplasmic reticulum stress may play a pivotal role in lipid metabolic disorders in a novel mouse model of subclinical hypothyroidism. Sci Rep (2016) 6:31381. doi: 10.1038/srep31381
95. Haji M, Kimura N, Hanaoka T, Aso Y, Takemaru M, Hirano T, et al. Evaluation of regional cerebral blood flow in alzheimer’s disease patients with subclinical hypothyroidism. Dement Geriatr Cognit Disord (2015) 39:360–7. doi: 10.1159/000375298
96. Krausz Y, Freedman N, Lester H, Newman JP, Barkai G, Bocher M, et al. Regional cerebral blood flow in patients with mild hypothyroidism. J Nucl Med (2004) 45:1712–5.
97. Park J-H, Hong J-H, Lee S-W, Ji HD, Jung J-A, Yoon K, et al. The effect of chronic cerebral hypoperfusion on the pathology of alzheimer's disease: A positron emission tomography study in rats. Sci Rep (2019) 9:14102. doi: 10.1038/s41598-019-50681-4
98. Livschitz J, Yen TWF, Evans DB, Wang TS, Dream S. Long-term quality of life after parathyroidectomy for primary hyperparathyroidism a systematic review. JAMA Surg (2022) 157(11):1051–8. doi: 10.1001/jamasurg.2022.4249
99. Fukui T, Hasegawa Y, Takenaka H. Hyperthyroid dementia: clinicoradiological findings and response to treatment. J Neurol Sci (2001) 184:81–8. doi: 10.1016/s0022-510x(00)00487-1
100. Kumar M, Singh S, Rana P, Kumar P, Sekhri T, Kanwar R, et al. Neurometabolite Changes in Hyperthyroid Patients before and after Antithyroid Treatment: An in Vivo (1)H Mrs Study. Front Hum Neurosci (2021) 15:739917. doi: 10.3389/fnhum.2021.739917
101. Kumar M, Singh S, Rana P, Modi S, Sekhri T, Kanwar R, et al. Brain functional connectivity in patients with hyperthyroidism after anti-thyroid treatment. J Neuroendocrinol (2022) 34:e13075. doi: 10.1111/jne.13075
102. Rutigliano G, Bertolini A, Grittani N, Frascarelli S, Carnicelli V, Ippolito C, et al. Effect of combined levothyroxine (L-T4) and 3-iodothyronamine (T1am) supplementation on memory and adult hippocampal neurogenesis in a mouse model of hypothyroidism. Int J Mol Sci (2023) 24:13845. doi: 10.3390/ijms241813845
103. Farag EA, Filobbos SA, Afifi NM, Mahmoud ST, Alghandour SM. Thyroxine restores hippocampal neurogenesis and synaptogenesis in a male rat model of carbimazole-induced hypothyroidism: A histological study. Beni Suef Univ J Basic Appl Sci (2023) 12:57. doi: 10.1186/s43088-023-00395-4
104. Uma D, Rabbani R, Lee JH, Gavini DR, Shah PH, Hamid P. Does hormone supplementation with levothyroxine improve hypothyroid impaired cognitive dysfunction? Cureus (2021) 13:e17885. doi: 10.7759/cureus.17885
105. Camandola S, Mattson MP. Aberrant subcellular neuronal calcium regulation in aging and alzheimer's disease. Biochim Biophys Acta (2011) 1813:965–73. doi: 10.1016/j.bbamcr.2010.10.005
Keywords: thyroid dysfunction, hyperthyroidism, hypothyroidism, Alzheimer’s disease, cognition
Citation: Li Z and Liu J (2024) Thyroid dysfunction and Alzheimer's disease, a vicious circle. Front. Endocrinol. 15:1354372. doi: 10.3389/fendo.2024.1354372
Received: 12 December 2023; Accepted: 01 February 2024;
Published: 14 February 2024.
Edited by:
Rauf Latif, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Olga Barca Mayo, University of Santiago de Compostela, SpainWei Peng, Chengdu University of Traditional Chinese Medicine, China
Copyright © 2024 Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Liu, bGl1X2ppYUBqbHUuZWR1LmNu