 Valentina Guarnotta
Valentina Guarnotta Fabrizio Emanuele
Fabrizio Emanuele Riccardo Salzillo
Riccardo Salzillo Carla Giordano
Carla Giordano
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 12 December 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1329082
This article is part of the Research Topic Rare Forms of Pediatric Adrenal Disorders: Beyond Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency View all 10 articles
Adrenal Cushing’s syndrome is a rare cause of endogenous hypercortisolism in neonatal and early childhood stages. The most common causes of adrenal CS are hyperfunctioning adrenal tumours, adenoma or carcinoma. Rarer causes are primary bilateral macronodular adrenal hyperplasia (PBAMH), primary pigmented adrenocortical disease (PPNAD) and McCune Albright syndrome. The diagnosis represents a challenge for clinicians. In cases of clinical suspicion, confirmatory tests of hypercortisolism should be performed, similarly to those performed in adults. Radiological imaging should be always combined with biochemical confirmatory tests, for the differential diagnosis of adrenal CS causes. Treatment strategies for adrenal CS include surgery and in specific cases medical drugs. An adequate treatment is associated to an improvement of growth, bone health, reproduction and body composition from childhood into and during adult life. After cure, lifelong glucocorticoid replacement therapy and endocrine follow-up are required, notably in patients with Carney’s complex disease.
Cushing’s syndrome (CS) is characterized by an excess of glucocorticoids and represents a diagnostic and therapeutic challenge for clinicians. The total incidence of CS is approximately 0.7-2.4 new cases per million people per year. Of these, only about 10% occur in paediatric age (1). Paediatric and adolescent CS is quite rare, especially when compared to other disorders of this age including growth, puberty, thyroid and metabolic diseases.
The aim of this review is to discuss the causes of adrenal CS and their pathogenesis, focusing on the clinical presentation, diagnostic approach and therapeutic management, showing advantages and disadvantages in a critical way.
As in adult, paediatric and adolescent CS can result from an exogenous or endogenous cause. Exogenous CS, probably underdiagnosed, generally occurs in children who need to take steroid therapy. Although the majority of cases result from administration of oral or parenteral glucocorticoids, in childhood the topical and inhaled administration of supraphysiological doses requires particular attention. Since they have a thinner dermis layer of epidermis than adults, children are more vulnerable to systemic effects of topically glucocorticoids. In iatrogenic CS, it is essential for clinicians to be aware of sudden withdrawal of steroids and to inform patients and/or caregivers to prevent adrenal crisis.
Endogenous CS can be divided into ACTH dependent forms such as ectopic CS and pituitary ACTH-secreting tumours or ACTH independent forms including adrenocortical tumours (adenoma or carcinoma) and primary adrenocortical hyperplasia such as primary pigmented nodular adrenocortical disease (PNAD), primary bilateral adrenal macronodular hyperplasia (PBAMH) and McCune-Albright syndrome.
A bimodal age distribution has been reported, with higher incidence of adrenal forms before 7 years and ACTH-dependent forms over 7 years (2–4). Ectopic CS is very rare in children and adolescents with a prevalence <1% although a recent systematic review reported a median prevalence of 7% (X). Cushing’s disease (CD) accounts for 75%, while adrenal causes of CS account for 15% of all cases of hypercortisolism in children.
The most common causes of adrenal CS are hyperfunctioning adrenal adenoma or carcinoma. Rarer causes are BAMH, PPNAD and McCune Albright syndrome.
Adrenocortical tumours (ACTs) are very rare in childhood although they are an important cause of CS in this age. The reported incidence is about 0.2-0.3% of all paediatric tumours and a very low percentage of them are adrenocortical carcinomas. The incidence is different depending on the geographic area, with a higher incidence in Brazil compared to the United States (5, 6).
Paediatric adrenocortical tumours notably affect children aged from 0 to 4 years old and adolescents. In addition, a gender difference has been reported, with a female predominance before 3 years and after 13 years (5, 6). Two large studies have collected paediatric adrenocortical cancer cases, the International Pediatric Adrenocortical Tumor Registry (IPACTR) (7), which includes 254 patients, and a Brazilian monocentric study including 73 children (8).
The higher incidence in Brazil is explained by a high prevalence of p53 tumour suppression (TP53) mutation, which is involved in ACT pathogenesis. In patients without TP53 mutation, 11p15 chromosome defects have been reported (Beckwith-Wideman syndrome) (6). ACTs are generally functional and are only associated with virilizing signs and symptoms including pubic hair, faster growth and skeletal maturation and external genitalia enlargement in about 50-60% of children (9–12). In some cases virilization is combined with hypercortisolism (about 30% of cases) (13). Less frequently (10-15% of all cases) ACTs tend to present with CS, even though CS secondary to adrenocortical cancer is more frequent in adolescents and is associated with a poor survival rate (3, 14). Less than 5% of patients show feminization or hyperaldosteronism (13).
PPNAD can be isolated or associated with Carney’s complex. It frequently occurs in adolescents, even though several cases have been described in childhood (15). PPNAD is characterized by black adrenocortical micronodules located in the adrenal gland that appears atrophic in the areas not involved by nodules. Further, non-pigmented adrenocortical nodular disease has also been described, characterized by a unilateral adrenal tumour and absence of pigmentation, caused by a myosin heavy chain 8 mutation (16, 17).
PPNAD is generally caused by a mutation of the protein kinase 1 regulatory subunit 1A (PRKAR1A) gene, leading to activation of the cAMP/PKA pathway associated with a glucocorticoid hypersecretion (18). However, second-line molecular events are involved in cortisol hypersecretion as shown in patients with armadillo-repeat containing 5 (ARMC5) gene mutations (19).
PPNAD is generally characterized by overt CS; it is rarely associated with a subclinical or cyclical CS (20–22). The clinical presentation can rarely be classical (23) and is more frequently characterized by hyperandrogenism and osteoporosis beyond typical symptoms of Carney complex, which can be combined, such as skin pigmentation, cardiac myxomas and endocrine and non-endocrine tumours (15).
Primary bilateral macronodular adrenal hyperplasia (PBMAH), is a bilateral adrenal hyperplasia characterized by large yellow-to brown cortisol secreting nodules, not associated with other disorders. It is very rare in childhood and adolescence, while it is more frequent in older patients. In pediatric/adolescent cases, PBMAH can be associated with dominantly inherited genetic condition. It can be sporadic or associated with genetic mutations, such as hyperexpression of the G-protein aberrant receptors and pathogenic variants of MC2R, GNAS, PRKAR1A, and PDE11A. However, the ARMC5 gene is believed to be a major genetic cause of PBMAH, accounting for more than 80% of the familial forms (24). In patients with an aberrant expression of the gastric inhibitory polypeptide receptor (GIPR) in the adrenal glands, cortisol hypersecretion can be food-dependent (25). Clinical presentation can be overt or subclinical CS (26).
McCune-Albright syndrome, a sporadic heterogeneous disorder caused by somatic or post-zygotic activating mutations in the α subunit of G protein (Gsα gene) is the main cause of CS in the neonatal period. McCune-Albright syndrome is generally characterized by peripheral precocious puberty, café-au-lait skin pigmentation and polyostotic fibrous dysplasia, which can be combined with other endocrine disorders including CS, hyperthyroidism and growth hormone excess (27). In addition, it generally presents with severe CS (28–30), typically in the neonatal period, and though severe, it could be also transient. However, milder cases have also been reported (31).
The clinical presentation of paediatric CS is characterized by many different symptoms. Contrary to what is observed in pediatric CD where there is a male prevalence, non-gender differences are observed in pediatric adrenal CS.
The two most common are weight gain and growth retardation. Other signs include faciotruncular fat distribution, skin fragility, arterial hypertension, violaceus skin striae, acanthosis nigricans, hypertensive encephalopathy (32), skin infection, recurrent infection (33), nephrocalcinosis and kidney stone (34), delayed bone mineralization and puberty (35), depression, fatigability, behaviour disorders (36), ion disorders, such as hypokalaemia, and hypercalcemia, glucose intolerance or diabetes, as shown in Figure 1.
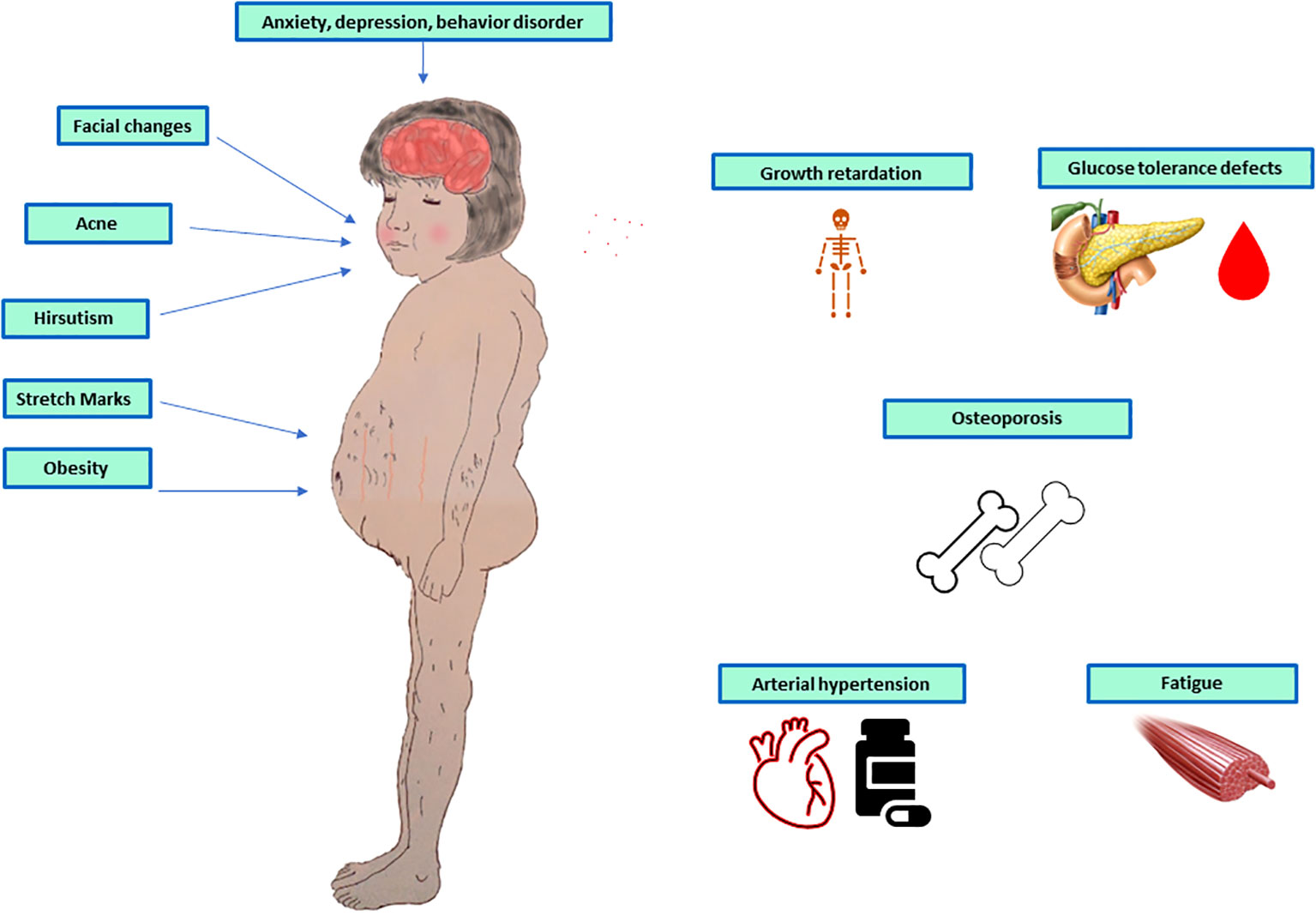
Figure 1 Symptoms and signs of paediatric adrenal Cushing’s syndrome.
In a recent retrospective study on a Chinese paediatric population with Cushing’s disease, growth retardation, weight gain, hirsutism and acne were the main clinical signs (37).
Chronic exposure to glucocorticoids results in an overall inhibition of the somatotropic axis with reduced production of GH and IGF-1. This factor, in association with some direct effects of glucocorticoids such as inhibition of collagen synthesis, cartilage sulphation and chondrocyte mitosis, contributes to growth arrest (38). Moreover, prolonged exposure to excess glucocorticoids can lead to pathological reduction of bone mass and in some cases osteoporosis, even in childhood. Another very significant aspect is pubertal development: a pseudo-precocious puberty and virilization were observed with gonadotrophin levels subnormal by a suppressive effect of chronic hypercortisolism.
In a large Brazilian study in 254 patients younger than 20 years of age with newly diagnosed or previously treated ACTs, Michalkiewicz et al. (7) reported a significant frequency of virilization (around 84% in the examined population) as a clinical onset sign in paediatric adrenal CS, either isolated or combined with other symptoms. Isolated Cushing’s syndrome was rare.
The impact of CS on the psychological sphere of children can be much more devastating. Indeed, in adults an improvement in quality of life and cognitive functions are generally described, while in children a worsening of cognitive functions and the appearance of psychopathological manifestations are observed even after the remission of hypercortisolism (39, 40).
Anyway, the clinical presentation of CS is different according to the age onset. In neonatal age it is generally associated with the classical skin marks; bone dysplasia and early puberty can also occur (41). In childhood the common causes of hypercortisolism are ACTs and the most frequent symptoms are hyperandrogenism, hirsutism and acne (early puberty). Another cause is PPNAD, which is not systematically associated with delayed growth (42, 43). In the peri-puberty period the typical symptoms are slow growth and weight gain, combined with delayed puberty, and behaviour disorder (aggressiveness, fatigability, poorer tolerance of physical exercises) (36).
After exogenous causes are excluded, a suspect pediatric patient should be referred to a third-level centre. Since there are no pediatric guidelines, the diagnosis and confirmation of hypercortisolism should be performed according to current guidelines for adult CS although there may be some difference (44, 45). It is important to note that none of these tests have not been used extensively in children population.
Three consecutive 24-hour urine collection, to evaluate urinary free cortisol (UFC), corrected for body surface area, are the first-level exams in older children (44). In younger children an adequate 24h urine collection could be difficult, leading to false negative results. False positive results could be obtained in cases of excessive water intake (more than 5L/day), severe obesity, anorexia, malnutrition, and physical and emotional stress, which could be indicators of pseudo-Cushing states (46).
The loss of circadian rhythm represents an hallmark of CS. Late-night salivary cortisol can be easily performed in children, notably in younger children and babies, even though it is characterized by greater inter-laboratory variability and normal levels in children are uncertain (47–49). Recently, in 320 healthy children (174 girls) between 4 and 16 years of age, bedtime and morning salivary cortisol and cortisone were measured by LC-MS/MS (49). The cutoff level for bedtime salivary cortisone was 12.0 nmol/L and salivary cortisol was 2.4 nmol/L, slightly lower than that of adults using the same method (2.8 nmol/L) supporting that age and gender-specific cutoff levels are not required for bedtime salivary cortisol or cortisone. Salivary cortisone and cortisol can be used interchangeably. In addition, exogenous hydrocortisone-contaminated samples can easily be identified by LC-MS/MS because cortisone/cortisol ration is very low.
An overnight suppression test can be performed by administering 25 μg/kg of dexamethasone at 11 p.m./midnight (maximum dose 1 mg) and serum cortisol measurement on the following day at 8 AM (50). Another biochemical test is the 2mg-2days dexamethasone suppression test (DST), which consists in the administration of 20-30 μg/kg/day of dexamethasone (maximum dose 2 mg/day) divided into 0.5 mg doses every 6 h, given at 09.00, 15.00, 21.00 and 03.00 h for 48 h (51, 52). The cortisol cut-off level should be <1.8 ug/dl (50 nmol/L).
Once the diagnosis is confirmed we should distinguish between ACTH dependent and ACTH independent forms, by assaying ACTH levels. If ACTH value is less than 5 pg/mL (1.1 pmol/L), it is strongly indicative of ACTH-independent CS. ACTH levels between 5 and 29 pg/mL (1.1-6.4 pmol/L) can be considered a grey zone, while ACTH values greater than 29 pg/mL (6.4 pmol/L) are strongly suggestive for ACTH-dependent conditions. See diagnostic work-up in Figure 2 (53).
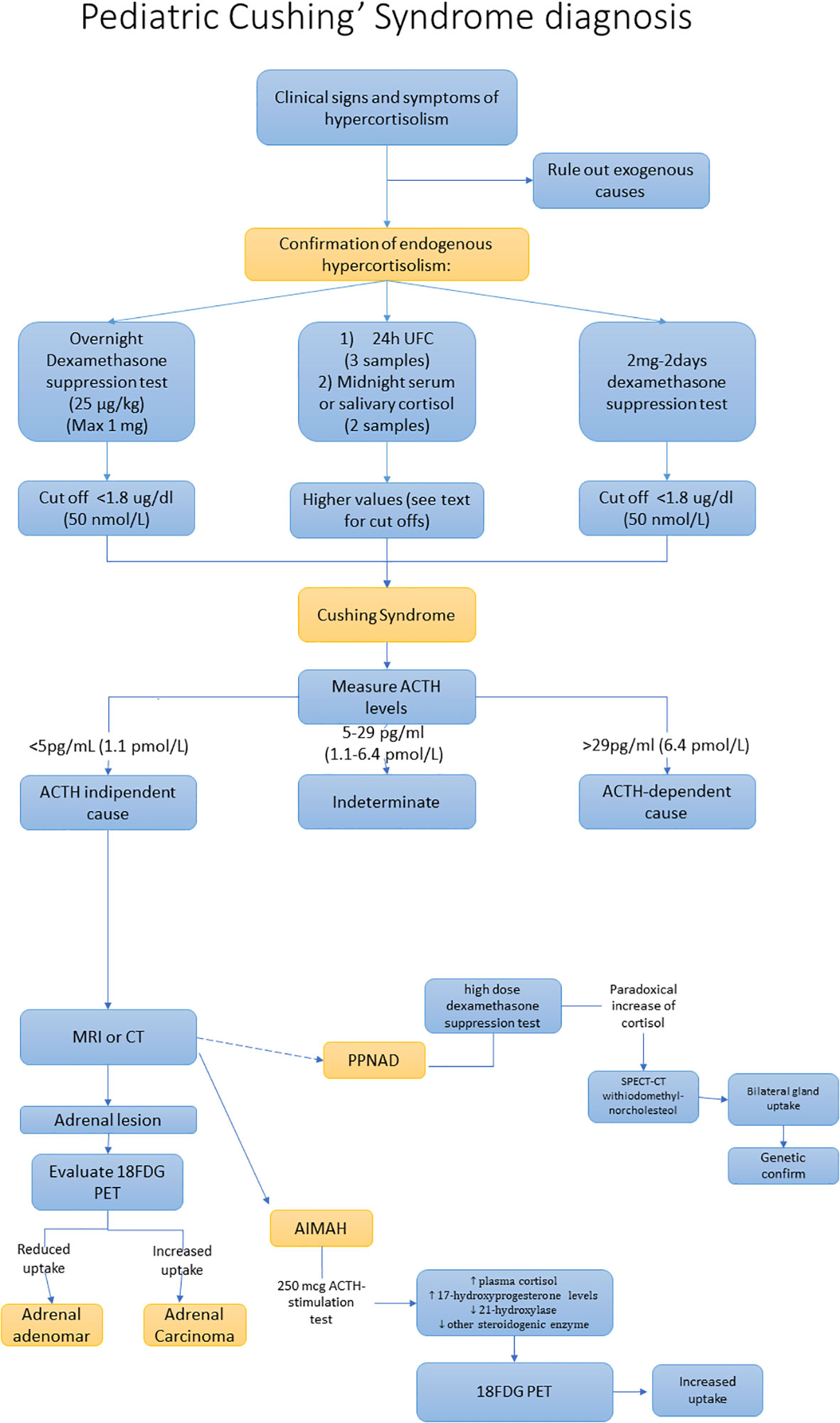
Figure 2 Diagnostic work-up of adrenal Cushing’s syndrome.
Additional exams are the high dose dexamethasone suppression test (HDDST) which can be performed for the PPNAD diagnosis. Indeed, in patients with PPNAD, HDDST can provide a paradoxical answer, with an increase of UFC/cortisol instead of a reduction. However, the sensitivity of the HDDST is quite low (39%) (12, 54, 55). Recently some studies identified a cut-off of UFC post HDDST/UFC pre HDDST of 1.08 reporting a sensitivity of 84% and a specificity of 75.6% (56). In patients with a high suspicion for PPNAD, the genetic analysis to identify mutations of the PKAR1A could confirm the diagnosis of PPNAD, notably in patients with combined Carney’s complex.
Modest elevations in plasma 17-hydroxyprogesterone or urinary 17-OH corticosteroids levels are frequently found in PBAMH (57, 58). In these patients, the 250 mcg ACTH-stimulation test leads to increased plasma cortisol and 17-hydroxyprogesterone levels reflecting the large hyperplastic adrenal glands with relative 21-hydoxylase and other steroidogenic enzyme deficiencies (57, 59). However, the suppressed ACTH levels allow one to distinguish PBAMH from congenital adrenal hyperplasia.
Adrenal imaging is essential for the definition of the adrenal CS cause. For the differential diagnosis, an upper abdomen ultrasound, which can be considered the first-line radiological exam, can be performed, followed by adrenal CT or MRI, which can show more clearly a single adrenal tumour or bilateral hyperplasia.
Adrenal CT scan and MRI show higher sensitivity and accuracy than ultrasound and ensure a better definition of fat richness, providing information on size, calcifications and heterogeneity of the lesion, and the anatomical relationship with adjacent organs. A CT scan using 3 mm or less in thickness can reveal micronodules inside the adrenal gland (60). MRI shows several advantages compared to CT scan, including absence of ionizing radiation, capability of imaging multiple planes, and improved tissue contrast differentiation. For this reason, an MR scan should be preferred in patients who need long-term follow-up.
High-resolution 18FDG PET could be useful to distinguish benign from malignant tumours (61, 62). However, an increased uptake of radiotracer has been reported in PBAMH, with an uptake similar to that of a malignant lesion (maximum standardized uptake value > 3.1), reflecting the high glycolytic activity of nodular lesions (63). Single-photon emission computed tomography (SPECT-CT) with the iodomethyl-norcholesterol (I-131) radiotracer can be useful in patients with PPNAD, showing bilateral gland uptake (64). However, considering the high radioactive exposure, the use of this method should be reserved for exceptional cases.
The therapeutic management of adrenal causes of CS depends on the primary cause. Adrenalectomy is the first-choice treatment. However, when surgery is contraindicated or refused, medical therapy should be recommended.
Unilateral adrenalectomy is indicated for unilateral benign cortisol-secreting adenomas.
Surgery is also the first-line therapeutic approach for adrenocortical cortisol-secreting cancers (10, 65). Contrasting findings are currently available on the usefulness of lymph node dissection (66). Mitotane is often used in adults as adjuvant therapy, but due to its neurotoxicity, in children it should be used with caution (67, 68). A nonrandomized single-arm study showed better survival in those children who achieved a mitotane level greater than 14 mg/L (69). In patients with unresectable adrenocortical cancers adjuvant chemotherapy could be used. Chemotherapy in children generally includes cisplatin and etoposide with or without doxorubicin combined with mitotane. Despite this multimodality approach, prognosis of paediatric adrenocortical cancer with metastatic disease remains poor, with an estimated 5-year survival below 20% (70, 71).
In cases of adrenocortical cancers with distant metastases, checkpoint inhibitors could be recommended. Notably, some encouraging findings have been reported with pembrolizumab (10, 72).
Bilateral adrenalectomy is strongly recommended in patients with PPNAD (73). Surgery is associated with biochemical remission, growth catch-up, weight loss and improvement of CS phenotype (74). Unilateral adrenalectomy can be associated with remission of symptoms without risk of adrenal insufficiency. However, in some patients recurrence of CS could occur (75, 76).
In patients with PBAMH the suggested treatment consists in surgical removal of one or both adrenal glands, and rarely in specific drugs inhibiting excessive cortisol secretion (77, 78). The best surgical approach is still on debate. Indeed, unilateral adrenalectomy has a remission rate of hypercortisolism reaching 96%, but with a recurrence rate of 23% (75, 79, 80). However, a post operative follow-up is strongly required in order to prevent adrenal insufficiency and CS recurrence (81, 82).
In patients with contraindications or refuse of surgery, drugs inhibiting adrenal cortisol synthesis (ketoconazole, metyrapone and recently osilodrostat) have been used in rare cases (83). At the moment, there are no medications approved by EMA or FDA for use in pediatric CS.
Recently, a child with mild, cyclical CS due to PBAMH, who carries a novel germline pathogenic variant in KCNJ5, was treated at 4 years 9 months with very low doses of ketoconazole (300 mg/day), increased to 400 mg/day at 8 years. She showed improved linear growth and normalization of BMI, along with resolution of behavior changes and normalization of blood pressure. However, the discontinuation of the drug for 6 weeks led to recurrence of CS (84).
Osilodrostat has not yet been approved for pediatric patients, but is currently being evaluated in a small Phase II trial (NCT03708900) in the pediatric population (<18 years) and the results are expected at the end of 2023. Recently, in a 14-year-old male with ectopic CS due to metastatic pancreatic tumor, osilodrostat (18 mg twice daily) was well tolerated and obtained a rapid improvement and normalization of UFC (85).
Glucocorticoid replacement therapy has a significant role in the management of patients with adrenal CS who have undergone adrenalectomy. Generally glucocorticoid replacement therapy should be recommended in the pre- and perioperative periods and after surgery for at least 6 months from the adrenalectomy (1). In patients who have undergone bilateral adrenalectomy, a chronic steroid replacement therapy is required, except for patients who have undergone adrenal sparing surgery with preservation of healthy cortical tissue.
Adrenal CS is a quite rare condition in childhood that occurs more frequently in children aged less than 7 years. Adrenal CS can result from adrenocortical tumours including adenoma or carcinoma or from primary adrenal hyperplasia including PPNAD, PBAMH or McCune Albright syndrome. The clinical presentation can vary according to the age of onset. However, weight gain, clinical hyperandrogenism and growth disorders are the most frequent signs and symptoms. Early diagnosis is essential to improve clinical symptoms and to better manage the primary cause. Indeed, in cases of adrenocortical cancer an early diagnosis is associated with a better survival. Unilateral or bilateral adrenalectomy is the first-line therapeutic approach. In patients who have undergone bilateral adrenalectomy lifelong glucocorticoid replacement therapy is required. The prognosis is generally good, except for patients with adrenocortical cancer, for which it depends greatly on the initial stage.
VG: Conceptualization, Writing – original draft, Writing – review & editing. FE: Writing – original draft. RS: Writing – original draft. CG: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lodish MB, Keil MF, Stratakis CA. Cushing’s syndrome in pediatrics. Endocrinol Metab Clin North Am (2018) 47:451–62. doi: 10.1016/j.ecl.2018.02.008
2. Libuit LG, KaraGeorgiadis AS, Sinaii N, Nguyen May NM, Keil MF, Lodish MB, et al. A gender-dependent analysis of Cushing’s disease in childhood: pre- and postoperative follow-up. Clin Endocrinol (Oxf) (2015) 83:72–7. doi: 10.1111/cen.12644
3. Stratakis CA. Cushing Syndrome Caused by Adrenocortical Tumors and Hyperplasias (Corticotropin- Independent Cushing Syndrome). In: Flück CE, Miller WL, editors. Endocrine Development. Basel: KARGER (2008). p. 117–32. doi: 10.1159/000134829
4. Pasternak-Pietrzak K, Moszczyńska E, Jurkiewicz E, Szalecki M. Paediatric Cushing’s disease — a literature review of epidemiology, pathogenesis, clinical symptoms, and diagnostics. Endokrynol Pol (2020) 71:87–95. doi: 10.5603/EP.a2019.0040
5. Gupta N, Rivera M, Novotny P, Rodriguez V, Bancos I, Lteif A. Adrenocortical carcinoma in children: A clinicopathological analysis of 41 patients at the mayo clinic from 1950 to 2017. Horm Res Paediatr (2018) 90:8–18. doi: 10.1159/000488855
6. Pinto EM, Rodriguez-Galindo C, Pounds SB, Wang L, Clay MR, Neale G, et al. Identification of clinical and biologic correlates associated with outcome in children with adrenocortical tumors without germline TP53 mutations: A st jude adrenocortical tumor registry and children’s oncology group study. J Clin Oncol (2017) 35:3956–63. doi: 10.1200/JCO.2017.74.2460
7. Michalkiewicz E, Sandrini R, Figueiredo B, Miranda ECM, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol (2004) 22:838–45. doi: 10.1200/JCO.2004.08.085
8. Sandrini R. Childhood adrenocortical tumors. J Clin Endocrinol Metab (1997) 82:2027–31. doi: 10.1210/jc.82.7.2027
9. Rodriguez-Galindo C, Figueiredo BC, Zambetti GP, Ribeiro RC. Biology, clinical characteristics, and management of adrenocortical tumors in children. Pediatr Blood Cancer (2005) 45:265–73. doi: 10.1002/pbc.20318
10. Ribeiro RC, Pinto EM, Zambetti GP, Rodriguez-Galindo C. The International Pediatric Adrenocortical Tumor Registry initiative: Contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol (2012) 351:37–43. doi: 10.1016/j.mce.2011.10.015
11. Federici S, Galli G, Ceccarelli P, Ferrari M, Cicognani A, Cacciari E, et al. Adrenocortical tumors in children: A report of 12 cases. Eur J Pediatr Surg (1994) 4:21–5. doi: 10.1055/s-2008-1066060
12. Chen Q-L, Su Z, Li Y-H, Ma H-M, Chen H-S, Du M-L. Clinical characteristics of adrenocortical tumors in children. J Pediatr Endocrinol Metab (2011) 24:535–41. doi: 10.1515/jpem.2011.175
13. Ribeiro RC, Figueiredo B. Childhood adrenocortical tumours. Eur J Cancer (2004) 40:1117–26. doi: 10.1016/j.ejca.2004.01.031
14. Gkourogianni A, Lodish MB, Zilbermint M, Lyssikatos C, Belyavskaya E, Keil MF, et al. Death in pediatric Cushing syndrome is uncommon but still occurs. Eur J Pediatr (2015) 174:501–7. doi: 10.1007/s00431-014-2427-y
15. Memon SS, Thakkar K, Patil V, Jadhav S, Lila AR, Fernandes G, et al. Primary pigmented nodular adrenocortical disease (PPNAD): single centre experience. J Pediatr Endocrinol Metab (2019) 32:391–7. doi: 10.1515/jpem-2018-0413
16. Majumder S, Chakraborty PP, Ghosh PC, Bera M. Cushing’s syndrome in early infancy due to isolated sporadic bilateral micronodular adrenocortical disease associated with myosin heavy chain 8 mutation: diagnostic challenges, too many! BMJ Case Rep (2020) 13:e236850. doi: 10.1136/bcr-2020-236850
17. Bourdeau I, Parisien-La Salle S, Lacroix A. Adrenocortical hyperplasia: A multifaceted disease. Best Pract Res Clin Endocrinol Metab (2020) 34:101386. doi: 10.1016/j.beem.2020.101386
18. Bram Z, Louiset E, Ragazzon B, Renouf S, Wils J, Duparc C, et al. PKA regulatory subunit 1A inactivating mutation induces serotonin signaling in primary pigmented nodular adrenal disease. JCI Insight (2016) 1:e87958. doi: 10.1172/jci.insight.87958
19. Maria AG, Tatsi C, Berthon A, Drougat L, Settas N, Hannah-Shmouni F, et al. ARMC5 variants in PRKAR1A-mutated patients modify cortisol levels and Cushing’s syndrome. Endocr Relat Cancer (2020) 27:509–17. doi: 10.1530/ERC-20-0273
20. Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med (1999) 131:585. doi: 10.7326/0003-4819-131-8-199910190-00006
21. Sandrini F, Stratakis C. Clinical and molecular genetics of primary pigmented nodular adrenocortical disease. Arq Bras Endocrinol Metabol (2004) 48:637–41. doi: 10.1590/S0004-27302004000500007
22. Gunther DF, Bourdeau I, Matyakhina L, Cassarino D, Kleiner DE, Griffin K, et al. Cyclical cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab (2004) 89:3173–82. doi: 10.1210/jc.2003-032247
23. Storr HL, Chan LF, Grossman AB, Savage MO. Paediatric Cushing’s syndrome: epidemiology, investigation and therapeutic advances. Trends Endocrinol Metab (2007) 18:167–74. doi: 10.1016/j.tem.2007.03.005
24. Charchar HLS, Fragoso MCBV. An overview of the heterogeneous causes of cushing syndrome resulting from primary macronodular adrenal hyperplasia (PMAH). J Endocr Soc (2022) 6:bvac041. doi: 10.1210/jendso/bvac041
25. Vaczlavik A, Bouys L, Violon F, Giannone G, Jouinot A, Armignacco R, et al. KDM1A inactivation causes hereditary food-dependent Cushing syndrome. Genet Med (2022) 24:374–83. doi: 10.1016/j.gim.2021.09.018
26. Christopoulos S, Bourdeau I, Lacroix A. Clinical and subclinical ACTH-independent macronodular adrenal hyperplasia and aberrant hormone receptors. Horm Res Paediatr (2005) 64:119–31. doi: 10.1159/000088818
27. Völkl TMK, Dörr HG. McCune-albright syndrome: clinical picture and natural history in children and adolescents. J Pediatr Endocrinol Metab (2006) 19(Suppl 2):551–9. doi: 10.1515/JPEM.2006.19.S2.551
28. Kirk JMW, Brain CE, Carson DJ, Hyde JC, Grant DB. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J Pediatr (1999) 134:789–92. doi: 10.1016/S0022-3476(99)70302-1
29. Yoshimoto M, Nakayama M, Baba T, Uehara Y, Niikawa N, Ito M, et al. A case of neonatal mcCune-albright syndrome with cushing syndrome and hyperthyroidism. Acta Paediatr (1991) 80:984–7. doi: 10.1111/j.1651-2227.1991.tb11769.x
30. Hamajima T, Maruwaka K, Homma K, Matsuo K, Fujieda K, Hasegawa T. Unilateral adrenalectomy can be an alternative therapy for infantile onset Cushing’ s syndrome caused by ACTHindependent macronodular adrenal hyperplasia with McCune-Albright syndrome. Endocr J (2010) 57:819–24. doi: 10.1507/endocrj.K10E-003
31. Paris F, Philibert P, Lumbroso S, Servant N, Kalfa N, Sultan C. Isolated cushing’s syndrome: an unusual presentation of mcCune-albright syndrome in the neonatal period. Horm Res Paediatr (2009) 72:315–9. doi: 10.1159/000245934
32. Lodish M, Patronas NJ, Stratakis CA. Reversible posterior encephalopathy syndrome associated with micronodular adrenocortical disease and Cushing syndrome. Eur J Pediatr (2010) 169:125–6. doi: 10.1007/s00431-009-0990-4
33. Tatsi C, Boden R, Sinaii N, Keil M, Lyssikatos C, Belyavskaya E, et al. Decreased lymphocytes and increased risk for infection are common in endogenous pediatric Cushing syndrome. Pediatr Res (2018) 83:431–7. doi: 10.1038/pr.2017.278
34. Rahman SH, Papadakis GZ, Keil MF, Faucz FR, Lodish MB, Stratakis CA. Kidney stones as an underrecognized clinical sign in pediatric cushing disease. J Pediatr (2016) 170:273–277.e1. doi: 10.1016/j.jpeds.2015.11.045
35. Lodish MB, Hsiao H-P, Serbis A, Sinaii N, Rothenbuhler A, Keil MF, et al. Effects of Cushing disease on bone mineral density in a pediatric population. J Pediatr (2010) 156:1001–5. doi: 10.1016/j.jpeds.2009.12.027
36. Keil MF, Zametkin A, Ryder C, Lodish M, Stratakis CA. Cases of psychiatric morbidity in pediatric patients after remission of cushing syndrome. Pediatrics (2016) 137:e20152234. doi: 10.1542/peds.2015-2234
37. Zheng X, Wang H, Zhang W, Feng S, Liu Y, Li S, et al. Diagnosis, manifestations, laboratory investigations, and prognosis in pediatric and adult cushing’s disease in a large center in China. Front Endocrinol (2021) 12:749246. doi: 10.3389/fendo.2021.749246
38. Hochberg Z. Mechanisms of steroid impairment of growth. Horm Res Paediatr (2002) 58:33–8. doi: 10.1159/000064764
39. Merke DP, Giedd JN, Keil MF, Mehlinger SL, Wiggs EA, Holzer S, et al. Children experience cognitive decline despite reversal of brain atrophy one year after resolution of cushing syndrome. J Clin Endocrinol Metab (2005) 90:2531–6. doi: 10.1210/jc.2004-2488
40. Keil MF. Quality of life and other outcomes in children treated for cushing syndrome. J Clin Endocrinol Metab (2013) 98:2667–78. doi: 10.1210/jc.2013-1123
41. Holbrook L, Brady R. McCune-albright syndrome, in: StatPearls (2023). Treasure Island (FL: StatPearls Publishing. Available at: http://www.ncbi.nlm.nih.gov/books/NBK537092/ (Accessed October 2, 2023).
42. Neary NM, Lopez-Chavez A, Abel BS, Boyce AM, Schaub N, Kwong K, et al. Neuroendocrine ACTH-producing tumor of the thymus—Experience with 12 patients over 25 years. J Clin Endocrinol Metab (2012) 97:2223–30. doi: 10.1210/jc.2011-3355
43. Tatsi C, Stratakis CA. Neonatal cushing syndrome: A rare but potentially devastating disease. Clin Perinatol (2018) 45:103–18. doi: 10.1016/j.clp.2017.10.002
44. Tabarin A, Assié G, Barat P, Bonnet F, Bonneville JF, Borson-Chazot F, et al. Consensus statement by the French Society of Endocrinology (SFE) and French Society of Pediatric Endocrinology & Diabetology (SFEDP) on diagnosis of Cushing’s syndrome. Ann Endocrinol (2022) 83:119–41. doi: 10.1016/j.ando.2022.02.001
45. Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2008) 93:1526–40. doi: 10.1210/jc.2008-0125
46. Stratakis CA. Diagnosis and clinical genetics of cushing syndrome in pediatrics. Endocrinol Metab Clin North Am (2016) 45:311–28. doi: 10.1016/j.ecl.2016.01.006
47. Elias PCL, Martinez EZ, Barone BFC, Mermejo LM, Castro M, Moreira AC. Late-night salivary cortisol has a better performance than urinary free cortisol in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab (2014) 99:2045–51. doi: 10.1210/jc.2013-4262
48. Gafni RI, Papanicolaou DA, Nieman LK. Nighttime salivary cortisol measurement as a simple, noninvasive, outpatient screening test for Cushing’s syndrome in children and adolescents. J Pediatr (2000) 137:30–5. doi: 10.1067/mpd.2000.106226
49. Ueland GÅ, Kellmann R, Jørstad Davidsen M, Viste K, Husebye ES, Almås B, et al. Bedtime salivary cortisol as a screening test for cushing syndrome in children. J Endocr Soc (2021) 5:bvab033. doi: 10.1210/jendso/bvab033
50. Wędrychowicz A, Hull B, Tyrawa K, Kalicka-Kasperczyk A, Zieliński G, Starzyk J. Cushing disease in children and adolescents – assessment of the clinical course, diagnostic process, and effects of the treatment – experience from a single paediatric centre. Pediatr Endocrinol Diabetes Metab (2019) 25:127–43. doi: 10.5114/pedm.2019.87179
51. Güemes M, Murray PG, Brain CE, Spoudeas HA, Peters CJ, Hindmarsh PC, et al. Management of Cushing syndrome in children and adolescents: experience of a single tertiary centre. Eur J Pediatr (2016) 175:967–76. doi: 10.1007/s00431-016-2727-5
52. Storr HL, Alexandraki KI, Martin L, Isidori AM, Kaltsas GA, Monson JP, et al. Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur J Endocrinol (2011) 164:667–74. doi: 10.1530/EJE-10-1120
53. Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics (2007) 120:e575–586. doi: 10.1542/peds.2006-2402
54. Stratakis CA. Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am (2012) 41:793–803. doi: 10.1016/j.ecl.2012.08.002
55. Chevalier B, Vantyghem M-C, Espiard S. Bilateral adrenal hyperplasia: pathogenesis and treatment. Biomedicines (2021) 9:1397. doi: 10.3390/biomedicines9101397
56. Chen S, Li R, Lu L, Duan L, Zhang X, Tong A, et al. Efficacy of dexamethasone suppression test during the diagnosis of primary pigmented nodular adrenocortical disease in Chinese adrenocorticotropic hormone-independent Cushing syndrome. Endocrine (2018) 59:183–90. doi: 10.1007/s12020-017-1436-9
57. Libé R, Coste J, Guignat L, Tissier F, Lefebvre H, Barrande G, et al. Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: a frequent finding in a prospective study of 32 patients with overt or subclinical Cushing’s syndrome. Eur J Endocrinol (2010) 163:129–38. doi: 10.1530/EJE-10-0195
58. Hsiao H-P, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab (2009) 94:2930–7. doi: 10.1210/jc.2009-0516
59. Antonini SR, Baldacchino V, Tremblay J, Hamet P, Lacroix A. Expression of ACTH receptor pathway genes in glucose-dependent insulinotrophic peptide (GIP)-dependent Cushing’s syndrome. Clin Endocrinol (Oxf) (2006) 64:29–36. doi: 10.1111/j.1365-2265.2005.02411.x
60. Courcoutsakis NA, Tatsi C, Patronas NJ, Lee C-CR, Prassopoulos PK, Stratakis CA. The complex of myxomas, spotty skin pigmentation and endocrine overactivity (Carney complex): imaging findings with clinical and pathological correlation. Insights Imaging (2013) 4:119–33. doi: 10.1007/s13244-012-0208-6
61. Sargar KM, Khanna G, Hulett Bowling R. Imaging of nonmalignant adrenal lesions in children. RadioGraphics (2017) 37:1648–64. doi: 10.1148/rg.2017170043
62. Chittiboina P, Montgomery BK, Millo C, Herscovitch P, Lonser RR. High-resolution(18)F-fluorodeoxyglucose positron emission tomography and magnetic resonance imaging for pituitary adenoma detection in Cushing disease. J Neurosurg (2015) 122:791–7. doi: 10.3171/2014.10.JNS14911
63. Alencar GA, Fragoso MCBV, Yamaga LYI, Lerario AM, Mendonca BB. 18 F-FDG-PET/CT imaging of ACTH-independent macronodular adrenocortical hyperplasia (AIMAH) demonstrating increased 18 F-FDG uptake. J Clin Endocrinol Metab (2011) 96:3300–1. doi: 10.1210/jc.2011-1397
64. Cyranska-Chyrek E, Filipowicz D, Szczepanek-Parulska E, Nowaczyk M, Ambroziak U, Toutounchi S, et al. Primary pigmented nodular adrenocortical disease (PPNAD) as an underlying cause of symptoms in a patient presenting with hirsutism and secondary amenorrhea: case report and literature review. Gynecol Endocrinol Off J Int Soc Gynecol Endocrinol (2018) 34:1022–6. doi: 10.1080/09513590.2018.1493101
65. Sandrini R, Ribeiro RC, DeLacerda L. Childhood adrenocortical tumors. J Clin Endocrinol Metab (1997) 82:2027–31. doi: 10.1210/jcem.82.7.4057
66. Rodriguez-Galindo C, Krailo MD, Pinto EM, Pashankar F, Weldon CB, Huang L, et al. Treatment of pediatric adrenocortical carcinoma with surgery, retroperitoneal lymph node dissection, and chemotherapy: the children’s oncology group ARAR0332 protocol. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:2463–73. doi: 10.1200/JCO.20.02871
67. De León DD, Lange BJ, Walterhouse D, Moshang T. Long-term (15 years) outcome in an infant with metastatic adrenocortical carcinoma. J Clin Endocrinol Metab (2002) 87:4452–6. doi: 10.1210/jc.2001-011978
68. Zancanella P, Pianovski MAD, Oliveira BH, Ferman S, Piovezan GC, Lichtvan LL, et al. Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma: mitotane monitoring and tumor regression. J Pediatr Hematol Oncol (2006) 28:513–24. doi: 10.1097/01.mph.0000212965.52759.1c
69. Paragliola RM, Torino F, Papi G, Locantore P, Pontecorvi A. Role of mitotane in adrenocortical carcinoma – review and state of the art. Eur Endocrinol (2018) 14:62. doi: 10.17925/EE.2018.14.2.62
70. Redlich A, Boxberger N, Strugala D, Frühwald M, Leuschner I, Kropf S, et al. Systemic treatment of adrenocortical carcinoma in children: data from the german GPOH-MET 97 trial. Klin Pädiatr (2012) 224:366–71. doi: 10.1055/s-0032-1327579
71. Wieneke JA, Thompson LDR, Heffess CS. Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol (2003) 27:867–81. doi: 10.1097/00000478-200307000-00001
72. Pinto EM, Rodriguez-Galindo C, Choi JK, Pounds S, Liu Z, Neale G, et al. Prognostic significance of major histocompatibility complex class II expression in pediatric adrenocortical tumors: A St. Jude and children’s oncology group study. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:6247–55. doi: 10.1158/1078-0432.CCR-15-2738
73. Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Endocrine society. Treatment of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2015) 100:2807–31. doi: 10.1210/jc.2015-1818
74. Liu X, Zhang S, Guo Y, Gang X, Wang G. Treatment of primary pigmented nodular adrenocortical disease. Horm Metab Res (2022) 54:721–30. doi: 10.1055/a-1948-6990
75. Xu Y, Rui W, Qi Y, Zhang C, Zhao J, Wang X, et al. The role of unilateral adrenalectomy in corticotropin-independent bilateral adrenocortical hyperplasias. World J Surg (2013) 37:1626–32. doi: 10.1007/s00268-013-2059-9
76. Kyrilli A, Lytrivi M, Bouquegneau MS, Demetter P, Lucidi V, Garcia C, et al. Unilateral adrenalectomy could be a valid option for primary nodular adrenal disease: evidence from twins. J Endocr Soc (2019) 3:129–34. doi: 10.1210/js.2018-00261
77. Bourdeau I, D’Amour P, Hamet P, Boutin JM, Lacroix A. Aberrant membrane hormone receptors in incidentally discovered bilateral macronodular adrenal hyperplasia with subclinical Cushing’s syndrome. J Clin Endocrinol Metab (2001) 86:5534–40. doi: 10.1210/jcem.86.11.8062
78. Guerin C, Taieb D, Treglia G, Brue T, Lacroix A, Sebag F, et al. Bilateral adrenalectomy in the 21st century: when to use it for hypercortisolism? Endocr Relat Cancer (2016) 23:R131–142. doi: 10.1530/ERC-15-0541
79. Albiger NM, Ceccato F, Zilio M, Barbot M, Occhi G, Rizzati S, et al. An analysis of different therapeutic options in patients with Cushing’s syndrome due to bilateral macronodular adrenal hyperplasia: a single-centre experience. Clin Endocrinol (Oxf) (2015) 82:808–15. doi: 10.1111/cen.12763
80. Debillon E, Velayoudom-Cephise F, Salenave S, Caron P, Chaffanjon P, Wagner T, et al. Unilateral adrenalectomy as a first-line treatment of cushing’s syndrome in patients with primary bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab (2015) 100:4417–24. doi: 10.1210/jc.2015-2662
81. Yoshiaki Tanno F, Srougi V, Almeida MQ, Ide Yamauchi F, Morbeck Almeida Coelho F, Nishi MY, et al. A new insight into the surgical treatment of primary macronodular adrenal hyperplasia. J Endocr Soc (2020) 4:bvaa083. doi: 10.1210/jendso/bvaa083
82. Zhang Y, Li H. Classification and surgical treatment for 180 cases of adrenocortical hyperplastic disease. Int J Clin Exp Med (2015) 8:19311–7.
83. Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol (2021) 9:847–75. doi: 10.1016/S2213-8587(21)00235-7
84. Tatsi C, Maria AG, Malloy C, Lin L, London E, Settas N, et al. Cushing syndrome in a pediatric patient with a KCNJ5 variant and successful treatment with low-dose ketoconazole. J Clin Endocrinol Metab (2021) 106:1606–16. doi: 10.1210/clinem/dgab118
Keywords: hypercortisolism, pediatric, childhood, adrenal tumors, adrenal hyperplasia
Citation: Guarnotta V, Emanuele F, Salzillo R and Giordano C (2023) Adrenal Cushing’s syndrome in children. Front. Endocrinol. 14:1329082. doi: 10.3389/fendo.2023.1329082
Received: 27 October 2023; Accepted: 29 November 2023;
Published: 12 December 2023.
Edited by:
Mariacarolina Salerno, University of Naples Federico II, ItalyReviewed by:
Marco Cappa, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2023 Guarnotta, Emanuele, Salzillo and Giordano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Guarnotta, dmFsZW50aW5hLmd1YXJub3R0YUB1bmlwYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.