 Vikas Bansal
Vikas Bansal Bernhard R. Winkelmann3
Bernhard R. Winkelmann3 Johannes W. Dietrich
Johannes W. Dietrich
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 20 February 2024
Sec. Clinical Diabetes
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1258982
This article is part of the Research Topic Insights in Epigenetic Regulation of Metabolic and Endocrine Disorders View all 4 articles
Genome-wide association studies have identified several hundred loci associated with type 2 diabetes mellitus (T2DM). Additionally, pathogenic variants in several genes are known to cause monogenic diabetes that overlaps clinically with T2DM. Whole-exome sequencing of related individuals with T2DM is a powerful approach to identify novel high-penetrance disease variants in coding regions of the genome. We performed whole-exome sequencing on four related individuals with T2DM – including one individual diagnosed at the age of 33 years. The individuals were negative for mutations in monogenic diabetes genes, had a strong family history of T2DM, and presented with several characteristics of metabolic syndrome. A missense variant (p.N2291D) in the type 2 ryanodine receptor (RyR2) gene was one of eight rare coding variants shared by all individuals. The variant was absent in large population databases and affects a highly conserved amino acid located in a mutational hotspot for pathogenic variants in Catecholaminergic polymorphic ventricular tachycardia (CPVT). Electrocardiogram data did not reveal any cardiac abnormalities except a lower-than-normal resting heart rate (< 60 bpm) in two individuals – a phenotype observed in CPVT individuals with RyR2 mutations. RyR2-mediated Ca2+ release contributes to glucose-mediated insulin secretion and pathogenic RyR2 mutations cause glucose intolerance in humans and mice. Analysis of glucose tolerance testing data revealed that missense mutations in a CPVT mutation hotspot region – overlapping the p.N2291D variant – are associated with complete penetrance for glucose intolerance. In conclusion, we have identified an atypical missense variant in the RyR2 gene that co-segregates with diabetes in the absence of overt CPVT.
Diabetes mellitus is a heterogeneous disorder that encompasses several distinct forms each with characteristic clinical presentation at onset and during follow-up (1–3). Type 2 diabetes mellitus is the most common form of diabetes that results from a combination of genetic and environmental risk factors (1, 3). Large-scale genome-wide association studies (GWAS) have identified several hundred distinct T2DM susceptibility loci in the human genome (4). However, disease-associated variants at these loci account for only a small fraction of the heritability of T2DM. The availability of low-cost DNA sequencing technologies has allowed researchers to investigate the contribution of rare variants - particularly in coding regions of the human genome – to risk for T2DM. A number of case-control studies for T2DM that utilize exome sequencing have been published (5, 6). These studies have identified several diabetes-associated rare variants (7, 8) and linked several genes (e.g. SLC30A8) with risk for T2DM (9, p. 30; 5).
In spite of large sample sizes, sequencing based case-control studies have limited ability to link very rare variants with disease due to the low number of carriers of such variants in the dataset. Exome or whole-genome sequencing studies in families with clustering of T2DM have the potential to identify high-penetrance risk variants for diabetes. In this approach, comparison of the DNA sequences of affected and unaffected family members can be used to identify high-penetrance disease variants that are very rare or absent in the population. (10) utilized this strategy to associate loss-of-function variants in the APPL1 gene with diabetes in two families with a high prevalence of diabetes. Similarly, a missense mutation in the beta-cell specific transcription factor MAFA was associated with autosomal dominant diabetes (and insulinomas) using the same approach (11). Such sequencing based studies have a greater chance of success in families with young-onset diabetes or with a multi-generation family history of diabetes. Targeted sequencing studies of monogenic diabetes genes in individuals diagnosed with T2DM have shown that individuals with a lower age at diagnosis have a higher prevalence of pathogenic mutations in MODY genes (12–14). Nevertheless, the vast majority of such individuals are negative for mutations in monogenic diabetes genes suggesting that novel high-penetrance risk variants for T2DM remain to be identified.
In this study, we used whole-exome sequencing to investigate the genetic basis of T2DM in a group of four related individuals identified from a large case-control study of diabetes. Notably, all individuals presented with high BMI and characteristics of metabolic syndrome suggesting the presence of classical T2DM. Additionally, two of the four sequenced individuals were diagnosed with diabetes at the age of 33 and 35 years.
The four individuals sequenced in this study were identified from a previous case-control sequencing study for T2D (12). All individuals were residents of Germany and clinical data was obtained from the treating physicians. All individuals were screened for the presence of complications of diabetes (neuropathy, retinopathy, nephropathy). Islet cell-specific autoantibodies (ICA, GADA, IA-2A), were also measured in all individuals to rule out the presence of autoimmune diabetes. Phenotyping included extensive clinical chemistry evaluation (Supplementary Information) 12-lead electrocardiogram was performed on all individuals.
Whole exome sequencing was performed on genomic DNA obtained from the four individuals (Novogene Corporation, Sacramento California) using Agilent SureSelect All Exon V6 exome capture kits and the Illumina sequencing technology. Paired-end sequence reads (150 base pairs in length) for each individual were aligned to the UCSC hg19 reference human genome sequence using the BWA aligner (v 0.7.16). The aligned reads were sorted using samtools and PCR duplicates were marked using Picard (http://broadinstitute.github.io/picard/). Variant calling was performed jointly for the four individuals using the Genome Analysis Toolkit (GATK v4.0.1.2, HaplotypeCaller). Variants showing skewed allele ratio for heterozygous genotypes that overlapped segmental duplications in the human genome were filtered out.
All identified variants were annotated using the Annovar annotation program and the RefSeq transcript database (15). Computational predictions for missense variants using in-silico prediction tools such as PolyPhen2(16), SIFT (17), MutationAssessor (18), FATHMM (19), Provean (20) and REVEL(21) were obtained from the dbNSFP database (22). Variant allele frequencies were annotated using the Genome Aggregation Database or gnomAD (v2.1.1) (23). Single-variant association statistics were obtained from the Genebass (24) web portal (app.genebass.org). For type 2 diabetes association analysis, we used the phenocode ‘T2D_custom’ with 20813 cases and 371952 controls.
Genetic relatedness was estimated using the KING tool (25) that performs identity-by-descent (IBD) analysis for pairs of individuals from unphased genotype data.
Oral glucose tolerance test (OGTT) data for CPVT patients was obtained from (26). The OGTT values for 27 individuals were divided into two groups based on the location of the mutations and statistical analysis was performed using Python (scipy package).
The four individuals selected for this study were identified from a targeted sequencing study that was conducted to search for pathogenic variants in genes associated with monogenic and type 2 diabetes (12). All four individuals had diabetes that was clinically classified as T2DM. Sharing of rare variants identified from the targeted sequencing data indicated that the individuals were related to each other. One of the four individuals – individual II - was diagnosed with diabetes at the age of 33 years (Table 1) and had a BMI of 24.8 at diagnosis. Individual III was diagnosed with diabetes at the age of 35 years and was obese (BMI = 32.1) at the time of diagnosis with increasing body weight since age 21. The two other individuals – I and IV - were diagnosed with diabetes at the ages of 45 and 47 respectively. All individuals reported a positive family history of diabetes (one or more siblings afflicted with diabetes). Hypertension was present in all four individuals and 3 of the 4 individuals also had low HDL cholesterol levels (Table 1). Therefore, the individuals presented with several clinical features of metabolic syndrome. None of the individuals were positive for islet cell antibodies, namely ICA, GADA and IA-2A ruling out a role of islet-cell autoimmunity as a driver of diabetes manifestation. Analysis of insulin-glucose homeostasis showed that the secretory capacity of beta-cells at the fasting state was higher in two of the four individuals (individuals II and IV) compared to normoglycemic subjects, suggesting a compensatory increase of insulin release (Supplementary Tables 1, 2). Individual III had evidence of glucose intolerance prior to diagnosis of diabetes (2 hour oral glucose tolerance test (OGTT) value equal to 193 mg/dl). At diagnosis, this individual had elevated C-peptide (3.8 ug/l) and proinsulin levels (30 pmol/l) – indicative of an insulin resistant state. The two hour OGTT also revealed an abnormal insulin response with high proinsulin to insulin ratio at 120 minutes (Supplementary Figure 1). In particular, the elevated two-hour proinsulin indicated a high level of ER stress at the level of the beta cells.
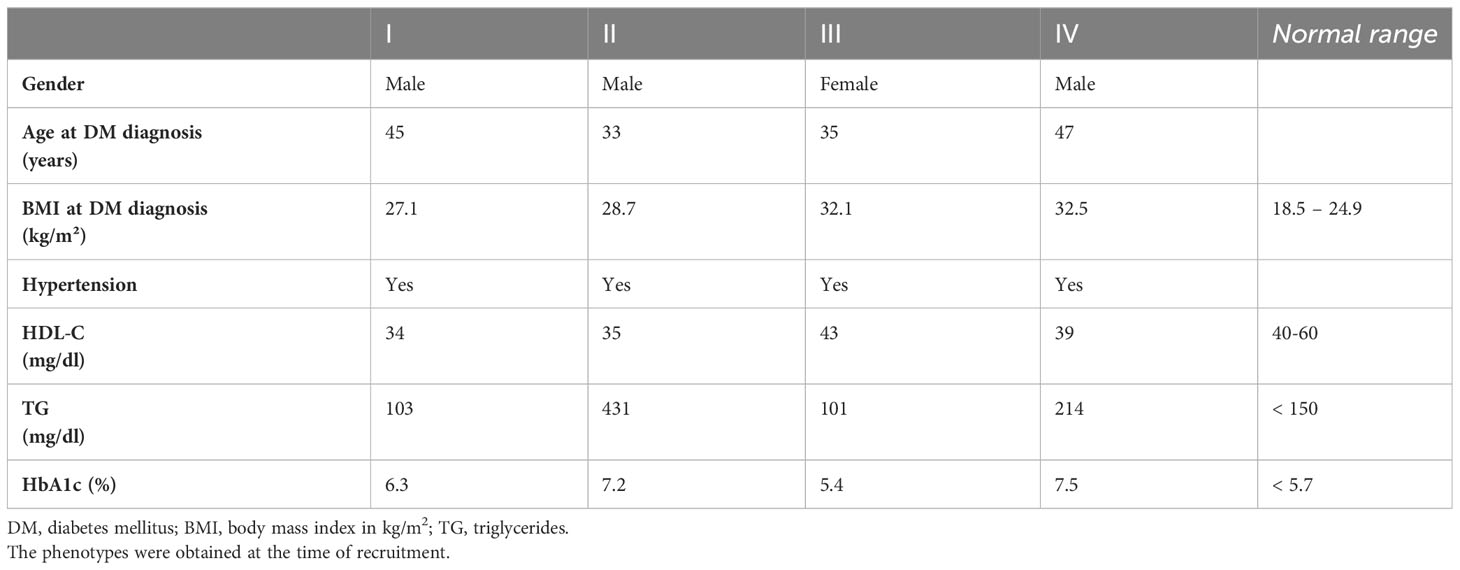
Table 1 Phenotypes for the four individuals sequenced in the study.
The individuals were previously observed to be negative for any potential pathogenic variants in commonly mutated MODY genes(12). Therefore, we chose to perform exome sequencing to search for rare coding variants that potentially contribute to the shared diabetic phenotype. Whole-exome sequencing on the four individuals using paired-end Illumina sequencing technology (Methods) resulted in 6-7 gigabases of aligned sequence data and 58-65x coverage of the targeted regions (Supplementary Table 3). Variant calling using the reads aligned to the reference human genome (hg19) – jointly for the four individuals - identified approximately 360,000 single nucleotide variants and short insertion/deletion variants (see Methods).
First, we performed identical-by-descent (IBD) analysis using the variants identified from the exome sequence data. IBD analysis using the KING tool for analyzing genetic relatedness (25) confirmed that the four individuals are related: individuals I, II and IV had pairwise IBD estimates (IBD1 = 0.43-0.45 and IBD2 = 0.24-0.26) that are typically seen in full siblings (Supplementary Table 4). Individual III also had high IBD sharing (IBD1 = 0.33 – 0.46) with the other three individuals indicative of a second-degree relationship. IBD analysis using another tool for estimating genetic relatedness (27) produced very similar results (data not shown). Next, we contacted the physicians for these four individuals and obtained family trees for the individuals. The family trees confirmed that individual III shared an avuncular relationship with individuals I and IV (Figure 1). Notably, individual III’s father (sibling of individuals I and IV) and paternal grandfather also has diabetes consistent with a dominant mode of inheritance for diabetes. Although we could not obtain full family data for individual II, IBD based inference clearly indicated that individual II is a sibling of individuals I and IV.
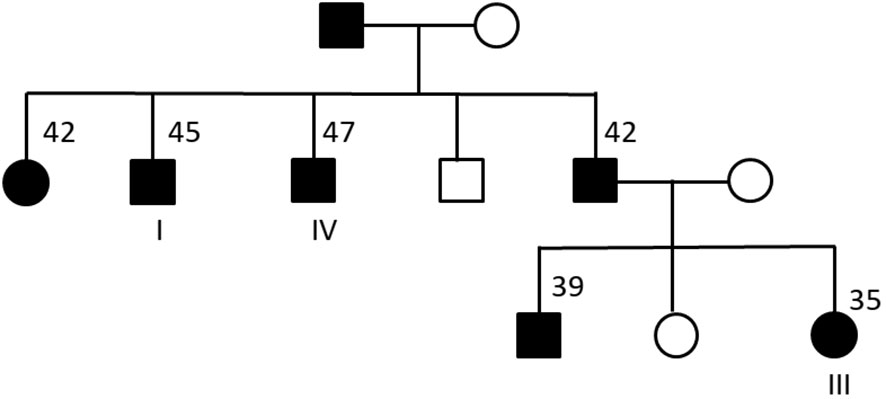
Figure 1 Family tree for three of the four sequenced individuals. Males are represented using squares and females by circles. Black filled symbols correspond to individuals with type 2 diabetes. For some of the individuals, the age at diagnosis of diabetes is shown. Individual II (not shown) was inferred to be a sibling of individuals I and IV based on IBD analysis.
To search for rare genetic variant(s) that could explain the presence of diabetes in the four individuals under a dominant model of inheritance, we used a standard filtering approach for prioritizing variants identified from the exome sequencing (28). First, we filtered out variants present at a minor allele frequency of 0.1% or greater in the gnomAD database (variant data from 125,748 exomes and 15,708 genomes). Next, we prioritized variants that were predicted to affect the protein coding sequence (missense, frameshift or stop-loss) or impact splicing. There were only eight such variants (Table 2) – all missense variants - that were shared by all four sequenced individuals. Two of these missense variants (p.N2291D in RyR2 and p.S33R in SLC30A9) were absent in the gnomAD database. Based on the family tree (Figure 1), we can infer that each of the eight missense variants is also present in the diabetic father of individual III and absent in the unaffected mother of individual III.
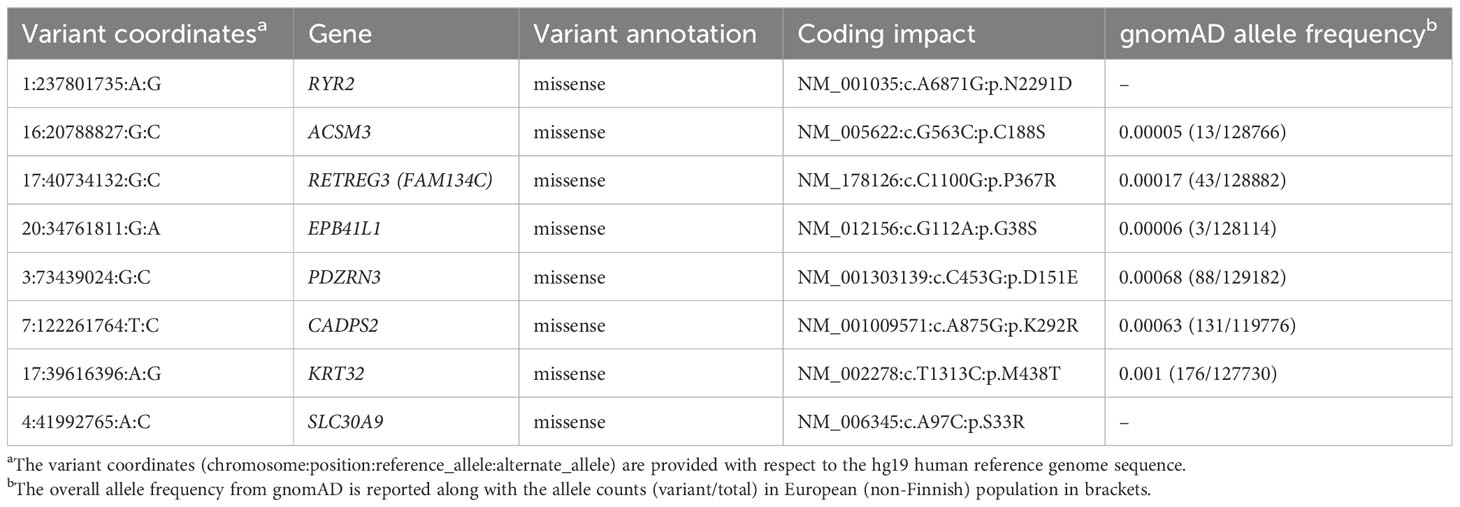
Table 2 List of rare protein-impacting variants shared by all sequenced individuals.
Three of the eight genes with missense variants – RYR2, ASMC3 and CADPS2 – had evidence of prior association with diabetes-relevant phenotypes or metabolic syndrome in humans or model organisms. RYR2 is expressed in the pancreatic beta cells and known to mediate Ca2+ release that plays an important role in glucose-mediated insulin secretion (29, 30). Similarly, CADPS2 is also expressed in the pancreatic beta cells and CADPS2-/- mice show reduced glucose-induced insulin secretion (31). ACSM3 (acyl-CoA synthetase medium chain family member 3) is highly expressed in the liver and kidneys and was identified as a candidate gene that may contribute to hypertension in rats and humans (32, 33). Another gene with a missense variant, SLC30A9, is known to cause a rare recessive disease with complex movement abnormalities and renal impairment (34, 35). SLC30A9 is a member of the SLC30 family of zinc transporters and variants in a different member of this family (SLC30A8) have been associated with risk of T2DM (9).
Next, we used predictions from several individual in-silico missense prediction tools (PolyPhen2, SIFT, MutationAssessor, Provean and FATHMM) and the ensemble predictor REVEL (21) to analyze the deleteriousness of the eight missense variants. REVEL combines predictions from 13 individual tools into a single score (ranging from 0 to 1 with higher scores corresponding to greater chance of pathogenicity) and has been shown to be highly effective in discriminating pathogenic variants from benign variants. The missense variant in RYR2 had the highest REVEL score equal to 0.783 followed by the missense variant in ACMS3 (0.304, Supplementary Table 5). The missense variants in RYR2 and ACMS3 were the only variants that were predicted to be deleterious by at least three of the five individual prediction tools (Supplementary Table 5). In contrast, the missense variants in the CADPS2 and SLC30A9 genes were predicted to be benign by at least four of the five tools and had low REVEL scores (< 0.1).
RYR2 is a well-known Mendelian disease gene that encodes for the cardiac ryanodine receptor - the major calcium release channel on the sarcoplasmic reticulum in cardiomyocytes (36, 37, p. 2). Heterozygous mutations in RYR2 cause cardiac arrhythmias including Catecholaminergic polymorphic ventricular tachycardia (CPVT) that are characterized by exercise-induced syncope, sudden cardiac arrest or sudden cardiac death. Pathogenic CPVT mutations in RYR2 cluster in four hotspots and the p.N2291D variant is located in the second mutational hotspot (AA 2246-2534, Figure 2A). The N2291 residue overlaps the RIH (RyR and IP3R Homology) domain of the protein and is conserved across species and across the paralogs of the ryanodine receptor gene family (Figure 2B). The p.N2291D variant has been not reported in individuals with CPVT or RyR2-related conditions, however, Clinvar contained a single report (rs1681101676) of the variant being detected in clinical testing for CPVT. The variant was classified as a Variant of Uncertain Significance (VUS) due to insufficient evidence.
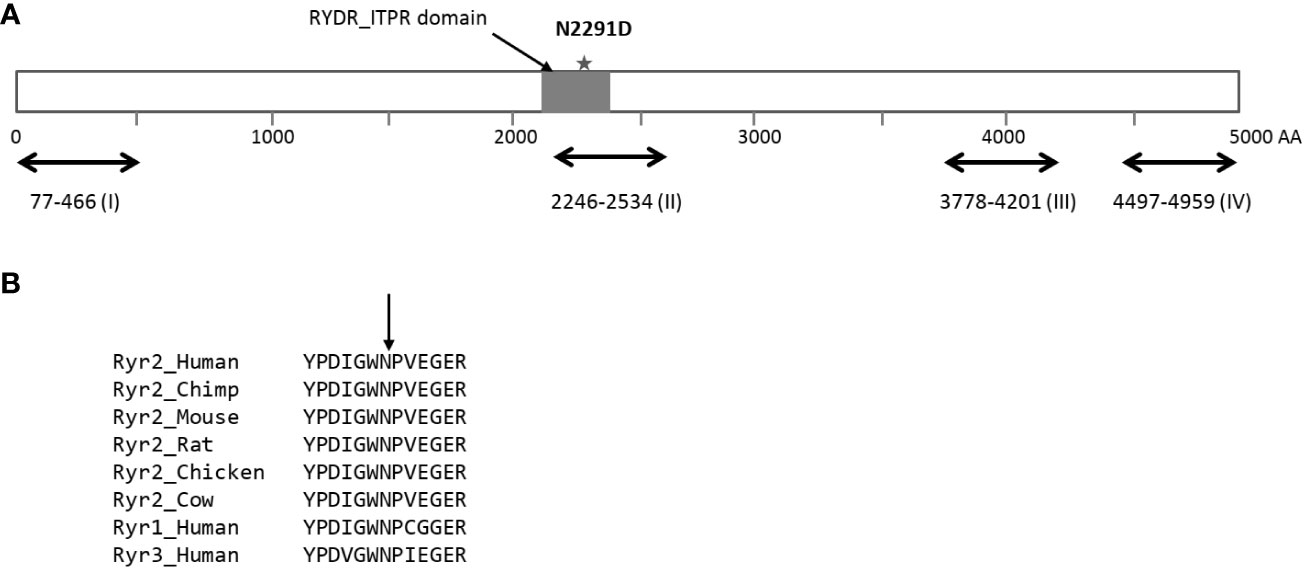
Figure 2 Location of the p.N2291D missense variant in the sequence of the RyR2 protein. (A) Schematic of the Ryr2 protein (4967 residues long) with the four hotposts for missense mutations observed in individuals with CPVT. The p.N2291D is located in the RIH (RyR and IP3R Homology) domain of the protein and also overlaps hotspot II. (B) The asparagine residue at position 2291 is conserved across the RyR2 protein sequence of multiple species and also across the RyR gene family in humans.
There was no reported history of any CPVT-relevant phenotypes such as syncope, sudden cardiac arrest or premature cardiac death triggered by exercise or stress in the four sequenced individuals or their family members. To search for the presence of any subtle cardiac abnormalities, we obtained resting 12-lead electrocardiogram (ECG) data for each of the four individuals. The ECG was normal for individuals I, III and IV (Table 3). For individual II, the ECG showed the presence of coronary artery disease (first diagnosed at age 52 with a stroke at age 55) but no other abnormalities. Individuals with CPVT typically have a normal resting ECG but male patients have a significantly lower heart rate compared to unaffected family members (37, 38). In addition, low-frequency variants at the RYR2 gene locus have been strongly associated with resting heart rate in a genome-wide association study with more than 450,000 individuals (39). The resting heart rate for individuals II and IV – both males - was 55 and 56 bpm respectively, lower than the normal range of 60-100 bpm. The resting heart rates for individual I (male) and III (female) were 60 and 75 bpm respectively. None of the individuals with a low heart rate (individuals I, II and IV) were using beta-blocker medications. Therefore, the lower-than-normal resting heart rate suggested the presence of a mild cardiac phenotype.
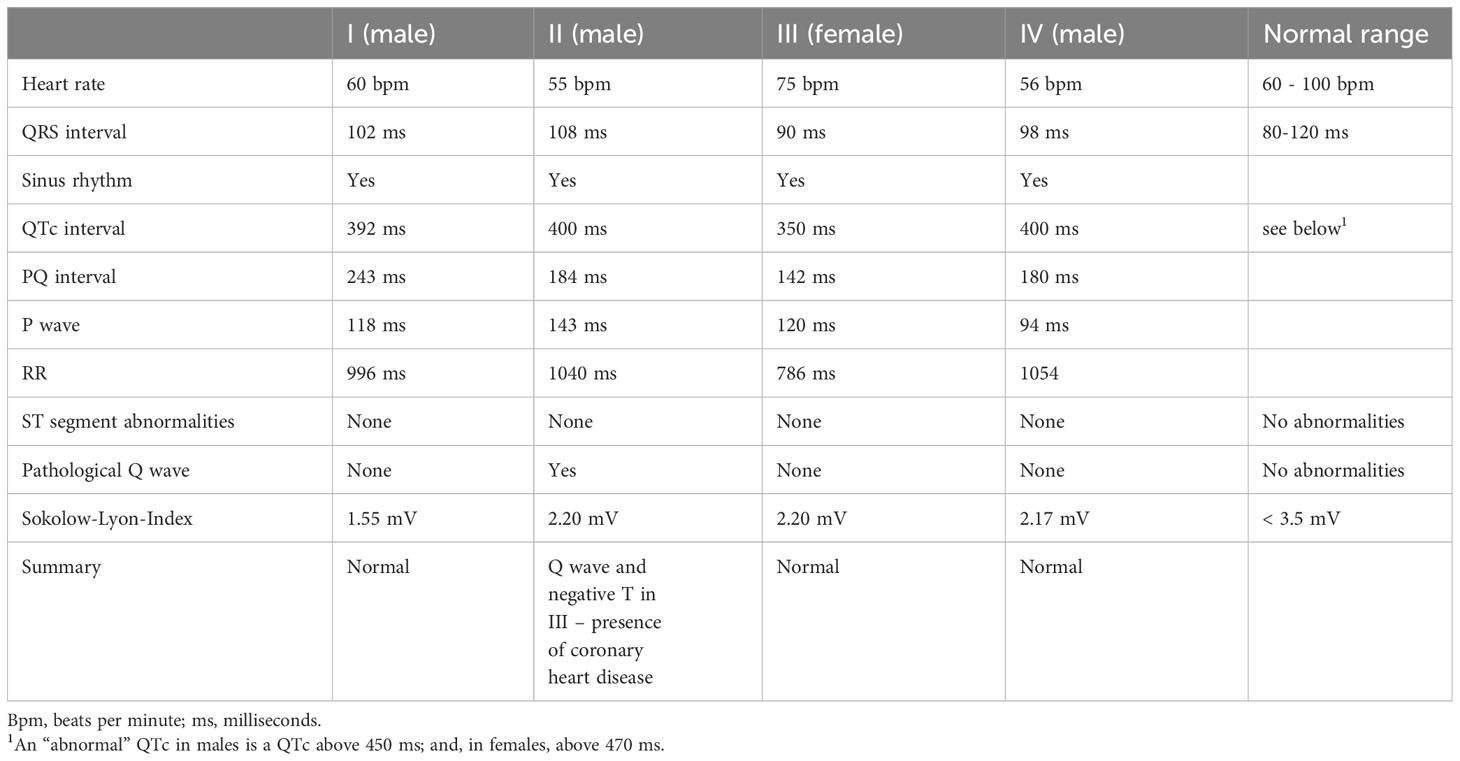
Table 3 Cardiac phenotypes for the four sequenced individuals obtained from a 12-lead electrocardiogram.
Heterozygous missense mutations in RYR2 that cause CPVT result in a leaky RyR2 channel that affects glucose homeostasis in both humans and mice (26). We re-analyzed OGTT data for 27 CPVT patients with heterozygous missense variants in RYR2 (26) to further dissect the impact of missense mutations in RYR2 on glucose homeostasis. Stratifying the data based on position of the missense mutations revealed that individuals with RYR2 mutations in the hotspot II region (Figure 3) had significantly higher 2-hour OGTT values (190 ± 6.20) compared to individuals with RYR2 mutations in the rest of the protein sequence (160.36 ± 25.36, Wilcoxon rank sum test p-value = 0.0048). Notably, 13 of 13 carriers with missense variants in hotspot II region had 2-hour OGTT values of 170 mg/dl or greater and the p.N2291D missense variant was located very close to a cluster of such variants (Supplementary Figure 2). This indicated that the second hotspot region of RYR2 has particular functional importance in pancreatic beta cells and provided additional evidence that the p.N2291D variant is deleterious. In addition, 4 of the 8 individuals with mutations in the C-terminal region (Supplementary Figure 2) had normal glucose tolerance (< 140 mg/dl) indicating incomplete penetrance for glucose intolerance due to CPVT-causing mutations.
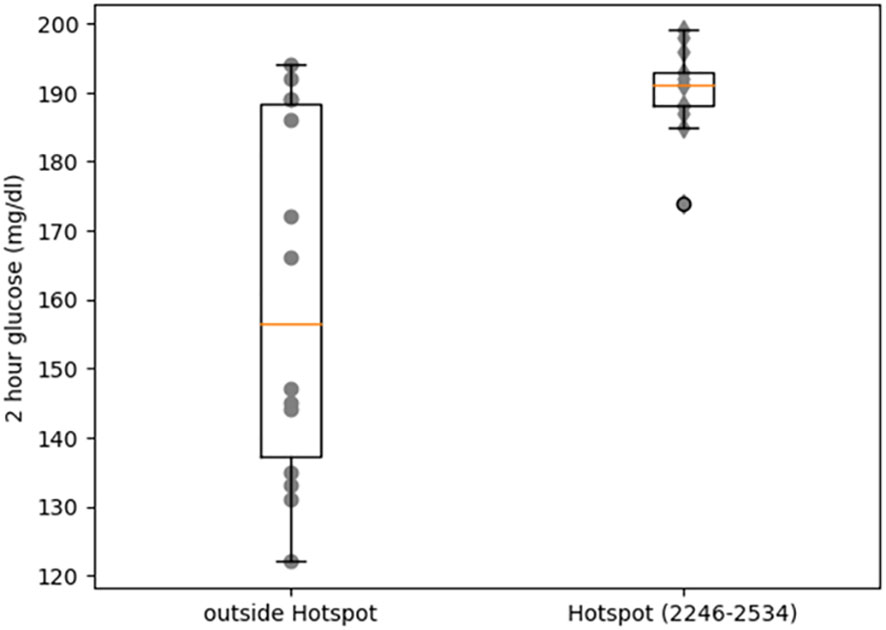
Figure 3 Comparison of blood glucose levels (2h hours after OGTT) between CPVT individuals with mutations in a CPVT hotspot region of RyR2 (residues 2246 to 2534) and individuals with mutations outside this region.
Finally, we explored the possibility that the ACSM3 missense variant is a genetic risk factor for diabetes and related phenotypes (e.g. hypertension) in the four individuals. For this, we utilized the Genebass resource of exome-based association statistics for 394,841 individuals from the UK Biobank with 4,529 phenotypes (see Methods). In this dataset, the minor allele frequency of the ACSM3 variant was 0.00015 similar to the minor allele frequency in the European ancestry individuals in the gnomAD database (0.0001). The variant showed modest association with type 2 diabetes (p-value = 0.0023) but no association with hypertension, HDL cholesterol or BMI (p-value > 0.05). There were 4517 single variant associations available for this variant and the association with type 2 diabetes was not reported to be statistically significant in Genebass. Nevertheless, we cannot rule out the possibility that the ACSM3 variant is a genetic risk factor for diabetes in the individuals in our study.
In this study, we used whole-exome sequencing to search for rare coding variants involved in susceptibility to diabetes in four related individuals that were clinically classified as T2DM. Several clinical features of metabolic syndrome including hypertension, high BMI, low HDL cholesterol and insulin resistance were also present in the four individuals. We identified a missense variant in the RyR2 gene that co-segregated with diabetes in six individuals – four sequenced individuals and two non-genotyped individuals (parents of one of the four sequenced individuals). Several lines of evidence indicate that this RyR2 missense variant is functionally deleterious and likely involved in the pathogenesis of T2DM. First, the variant affects a highly conserved amino acid and is predicted to be deleterious by multiple computational prediction tools. Second, the variant is located in a hotspot for CPVT mutations in RYR2 that is associated with complete penetrance for glucose intolerance. Finally, the known association of common and rare RYR2 variants with resting heart rate and the presence of a lower-than-normal heart rate in two of the four individuals suggests that the missense variant is likely functional.
Ryanodine receptors are a family of intracellular Ca2+ permeable channels that provide the sarcoplasmic reticulum Ca2+ release in various tissues such as skeletal and cardiac muscle. Humans have three such receptors (RyR1-3) and the mutations in all three receptors have been shown to cause Mendelian diseases – both dominant and recessive (30, 40). The type 2 ryanodine receptor is expressed mostly in the heart, brain and pancreas and is expressed at much higher levels than RyR1 and RyR3 in islet cells (29). Consistent with these expression patterns, RyR2 mutations have been linked to phenotypes in each of these tissues. The association of gain-of-function and loss-of-function variants in RyR2 with cardiac arrhythmias and sudden death is well established (41, p. 2). A recent study linked a RyR2 missense variant with epilepsy in a 32-year old female without cardiac abnormalities while her brother – carrying the same mutation – had CPVT (42). Another study found an association between missense variants in Ryr2 and epilepsy with or without arrhythmia (43). Dixit et al. used a mouse model to demonstrate that a gain-of-function missense mutation in RyR2 that results in constitutive phosphorylation led to basal hyperinsulinemia and other known hallmarks of pre-diabetes (29). Santulli et al. demonstrated that CPVT mutations in RyR2 result in glucose intolerance and impaired insulin secretion in humans and mice (26).
The aforementioned study is the only study that has investigated the association of RyR2 missense variants with human phenotypes relevant to diabetes (26). 85% of individuals with CPVT and RyR2 mutations in this study were found to have 2-hour impaired glucose levels. Notably, none of the individuals in this study had abnormal fasting glucose or met the criteria for being diabetic (glucose level > 200 mg/dl at 2-hours post OGTT). Our analysis of this data revealed positional heterogeneity in the penetrance of RyR2 mutations where missense mutations located in one of the hotspots for CPVT mutations (residues 2246 to 2534) had complete penetrance for glucose intolerance while mutations in the C-terminal region had incomplete penetrance. RyR2 is a large protein (4967 residues long) and several hundred pathogenic missense mutations in the gene have been reported. A recent study found that pathogenic variants in RyR2 associated with sudden death during sleep were enriched in the C-terminal region of the protein (41). Another study that analyzed the prevalence of cardiac phenotypes in relatives of individuals with CPVT found that mutation location was associated with severity of cardiac phenotypes (44). Our results show that this variation in phenotype severity as a function of mutation location for the RYR2 gene is not limited to cardiac phenotypes.
Type 2 diabetes is known to result from gene-environment interplay. Unlike mutations in monogenic diabetes genes, pathogenic missense variants in RyR2 result in normal fasting glucose levels with impaired glucose tolerance at an early age (26). However, it is well known that not all individuals with impaired glucose tolerance progress to diabetes at a later age (45). This may explain why diabetes has not been reported to aggregate in families with CPVT. The four individuals with the RyR2 missense variant presented with a high BMI (28-33 kg/m2) and hypertension from an early age. One of the four individuals had impaired glucose tolerance before diagnosis of diabetes and clear evidence of ER stress at the level of the beta cells. Therefore, it is likely that the genetic predisposition in the form of glucose intolerance combined with environmental factors played an important role in progression to clinically overt diabetes.
Our study shows the great potential of combining genetic analysis and phenotyping of subjects clinically classified as type 2 diabetics to pinpoint the heterogeneity of chronic hyperglycemia as an example of precision diabetes medicine in advanced clinical practice (46, 47). Individuals in our study were not recruited as a family but identified from genetic analysis of relatedness among individuals in a case-control study of T2DM. None of the individuals presented at diagnosis as a normal weight individual. All of them had additional phenotypes observed in metabolic syndrome and such individuals are less likely to be included in studies of monogenic diabetes.
This study has a number of limitations. First, we did not evaluate the impact of the missense variant identified in this study on RyR2 channel function. Second, the individuals sequenced in our study were ascertained from a case-control study of diabetes and additional family members could not be included for genotype-phenotype analysis. Finally, we did not evaluate the contribution of common variants associated with risk of T2D or non-coding variants that are not identified in exome sequencing. Further studies are needed to investigate the contribution of rare RyR2 missense mutations to diabetes and related phenotypes.
The data analyzed in the study have been deposited in the Figshare repository, accession number 10.6084/m9.figshare.25044389.
The studies involving humans were approved by Institutional Review Board (IRB) of Ulm University, Ulm, Germany; (registration numbers 42/2004 and 189/2007) and the Chamber of Physicians, State Baden-Wuerttemberg, Germany (registration number 133-2002). The studies were conducted in accordance with the local legislation and institutional requirements. The human DNA samples used in this study were acquired from our previous studies for which ethical approval was obtained. All individuals provided written informed consent for using their DNA samples for genetic studies.
VB: Conceptualization, Investigation, Methodology, Writing – original draft. BW: Data curation, Resources, Writing – review & editing. JD: Data curation, Resources, Writing – review & editing. BB: Data curation, Investigation, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by start-up funds from the Department of Pediatrics at University of California San Diego to VB. BB is supported by an Ong Tiong Tat Professorship from the Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore and a Visiting Professorship by Ulm University, Ulm, Germany.
We thank Dr Simone Claudi-Boehm and Dr Burkhard Manfras for patient recruitment and providing clinical data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1258982/full#supplementary-material
1. Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L. The many faces of diabetes: a disease with increasing heterogeneity. Lancet (2014) 383:1084–94. doi: 10.1016/S0140-6736(13)62219-9
2. Pearson ER. Type 2 diabetes: a multifaceted disease. Diabetologia (2019) 62:1107–12. doi: 10.1007/s00125-019-4909-y
3. Philipson LH. Harnessing heterogeneity in type 2 diabetes mellitus. Nat Rev Endocrinol (2020) 16:79–80. doi: 10.1038/s41574-019-0308-1
4. DeForest N, Majithia AR. Genetics of type 2 diabetes: implications from large-scale studies. Curr Diab Rep (2022) 22:227–35. doi: 10.1007/s11892-022-01462-3
5. Gusarova V, O’Dushlaine C, Teslovich TM, Benotti PN, Mirshahi T, Gottesman O, et al. Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun (2018) 9:2252. doi: 10.1038/s41467-018-04611-z
6. Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature (2019) 570:71–6. doi: 10.1038/s41586-019-1231-2
7. Estrada K, Aukrust I, Bj?rkhaug L, Burtt NP, Mercader JM, Garcia-Ortiz H, et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA (2014) 311:2305–14. doi: 10.1001/jama.2014.6511
8. Mercader JM, Liao RG, Bell AD, Dymek Z, Estrada K, Tukiainen T, et al. A loss-of-function splice acceptor variant in IGF2 is protective for type 2 diabetes. Diabetes (2017) 66:2903–14. doi: 10.2337/db17-0187
9. Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet (2014) 46:357–63. doi: 10.1038/ng.2915
10. Prudente S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet (2015) 97:177–85. doi: 10.1016/j.ajhg.2015.05.011
11. Iacovazzo D, Flanagan SE, Walker E, Quezado R, de Sousa Barros FA, Caswell R, et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci U. S. A. (2018) 115:1027–32. doi: 10.1073/pnas.1712262115
12. Bansal V, Gassenhuber J, Phillips T, Oliveira G, Harbaugh R, Villarasa N, et al. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med (2017) 15:213. doi: 10.1186/s12916-017-0977-3
13. Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, et al. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis. BMC Med (2019) 17:132. doi: 10.1186/s12916-019-1363-0
14. Bonnefond A, Boissel M, Bolze A, Durand E, Toussaint B, Vaillant E, et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat Metab (2020) 2:1126–34. doi: 10.1038/s42255-020-00294-3
15. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
16. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7:248–9. doi: 10.1038/nmeth0410-248
17. Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res (2012) 40:W452–7. doi: 10.1093/nar/gks539
18. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res (2011) 39:e118. doi: 10.1093/nar/gkr407
19. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat (2013) 34:57–65. doi: 10.1002/humu.22225
20. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinforma Oxf Engl (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
21. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
22. Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med (2020) 12:103. doi: 10.1186/s13073-020-00803-9
23. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
24. Karczewski KJ, Solomonson M, Chao KR, Goodrich JK, Tiao G, Lu W, et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genomics (2022) 2:100168. doi: 10.1016/j.xgen.2022.100168
25. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinforma Oxf Engl (2010) 26:2867–73. doi: 10.1093/bioinformatics/btq559
26. Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest (2015) 125:1968–78. doi: 10.1172/JCI79273
27. Dimitromanolakis A, Paterson AD, Sun L. Fast and accurate shared segment detection and relatedness estimation in un-phased genetic data via TRUFFLE. Am J Hum Genet (2019) 105:78–88. doi: 10.1016/j.ajhg.2019.05.007
28. Ng SB, Nickerson DA, Bamshad MJ, Shendure J. Massively parallel sequencing and rare disease. Hum Mol Genet (2010) 19:R119–124. doi: 10.1093/hmg/ddq390
29. Dixit SS, Wang T, Manzano EJQ, Yoo S, Lee J, Chiang DY, et al. Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PloS One (2013) 8:e58655. doi: 10.1371/journal.pone.0058655
30. Kushnir A, Wajsberg B, Marks AR. Ryanodine receptor dysfunction in human disorders. Biochim Biophys Acta Mol Cell Res (2018) 1865:1687–97. doi: 10.1016/j.bbamcr.2018.07.011
31. Speidel D, Salehi A, Obermueller S, Lundquist I, Brose N, Renström E, et al. CAPS1 and CAPS2 regulate stability and recruitment of insulin granules in mouse pancreatic beta cells. Cell Metab (2008) 7:57–67. doi: 10.1016/j.cmet.2007.11.009
32. Iwai N, Ohmichi N, Hanai K, Nakamura Y, Kinoshita M. Human SA gene locus as a candidate locus for essential hypertension. Hypertens Dallas Tex 1979 (1994) 23:375–80. doi: 10.1161/01.hyp.23.3.375
33. Iwai N, Katsuya T, Mannami T, Higaki J, Ogihara T, Kokame K, et al. Association between SAH, an acyl-CoA synthetase gene, and hypertriglyceridemia, obesity, and hypertension. Circulation (2002) 105:41–7. doi: 10.1161/hc0102.101780
34. Perez Y, Shorer Z, Liani-Leibson K, Chabosseau P, Kadir R, Volodarsky M, et al. SLC30A9 mutation affecting intracellular zinc homeostasis causes a novel cerebro-renal syndrome. Brain J Neurol (2017) 140:928–39. doi: 10.1093/brain/awx013
35. Kleyner R, Arif M, Marchi E, Horowitz N, Haworth A, King B, et al. Autosomal recessive SLC30A9 variants in a proband with a cerebrorenal syndrome and no parental consanguinity. Cold Spring Harb Mol Case Stud (2022) 8:a006137. doi: 10.1101/mcs.a006137
36. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation (2001) 103:196–200. doi: 10.1161/01.cir.103.2.196
37. Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff J-M, Vaksmann G, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet (2005) 42:863–70. doi: 10.1136/jmg.2004.028993
38. Miyata K, Ohno S, Itoh H, Horie M. Bradycardia is a specific phenotype of catecholaminergic polymorphic ventricular tachycardia induced by RYR2 mutations. Intern Med Tokyo Jpn (2018) 57:1813–7. doi: 10.2169/internalmedicine.9843-17
39. Zhu Z, Wang X, Li X, Lin Y, Shen S, Liu C-L, et al. Genetic overlap of chronic obstructive pulmonary disease and cardiovascular disease-related traits: a large-scale genome-wide cross-trait analysis. Respir Res (2019) 20:64. doi: 10.1186/s12931-019-1036-8
40. Betzenhauser MJ, Marks AR. Ryanodine receptor channelopathies. Pflugers Arch (2010) 460:467–80. doi: 10.1007/s00424-010-0794-4
41. Olubando D, Hopton C, Eden J, Caswell R, Lowri Thomas N, Roberts SA, et al. Classification and correlation of RYR2 missense variants in individuals with catecholaminergic polymorphic ventricular tachycardia reveals phenotypic relationships. J Hum Genet (2020) 65:531–9. doi: 10.1038/s10038-020-0738-6
42. Yap SM, Smyth S. Ryanodine receptor 2 (RYR2) mutation: A potentially novel neurocardiac calcium channelopathy manifesting as primary generalised epilepsy. Seizure (2019) 67:11–4. doi: 10.1016/j.seizure.2019.02.017
43. Ma M-G, Liu X-R, Wu Y, Wang J, Li B-M, Shi Y-W, et al. RYR2 mutations are associated with benign epilepsy of childhood with centrotemporal spikes with or without arrhythmia. Front Neurosci (2021) 15:629610. doi: 10.3389/fnins.2021.629610
44. van der Werf C, Nederend I, Hofman N, van Geloven N, Ebink C, Frohn-Mulder IME, et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol (2012) 5:748–56. doi: 10.1161/CIRCEP.112.970517
45. Engberg S, Vistisen D, Lau C, Glümer C, Jørgensen T, Pedersen O, et al. Progression to impaired glucose regulation and diabetes in the population-based Inter99 study. Diabetes Care (2009) 32:606–11. doi: 10.2337/dc08-1869
46. Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia (2017) 60:769–77. doi: 10.1007/s00125-017-4226-2
Keywords: type 2 diabetes, RyR2, monogenic diabetes, pathogenic variant, metabolic syndrome, CPVT
Citation: Bansal V, Winkelmann BR, Dietrich JW and Boehm BO (2024) Whole-exome sequencing in familial type 2 diabetes identifies an atypical missense variant in the RyR2 gene. Front. Endocrinol. 15:1258982. doi: 10.3389/fendo.2024.1258982
Received: 14 July 2023; Accepted: 10 January 2024;
Published: 20 February 2024.
Edited by:
Nady El Hajj, Hamad bin Khalifa University, QatarReviewed by:
Wenjun Xie, Xi’an Jiaotong University, ChinaCopyright © 2024 Bansal, Winkelmann, Dietrich and Boehm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vikas Bansal, dmliYW5zYWxAdWNzZC5lZHU=; Bernhard O. Boehm, YmVybmhhcmQuby5ib2VobUBtYWlsYm94Lm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.