 Tiffany K. Miles1*
Tiffany K. Miles1* Melody L. Allensworth-James1
Melody L. Allensworth-James1 Angela K. Odle1Ana Rita Silva Moreira1Anessa C. Haney1Alex N. LaGasse1
Angela K. Odle1Ana Rita Silva Moreira1Anessa C. Haney1Alex N. LaGasse1 Allen J. Gies1
Allen J. Gies1 Stephanie D. Byrum1
Stephanie D. Byrum1 Angelica M. Riojas2
Angelica M. Riojas2 Melanie C. MacNicol1
Melanie C. MacNicol1 Angus M. MacNicol1
Angus M. MacNicol1 Gwen V. Childs1
Gwen V. Childs1- 1Department of Neurobiology & Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 2Department of Radiology, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
Maternal nutrition during embryonic development and lactation influences multiple aspects of offspring health. Using mice, this study investigates the effects of maternal caloric restriction (CR) during mid-gestation and lactation on offspring neonatal development and on adult metabolic function when challenged by a high fat diet (HFD). The CR maternal model produced male and female offspring that were significantly smaller, in terms of weight and length, and females had delayed puberty. Adult offspring born to CR dams had a sexually dimorphic response to the high fat diet. Compared to offspring of maternal control dams, adult female, but not male, CR offspring gained more weight in response to high fat diet at 10 weeks. In adipose tissue of male HFD offspring, maternal undernutrition resulted in blunted expression of genes associated with weight gain and increased expression of genes that protect against weight gain. Regardless of maternal nutrition status, HFD male offspring showed increased expression of genes associated with progression toward nonalcoholic fatty liver disease (NAFLD). Furthermore, we observed significant, sexually dimorphic differences in serum TSH. These data reveal tissue- and sex-specific changes in gene and hormone regulation following mild maternal undernutrition, which may offer protection against diet induced weight gain in adult male offspring.
1 Introduction
Maternal levels of nutrition during gestation and lactation play a critical role in offspring development and adult metabolic health (1, 2). The earliest studies of maternal undernutrition affecting offspring metabolic function was from the Dutch “Hunger Winter”, in which a short-time famine occurred from 1944-1945. A review of the Dutch “Hunger Winter” cohort study found that in offspring born to mothers exposed to the famine in late gestation weighed less and were shorter, had smaller head circumferences at birth and that the placenta was less efficient (3, 4). These characteristics represent the hallmarks of intrauterine growth restriction (IUGR) (4–6).
The adult offspring of famished mothers were at an increased risk for metabolic syndrome compared to people not exposed to the maternal nutrient restriction (7, 8). Men exposed to the famine during late gestation were less likely to become obese compared to those who were exposed in early gestation (9). Studies have demonstrated that the prenatal environment affects DNA methylation and is dependent on gestational time of exposure and sex (10, 11).
These studies in humans have led to a fundamental hypothesis called the “thrifty phenotype”, initially proposed by Hales and Barker in 1992, and associates poor fetal growth with the development of impaired glucose tolerance and the metabolic syndrome in adulthood (12, 13). Undernutrition in late gestation or during lactation disrupts the neuroendocrine axes and alters the timing of pubertal development, resulting in altered neonatal IGF-1 and GH serum levels and decreased glucose tolerance in adulthood (14). IUGR influences growth, cognitive development, cardiac function, and metabolic homeostasis into adulthood (15, 16).
Researchers are still defining the role of the metabolic signaling adipokine, leptin, in embryonic development and adult health. During the rodent postnatal period and third trimester of humans, there is a leptin surge that occurs independent of adiposity (17). This postnatal leptin surge plays a vital role in the development of the hypothalamic wiring and multiple other organs, including browning of adipose tissue (18–21). The timing and amplitude of the leptin surge was found to be altered by nutrient availability during lactation, through altering litter size (22).
In 2005, the Yura et al. study was the first to demonstrate a premature shift in the neonatal leptin surge as a result of a 30% maternal undernutrition in C57BL/6 mice (23). The undernutrition started in mid-gestation and the neonatal leptin surge was characterized by combining male and female data, since there were no observable sex differences in the leptin surge. Male offspring were only used after weaning and challenged with the 60% high fat diet (HFD) as adults. They had increased weight gain on the 60% HFD and exogenous leptin administered at the peak of the premature surge was shown to replicate the increased HFD induced weight gain, as seen by the maternal undernutrition model. Using the C57BL/6 mouse, this study demonstrated that maternal undernutrition connected the premature shift in the neonatal leptin surge to adult weight gain following HFD exposure.
Our ongoing studies, using leptin receptor (LEPR) knockout “sighted” FVB mice, demonstrate the significance of somatotrope regulation by leptin and the necessity for determining the effect of a neonatal leptin surge on adult pituitary function (24–26). The focus of this study was to use a maternal undernutrition model (a non-genetic method) to shift the offspring neonatal leptin surge and determine the impact on offspring pituitary and metabolic function in adulthood, especially when challenged by nutrient stress (excess caloric fat). In this study, we show that a mild maternal murine undernutrition, starting in late gestation, caused a 3-day advance in the postnatal leptin surge and affected the way in which these offspring responded to a HFD as adults. We report sex differences in responses, including the important finding that males from underfed dams did not gain weight on the HFD, unlike controls (from ad libitum fed dams), which differs from what was expected by the “thrifty phenotype” hypothesis and the Yura et al. study (23). RNA-sequencing analysis (RNA-seq) of epididymal white adipose tissue (eWAT), liver and pituitaries from males born to undernourished and ad libitum fed dams reveal distinct tissue responses to the HFD and maternal undernutrition. These data offer insight into tissue specific regulation that affords protection from HFD-induced weight gain following a nutrient deficient developmental condition.
2 Materials and methods
2.1 Animal husbandry
All animal care protocols were approved by the UAMS Institutional Animal Care and Use Committee. FVB.129P2-Pde6b+ Tyrc-ch/AntJ (sighted FVB) mice were housed 1-4 animals/cage at 22.5°C with a 14-hour light (06:00-20:00) and 10-hour dark cycle. Dams for maternal undernutrition breeding were 8-11 weeks of age and bred with 3–4-month-old males. Breeder dams were fed a diet where 20.2% kilocalories (kcal) derived from protein, 21.4% kcal from fat, and 58.4% kcal from carbohydrates (5V5M, PicoLab). At postnatal day (PND) 21, offspring were weaned. Non-breeding mice were fed a diet consisting of 23.086% protein, 14.787% fat, and 62.127% carbohydrates (5V5R, PicoLab).
2.2 Establishing the maternal undernutrition model
Figure 1 summarizes the steps involved with establishing the 20% calorie restriction model. Male FVB.129P2 mice were singly housed for at least 24 hours prior to mating. Female FVB.129P2 mice were introduced to males in the evening of proestrus (embryonic day -1), which was determined by vaginal cytology. Vaginal plugs were visualized the next morning to confirm that mating occurred. Embryonic day 0 (E0) was established as the day after mating, when the vaginal plug was visualized. On E0, mated females were singly housed and weighed. At E10, females were again weighed to verify pregnancy with an expected 3 g increase in weight from E0. Pregnant females, or dams, received ~25 g of food daily starting at E12 to visually acclimate to receiving less food. Dams were paired into two feeding groups: ad libitum (FED) and 20% calorie restricted (CR20). At E15, the caloric restriction began, with CR20 dams receiving 80% of the food the paired FED dam consumed from the previous day. Food intake was measured daily around 0930. The undernutrition continued until the offspring were euthanized or weaned. There was one exception to this protocol: The day before parturition, the FED dams consumed less than 3 g of food. To prevent CR20 dams from consuming young at any point in the study, 3 g of food was the minimum made available to CR20 dams. Therefore, if the FED dams consumed less than 3 g of food before parturition, available food to undernourished dams was not reduced below 3 g. Pups were born around E19, with the average litter size being 8 ± 1 pups.
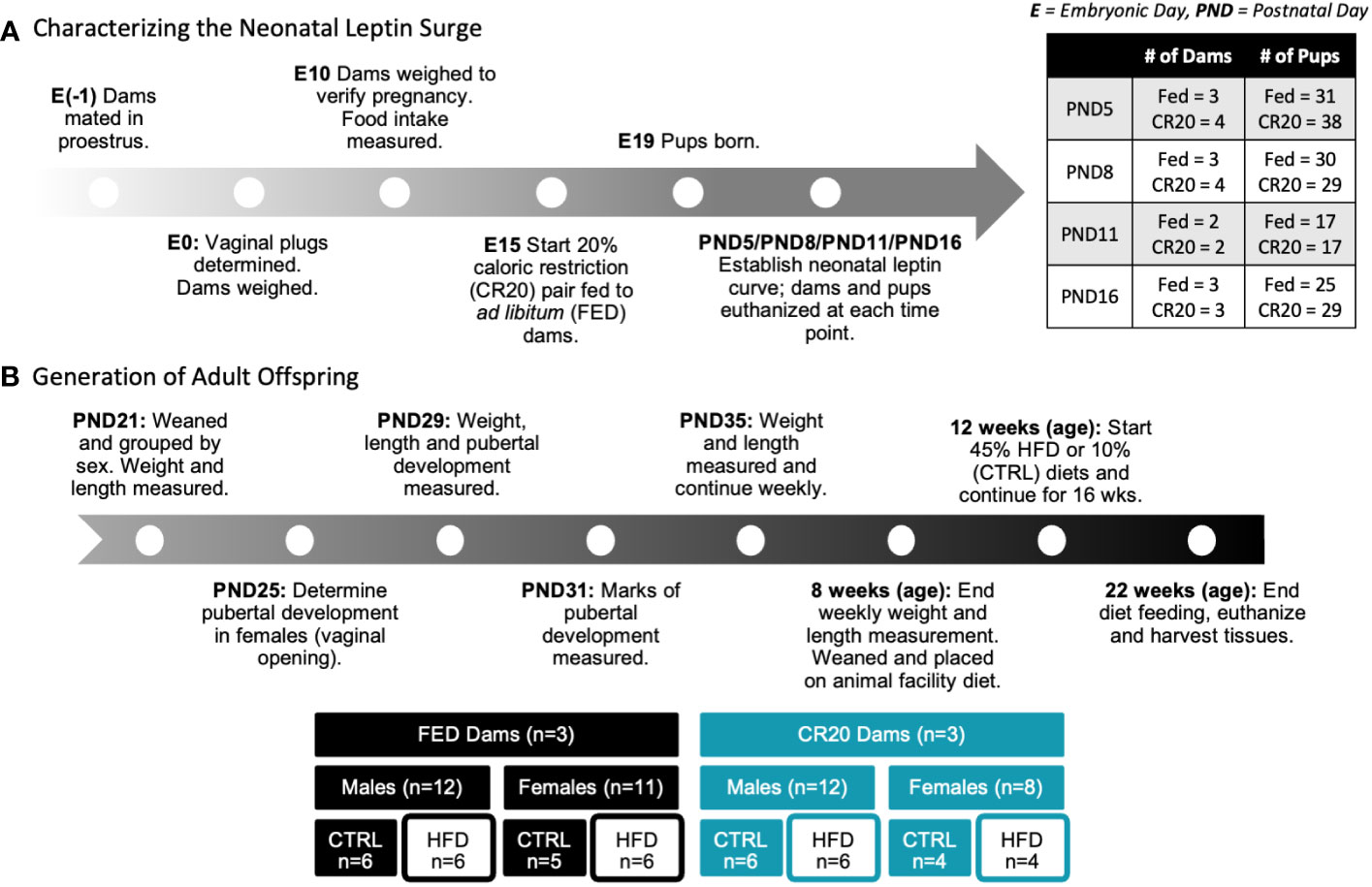
Figure 1 Schematic of experimental design. (A) Dam matched feeding, either ad libitum (FED) or calorically restricted by 20% compared to ad libitum mice (CR20), began at embyronic day 15 (E15). Pups were either euthanized at time points (PND 5, 8, 11 or 16) or continued to weaning. (B) Adult offspring were weight matched at weaning (PND21) within treatment groups and administered either the 10% fat diet (CTRL) or 45% high fat diet (HFD) for 10- or 16-weeks.
2.3 Characterizing FVB neonatal leptin curve induced by CR20 model
The neonatal leptin curve was determined by measuring leptin in the serum of pups at the following time points: PND1, PND5, PND8, PND11, and PND16. To generate a significant number of pups, 3 dams for each nutrient condition (FED or CR20) were used for each time point, except for PND1 where one dam was used for each condition. This resulted in ~28 pups/nutrient condition/time point. At euthanasia, dams and pups were anesthetized with isoflurane (Piramal Critical Care) and decapitated by guillotine (dams) or scissors (pups). Dams were euthanized prior to pups to prevent their stress response caused by pup removal. Trunk serum was collected into ice cold Eppendorf tubes (pups) or conical tubes (dams), and pituitaries collected in 150 μL RIPA (Sigma Aldrich, R0278) containing 10 μL/mL protease inhibitor cocktail (Sigma-Aldrich, P8340). To confirm sex, pup tail snips were collected and genotyped to determine the presence or absence of the male-specific “sex-determining region Y” gene, Sry, as previously described (27).
2.4 Diet induced obesity of adult CR20 offspring
For the diet induced obesity (DIO) studies, additional offspring were generated using the CR20 model as described above (see 2.2). FED and CR20 offspring were weaned at PND21 and separated by sex. Offspring weight and length (nose to anus) were measured weekly from weaning to 8 weeks of age. Female pups were inspected daily from PND 21 until the day of vaginal opening to determine the timing of puberty.
At 8 weeks of age, pups from each dam were blindly assigned to either a CTRL or HFD feeding group. Offspring were introduced to either a 10% fat (CTRL) diet (Envigo, TD.110675) or a 45% high fat diet (HFD) (Envigo, TD.06415) which they consumed for 10 weeks or 16 weeks. For each sex, this resulted in four experimental groups with 5 ± 1 mice/group: FED control (FED-CTRL); FED high fat diet (FED-HFD) and calorie restricted control (CR20-CTRL) and HFD (CR20-HFD). Food intake, an average of food consumed divided by the number of mice per cage, and individual mouse weights were measured weekly for the duration of the experiment. At euthanasia, trunk serum, pituitaries, eWAT, and liver were collected.
2.5 Pituitary, eWAT and liver collection
Pituitaries were collected in RIPA buffer with 10% protease inhibitor on ice and homogenized for 20 sec with a handheld homogenizer. A 20-30 μL aliquot was collected for RNA extraction, and the remaining homogenate was incubated overnight at 4°C. The next morning, the homogenate was centrifuged at 19283 x g for 20 minutes at 4°C. The supernatant was collected and stored at -20°C for later use. The left lateral lobe of the liver and epidydimal white adipose tissue (eWAT) were extracted and flash frozen in liquid nitrogen. For eWAT and liver, a 20-30 mg selection was excised from frozen tissue and homogenized in RIPA buffer with 10% protease inhibitor. RNA was extracted using the Maxwell 16 LEV simplyRNA Tissue Kit (liver samples, Promega, AS1280) or RNAzol RT (eWAT and pituitary samples, Sigma-Aldrich, R4533) and stored at -80°C. Liver samples to be used specifically for RNA-seq were lysed in Zymo DNA/RNA Shield buffer and RNA extracted using a Zymo column-based protocol.
2.6 Serum preparation and analyses
The postnatal leptin surge in our FVB mice was initially characterized with serum collected from pups aged postnatal day (PND) 1, 5, 8, 11, and 16. Ahima et al. (17) and results from a pilot study dictated our selection of PND 1, 5, 8, 11, and 16 for our CR20 study (see Methods 2.3). Whole blood was incubated on ice for 1 hour and centrifuged at 3200 x g for 20 minutes at 4°C. Serum was collected on ice and stored at -20°C until use. Serum leptin levels were measured at a 1:10 dilution using the Mouse/Rat Leptin Quantikine ELISA Kit (RNDSystems, MOB00). Serum IGF-1 was measured at a 1:500 dilution using R&D Systems Mouse/Rat IGF-I/IGF-1 Quantikine ELISA Kit (MG100). Serum pituitary hormones, LH, PRL, ACTH, FSH, and TSH, were measured with at 1:2.5 dilution with Milliplex’s MAP Mouse Pituitary Magnetic Bead Panel - Endocrine Multiplex Assay (MPTMAG-49K). Serum adipokines (IL-6, Insulin, MCP-1, PAI-1, Resistin, TNF-α) were measured undiluted with Milliplex’s MAP Mouse Adipokine Magnetic Bead Panel (MADKMAG-71K). Serum GH was measured at 1:2.5 dilution with Milliplex’s MAP Rat Pituitary Magnetic Bead Panel (RPTMAG-86K-01).
2.7 RNA sequencing and bioinformatics analysis
RNA extracted from pituitary, liver, and eWAT were submitted for RNAseq. Libraries for RNAseq were prepared using the TruSeq Stranded Total RNA Library Prep Gold kit (Illumina Inc) according to the manufacturer’s protocol. The resulting libraries were sequenced on the NovaSeq; paired-end 2X100. Libraries were prepared with an Illumina TruSeq Stranded mRNA kit using unique dual-indexing. Quality of sequencing for RNA reads were checked using FastQC. The adaptors and low-quality bases (Q < 20) were trimmed to a minimum of 36 base pairs using Trimmomatic (28). STAR was used to align Mus musculus GRCm39.104 reference genome with reads that passed quality control (29). To obtain raw counts, BAM files using Subread’s features Count function were transformed to log2 counts per million (CPM) (30). Genes with a low expression were filtered out and libraries normalized by trimmed mean of M values (31). R package, Limma, using the voom “With Quality Weights” function was used for differential expression and linear modeling (32). We did both Principal Component Analysis (PCA) and cluster analysis as part of the quality control (Supplementary Figure 6). Genes with an FDR adjusted p-values < 0.055 and a Log2FC ≥ 0.58 (1.5-fold increase) were considered significant and designated as differentially expressed genes (DEGs). Protocol was modified from previously described work (33).
Qiagen Ingenuity Pathway Analysis was used to generate potential upstream regulators from DEGs using the “Comparison Analysis” feature. Activation z-score, with associated p-value, was used to generate heatmaps of potential upstream regulators.
2.8 Quantitative real time polymerase chain reaction
For qRT-PCR verification of RNA-seq findings, complementary DNA was synthesized from pituitary, liver, and eWAT lysate (iScript, Bio-Rad, 170-8890), and qRT-PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659) and the QuantStudio 12K Flex system (Applied Biosystems, Life Technologies) as previously published (33). qPCR primers are listed in Table 1. Expression of each target was normalized to cyclophilin (Ppia), and fold-change was quantified using the delta-delta-CT method.
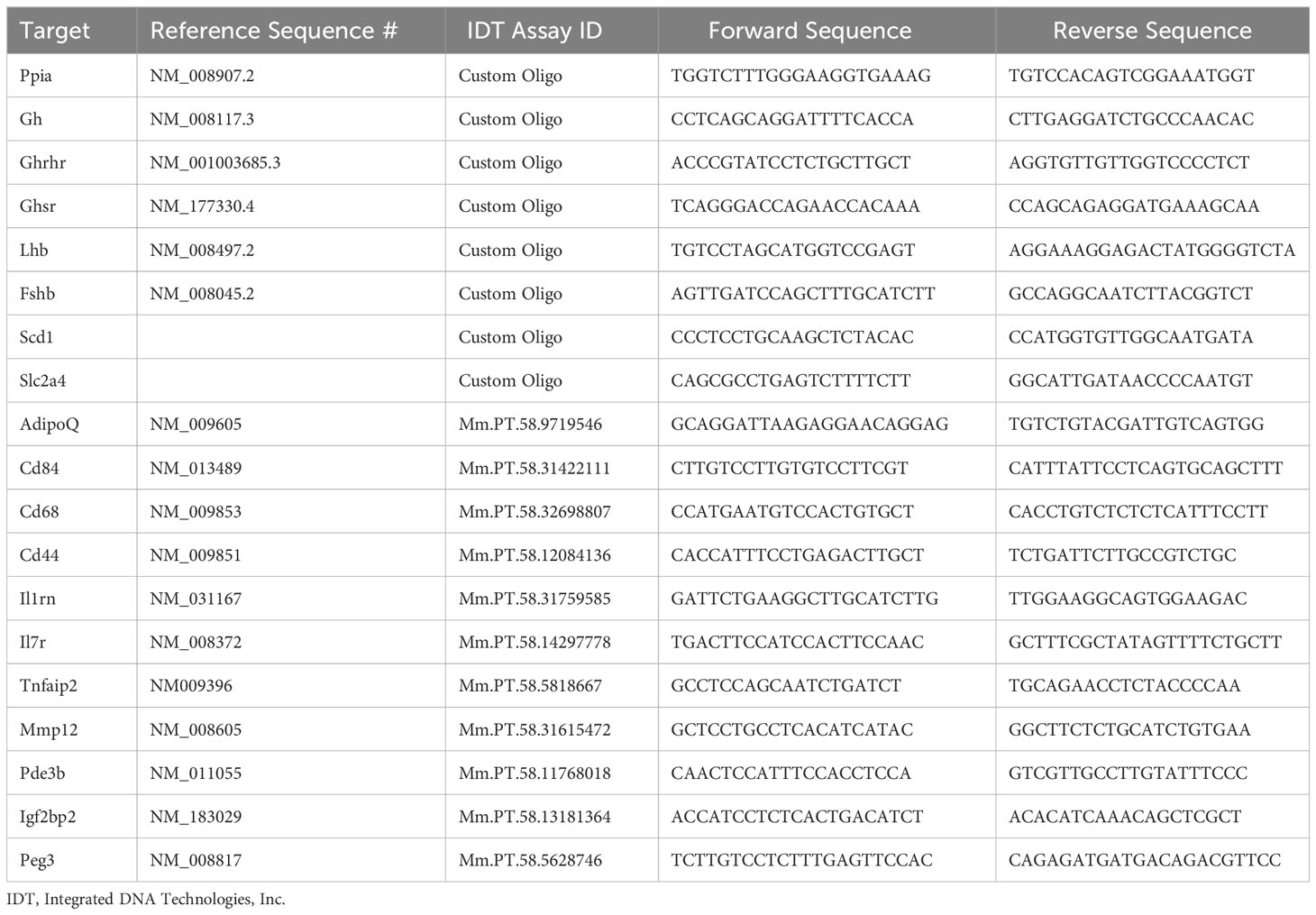
Table 1 Primers used for quantitative real time PCR.
2.9 Statistics
Statistical analyses of mRNA and pituitary hormone levels were performed on 6 male mice and 5 ± 1 female mice per condition. For most analyses, a Two-Way ANOVA followed by Newman-Keul’s, Bonferroni’s, or Fisher’s Least Significant Difference post hoc tests (p<0.05 was considered significant) was used, unless otherwise indicated. Three-way ANOVA followed by Fisher’s LSD test or Student’s t test was used when appropriate. All statistical methods are described in the individual figure legends. Power analyses have been published (34).
3 Results
3.1 Impact of 20% maternal calorie restriction on weight and leptin of dams and progeny
This study, to our knowledge, is the first to characterize a model of maternal caloric restriction and subsequent effects on offspring in the “sighted” FVB mouse strain. Therefore, we undertook a comprehensive characterization of food intake, weight gain, and serum markers to determine the metabolic impact of our design on this strain.
Dam weight gain at PND1 indicated weight gain from E15 to PND1 and showed that FED and caloric restricted (CR20) dams had similar weight gain during pregnancy (Figure 2A). Litter size was not significantly decreased by mild maternal undernutrition at each neonatal time point (data not shown). After birth, CR20 dams gained significantly less weight than FED dams at PND5 and PND16 (Figure 2A). This lack of weight gain correlated with significantly lower levels of serum leptin in CR20 dams (Figure 2C; Supplementary Figure 1).
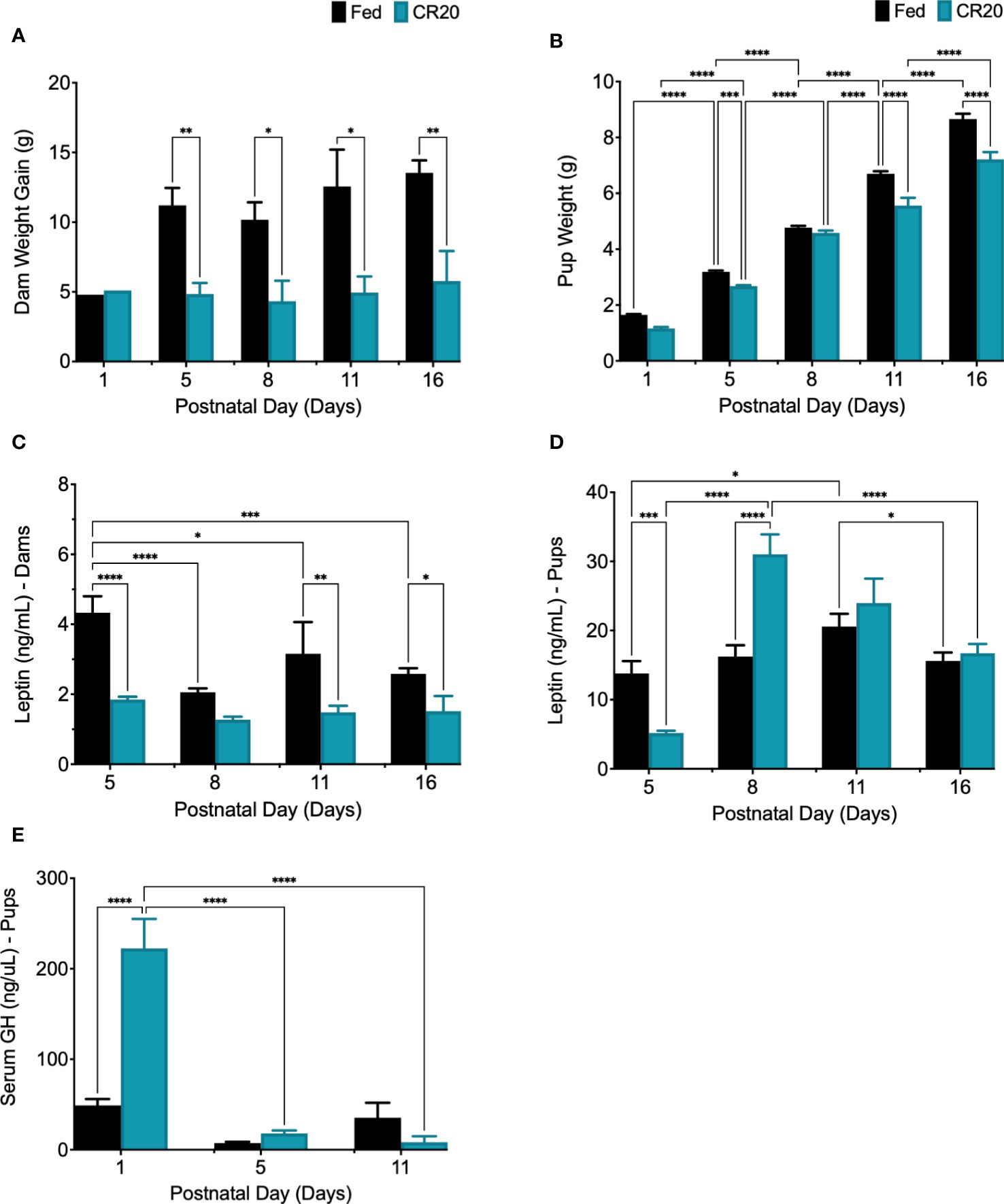
Figure 2 Characterizing postnatal leptin curve in offspring of FED and CR20 dams. (A) Dam weight gain (g) from D0 (mating) to euthanasia, PND5 (-6.35g, p = 0.0386) and PND16 (-7.76g, p = 0.0092). (B) Pup weight at euthanasia was less in CR20 pups: PND1 (-0.4875g, p<0.0001), PND5 (-0.514g, p = 0.0008), PND11 (-1.141g, p<0.0001), and PND16 (-1.445g, p<0.0001). (C) Dam serum leptin. (D) Fed pup serum leptin levels peaked at PND11 (+7070pg/mL from PND5 to PND11, p = 0.0260). (E) Serum GH was measured in PND1 (+173589pg/mL, p<0.0001), PND5, and PND11 pups by ELISA. For dam, n = 2-4 per condition and time point; error bars are SEM. For pups, n = 17-38 per condition and time point; error bars are SEM. Values that differ significantly between conditions: *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001. Dam Weight: Two-way ANOVA F (4, 17) = 0.9019. Pup Weight: Two-way ANOVA F (4, 202) = 8.913 and Student’s t-test was used for pup weight at PND1. Leptin Dams: Two Way ANOVA F (3, 16) = 4.906. Leptin Pups: Two-Way ANOVA F (3, 130) = 16.40, among FED pup time points: One-way ANOVA F (3, 58) = 3.022, and among CR20 pup time points: One-way ANOVA F (3, 72) = 46.46. Serum GH: Two-way ANOVA F (2, 37) = 19.69; Serum IGF-1: Student’s t test, one-tailed, *p<0.02, t = 2.198, df = 9.
Pups from CR20 dams weighed significantly less than pups from FED dams at PND1, PND5, PND11, and PND16 (Figure 2B). Analysis of the postnatal leptin surge revealed that serum leptin peaked at PND11, in both male and female FED pups (Figure 2D). Since there were no sex differences for postnatal serum leptin among experimental groups, postnatal male and female data were combined. In pups from CR20 dams, the peak leptin expression was three days earlier (PND8) than that of the FED dam’s offspring (PND11) (Figure 2D). Serum leptin levels were significantly lower in CR20 dam pups on PND5 than in FED pups (-9777pg/mL, p<0.0001). On PND8, serum leptin levels from CR20 dam pups elevated by +11656pg/mL (p<0.0001) compared to FED dam pups (Figure 2D). This was followed by a decrease at PND11 and PND16 to levels not different from those in FED dam pups (Figure 2D). Although serum GH levels were elevated in CR20 dam pups on PND1 (Figure 2E), neither postnatal nor adult mice from any group had significant changes in serum GH (Supplementary Figure 3). Serum IGF-1 was measured in serum from PND16 mice, but there was no significant difference between FED and CR20 offspring (Supplementary Figure 1B).
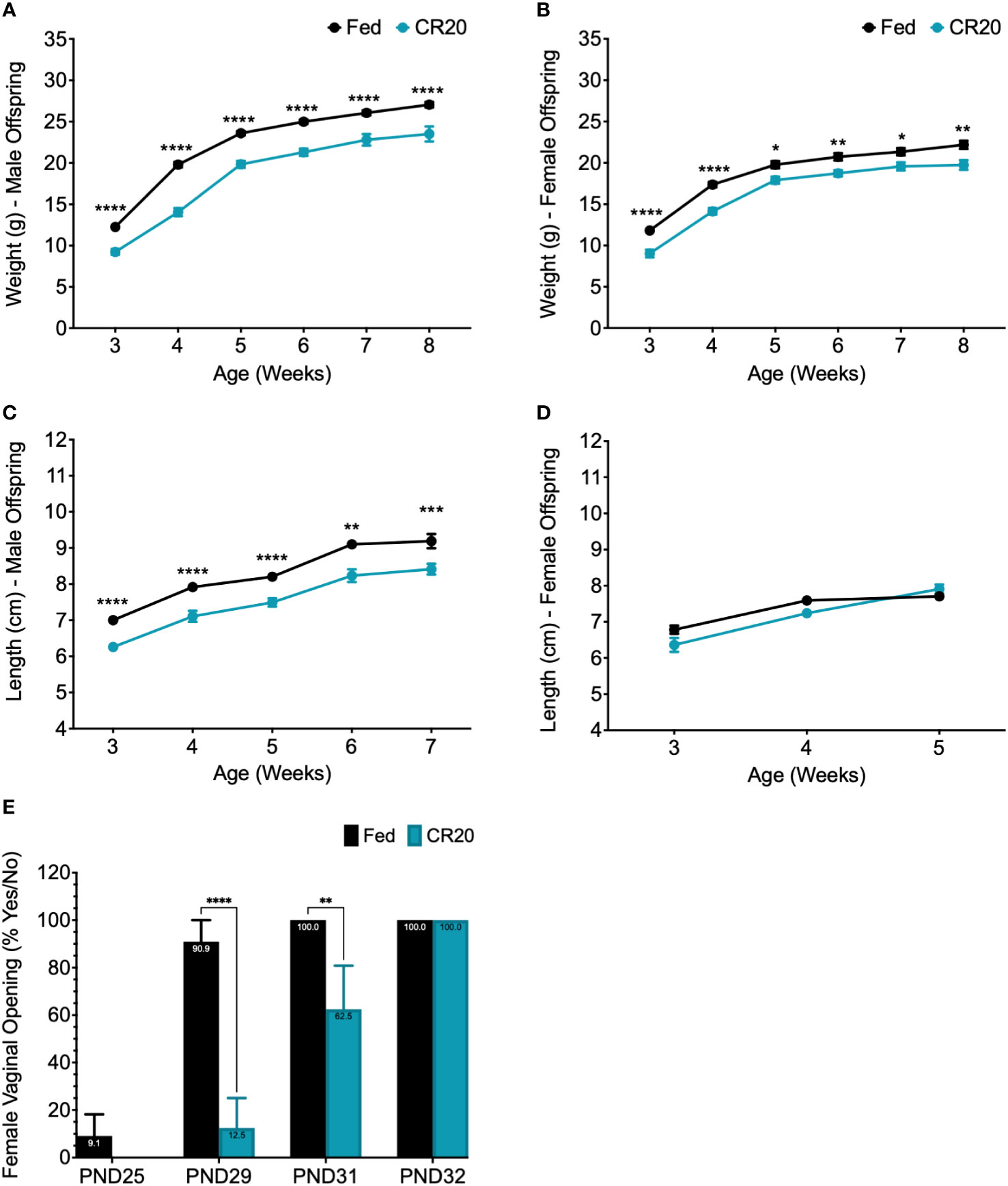
Figure 3 Weight and length of maturing offspring from CR20 and FED dams. The weight (g) of male (A) and female (B) offspring measured weekly starting at 3 weeks (weaning) to 8 weeks of age. Length (cm), from nose to anus, of male (C) and female (D) offspring starting at 3 weeks of age. Females were measured until 5 weeks, when length was shown to be not significant among conditions. (E) Female vaginal opening was determined (PND25-PND32) and numerical value given for “yes” = 100 and "no" = 0 for each mouse. For male offspring of each condition, n = 12. For female offspring, n = 11 from FED dams and n = 8 from CR20 dams. Values that differ significantly between conditions: *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001. Two-way ANOVA: male weight - F (5, 122) = 3.181, female weight - F (5, 100) = 0.8753, male length - F (4, 86) = 0.09828, and female length - F (2, 51) = 4.641.
3.2 Impact of maternal undernutrition on pup maturation
Offspring weight and length were measured weekly from weaning (3 weeks of age) to adult (8 weeks of age). Weight of male and female offspring of CR20 dams increased with age but did not catch up in weight to FED dam offspring (Figures 3A, B). This difference in weight was maintained until 8 weeks of age in both sexes (Figures 3A, B). Male CR20 offspring were significantly shorter than FED dam pups from weaning to adulthood (Figure 3C). In contrast, nose to anus length of female offspring were not significantly different at any time point between CR20 and FED groups (Figure 3D). Vaginal opening in the CR20 dam pups was delayed by 2-3 days compared to FED dam pups (Figure 3E).
3.3 Responses to 10-week high fat diet by progeny from FED and CR20 dams
The “thrifty phenotype” hypothesis suggests that maternal undernutrition programs offspring for metabolic dysfunction (35, 36) and C57BL/6 male offspring were shown to gain more weight on a high fat diet as adults if born to undernourished dams (23). Our study shows a sex-specific response by underfed progeny to a 10-week (Figure 4) treatment of a 45% high fat diet starting at 8 weeks of age.
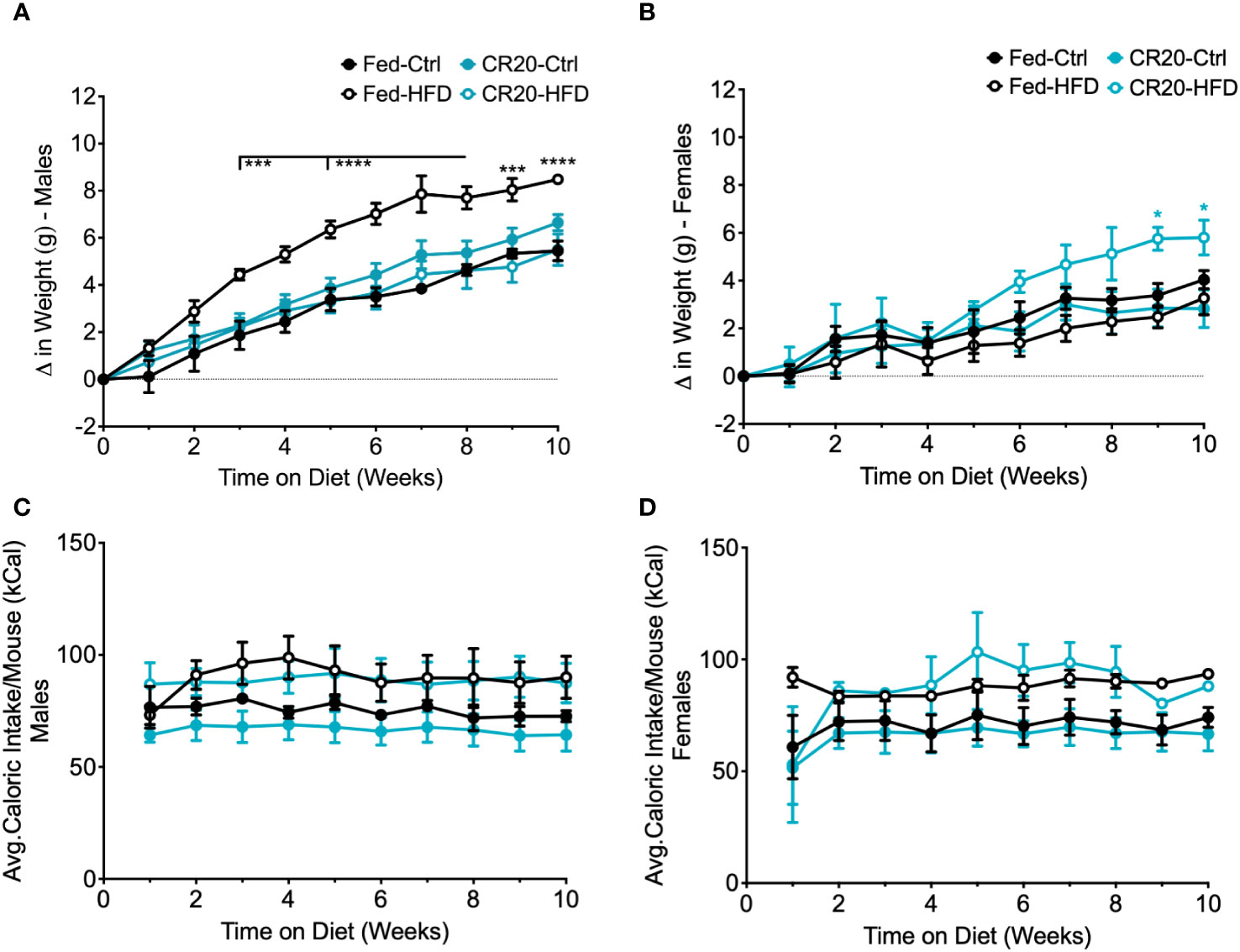
Figure 4 Weekly food intake, weight gain, and serum leptin of CR20 and FED offspring indicate that CR20 male offspring may be protected from diet induced weight gain on 10-week HFD. Weekly difference in weight gain in males (A) and female (B) due to diet and maternal nutrition. Average caloric food intake (kcal) per mouse for each week on the diet for males (C) and females (D). n = 6 per condition for males, n = 5-6 for FED females per condition and n = 4 for CR20 females per condition. Significant difference between HFD vs. CTRL among FED group: ***p<0.001, ****p<0.0001.
Among the adult males, only FED dam progeny on the high fat diet (FED-HFD) showed a significant increase in weight (Figure 4A). Adult males from the CR20 dams did not gain significantly more weight due to the HFD and thus appeared to be resistant to HFD induced weight gain (Figure 4A). In contrast, CR20-HFD females gradually increased in weight starting at 6 weeks on the HFD, and by 8 weeks, weight was significantly different than all three other groups (Figure 4B). The average food intake for adult offspring, irrespective of diet, was equivalent in term of kilocalories (Three-way ANOVA, Figures 4C, D).
3.4 Changes in serum hormone and adipokines in progeny from FED and CR20 dams on 10-week diet
The adipokine, leptin, is known to correlate with adiposity and assessed to further characterize the effect of maternal diet combined with nutrient stress. We found that correlation with their significant weight gain, FED-HFD males had higher levels of serum leptin compared to FED-CTRL males at 10 weeks of treatment (Figure 5A). The lack of weight difference between CTRL and HFD adult males from CR20 dams corresponded with the lack of change in serum leptin (Figure 5A). Despite the significant increase in CR20-HFD female weight gain, serum leptin levels were not significantly different among the HFD female groups following 10 weeks (Figure 5B).
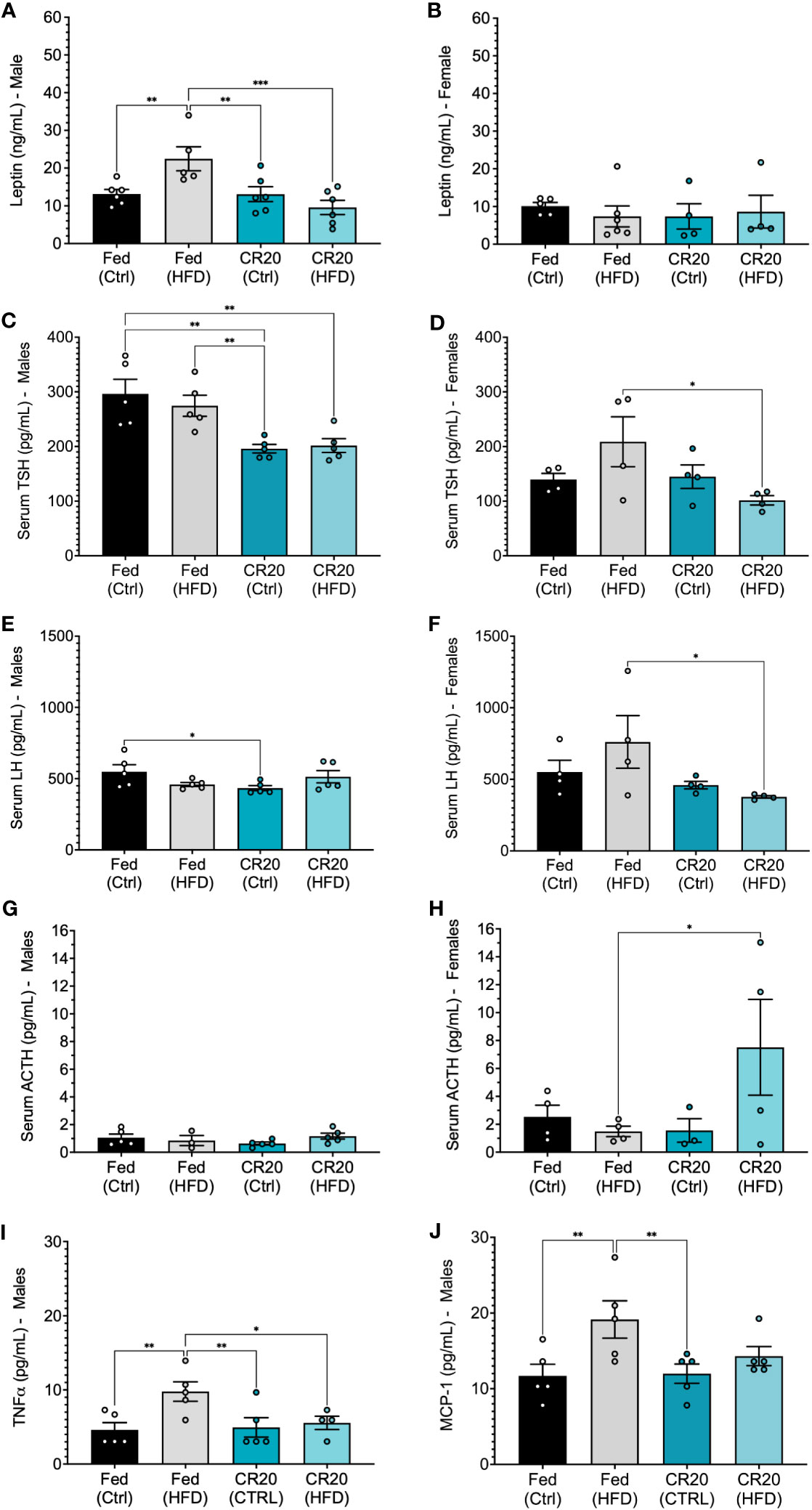
Figure 5 Serum hormone expression in male and female offspring, and male serum adipokines of 10-week treated mice. Leptin levels (ng/mL) in (A) male (FED-HFD vs FED-CTRL, +59%, p=0.0365) and (B) female CR20 and FED offspring at the conclusion of feeding on the diet. Serum TSH (C, D), LH (E, F), and ACTH (G, H) expression in male and female offspring of FED and CR20 dams. Serum adipokines, TNFα (I) and MCP-1 (J) in male offspring. n = 4-5 per condition for males and n = 4 for females per condition. Error bars are SEM. ANOVA followed by Fisher’s LSD test. *p<0.05; **p<0.01, ***p<0.001.
Since, pituitary products have been shown to be sensitive to nutritional status, we quantified serum hormone changes in the CTRL and HFD adult mice born to either FED or CR20 dams. Serum levels of all major pituitary hormones were quantified in all groups, but only TSH, LH and ACTH were significantly altered (Figure 5). TSH is involved in obesity through 1) insulin inhibited deodinase (less T3, high T4), which provides no negative feedback and resulting in high TSH, and 2) leptin is a stimulator of TRH (hypothalamus) which could increase TSH (37). Maternal caloric restriction significantly suppressed TSHß secretion in males regardless of offspring diet (Figure 5C). The maternal CR also significantly decreased serum LH levels in CR20-CTRL males (Figure 5E) and had no effect on serum ACTH (Figure 5G). Adult female offspring showed similar, significant trends in serum levels of both TSHß and LHß; FED-HFD females had slightly higher TSHß and LHß than FED-CTRL females, while CR20-HFD females had slightly lower serum TSHß and LHß compared to the CR20 controls. This results in statistically significant differences between the FED-HFD and CR20-HFD for both hormones (Figures 5D, F). In addition, the female CR20-HFD offspring had significantly increased serum ACTH levels compared to FED-HFD females (Figure 5H). No significant differences were found in serum PRL levels across any of the groups.
Since male, but not female, CR20 offspring showed resistance to weight gain in response to the HFD after 10 weeks, male serum was further probed for levels of adipokines and insulin. TNFα and MCP-1 were elevated among FED-HFD males, corresponding with their weight gain and suggestive of adipose tissue inflammation (Figures 5I, J). Insulin trended lower in the CR20-HFD males, but this was not statistically significant. The serum adipokines, resistin, IL-6, and PAI-1, were not changed among any of the groups (Supplementary Figure 3).
3.5 Response by male pituitary, eWAT, and liver transcriptome to maternal undernutrition and 10-week HFD challenge
Though our targeted approach revealed interesting hormonal changes, we next wanted to utilize an unbiased approach to determine the impact of the maternal nutrition-adult nutrient stress model on the global pituitary transcriptome. Since the adult male FED offspring were responsive to the HFD and CR20 offspring showed resistance to weight gain, we focused on males for this unbiased ‘omics’ study. Since, the adipose tissue and liver are central in whole body energy homeostasis, in addition to the pituitary, we included liver and epididymal white adipose tissue (eWAT) in our analyses to discern the genes and pathways involved in the protection against weight gain on a HFD in the CR20-HFD group. All three tissues (pituitary, eWAT, and liver) were assessed by RNA sequencing (RNAseq). Four differential expression comparisons were performed for each tissue’s RNA-seq datasets: CR20-CTRL vs FED-CTRL, CR20-HFD vs FED-HFD, FED-HFD vs FED-CTRL, and CR20-HFD vs CR20-CTRL. Differential expression analysis of the pituitary RNA-sequencing data revealed no significant differences in gene expression among any of the groups with the adj. p-value threshold set at <0.05. (Supplementary Tables 1, 2). This data is supported by modest transcriptomic changes observed by single-cell RNAseq analysis of FVB males exposed to a 60% HFD for 10 and 15 weeks at thermoneutral temperatures (38).
3.5.1 eWAT transcriptomics
Out of the three tissues analyzed, the eWAT had the largest total number of differentially expressed genes. The RNAseq analysis of eWAT revealed statistically significant differentially expressed genes (DEGs) (-log10adj. p-value>1.3, log2FC>1) from the following comparisons: CR20-HFD vs FED-HFD (140 genes upregulated, 642 downregulated) and FED-HFD vs FED-CTRL (517 upregulated, 135 downregulated) (Figures 6A, B). Notably, there were no significant changes when CR20-HFD eWAT gene expression was compared with that of the CR20-CTRL males (Supplementary Table 3). This corresponds well with the lack of weight gain in the CR20-HFD group.
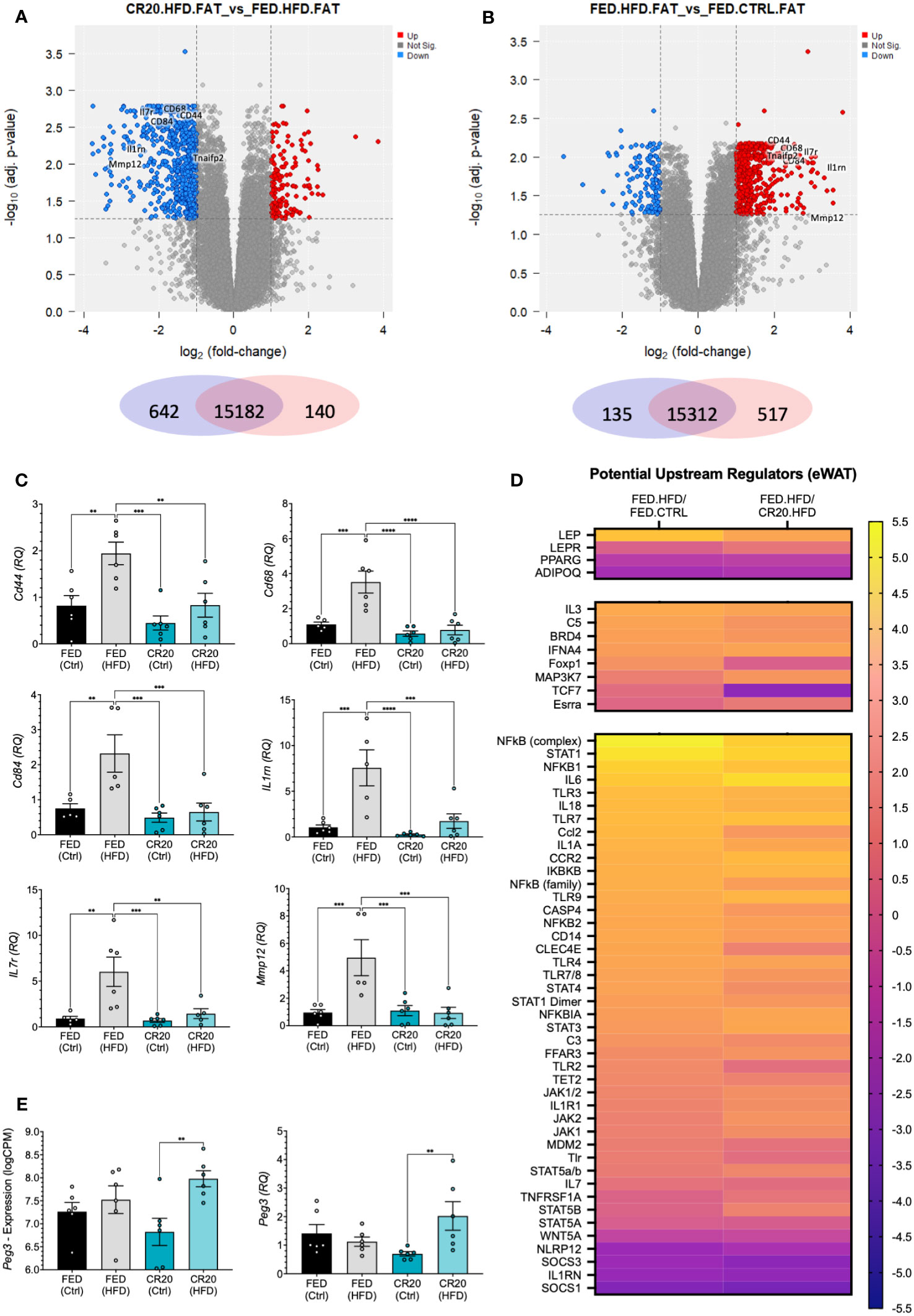
Figure 6 eWAT transcript expression by RNAseq of FED and CR20 male offspring fed either a CTRL or HFD for 10-weeks. Volcano plots of differentially expressed genes (logFC >= 1, adj. p-value <= 0.055) CR20.HFD vs. FED.HFD (A) and FED.HFD vs. FED.CTRL (B) comparisons. (C) qRT-PCR validation of identified cytokines in RNAseq data: Cd44, Cd68, Cd84, Il1rn, Il7r, and Mmp12. (D) Heatmap of potentially upstream regulators derived from IPA comparison analysis of DEG’s, separated by genes most associated with obesity, also associated with obesity, and considered protective against obesity. (E) Individual mouse RNAseq expression (logCPM, logFC = 1.156, adj. p=0.0018), and qRT-PCR (RQ, FC=2.912, p=0.0064) expression of Peg3. n = 6. Error bars are SEM. ANOVA followed by Fisher’s LSD test. Values that differ significantly between conditions: **p<0.01, ***p<0.001, ****p<0.0001.
To discern why the CR20 males responded differently to the HFD as compared to the FED-HFD, we identified inflammatory cytokines that were differentially expressed in the CR20-HFD vs FED-HFD and FED-HFD vs FED-CTRL comparisons (Figures 6A, B; Supplementary Tables 3, 4). Identified cytokines (Cd44, Cd68, Cd84, Il1rn, Il7r, Tnfaip2, and Mmp12) were upregulated in the FED-HFD vs FED-CTL but downregulated in the CR20 males when both HFD groups were compared. We confirmed these results with qRT-PCR of each target (Figure 6C). These same cytokines were unchanged in the CR20-HFD compared to CR20-CTRL (Figure 6C). Thus, the high fat diet did induce pro-inflammatory gene expression in the eWAT of normal FED dam male offspring, but not in the CR20 male offspring.
The Ingenuity Pathway Analysis (IPA) feature “Comparison Analysis” was used to determine correlations among significantly changed genes (adj. p-value<0.05, logFC>0.58) in the CR20-HFD vs FED-HFD and FED-HFD vs FED-CTRL comparisons. A subsequent upstream regulator analysis inferred the effects that various upstream regulators may be imparting on genes/pathways in our data sets. Predicted upstream regulators effects are designated as either “activated” or “inhibited”, with either a positive or negative activation z-score indicating whether there is increased activation, decreased activation, increased inhibition, or decreased inhibition. Based on existing literature, we identified upstream regulators “associated with obesity”, “closely associated with obesity”, and “protective against obesity when reduced”, all listed in Supplementary Table 5 (39, 40). Using the z-score we generated a heat map. Initial CR20-HFD vs FED-HFD comparison appeared to directly go the opposite of the FED-HFD vs FED-CTRL regulators. We inverted the analysis, instead comparing FED-HFD vs CR20-HFD, to determine if the CR20-HFD differed from the FED-CTRL. Figure 6D illustrates the activation z-score for each of these upstream regulators, and in most cases, the FED-HFD vs CR20-HFD comparison is predicted to change in the same direction as potential upstream regulator with the FED-HFD vs FED-CTRL comparison. This indicates that, when compared to FED-HFD, the CR20-HFD males are not unlike the FED-CTRL males, corresponding with the failure of CR20 males to gain weight on the HFD.
As mentioned, there were no DEGs identified in the CR20-HFD vs CR20-CTRL comparison when our standard constraints were applied (adj. p-value<0.05, logFC>0.58). This is highly significant, as the HFD had seemingly no major effect on gene expression in the eWAT. This is quite the opposite of what was seen with the FED groups, wherein 652 genes were differentially expressed with the HFD treatment. When statistical constraints were relaxed to consider DEGs with a non-adjusted p-value<0.05, we were able to identify a small number of DE genes in the CR20 FED vs HFD comparison (Figure 6E). Out of these genes, the most relevant to our study was paternally expressed 3 (Peg3), a transcription factor negatively correlated with adiposity (41). In CR20-HFD males, Peg3 was significantly increased and was the only gene changed with HFD treatment that was specific to the CR20 males. This finding was confirmed with qPCR (Figure 6E).
3.5.2 Liver transcriptomics
Maternal undernutrition did not affect how the liver responded to the HFD. RNAseq analysis revealed DEGs (-log10adj. p-value>1.3, log2FC>1) only in the FED-HFD vs FED-CTRL (2 upregulated, 10 downregulated) and the CR20-HFD vs CR20-CTRL (2 upregulated, 21 downregulated) comparisons (Figures 7A, B; Supplementary Tables 6, 7). Scd1 and Scl2a4, markers of lipid synthesis, were significantly decreased in both HFD groups, as shown by RNAseq and qPCR, which is expected with obesity progression (Figures 7A–C) (42, 43). IPA upstream regulator analysis, where HFD was compared to CTRL for each maternal condition, revealed upstream regulators with similar z-scores for the HFD data sets compared to their respective controls (Figure 7D; Supplementary Table 8). These potential regulators are involved in non-alcoholic liver disease (NAFLD) progression or inhibition (44–47).
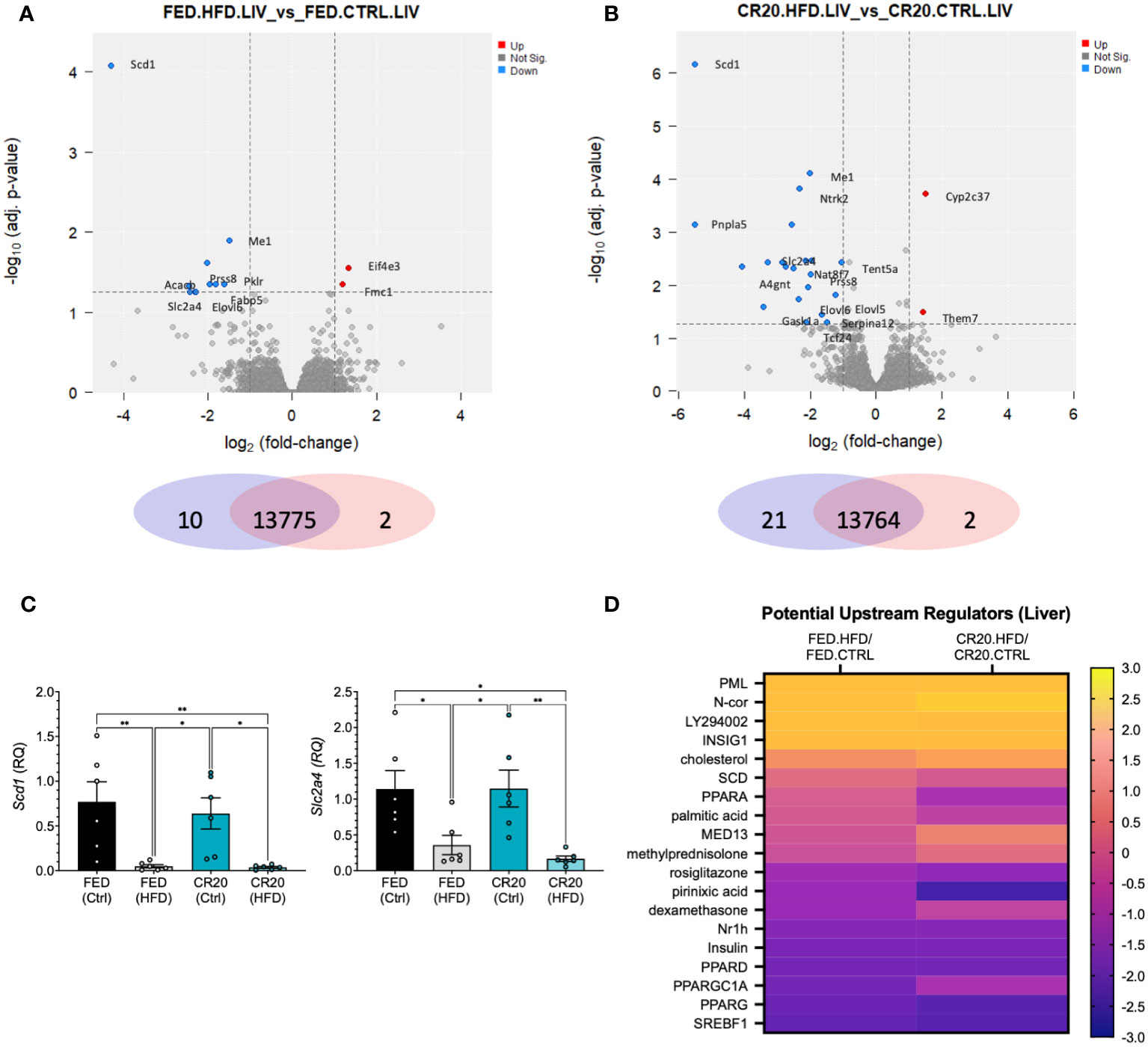
Figure 7 RNAseq liver transcript expression of the same adult male offspring born to FED and CR20 dams that were treated for 10-weeks. Volcano plots of differentially expressed genes (logFC >= 1, adj. p-value <= 0.055) FED.HFD vs FED.CTRL (A) and CR20.HFD vs. CR20.CTRL (B) comparisons. (C) qRT-PCR of Scd1 and Slc2a4, known liver markers of HFD induced obesity, validate RNAseq data. (D) Heatmap of potentially upstream regulators derived from IPA comparison analysis of DEG’s associated with NAFLD. n = 6. Error bars are SEM. ANOVA followed by Fisher’s LSD test. Significance designated by: *p<0.05 and **p<0.01.
3.6 Responses to 16-week high fat diet from FED and CR20 progeny
The lack of weight gain by CR20 males differed from the expected increased weight gain suggested by the “thrifty phenotype” hypothesis. We did an initial pilot study to verify findings. More CR20 offspring were generated and subject to a longer treatment time, 16-weeks, on the HFD.
Male and female offspring of FED dams showed significant weight gain with the increased treatment time on the high fat diet (Figures 8A, B). As observed in the 10-week treatment time, adult males from the CR20 dams did not gain significantly more weight with HFD, appearing again to be resistant to HFD induced weight gain (Figure 8A). With the longer treatment time, we were able to observe the FED-HFD females have significant weight gain by 12 weeks on the diet (Figure 8B). CR20-HFD females significant weight gain was seen as early as 9 weeks on the diet (Figure 8B) and did not rise to the same extent of the FED-HFD females, appearing to be slightly blunted. Again, the average food intake for adult offspring, irrespective of diet, was equivalent in term of kilocalories (Figures 8C, D).
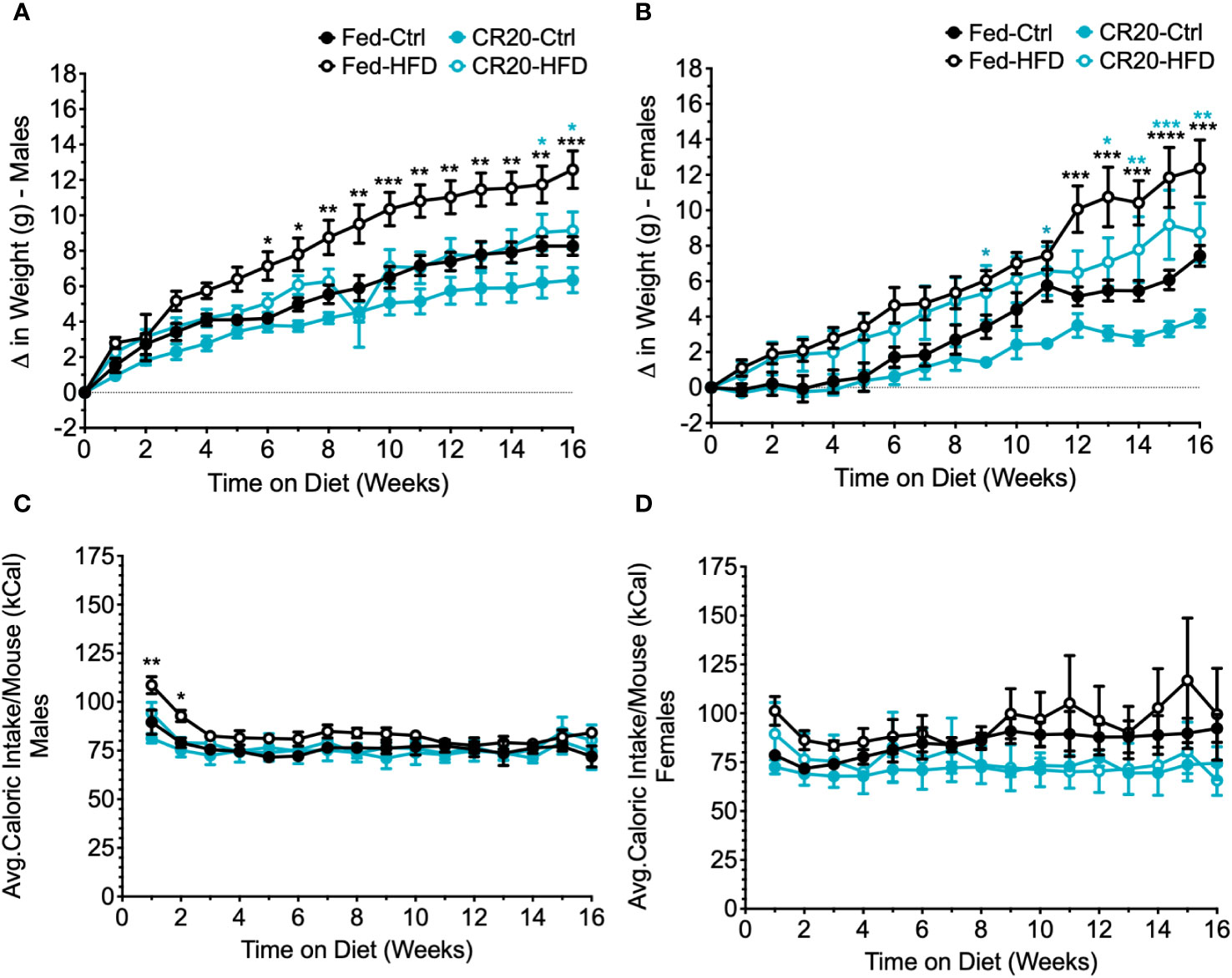
Figure 8 Weekly food intake, weight gain, and serum leptin of CR20 and Fed offspring treated for 16 weeks further indicate that CR20 male offspring are protected from weight gain on 45% fat diet diet and female offspring are partially protected. Average food intake per mouse for each week on the diet for males (A) and females (B). Weekly difference in weight from the start of the diet in male (C) and female (D) offspring. n = 7-8 for males and n = 5-8 for females per condition. Two-Way ANOVA. Values that differ significantly between HFD vs. CTRL of FED group (black asterisks) and HFD vs. CTRL of CR20 group (teal asterisks): *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Initial hormone and adipokine analysis of 16-week fed males, supported what was observed with the 10-week feeding. Serum TSH was lower with CR20-CTRL 16-week males (Figure 9A), but not significant among females (Figure 9B), although trending the same as with the 10-week feeding. Serum leptin was elevated among FED-HFD males, and the longer HFD feeding time caused an increase in serum leptin among the CR20 group, but not to the same extent as the FED (Figure 9C). Female serum leptin was not significantly altered but appeared to be shifted in the same direction as with the males (Figure 9D). Serum insulin was only elevated in the FED-HFD males at 16-weeks (Figure 9E), which was not observed among the 10-week fed group, and insulin was unchanged in females among all conditions (Figure 9F). In addition, IL-6 was also only significantly elevated among the FED-HFD males (Figure 9G), but not among females (Figure 9H).
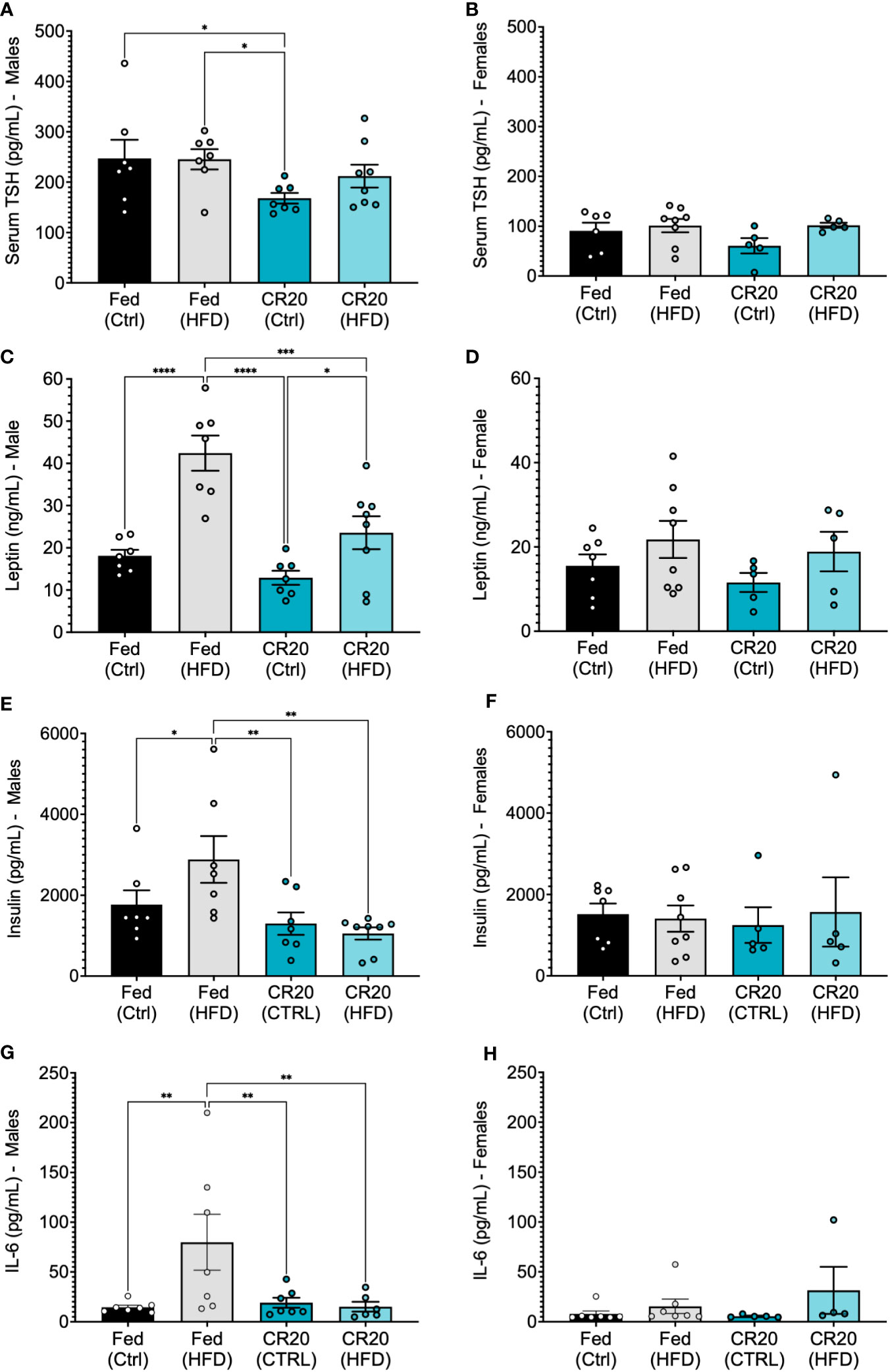
Figure 9 Serum expression of anterior pituitary hormones and adipokines from adult CR20 and FED offspring on the CTRL or HFD for 16-weeks. Serum expression of thyroid stimulating hormone (A, B). Leptin levels (ng/mL) in male (C) and female (D) CR20 and Fed offspring at the conclusion of diet treatment. Male (E) and female (F) serum insulin (pg/mL) and IL-6 (G, H). n = 7-8 for males and n = 5-8 for females per condition. ANOVA followed by Fisher’s LSD test. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
4 Discussion
We have previously characterized the role of leptin signaling to pituitary somatotropes and gonadotropes using selective deletion of LepR in sighted FVB mouse lines (24, 26, 34, 48–51). To better allow conclusions across studies and increase knowledge among diverse strains, we wanted to establish a model of maternal undernutrition in sighted FVB mice and investigate the effect of this undernutrition on the physiology of adult offspring challenged by a high fat diet. The mild (20%) caloric restriction model (CR20), starting in late gestation at embryonic day 15 (E15) and continuing through lactation, imparted expected and unexpected consequences upon offspring development and adult health.
4.1 Maternal caloric restriction caused delays in growth and pubertal development
“Catch-up growth” refers to an acceleration in growth (measuring body length, weight, organ weight, etc.), possibly due to endocrine or growth plate mechanisms, that permits normal growth progression after early growth retardation (52). The offspring of CR20 dams were born smaller compared to offspring of FED dams, in terms of both length and weight, and the female offspring had delayed pubertal development. These results are consistent with the effects of maternal undernutrition in the literature (53, 54). Decreases in birth length and weight are signs of intrauterine growth restriction (IUGR), a disorder that can be caused by maternal undernutrition (55, 56). Previous studies have also demonstrated that the timing of maternal undernutrition during gestation or lactation affects offspring’s propensity for obesity (22, 57). The present study was designed to initiate maternal undernutrition at late gestation and continue through lactation, a paradigm known to cause IUGR and affect the neonatal leptin curve (23, 54). Our CR20-CTRL male mice never showed “catch-up growth” in weight or length and our CR20 females continued to show reduced weight into adulthood.
We were intrigued by our observation of an early surge of leptin in the CR20 dam offspring, suggesting that maternal undernutrition by 20% resulted in an accelerated expression in the offspring of factors that drive adipocytes to secrete the leptin surge. At this point, the identity of the factors that regulate the normal neonatal leptin surge is poorly understood. It is known that the neonatal leptin surge is dependent on maternal nutrient status. In the study presented here, postnatal offspring of undernourished dams had significant alterations in both timing of the leptin surge and levels of leptin throughout the surge period. At PND5, serum leptin levels were lower in CR20 dam offspring than those of FED dam offspring. CR20 dam offspring had an early leptin surge peak, occurring at PND8, which was 3 days earlier than that of the FED dam offspring. However, by the time of the leptin surge peak in the FED dam offspring (PND11), the leptin levels in the CR20 offspring were significantly lower than at PND8 and to the same level as FED PND11.
Our CR20 female offspring had delayed puberty (males were not assessed). Previous studies have shown pubertal delay in both sexes when the neonatal leptin surge was ablated through a 50% maternal undernutrition model in rats (58), decreased hypothalamic regulation of pubertal maturation, and a delay in puberty when the neonatal leptin surge was blocked with a leptin antagonist (59, 60), indicating that pre-pubertal leptin levels can affect the timing of puberty onset in rodents.
4.2 Maternal caloric restriction resulted in a premature neonatal leptin surge and male offspring that did not show a high fat diet induced weight gain
In our CR20 model, offspring have a shifted leptin curve, with a peak at PND8, whereas the offspring from FED dams peaked at PND11. A shift in the peak of the leptin surge has been previously shown to have an impact on adult metabolism. Yura et al. (2005) demonstrated that a 30% maternal caloric restriction model resulted in a premature leptin surge which was associated with hypothalamic changes that rendered the animals less sensitive to the anorexigenic effects of leptin (23). The male mice gained more weight on a HFD, supporting the idea that the timing of the leptin surge is important to adult metabolic health. Further examination of the “thrifty phenotype” hypothesis suggests that since fetal development is sensitive to the nutritional environment, the resulting adaptations should improve fetal survival but in the long run may result in metabolic dysfunction in adulthood (52).
Our mild maternal nutrition model reveals responses to maternal undernutrition +/- HFD in adult offspring that are sex-specific. Weight gain to a HFD by female mice born to calorically restricted dams is lacking and could be due to the extensive time (27 weeks) needed to report weight response to the HFD (61). The novelty of this study is that changes in weight, or lack thereof, due to a HFD are reported in “sighted” FVB male and female offspring born to calorically restricted dams. Females born to FED dams did not gain significant weight on the HFD at 10 weeks, while the males did. We reasoned that FVB females on the 45% HFD used in our study would gain excessive weight with a longer treatment time, and our pilot study of the prolonged (16-week) HFD exposure supports this. The female FED-HFD had consistent, significant weight gain starting at 12 weeks, with a final delta weight of 12.4 g. In our studies of the somatotrope Lepr-null model, weight gain for females was slower than that of males and not significantly different from control females until 6 months of age (26). We did, however, observe increased adiposity in somatotrope Lepr-null females as young as 3-4 months of age (27). We did not assess adiposity in the current study; however, based on experience with the Lepr-null model, we conjecture that adiposity may increase before the onset of significant weight gain in FED-HFD females. Ongoing studies will address this question.
The CR20-HFD females gained significant weight earlier (10 weeks) than FED-HFD females. This finding supports studies where maternal undernutrition produced offspring that gained more weight or were more metabolically dysfunctional compared to control diet fed dam offspring (23) and supports the “thrifty phenotype” hypothesis. Thus, the maternal undernutrition may alter metabolic responses in females, so they become more sensitive to a HFD than their FED-HFD counterparts.
However, in sharp contrast, the FED-HFD males, but not the CR20-HFD males, gained significantly more weight, which was detected as early as 3 weeks on the diet. Unexpectedly, male CR20 dam offspring, on either diet, did not gain more weight than FED-FED males at 10 weeks. This indicates that the shift in the leptin surge did not compromise their responses to orexigenic or anorexigenic hormones. Indeed, unlike the FED-HFD males, the CR20-HFD males appeared resistant to the HFD. This differs from the Yura et al. (23) findings and could be due to 1) strain difference, C57BL/6 vs “sighted” FVB mouse and 2) start time of the maternal caloric restrictions, embryonic day 10.5 compared to e15 used in this study. Serum leptin was increased with HFD feeding in FED-HFD males, as expected with increased adiposity and weight gain seen in this group. However, leptin levels appeared normal in all other groups, which corresponded with the absence of significant weight gain.
In the serum, we observed significant yet distinct changes in TSH and LH with both sexes. As serum TSH is involved in regulating adipose tissue and found to be high in obese mice (62, 63), reduced serum TSH in male CR20 offspring indicates a TSH mediated mechanism involved in reduced adipose tissue accumulation. Female CR20 on the HFD had reduced TSH levels compared to FED-HFD, which may benefit long term adipose accumulation. Serum LH was reduced in the CR20 groups compared to control counterpart by both sexes, which may indicate prolonged reproductive function that was initially indicated by delayed puberty (64, 65). Elevated female CR20-HFD ACTH may be one of the comorbidities of obesity and is reflected by the advanced weight gain of this group (66, 67). Elevated TNFα and MCP-1, contributors to metabolic disease progression (68, 69), classify the increased weight gain in FED-HFD males as an obese state. We did not observe changes in offspring serum GH, therefore, we believe that ghrelin secretion was not altered due to maternal undernutrition.
4.3 Males born to undernourished dams had tissue specific responses to the high fat diet
We observed that weight gain of CR20-HFD males was blunted weight with 10-week of HFD treatment. Therefore, we wanted to determine tissue specific mechanisms that may contribute to this observation. In epidydimal white adipose tissue, significant transcript changes were found with HFD among the FED group and not the CR20 offspring, which corresponded with observed phenotype. Lack of increased inflammatory cytokines by the CR20-HFD mice supported the lack of progression toward obesity and metabolic disease that was seen in the FED-HFD group. Upstream regulator analysis of obesity associated genes and genes protective against obesity illustrate that the adipose tissue of FED-HFD males is poised for obesity development, while CR20-HFD males lack the changes in the genes that promote disease formation and thus are similar to the HFD-CTRL group. For example, the prevalence of Brd4 promotes obesity associated inflammation (70) and overexpression of proinflammatory cytokines, such as interferons (IFNA4), are associated with obesity pathogenesis (71), which are both elevated only in the FED group. Foxp1-deficient mice were reported to augment energy expenditure and be protected from diet-induced obesity (72), which is predicted to be slightly reduced in CR20-HFD males compared to the FED-HFD males. TCF7, a transcription factor involved in the canonical WNT/β-catenin pathway, was reported to regulate promoter activation of Ucp1 and Gprc6a in brown adipose tissue independent of the Wnt/β-Catenin Pathway (73). TCF7 is proposed to be reduced in CR20-HFD males RNAseq data, which suggests that the lack of weight gain on the HFD may not be due to eWAT browning. TLR3 and TLR7 KOs were found to be protective against HFD induced obesity and weight gain and IL1a-KO reduces adiposity, which reflects predicted gene changes in CR20-HFD males compared to FED-HFD (39).
The transcription factor Peg3 is a unique gene, as it was specifically changed in the CR20-HFD males, according to p-value, and validated by qRT-PCR. It is reported that Peg3+/− mutants develop excess adiposity, despite low food intake, have delays in pubertal development, and are leptin resistant (41). Peg3 is strongly expressed in the hypothalamus and pituitary during development (41). Peg3 is primarily expressed by the paternal allele due to repression of the maternal allele by DNA methylation. When the imprinting control region was deleted on the maternal allele, transcriptional regulation of Peg3 paternal allele was upregulated and resulted in increased body weight (74). Prolonged caloric restriction has been shown to alter DNA methylation and increase healthy lifespan (75). Therefore, epigenetic regulation of Peg3 maternal allele could potentially alter the action of Peg3. Further studies are needed to determine if nutrient stress alters the epigenetic regulation of Peg3 that would alter changes in weight.
Interestingly, the liver of HFD mice, regardless of maternal nutrition, responded equally to nutrient stress and showed indications of NAFLD progression. The liver and adipose tissue form a metabolic axis involving crosstalk, maintenance of energy homeostasis and having essential roles in nutrient uptake, processing, and storage (76–78). “Adipose tissue failure” is a term used to describe the failure of lipid storage and mobility by adipose tissue that results in lipid overload to the liver (79). Although lean, these individuals have NALFD progression but thought to be “metabolically unhealthy lean”, which is approximately 20% of the normal weight population and can have a three-fold greater risk for mortality (80). This observation in humans may account for the dysregulated lipid progression in the liver and eWAT of the CR20-HFD males, and if so, then a mild maternal undernutrition may be involved in metabolically unhealthy progression.
5 Conclusion
This study reports that a mild 20% maternal undernutrition in late gestation of mice affects the timing of the offspring neonatal leptin surge so that it peaks prematurely. The timing of this peak is like that reported by Yura et al. (2005) who used a 30% undernutrition model and reported that the adult progeny gained more weight on a HFD than their counterparts born to Fed dams (23). Female progeny of the 20% calorically restricted dams showed hallmarks of the “thrifty phenotype” hypothesis, with being smaller in size, having a delay in pubertal development, and gaining weight on the HFD as adults, but not more so than the control group. In contrast, male progeny from our 20% maternal undernutrition model had reduced weight throughout adult life and were resistant to the 45% HFD induced weight gain yet may have been metabolically unhealthy due to indications of NAFLD progression. Thus, this model shows distinct sex differences in developmental programming. In females, a premature leptin surge may have programmed the hypothalamus and other sensitive organs to become more sensitive to obesogenic regulators, while in males, this premature leptin surge may have conferred protection against adiposity at the expense of liver lipid toxicity.
Data availability statement
Original datasets are available in the Gene Expression Omnibus under accession number GSE247985. The original contributions presented in the study are publicly available. This data can be found here: https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE247985__;!!LFqOYw!pMmVRDxBAtsYN2f-zzT0NrwKrbz4T0JYw1GWHmxOWnhYLcQna-4A0xBnHmhUzwQR1YhyZIIBixVZNgehkiSb$.
Ethics statement
The animal study was approved by UAMS Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. MA-J: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Writing – review & editing, Validation. AO: Conceptualization, Writing – review & editing. AM: Investigation, Writing – review & editing. AH: Investigation, Writing – review & editing. AL: Investigation, Writing – review & editing. AG: Data curation, Formal analysis, Software, Writing – review & editing. SB: Data curation, Formal analysis, Software, Writing – review & editing. AR: Conceptualization, Methodology, Writing – review & editing. MM: Conceptualization, Funding acquisition, Resources, Writing – review & editing. AM: Conceptualization, Funding acquisition, Resources, Writing – review & editing. GC: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was funded in part by R01 HD087057 (GC, AM), R01HD093461 (AM, GC, MM), R01DK113776 (GC, MM, AM), 5 R01 DK127723 (GC, MM, AM), NCATS TRI: UL1 TR003107 and TL1 TR003109 (TKM), Arkansas Children’s Research Institute and the Arkansas Biosciences Institute: ABIPG4394 (TKM), TL1 TR002647 (AMR), and core facilities: NIGMS P20 GM103425 and P30 GM110702 (PI: Garcia-Rill).
Acknowledgments
Special thanks to Jewel Banik, Juchan Lim and Katherine Bronson for support during this project. Would also like to thank the University of Arkansas for Medical Sciences Division of Laboratory Animal Medicine, Genomics Core, and Bioinformatics Core.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1332959/full#supplementary-material
Supplementary Figure 1 | Dam correlation analysis of leptin to weight and serum IGF-1 in PND16 pups. (A) Pearson correlation analysis (two-tailed) of weight to leptin for dams revealed a moderate positive correlation for the FED (r=0.6091, p=0.0467) and CR20 (r=0.6794, p=0.0075) groups. (B) Serum IGF1 protein levels in PND16 pups was quantified by ELISA. Student’s t test.
Supplementary Figure 2 | Pituitary mRNA expression by qRT-PCR of Gh, Ghrhr, and Ghsr in adult CR20 and FED offspring on the Ctrl or HFD for 10-weeks. (A, B) Gh, (C, D) Ghrhr, and (E, F) Ghsr expression in male and female offspring of Fed and CR20 dams. n = 6 per condition for males, n = 5-6 for FED females per condition and n = 4 for CR20 females per condition. Error bars are SEM. ANOVA followed by Fisher's LSD test: *p<0.05.
Supplementary Figure 3 | Serum protein expression of GH and adipokines from adult CR20 and FED offspring on the Ctrl or HFD for 16-weeks. GH serum expression in males (A) and females (B). The adipokines resistin (C), IL-6 (D), and PAI-1 (E) in male offspring of Fed and CR20 dams. n = 7-8 for males and n = 5-8 for females per condition. ANOVA followed by Fisher's LSD test.
Supplementary Figure 4 | Pituitary mRNA expression by qRT-PCR of Gh, Lh and Fshb in 16-week treated mice. Pituitary transcript expression of (A, B) Gh, (C, D) Lh, and (E, F) Fshb in male and female offspring of Fed and CR20 dams. n = 4-7 for males and n = 3-4 for females per condition. Error bars are SEM. ANOVA followed by Fisher's LSD test. *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001.
Supplementary Figure 5 | Serum protein expression of anterior pituitary hormones and adipokines from adult CR20 and FED offspring on the Ctrl or HFD for 16-weeks. Serum expression of (A, B) LH, (C, D) ACTH, (E, F) PAI-1, and resistin (G, H) in male and female offspring of Fed and CR20 dams. n = 7-8 for males and n = 5-8 for females per condition. ANOVA followed by Fisher's LSD test. *p<0.05.
Supplementary Figure 6 | PCA plots and clustered dendrograms to evaluate overall correlation structure of RNAseq data. PCA plot and dendogram for pituitary (A, B), eWAT (C, D), and liver (E, F).
Supplementary Table 1 | Pituitary RNAseq data of males on 10-week diet. Gene list, log2 counts-per-millions for each sample and comparison analysis of HFD vs. CTRL for Fed and CR20 offspring.
Supplementary Table 2 | Pituitary RNAseq data of males on 10-week diet. Gene list, log2 counts-per-millions for each sample and HFD vs. HFD and CTRL vs. CTRL comparison analysis for Fed and CR20 offspring.
Supplementary Table 3 | eWAT RNAseq gene list of 10-week treated males, log2 counts-per-millions for each sample and comparison analysis of HFD vs. CTRL for Fed and CR20 offspring, and FED.HFD vs. CR20.HFD comparison.
Supplementary Table 4 | eWAT RNAseq gene list of 10-week treated males, log2 counts-per-millions for each sample and HFD vs HFD and CTRL vs CTRL comparison analysis for Fed and CR20 offspring.
Supplementary Table 5 | eWAT upstream regulators derived from 10-week treated males RNAseq data set using the Ingenuity Pathway Analysis Comparison Analysis feature that are associated with fat accumulation and protection.
Supplementary Table 6 | Liver RNAseq data of 10-week treated males. Gene list, log2 counts-per-millions for each sample and comparison analysis of HFD vs. CTRL for Fed and CR20 offspring.
Supplementary Table 7 | Liver RNAseq data of 10-week treated males. Gene list, log2 counts-per-millions for each sample and HFD vs. HFD and CTRL vs. CTRL comparison analysis for Fed and CR20 offspring.
Supplementary Table 8 | Liver upstream regulators derived from 10-week treated males RNAseq data set using the Ingenuity Pathway Analysis Comparison Analysis feature that are associated with NAFLD.
References
1. Franke K, Van den Bergh BRH, de Rooij SR, Kroegel N, Nathanielsz PW, Rakers F, et al. Effects of maternal stress and nutrient restriction during gestation on offspring neuroanatomy in humans. Neurosci Biobehav Rev (2020) 5–25. doi: 10.1016/j.neubiorev.2020.01.031
2. Ellis PJ, Morris TJ, Skinner BM, Sargent CA, Vickers MH, Gluckman PD, et al. Thrifty metabolic programming in rats is induced by both maternal undernutrition and postnatal leptin treatment, but masked in the presence of both: implications for models of developmental programming. BMC Genomics (2014) 15:49. doi: 10.1186/1471-2164-15-49
3. De Rooij SR, Bleker LS, Painter RC, Ravelli AC, Roseboom TJ. Lessons learned from 25 Years of Research into Long term Consequences of Prenatal Exposure to the Dutch famine 1944-45: The Dutch famine Birth Cohort. Int J Environ Health Res (2022) 32(7):1432–46. doi: 10.1080/09603123.2021.1888894
4. Belkacemi L, Nelson DM, Desai M, Ross MG. Maternal undernutrition influences placental-fetal development. Biol Reprod (2010) 83(3):325–31. doi: 10.1095/biolreprod.110.084517
5. Woodall SM, Johnston BM, Breier BH, Gluckman PD. Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatr Res (1996) 40(3):438–43. doi: 10.1203/00006450-199609000-00012
6. Reynolds CM, Perry JK, Vickers MH. Manipulation of the growth hormone-insulin-like growth factor (GH-IGF) axis: A treatment strategy to reverse the effects of early life developmental programming. Int J Mol Sci (2017) 18(8). doi: 10.3390/ijms18081729
7. Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr (1999) 70(5):811–6. doi: 10.1093/ajcn/70.5.811
8. Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med (1976) 295(7):349–53. doi: 10.1056/NEJM197608122950701
9. Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev (2006) 82(8):485–91. doi: 10.1016/j.earlhumdev.2006.07.001
10. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA (2008) 105(44):17046–9. doi: 10.1073/pnas.0806560105
11. Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet (2009) 18(21):4046–53. doi: 10.1093/hmg/ddp353
12. Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia (1992) 35(7):595–601. doi: 10.1007/BF00400248
13. Correia-Branco A, Keating E, Martel F. Maternal undernutrition and fetal developmental programming of obesity: the glucocorticoid connection. Reprod Sci (2015) 22(2):138–45. doi: 10.1177/1933719114542012
14. Harding JE, Derraik JG, Bloomfield FH. Maternal undernutrition and endocrine development. Expert Rev Endocrinol Metab (2010) 5(2):297–312. doi: 10.1586/eem.09.62
15. Salam RA, Das JK, Ali A, Lassi ZS, Bhutta ZA. Maternal undernutrition and intrauterine growth restriction. Expert Rev Obstet Gynecol (2013) 8(6):559–67. doi: 10.1586/17474108.2013.850857
16. Priante E, Verlato G, Giordano G, Stocchero M, Visentin S, Mardegan V, et al. Intrauterine growth restriction: new insight from the metabolomic approach. Metabolites (2019) 9(11). doi: 10.3390/metabo9110267
17. Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest (1998) 101(5):1020–7. doi: 10.1172/JCI1176
18. Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science (2004) 304(5667):108–10. doi: 10.1126/science.1095004
19. Wu R, Yu W, Fu L, Li F, Jing J, Cui X, et al. Postnatal leptin surge is critical for the transient induction of the developmental beige adipocytes in mice. Am J Physiol Endocrinol Metab (2020) 318(4):E453–E61. doi: 10.1152/ajpendo.00292.2019
20. Coupe B, Amarger V, Grit I, Benani A, Parnet P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology (2010) 151(2):702–13. doi: 10.1210/en.2009-0893
21. Delahaye F, Breton C, Risold PY, Enache M, Dutriez-Casteloot I, Laborie C, et al. Maternal perinatal undernutrition drastically reduces postnatal leptin surge and affects the development of arcuate nucleus proopiomelanocortin neurons in neonatal male rat pups. Endocrinology (2008) 149(2):470–5. doi: 10.1210/en.2007-1263
22. Skowronski AA, Shaulson ED, Leibel RL, LeDuc CA. The postnatal leptin surge in mice is variable in both time and intensity and reflects nutritional status. Int J Obes (Lond) (2022) 46(1):39–49. doi: 10.1038/s41366-021-00957-5
23. Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H, Nakao K, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab (2005) 1(6):371–8. doi: 10.1016/j.cmet.2005.05.005
24. Allensworth-James ML, Odle AK, Lim J, LaGasse AN, Miles TK, Hardy LL, et al. Metabolic signalling to somatotrophs: Transcriptional and post-transcriptional mediators. J Neuroendocrinol (2020) 32(11):e12883. doi: 10.1111/jne.12883
25. Allensworth-James M, Odle AK, Haney A, Childs GV. Sex differences in somatotrope dependency on leptin receptors in young mice: ablation of LEPR causes severe growth hormone deficiency and abdominal obesity in males. Endocrinology (2015) 156(9):3253–64. doi: 10.1210/EN.2015-1198
26. Childs GV, Akhter N, Haney A, Syed M, Odle A, Cozart M, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology (2011) 152(1):69–81. doi: 10.1210/en.2010-0498
27. Allensworth-James ML, Odle A, Haney A, MacNicol M, MacNicol A, Childs G. Sex-specific changes in postnatal GH and PRL secretion in somatotrope LEPR-null mice. J Endocrinol (2018) 238(3):221–30. doi: 10.1530/JOE-18-0238
28. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
29. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (2013) 29(1):15–21. doi: 10.1093/bioinformatics/bts635
30. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (2014) 30(7):923–30. doi: 10.1093/bioinformatics/btt656
31. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol (2010) 11(3):R25. doi: 10.1186/gb-2010-11-3-r25
32. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res (2015) 43(7):e47. doi: 10.1093/nar/gkv007
33. Moreira ARS, Lim J, Urbaniak A, Banik J, Bronson K, Lagasse A, et al. Musashi exerts control of gonadotrope target mRNA translation during the mouse estrous cycle. Endocrinology (2023) 164(9). doi: 10.1210/endocr/bqad113
34. Allensworth-James M, Banik J, Odle A, Hardy L, Lagasse A, Moreira ARS, et al. Control of the anterior pituitary cell lineage regulator POU1F1 by the stem cell determinant musashi. Endocrinology (2020) 162(3). doi: 10.1210/endocr/bqaa245
35. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull (2001) 60:5–20. doi: 10.1093/bmb/60.1.5
36. Vaag AA, Grunnet LG, Arora GP, Brons C. The thrifty phenotype hypothesis revisited. Diabetologia (2012) 55(8):2085–8. doi: 10.1007/s00125-012-2589-y
37. Flier JS, Harris M, Hollenberg AN. Leptin, nutrition, and the thyroid: the why, the wherefore, and the wiring. J Clin Invest (2000) 105(7):859–61. doi: 10.1172/JCI9725
38. Miles TK, Odle AK, Byrum SD, Lagasse AN, Haney A, Ortega VG, et al. Anterior Pituitary Transcriptomics Following a High Fat Diet: Impact of oxidative stress on cell metabolism. Endocrinology (2023). (Preprint). doi: 10.1210/endocr/bqad191
39. Mikhailova SV, Ivanoshchuk DE. Innate-immunity genes in obesity. J Pers Med (2021) 11(11). doi: 10.3390/jpm11111201
40. Liu S, Kim TH, Franklin DA, Zhang Y. Protection against High-Fat-Diet-Induced Obesity in MDM2(C305F) Mice Due to Reduced p53 Activity and Enhanced Energy Expenditure. Cell Rep (2017) 18(4):1005–18. doi: 10.1016/j.celrep.2016.12.086
41. Curley JP, Pinnock SB, Dickson SL, Thresher R, Miyoshi N, Surani MA, et al. Increased body fat in mice with a targeted mutation of the paternally expressed imprinted gene Peg3. FASEB J (2005) 19(10):1302–4. doi: 10.1096/fj.04-3216fje
42. Silbernagel G, Kovarova M, Cegan A, Machann J, Schick F, Lehmann R, et al. High hepatic SCD1 activity is associated with low liver fat content in healthy subjects under a lipogenic diet. J Clin Endocrinol Metab (2012) 97(12):E2288–92. doi: 10.1210/jc.2012-2152
43. Stefan N, Peter A, Cegan A, Staiger H, Machann J, Schick F, et al. Low hepatic stearoyl-CoA desaturase 1 activity is associated with fatty liver and insulin resistance in obese humans. Diabetologia (2008) 51(4):648–56. doi: 10.1007/s00125-008-0938-7
44. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol (2006) 87(1):1–16. doi: 10.1111/j.0959-9673.2006.00465.x
45. Flessa CM, Nasiri-Ansari N, Kyrou I, Leca BM, Lianou M, Chatzigeorgiou A, et al. Genetic and diet-induced animal models for non-alcoholic fatty liver disease (NAFLD) research. Int J Mol Sci (2022) 23(24). doi: 10.3390/ijms232415791
46. Musso G, Bo S, Cassader M, De Michieli F, Gambino R. Impact of sterol regulatory element-binding factor-1c polymorphism on incidence of nonalcoholic fatty liver disease and on the severity of liver disease and of glucose and lipid dysmetabolism. Am J Clin Nutr (2013) 98(4):895–906. doi: 10.3945/ajcn.113.063792
47. Wang Y, Nakajima T, Gonzalez FJ, Tanaka N. PPARs as metabolic regulators in the liver: lessons from liver-specific PPAR-null mice. Int J Mol Sci (2020) 21(6). doi: 10.3390/ijms21062061
48. Akhter N, CarlLee T, Syed MM, Odle AK, Cozart MA, Haney AC, et al. Selective deletion of leptin receptors in gonadotropes reveals activin and GnRH-binding sites as leptin targets in support of fertility. Endocrinology (2014) 155(10):4027–42. doi: 10.1210/en.2014-1132
49. Odle AK, Allensworth-James ML, Akhter N, Syed M, Haney AC, MacNicol M, et al. A sex-dependent, tropic role for leptin in the somatotrope as a regulator of POU1F1 and POU1F1-dependent hormones. Endocrinology (2016) 157(10):3958–71. doi: 10.1210/en.2016-1472
50. Odle AK, Akhter N, Syed MM, Allensworth-James ML, Benes H, Melgar Castillo AI, et al. Leptin regulation of gonadotrope gonadotropin-releasing hormone receptors as a metabolic checkpoint and gateway to reproductive competence. Front Endocrinol (Lausanne) (2017) 8:367. doi: 10.3389/fendo.2017.00367
51. Odle AK, Benes H, Melgar Castillo A, Akhter N, Syed M, Haney A, et al. Association of gnrhr mRNA with the stem cell determinant musashi: A mechanism for leptin-mediated modulation of gnRHR expression. Endocrinology (2018) 159(2):883–94. doi: 10.1210/en.2017-00586
52. Hales CN, Ozanne SE. The dangerous road of catch-up growth. J Physiol (2003) 547(Pt 1):5–10. doi: 10.1113/jphysiol.2002.024406
53. Wu G, Bazer FW, Cudd TA, Meininger CJ, Spencer TE. Maternal nutrition and fetal development. J Nutr (2004) 134(9):2169–72. doi: 10.1093/jn/134.9.2169
54. Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology (2001) 142(5):1692–702. doi: 10.1210/endo.142.5.8139
55. Howie GJ, Sloboda DM, Vickers MH. Maternal undernutrition during critical windows of development results in differential and sex-specific effects on postnatal adiposity and related metabolic profiles in adult rat offspring. Br J Nutr (2012) 108(2):298–307. doi: 10.1017/S000711451100554X
56. Engelbregt MJ, Houdijk ME, Popp-Snijders C, Delemarre-van de Waal HA. The effects of intra-uterine growth retardation and postnatal undernutrition on onset of puberty in male and female rats. Pediatr Res (2000) 48(6):803–7. doi: 10.1203/00006450-200012000-00017
57. Pico C, Palou M, Priego T, Sanchez J, Palou A. Metabolic programming of obesity by energy restriction during the perinatal period: different outcomes depending on gender and period, type and severity of restriction. Front Physiol (2012) 3:436. doi: 10.3389/fphys.2012.00436
58. Leonhardt M, Lesage J, Croix D, Dutriez-Casteloot I, Beauvillain JC, Dupouy JP. Effects of perinatal maternal food restriction on pituitary-gonadal axis and plasma leptin level in rat pup at birth and weaning and on timing of puberty. Biol Reprod (2003) 68(2):390–400. doi: 10.1095/biolreprod.102.003269
59. Mela V, Diaz F, Lopez-Rodriguez AB, Vazquez MJ, Gertler A, Argente J, et al. Blockage of the neonatal leptin surge affects the gene expression of growth factors, glial proteins, and neuropeptides involved in the control of metabolism and reproduction in peripubertal male and female rats. Endocrinology (2015) 156(7):2571–81. doi: 10.1210/en.2014-1981
60. Mela V, Jimenez S, Freire-Regatillo A, Barrios V, Marco EM, Lopez-Rodriguez AB, et al. Blockage of neonatal leptin signaling induces changes in the hypothalamus associated with delayed pubertal onset and modifications in neuropeptide expression during adulthood in male rats. Peptides (2016) 86:63–71. doi: 10.1016/j.peptides.2016.10.003
61. Yang Y, Smith DL Jr., Keating KD, Allison DB, Nagy TR. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obes (Silver Spring) (2014) 22(10):2147–55. doi: 10.1002/oby.20811
62. Lee MH, Lee JU, Joung KH, Kim YK, Ryu MJ, Lee SE, et al. Thyroid dysfunction associated with follicular cell steatosis in obese male mice and humans. Endocrinology (2015) 156(3):1181–93. doi: 10.1210/en.2014-1670
63. Walczak K, Sieminska L. Obesity and thyroid axis. Int J Environ Res Public Health (2021) 18(18). doi: 10.3390/ijerph18189434
64. Sanchez-Garrido MA, Castellano JM, Ruiz-Pino F, Garcia-Galiano D, Manfredi-Lozano M, Leon S, et al. Metabolic programming of puberty: sexually dimorphic responses to early nutritional challenges. Endocrinology (2013) 154(9):3387–400. doi: 10.1210/en.2012-2157
65. Rae MT, Kyle CE, Miller DW, Hammond AJ, Brooks AN, Rhind SM. The effects of undernutrition, in utero, on reproductive function in adult male and female sheep. Anim Reprod Sci (2002) 72(1-2):63–71. doi: 10.1016/s0378-4320(02)00068-4
66. Iwen KA, Senyaman O, Schwartz A, Drenckhan M, Meier B, Hadaschik D, et al. Melanocortin crosstalk with adipose functions: ACTH directly induces insulin resistance, promotes a pro-inflammatory adipokine profile and stimulates UCP-1 in adipocytes. J Endocrinol (2008) 196(3):465–72. doi: 10.1677/JOE-07-0299
67. Lucassen EA, Cizza G. The hypothalamic-pituitary-adrenal axis, obesity, and chronic stress exposure: sleep and the HPA axis in obesity. Curr Obes Rep (2012) 1(4):208–15. doi: 10.1007/s13679-012-0028-5
68. Sethi JK, Hotamisligil GS. Metabolic Messengers: tumour necrosis factor. Nat Metab (2021) 3(10):1302–12. doi: 10.1038/s42255-021-00470-z
69. Panee J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine (2012) 60(1):1–12. doi: 10.1016/j.cyto.2012.06.018
70. Hu X, Dong X, Li G, Chen Y, Chen J, He X, et al. Brd4 modulates diet-induced obesity via PPARgamma-dependent Gdf3 expression in adipose tissue macrophages. JCI Insight (2021) 6(7). doi: 10.1172/jci.insight.143379
71. Huang LY, Chiu CJ, Hsing CH, Hsu YH. Interferon family cytokines in obesity and insulin sensitivity. Cells (2022) 11(24). doi: 10.3390/cells11244041
72. Liu P, Huang S, Ling S, Xu S, Wang F, Zhang W, et al. Foxp1 controls brown/beige adipocyte differentiation and thermogenesis through regulating beta3-AR desensitization. Nat Commun (2019) 10(1):5070. doi: 10.1038/s41467-019-12988-8
73. Li Q, Hua Y, Yang Y, He X, Zhu W, Wang J, et al. T cell factor 7 (TCF7)/TCF1 feedback controls osteocalcin signaling in brown adipocytes independent of the wnt/beta-catenin pathway. Mol Cell Biol (2018) 38(7). doi: 10.1128/MCB.00562-17
74. He H, Perera BP, Ye A, Kim J. Parental and sexual conflicts over the Peg3 imprinted domain. Sci Rep (2016) 6:38136. doi: 10.1038/srep38136
75. Waziry R, Ryan CP, Corcoran DL, Huffman KM, Kobor MS, Kothari M, et al. Effect of long-term caloric restriction on DNA methylation measures of biological aging in healthy adults from the CALERIE trial. Nat Aging (2023) 3(3):248–57. doi: 10.1038/s43587-022-00357-y
76. Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose tissue-liver cross talk in the control of whole-body metabolism: implications in nonalcoholic fatty liver disease. Gastroenterology (2020) 158(7):1899–912. doi: 10.1053/j.gastro.2019.12.054
77. Duwaerts CC, Maher JJ. Macronutrients and the adipose-liver axis in obesity and fatty liver. Cell Mol Gastroenterol Hepatol (2019) 7(4):749–61. doi: 10.1016/j.jcmgh.2019.02.001
78. Vargas-Bello-Perez E, Du X, Liu G, Wang J, Gonzalez-Ronquillo M. Editorial: Functions of liver and adipose tissue in metabolic disorder diseases of ruminants. Front Vet Sci (2022) 9:1009112. doi: 10.3389/fvets.2022.1009112
79. Lee E, Korf H, Vidal-Puig A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J Hepatol (2023) 78(5):1048–62. doi: 10.1016/j.jhep.2023.01.024
Keywords: maternal nutrition, diet induced obesity, developmental programming, IUGR (Intrauterine growth restriction), thrifty phenotype hypothesis, transcriptome (RNA-seq), neonatal leptin surge, metabolism and endocrinology
Citation: Miles TK, Allensworth-James ML, Odle AK, Silva Moreira AR, Haney AC, LaGasse AN, Gies AJ, Byrum SD, Riojas AM, MacNicol MC, MacNicol AM and Childs GV (2024) Maternal undernutrition results in transcript changes in male offspring that may promote resistance to high fat diet induced weight gain. Front. Endocrinol. 14:1332959. doi: 10.3389/fendo.2023.1332959
Received: 03 November 2023; Accepted: 14 December 2023;
Published: 17 January 2024.
Edited by:
Hiroyasu Kamei, Kanazawa University, JapanReviewed by:
Takahiro Sato, Kurume University, JapanLeandro Martin Velez, University of California, Irvine, United States
Copyright © 2024 Miles, Allensworth-James, Odle, Silva Moreira, Haney, LaGasse, Gies, Byrum, Riojas, MacNicol, MacNicol and Childs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiffany K. Miles, dGttaWxlc0B1YW1zLmVkdQ==