 Alejandro García-Castaño1†
Alejandro García-Castaño1† Leire Madariaga2†
Leire Madariaga2† Sara Gómez-Conde3Pedro González4Gema Grau5Itxaso Rica6Gustavo Pérez de Nanclares1
Sara Gómez-Conde3Pedro González4Gema Grau5Itxaso Rica6Gustavo Pérez de Nanclares1 Ana Belén De la Hoz1Aníbal Aguayo1Rosa Martínez1Inés Urrutia1Sonia Gaztambide7 Calcium Phosphorus Metabolism Molecular Biology GroupLuis Castaño3*
Ana Belén De la Hoz1Aníbal Aguayo1Rosa Martínez1Inés Urrutia1Sonia Gaztambide7 Calcium Phosphorus Metabolism Molecular Biology GroupLuis Castaño3*- 1Biobizkaia Health Research Institute, Hospital Universitario Cruces, CIBERDEM, CIBERER, EndoERN, Barakaldo, Bizkaia, Spain
- 2Pediatric Nephrology Department, Biobizkaia Health Research Institute, Hospital Universitario Cruces, University of the Basque Country (UPV/EHU), CIBERDEM, CIBERER, EndoERN, Barakaldo, Bizkaia, Spain
- 3Biobizkaia Health Research Institute, Hospital Universitario Cruces, University of the Basque Country (UPV/EHU), CIBERDEM, CIBERER, EndoERN, Barakaldo, Bizkaia, Spain
- 4Endocrinology and Nutrition Department, Biobizkaia Health Research Institute, Hospital Universitario Cruces, EndoERN, Barakaldo, Bizkaia, Spain
- 5Pediatric Endocrinology Department, Biobizkaia Health Research Institute, Hospital Universitario Cruces, EndoERN, Barakaldo, Bizkaia, Spain
- 6Pediatric Endocrinology Department, Biobizkaia Health Research Institute, Hospital Universitario Cruces, CIBERDEM, CIBERER, EndoERN, Barakaldo, Bizkaia, Spain
- 7Endocrinology and Nutrition Department, Biobizkaia Health Research Institute, Hospital Universitario Cruces, University of the Basque Country (UPV/EHU), CIBERDEM, CIBERER, EndoERN, Barakaldo, Bizkaia, Spain
Introduction: The disorders in the metabolism of calcium can present with manifestations that strongly suggest their diagnosis; however, most of the time, the symptoms with which they are expressed are nonspecific or present only as a laboratory finding, usually hypercalcemia. Because many of these disorders have a genetic etiology, in the present study, we sequenced a selection of 55 genes encoding the principal proteins involved in the regulation of calcium metabolism.
Methods: A cohort of 79 patients with hypercalcemia were analyzed by next-generation sequencing.
Results: The 30% of our cohort presented one pathogenic or likely pathogenic variant in genes associated with hypercalcemia. We confirmed the clinical diagnosis of 17 patients with hypocalciuric hypercalcemia (pathogenic or likely pathogenic variants in the CASR and AP2S1 genes), one patient with neonatal hyperparathyroidism (homozygous pathogenic variant in the CASR gene), and another patient with infantile hypercalcemia (two pathogenic variants in compound heterozygous state in the CYP24A1 gene). However, we also found variants in genes associated with primary hyperparathyroidism (GCM2), renal hypophosphatemia with or without rickets (SLC34A1, SLC34A3, SLC9A3R1, VDR, and CYP27B1), DiGeorge syndrome (TBX1 and NEBL), and hypophosphatasia (ALPL). Our genetic study revealed 11 novel variants.
Conclusions: Our study demonstrates the importance of genetic analysis through massive sequencing to obtain a clinical diagnosis of certainty. The identification of patients with a genetic cause is important for the appropriate treatment and identification of family members at risk of the disease.
Introduction
The third cause of consultation with the endocrinologist, after diabetes and thyroid diseases, is the disorders in the metabolism of calcium. A dynamic balance between intestinal absorption, bone resorption, and renal excretion maintains the calcium metabolism. Calcium is required for many intracellular functions (signal transmission and many enzymatic reactions) and extracellular functions (blood coagulation, muscle contraction, nerve conduction, hormone release, and mineralization of bone) and is regulated primarily by the actions of vitamin D and parathyroid hormone (PTH). Furthermore, calcitonin, a hormone that is produced and released by the C cells of the thyroid gland, opposes the actions of the PTH by decreasing calcium levels in blood (1). The calcium-sensing receptor (CaSR) plays a central role in the regulation of extracellular calcium homeostasis. Thus, in the presence of a high extracellular calcium concentration [Ca+2], the activation of CaSR inhibits the secretion of PTH, whereas the effect is reversed under low [Ca+2] (2). Moreover, hyperphosphatemia, hypomagnesemia, and the adrenergic action also contribute to PTH secretion. On the other hand, the 1,25-dihydroxyvitamin D3 inhibits PTH secretion.
The hypercalcemia can be an incidental finding, but, sometimes, patients have symptoms as lethargy, hypotonia, anorexia, weight loss, polyuria, polydipsia, vomiting, abdominal pain, and constipation. In long-term or severe cases, kidney failure, pancreatitis, arrhythmias, seizures, and psychiatric condition could be present. Because many of these disorders have a genetic etiology, in the present study, we sequenced a selection, carried out in 2017, of 55 genes encoding the principal proteins involved in the regulation of calcium metabolism to perform a genetic characterization of a cohort of 79 patients diagnosed with hypercalcemia.
Materials and methods
Ethics statement
The study was approved by the Ethics Committee for Clinical Research of Euskadi (CEIC-E, code E20/31). A written informed consent was obtained from the patients, as well as their participating relatives and minors’ legal guardian for the publication of any potentially identifiable images or data included in this article. Patients and their participating relatives provided a written informed consent for the genetic study. The research was carried out in accordance with the Declaration of Helsinki on human experimentation of the World Medical Association.
Patients
A cohort of 79 patients with hypercalcemia from 79 different unrelated families was included in this study. Patients were referred to our laboratory between 2003 and 2023. The main reason for this study was the genetic analysis of patients clinically diagnosed with familial hypocalciuric hypercalcemia (66 patients). Moreover, we included 13 patients who had neonatal or familial hypercalcemia. Patients with primary hyperparathyroidism were excluded. First-degree family members (mother and father) in 18 families with a genetic variant were analyzed. Clinical diagnoses were made by adult and pediatric endocrinologists or nephrologists working in 25 different hospitals. The molecular analysis was done in the Molecular Genetic Laboratory at Biobizkaia Health Research Institute, Barakaldo, Spain.
Gene selection and DNA analysis
A selection of 55 genes identified as potentially associated with disorders of calcium metabolism was screened for pathogenic variants (Table 1). For next-generation sequencing (NGS), a targeted panel was designed using the computer tool Ion AmpliSeq Designer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) with an expected coverage of 98%. This panel included the exon regions and flanking intronic (at least 40 bases) sequences of the 55 genes.
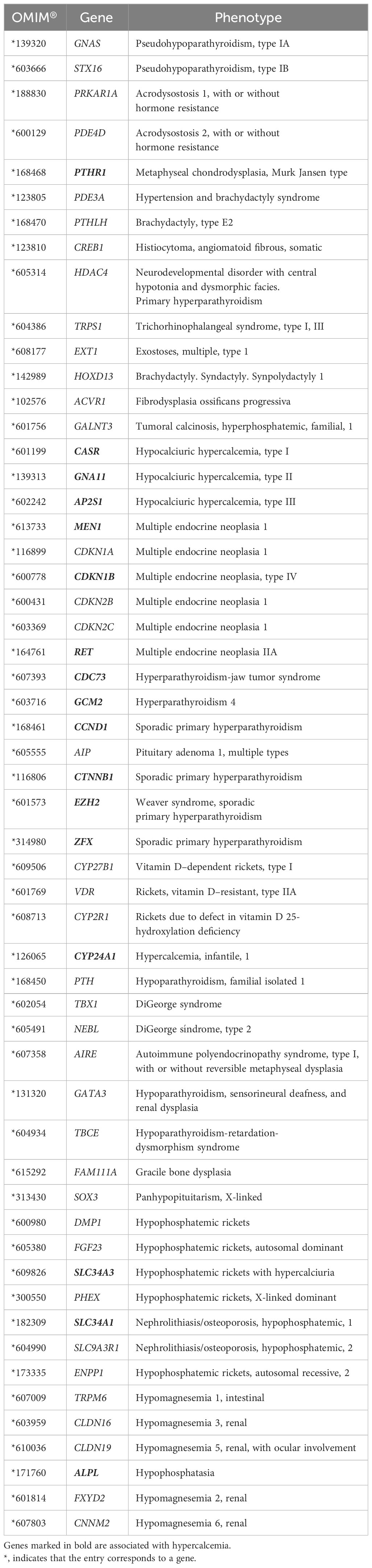
Table 1 Genes involved in the regulation of calcium metabolism included in the panel for analysis.
Genomic DNA was extracted from peripheral blood leukocytes using the MagPurix instrument (Zinexts Life Science Corp., New Taipei City, Taiwan, R.O.C.). DNA purity and concentration were then determined using Qubit 2.0 fluorometer (Thermo Fisher Scientific). Library preparation was done using the Ion AmpliSeq Library Kit v2.0 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Samples were then sequenced using the Ion GeneStudio S5 System (Thermo Fisher Scientific). Base calling, read filtering, alignment to the reference human genome GRCh37/hg19, and variant calling were done using Ion Torrent Suite and Ion Reporter Software (Thermo Fisher Scientific). Not appropriately covered amplicons (<20×) and candidate variants were assessed by Sanger sequencing after polymerase chain reaction, sequenced with fluorescent dideoxynucleotides (BigDye Terminator v3.1 Cycle Sequencing Kit, Life Technologies, Grand Island, NY, USA), and loaded onto an ABI3130xl Genetic Analyzer (Thermo Fisher Scientific).
In order to confirm the deletion detected by NGS in the TBX1 gene, a commercially available MLPA (Multiplex Ligation-dependent Probe Amplification) kit, SALSA MLPA Probemix P250 DiGeorge (MRC Holland, Amsterdam, The Netherlands), was used.
Novel DNA variants were named according to the Human Genome Variation Society guidelines (www.hgvs.org) and classified according to ACMG-AMP (American College of Medical Genetics and Genomics and the Association for Molecular Pathology) guidelines (3).
Results
Thirty percent of our cohort (24 out of the 79 index cases) presented one pathogenic or likely pathogenic variant in genes associated with hypercalcemia. In total, we found 15 pathogenic variants, nine likely pathogenic variants, and 12 variants of uncertain significance (10 variants in patients with hypocalciuric hypercalcemia and two variants in patients with hypercalcemia). Importantly, our genetic study revealed 11 variants not described so far (Table 2).
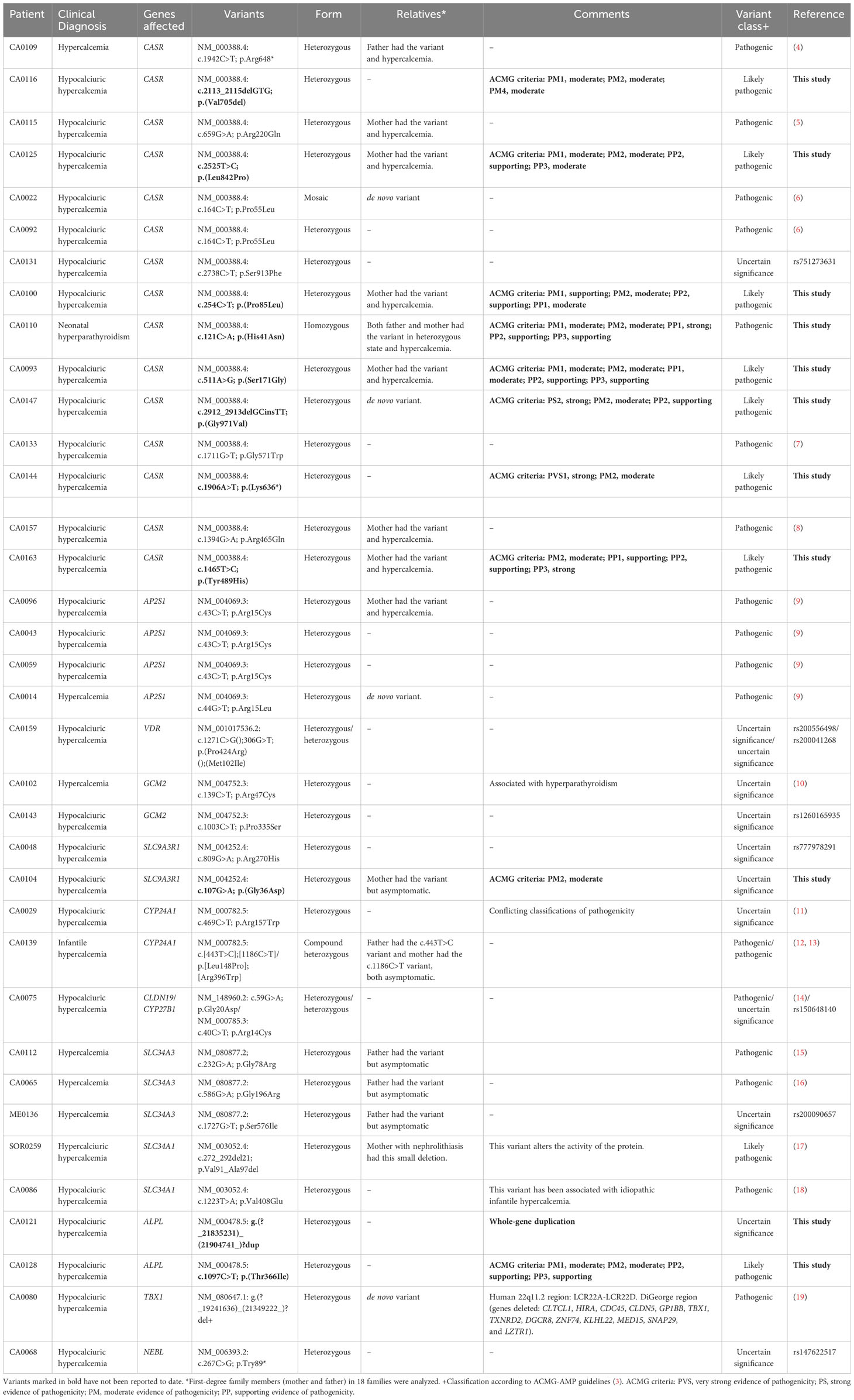
Table 2 Molecular results in patients with a genetic variant.
We confirmed the initial clinical diagnosis (biochemical data are shown in Table 3) of familial hypocalciuric hypercalcemia in 25% of the patients (17 out of the 66 patients had pathogenic or likely pathogenic variants in the CASR and AP2S1 genes). Moreover, we confirmed the initial clinical diagnosis of neonatal hyperparathyroidism in one patient (index case CA0110 had a homozygous pathogenic variant in the CASR gene) and infantile hypercalcemia in another patient (index case CA0139 had two pathogenic variants in compound heterozygous state in the CYP24A1 gene).
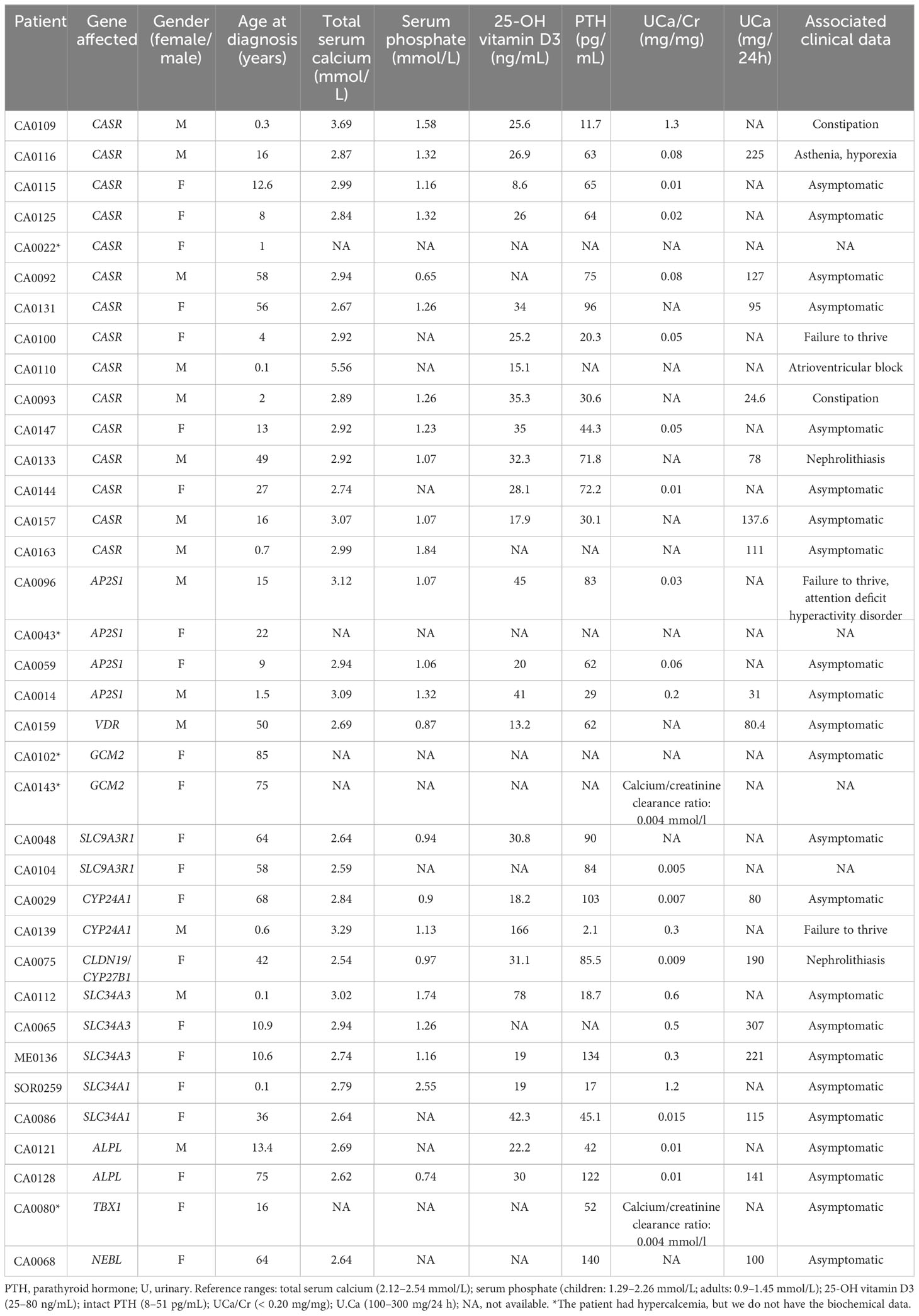
Table 3 Laboratory findings of index cases with a genetic variant.
As expected, most patients had suspected variants in the CASR gene (15 patients) or AP2S1 gene (four patients) that have been associated with hypocalciuric hypercalcemia type 1 (Online Mendelian Inheritance in Man (OMIM), #145980) and hypocalciuric hypercalcemia type 3 (OMIM, #600740), respectively (biochemical data are shown in Table 3).
Two index cases (CA0143 and CA0102) had variants of uncertain significance in the GCM2 gene, and gain-of function mutations in the GCM2 gene cause autosomal dominant hyperparathyroidism type 4 (OMIM, #617343), a disorder characterized by hypercalcemia and elevated PTH secretion by parathyroid glands. These patients had hypercalcemia but not hyperparathyroidism. One of the limitations of our study is the lack of functional studies. Therefore, we cannot assure that the hypercalcemia observed is due to these variants.
Furthermore, many patients had variants in genes usually associated with hypophosphatemia. Thus, five patients had suspected variants in sodium/phosphate cotransporters. Three families (CA0112, CA0065, and ME0136) had variants in the SLC34A3 gene, two previously described as pathogenic and one of uncertain significance. This gene encodes the sodium/phosphate cotransporter 2C, and pathogenic variants in this gene have been associated with hypophosphatemic rickets with hypercalciuria (OMIM, #241530). Moreover, index cases CA0086 and SOR0257 had a pathogenic variant (p.Val408Glu) and a likely pathogenic variant (p.Val91_Ala97del), respectively, in the SLC34A1 gene (sodium/phosphate cotransporter 2A). Pathogenic variants in the SLC34A1 gene have been associated with infantile hypercalcemia type 2 (OMIM, #616963) and nephrolithiasis/osteoporosis hypophosphatemic type 1 (OMIM, #612286). Another two patients (CA0048 and CA0104) had variants of uncertain significance in the SLC9A3R1 gene. This gene encoded a sodium/hydrogen exchanger regulatory cofactor (NHERF1). Pathogenic variants in the SLC9A3R1 gene have been associated with autosomal dominant nephrolithiasis/osteoporosis hypophosphatemic type 2 (OMIM, #612287).
On the other hand, we found variants in genes associated with vitamin D metabolism; one index case (CA0139) had two pathogenic variants in compound heterozygous (p.[Leu148Pro];[Arg396Trp]) in the CYP24A1 gene that encodes the vitamin D 24-hydroxylase; in the same gene, index case CA0029 had a variant of uncertain significance (p.Arg157Trp) in heterozygous state. Pathogenic variants in the CYP24A1 gene have been associated with infantile hypercalcemia type 1 (OMIM, #143880). Finally, in one patient (CA0159), we found two variants of uncertain significance in the VDR gene (vitamin D receptor). Unfortunately, we have not been able to check if the variants are on different alleles. Pathogenic variants in the VDR gene have been associated with autosomal recessive vitamin D dependent rickets type 2A (OMIM, #277440).
In addition, we found suspected variants in other five genes not associated with the phenotype (TBX1, ALPL, NEBL, CLDN19, and CYP27B1) (Table 2). Some of these variants were found in heterozygous state (CLDN19 and CYP27B1), in diseases described previously as recessively inherited.
Regarding patients without a genetic diagnosis, more studies should be carried out, for example, exome or genome sequencing, because they presented phenotypic characteristics similar to patients with a genetic variant.
Discussion
In this study, we examined the presence of genetic alterations in genes related to calcium metabolism in a cohort of 79 patients with hypercalcemia. We found pathogenic or likely pathogenic variants in 30% of patients in our cohort (24 out of 79 patients) that could explain the phenotype observed in those patients. Moreover, we have genetically confirmed the clinical diagnosis given by the clinician in the 24% of our cohort (17 patients with familial hypocalciuric hypercalcemia, one patient with neonatal hyperparathyroidism, and another patient with infantile hypercalcemia). Our complete genetic study revealed 15 pathogenic variants, nine likely pathogenic variants, and 12 of uncertain significance variants. Importantly, our genetic study revealed 11 novel variants.
We confirmed the clinical diagnosis of hypocalciuric hypercalcemia in 17 patients. Our high diagnostic yield respect others cohorts analyzed (20) may be due to a better clinical and biochemical characterization of the patients, to be restrict in the selection of the patients and the division in two groups (in one group, all the patients have hypercalcemia and hypocalciuria). Importantly, one of these patients, index case CA0022, had a de novo pathogenic variant in the CASR gene in mosaic (variant found in a 20% of reads), assuming that, it is possible that a minimal amount of defective protein is enough to develop the disease. To our knowledge, familial hypocalciuric hypercalcemia due to mosaicism in CASR has not been described before. Moreover, we found a pathogenic variant (p.Arg648*) in the CASR gene in another patient (index case CA0109) who had hypercalcemia but with hypercalciuria. It has been previously described that, in some cases, urinary calcium levels are normal or even high. Furthermore, the biochemical profile varies considerably and this variability is thought to be mutation dependent (21).
On the other hand, in four patients initially diagnosed with familial hypocalciuric hypercalcemia, we found suspected variants in other genes (TBX1, NEBL, and ALPL) not associated with the phenotype studied, and the definitive diagnosis could be changed. Thus, index case CA0080 had hypercalcemia, parathyroid hyperplasia, and hypocalciuria with a family history of hypocalciuria (mother and brother). Moreover, she presented with autoimmune hypothyroidism. In this patient, we found a pathogenic deletion in the DiGeorge syndrome chromosome region 22q11.2 (TBX1 gene included). It is difficult to assess how this deletion is influencing the patient’s phenotype because DiGeorge syndrome comprises hypocalcemia and hypoparathyroidism with parathyroid hypoplasia (OMIM, #188400). Furthermore, her mother and brother with hypocalciuria do not have the deletion. It is important to note that some patients with the deletion have minimal clinical expression. Thus, it has been described that 10% to 25% of parents of patients with DiGeorge exhibit the deletion and have no symptoms (22), and hypocalcemia has been described in up to 80% of adults with a 22q11.2 deletion sometime during their lifetime (23). The patient could have another alteration because the hypocalciuria detected is inherited. Moreover, environmental factors such as hydration and sodium intake may explain why a patient with DiGeorge syndrome might have hypercalcemia. On the other hand, patient CA0080 presented an autoimmune disease, and this disease is observed in most age groups with this large deletion in the chromosome region 22q11.2 (24). In another study, the authors described the phenotypic features of 78 adults with this deletion, and 20.5% presented with hypothyroidism (25). Importantly, another index case with hypocalciuric hypercalcemia (CA0068) of our cohort had a nonsense variant of uncertain significance (p.Try89*) in the NEBL gene. The NEBL gene has been found deleted in two patients with DiGeorge syndrome type 2, who showed cardiac defects, but not in two patients with the more distal deletion, which is associated with hypoparathyroism, deafness, and renal dysplasia (26).
Among patients with hypocalciuric hypercalcemia, two index cases (CA0121 and CA0128) had novel variants in the ALPL gene (one large duplication and one missense, respectively). The ALPL gene is known to cause autosomal recessive infantile hypophosphatasia (OMIM, #241500) and dominant or recessive adult odontohypophosphatasia (OMIM, #146300). However, index case CA0121 had high levels of alkaline phosphatase (655 IU/L; normal range, 44 IU/L to 147 IU/L). Therefore, as far as we know, this duplication could be the first variant reported in the ALPL gene with a gain of function effect and associated with high levels of alkaline phosphatase. He had the whole-gene ALPL duplicated, and this is the first whole-gene duplication detected in literature. On the other hand, patient CA0128 had osteopenia and weakness of the femoral head, clinical characteristic of hypophosphatasia. Unfortunately, we do not have the alkaline phosphatase value of patient CA0128. Administration of drugs that inhibit bone resorption is contraindicated in patients with hypophosphatasia. She was treated with vitamin D, which could have caused hypercalcemia in this patient. Further studies aimed at the functional characterization of these variants will be of help in defining the hypothesized pathogenic roles of these two variants in the ALPL gene.
Seven patients diagnosed with hypercalcemia had a variant in heterozygous state in genes associated with hypophosphatemia (SLC34A3, SLC34A1, and SLC9A3R1 genes). Minor symptoms such as mild hypophosphatemia and bone demineralization over time or kidney stones have been described in patients carrying heterozygous variants in these genes (27). Probably, in these patients, hypercalcemia may be justified because the renal phosphate wasting activates the secretion of calcitriol, which, in turn, increases calcium intestinal absorption (28). Moreover, we identified a heterozygous variant in the CYP24A1 gene in one patient (CA0029) with hypocalciuric hypercalcemia. Figueres et al. found the p.(Arg157Trp) variant in compound heterozygous state with a second potentially causative variant in two patients with hypercalcemia (11). On the other hand, allele frequency is greater than expected for disorder (0.33% of alleles in individuals of European descent in Genome Aggregation Database). Due to the absence of functional analysis the clinical significance of this variant is uncertain. Although an autosomal dominant disease inheritance has been proposed (29), we cannot exclude a second undetected variant in this patient. We recommended studying the genes associated with genetic rickets (SLC34A1, SLC34A3, and SLC9A3R1) in those patients with hypercalcemia of suspected genetic cause but without a confirmatory genetic diagnosis, because they could have a heterozygous pathogenic variant.
Finally, one patient (CA0159) had two variants of uncertain significance in the VDR gene. Although, patient CA0159 had hypophosphatemia and high PTH levels, it does not present the typical characteristics of the disease such as rickets, alopecia, cutaneous cysts, or hypocalcemia. Therefore, these two variants are probably located on the same allele or have no clinical relevance.
In conclusion, our study shows the utility of NGS in unraveling the genetic origin of disorders in the calcium and phosphorus metabolism and reveled interesting findings that demonstrate the importance of genetic analysis through massive sequencing (panels, exome, and genome) to obtain a clinical diagnosis of certainty. The identification of patients with a genetic cause is important for the appropriate treatment and identification of family members at risk of the disease.
Calcium and phosphorus metabolism molecular biology group
Idoia Martínez de LaPiscina, Laura Saso, Mirian Sánchez, Blanca González, Amaia Rodríguez, Olaia Velasco, Josu Aurrekoetxea, Begoña Calvo, Leire Gondra, June Corcuera, Ainhoa Camille Aranaga, Candela Baquero (Biobizkaia Health Research Institute, Hospital Universitario Cruces, Barakaldo, Bizkaia, Spain), María López-Iglesias (Endocrinology Department, Hospital General La Mancha Centro, Ciudad Real, Spain), Julia Sastre, María Ángeles Fernández (Hospital Virgen de la Salud, Toledo, Spain), Marta Murillo (UAB, Servei de Pediatria, Unitat d’Endocrinologia Pediàtrica, Hospital Universitari Germans Trias i Pujol, Badalona, Spain), Laura Martínez, Héctor Galbis, Luz Martínez, María Soledad Marín (Hospital Rafael Méndez, Lorca, Spain), Carla Criado, María Isidoro (Unidad de Referencia Regional de Enfermedades Raras DiERCyL, Centro de Referencia Nacional de Cardiopatías Familiares CSUR, Complejo Asistencial Universitario de Salamanca, Salamanca, Spain), Tommaso Matteucci, Ismene Bilbao (Hospital Universitario Donostia, Donostia, Spain), Javier Lumbreras, Diego de Sotto, Jordi Rosell (Hospital Universitario Son Espases, Palma de Mallorca, Spain), Rosa María Sánchez-Dehesa (Hospital Universitario Príncipe de Asturias, Madrid, Spain), María Pilar Bahillo (Hospital Clínico Universitario de Valladolid, Valladolid, Spain), Juan Cruz Len (Hospital de Txagorritxu, Vitoria, Spain), Raquel Barrio (Hospital Universitario Ramón y Cajal, Madrid, Spain), Sara Marsal, Olga Simó Servat, Gema Ariceta (Hospital Universitario Vall d’Hebron, Barcelona, Spain), Raúl Calzada (Instituto Nacional de Pediatría, Ciudad de México, México), Carmen Lourdes Rey Cordo (Pediatric Endocrinology Department, Hospital Álvaro Cunqueiro, EOXI, Vigo, Spain), Juan Marín (Hospital Clinic Universitari, Valencia, Spain), Jesús Barreiro (Hospital de Santiago de Compostela, Santiago de Compostela, Spain), Joaquín Ramírez (Hospital Príncipe de Asturias, Madrid, Spain), Jessica Ares, Cecilia Sánchez (Hospital Universitario Central de Asturias, Oviedo, Spain), Dulce Calderón (Hospital Virgen de la luz, Cuenca, Spain), Esteban Jodar (Hospital Quirón, Madrid, Spain), Jesús Manuel Morán (Hospital Virgen del Puerto, Plasencia, Spain), Cristina Castro (Hospital Universitari Doctor Peset, Valencia, Spain), José Manuel Rial (Hospital Universitario Hospiten Rambla, Santa Cruz de Tenerife, Spain) and Teresa Molins (Hospital Universitario de Navarra, Navarra, Spain).
Data availability statement
The data presented in the study are deposited in the ClinVar repository, accession numbers SCV004708174 - SCV004708203 and SCV004708204 -- SCV004708209.
Ethics statement
The studies involving humans were approved by Ethics Committee for Clinical Research of Euskadi (CEIC-E, code E20/31). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AG: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. LM: Conceptualization, Funding acquisition, Investigation, Visualization, Writing – original draft, Writing – review & editing. SG: Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. PG: Investigation, Resources, Validation, Visualization, Writing – review & editing. GG: Investigation, Resources, Validation, Visualization, Writing – review & editing. IR: Investigation, Resources, Validation, Visualization, Writing – review & editing. Gd: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. AD: Methodology, Writing – review & editing. AA: Investigation, Resources, Validation, Writing – review & editing. RM: Investigation, Validation, Visualization, Writing – review & editing. IU: Investigation, Validation, Visualization, Writing – review & editing. SG: Conceptualization, Data curation, Validation, Visualization, Writing – original draft, Writing – review & editing. LC: Conceptualization, Data curation, Funding acquisition, Investigation, Resources, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Institute of Health Carlos III (PI21/01419); the Department of Health (2019111052); and the Department of Education (IT-1281-19) of the Basque Government. This work is generated within the Endocrine European Reference Network (Project ID number of Endo-ERN: 739527). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Moe SM. Calcium homeostasis in health and in kidney disease. Compr Physiol. (2016) 6:1781–800. doi: 10.1002/cphy.c150052
2. Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Practice and Research. Clin Endocrinol Metab. (2013) 27:333–43. doi: 10.1016/j.beem.2013.02.006
3. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
4. Jap TS, Wu YC, Jenq SF, Won GS. A novel mutation in the calcium-sensing receptor gene in a Chinese subject with persistent hypercalcemia and hypocalciuria. J Clin Endocrinol Metab. (2001) 86:13–5. doi: 10.1210/jcem.86.1.7149
5. Pearce SH, Wooding C, Davies M, Tollefsen SE, Whyte MP, Thakker RV. Calcium-sensing receptor mutations in familial hypocalciuric hypercalcaemia with recurrent pancreatitis. Clin Endocrinol (Oxf). (1996) 45:675–80. doi: 10.1046/j.1365-2265.1996.750891.x
6. Pearce SH, Trump D, Wooding C, Besser GM, Chew SL, Grant DB, et al. Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest. (1995) 96:2683–92. doi: 10.1172/JCI118335
7. Kim ES, Kim SY, Lee JY, Han JH, Sohn TS, Son HS, et al. Identification and functional analysis of a novel CaSR mutation in a family with familial hypocalciuric hypercalcemia. J Bone Miner Metab. (2016) 34:662–7. doi: 10.1007/s00774-015-0713-z
8. Leech C, Lohse P, Stanojevic V, Lechner A, Göke B, Spitzweg C. Identification of a novel inactivating R465Q mutation of the calcium-sensing receptor. Biochem Biophys Res Commun. (2006) 342:996–1002. doi: 10.1016/j.bbrc.2006.02.018
9. Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. (2013) 45:93–7. doi: 10.1038/ng.2492
10. Coppin L, Dufosse M, Romanet P, Giraud S, North MO, Cardot Bauters C, et al. Should the GCM2 gene be tested when screening for familial primary hyperparathyroidism? Eur J Endocrinol. (2020) 182:57–65. doi: 10.1530/EJE-19-0641
11. Figueres ML, Linglart A, Bienaime F, Allain-Launay E, Roussey-Kessler G, Ryckewaert A, et al. Kidney function and influence of sunlight exposure in patients with impaired 24-hydroxylation of vitamin D due to CYP24A1 mutations. Am J Kidney Dis. (2015) 65:122–6. doi: 10.1053/j.ajkd.2014.06.037
12. Ertl DA, Raimann A, Csaicsich D, Patsch JM, Laccone F, Haeusler G. A pediatric patient with a CYP24A1 mutation: four years of clinical, biochemical, and imaging follow-up. Horm Res Paediatr. (2017) 87:196–204. doi: 10.1159/000450947
13. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. (2011) 365:410–21. doi: 10.1056/NEJMoa1103864
14. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. (2006) 79:949–57. doi: 10.1086/508617
15. Mejia-Gaviria N, Gil-Peña H, Coto E, Pérez-Menéndez TM, Santos F. Genetic and clinical peculiarities in a new family with hereditary hypophosphatemic rickets with hypercalciuria: a case report. Orphanet J Rare Dis. (2010) 5:1. doi: 10.1186/1750-1172-5-1
16. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. (2006) 78:179–92. doi: 10.1086/499409
17. Lapointe JY, Tessier J, Paquette Y, Wallendorff B, Coady MJ, Pichette V, et al. NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int. (2006) 69:2261–7. doi: 10.1038/sj.ki.5000437
18. Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, et al. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. (2016) 27:604–14. doi: 10.1681/ASN.2014101025
19. Burnside RD. 22q11.21 deletion syndromes: A review of proximal, central, and distal deletions and their associated features. Cytogenet Genome Res. (2015) 146:89–99. doi: 10.1159/000438708
20. Hureaux M, Ashton E, Dahan K, Houillier P, Blanchard A, Cormier C, et al. High-throughput sequencing contributes to the diagnosis of tubulopathies and familial hypercalcemia hypocalciuria in adults. Kidney Int. (2019) 96:1408–16. doi: 10.1016/j.kint.2019.08.027
21. García-Castaño A, Madariaga L, Pérez de Nanclares G, Ariceta G, Gaztambide S, Castaño L. Novel mutations associated with inherited human calcium-sensing receptor disorders: A clinical genetic study. Eur J Endocrinol. (2019) 180:59–70. doi: 10.1530/EJE-18-0129
22. Lévy A, Michel G, Lemerrer M, Philip N. Idiopathic thrombocytopenic purpura in two mothers of children with DiGeorge sequence: a new component manifestation of deletion 22q11? Am J Med Genet. (1997) 69:356–9. doi: 10.1002/(sici)1096-8628(19970414)69:4<356::aid-ajmg4>3.0.co;2-j
23. Cheung EN, George SR, Costain GA, Andrade DM, Chow EW, Silversides CK, et al. Prevalence of hypocalcaemia and its associated features in 22q11·2 deletion syndrome. Clin Endocrinol (Oxf). (2014) 81:190–6. doi: 10.1111/cen.12466
24. Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. (2001) 139:715–23. doi: 10.1067/mpd.2001.118534
25. Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, et al. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A. (2005) 138:307–13. doi: 10.1002/ajmg.a.30984
26. Villanueva MP, Aiyer AR, Muller S, Pletcher MT, Liu X, Emanuel B, et al. Genetic and comparative mapping of genes dysregulated in mouse hearts lacking the Hand2 transcription factor gene. Genomics. (2002) 80:593–600. doi: 10.1006/geno.2002.7009
27. Wagner CA, Rubio-Aliaga I, Hernando N. Renal phosphate handling and inherited disorders of phosphate reabsorption: an update. Pediatr Nephrol. (2019) 34:549–59. doi: 10.1007/s00467-017-3873-3
28. Molin A, Lemoine S, Kaufmann M, Breton P, Nowoczyn M, Ballandonne C, et al. Overlapping phenotypes associated with CYP24A1, SLC34A1, and SLC34A3 mutations: A cohort study of patients with hypersensitivity to vitamin D. Front Endocrinol (Lausanne). (2021) 12:736240. doi: 10.3389/fendo.2021.736240
29. Tebben PJ, Milliner DS, Horst RL, Harris PC, Singh RJ, Wu Y, et al. Hypercalcemia, hypercalciuria, and elevated calcitriol concentrations with autosomal dominant transmission due to CYP24A1 mutations: effects of ketoconazole therapy. J Clin Endocrinol Metab. (2012) 97:E423–7. doi: 10.1210/jc.2011-1935
Keywords: NGS, calcium, hypercalcemia, hypocalciuria, hyperparathyroidism
Citation: García-Castaño A, Madariaga L, Gómez-Conde S, González P, Grau G, Rica I, de Nanclares GP, De la Hoz AB, Aguayo A, Martínez R, Urrutia I, Gaztambide S, Calcium Phosphorus Metabolism Molecular Biology Group and Castaño L (2024) Genetic profile of a large Spanish cohort with hypercalcemia. Front. Endocrinol. 15:1297614. doi: 10.3389/fendo.2024.1297614
Received: 20 September 2023; Accepted: 27 February 2024;
Published: 22 March 2024.
Edited by:
Vito Guarnieri, Home for Relief of Suffering (IRCCS), ItalyReviewed by:
Hureaux Marguerite, Assistance Publique Hopitaux De Paris, FranceAnna Papadopoulou, University General Hospital Attikon, Greece
Copyright © 2024 García-Castaño, Madariaga, Gómez-Conde, González, Grau, Rica, de Nanclares, De la Hoz, Aguayo, Martínez, Urrutia, Gaztambide, Calcium Phosphorus Metabolism Molecular Biology Group and Castaño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Castaño, bHVpc2FudG9uaW8uY2FzdGFub2dvbnphbGV6QG9zYWtpZGV0emEuZXVz
†These authors have contributed equally to this work