 Yuxin Jiang
Yuxin Jiang Yin Cai
Yin Cai Ronghui Han3
Ronghui Han3 Youhua Xu
Youhua Xu Weiyi Xia
Weiyi Xia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 12 January 2024
Sec. Cardiovascular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1322474
This article is part of the Research TopicThe complex phenotype of Diabetic Cardiomyopathy: clinical indicators and novel treatment targetsView all 10 articles
The incidence of diabetes and related mortality rate increase yearly in modern cities. Additionally, elevated glucose levels can result in an increase of reactive oxygen species (ROS), ferroptosis, and the disruption of protective pathways in the heart. These factors collectively heighten the vulnerability of diabetic individuals to myocardial ischemia. Reperfusion therapies have been effectively used in clinical practice. There are limitations to the current clinical methods used to treat myocardial ischemia-reperfusion injury. As a result, reducing post-treatment ischemia/reperfusion injury remains a challenge. Therefore, efforts are underway to provide more efficient therapy. Salvia miltiorrhiza Bunge (Danshen) has been used for centuries in ancient China to treat cardiovascular diseases (CVD) with rare side effects. Salvianolic acid is a water-soluble phenolic compound with potent antioxidant properties and has the greatest hydrophilic property in Danshen. It has recently been discovered that salvianolic acids A (SAA) and B (SAB) are capable of inhibiting apoptosis by targeting the JNK/Akt pathway and the NF-κB pathway, respectively. This review delves into the most recent discoveries regarding the therapeutic and cardioprotective benefits of salvianolic acid for individuals with diabetes. Salvianolic acid shows great potential in myocardial protection in diabetes mellitus. A thorough understanding of the protective mechanism of salvianolic acid could expand its potential uses in developing medicines for treating diabetes mellitus related myocardial ischemia-reperfusion.
Myocardial ischemia is one of the most common types of cardiovascular disease that increases morbidity and mortality worldwide (1). Effective limitation of infarct size through timely restoration of blood flow to ischemic myocardium is the standard treatment to rescue ischemic myocardium and thus to improve the patient outcomes. Paradoxically, reperfusion itself causes cardiac injury, which is known as myocardial ischemia/reperfusion injury (MI/RI). Moreover, patients with diabetes are more vulnerable to MI/RI than those without diabetes (2), yet the underlying mechanism is incompletely understood. The complicities of the MI/RI which includes oxidative stress, calcium overload, inflammatory response, energy metabolism disorder, mitochondrial dysfunction, and apoptosis were shown in many studies (3, 4). Oxidative stress is known as an essential factor in myocardial ischemia reperfusion (I/R) (5, 6), and oxidative stress levels in the myocardium of diabetic patients were found to be significantly higher than that in non-diabetes. This could be one of the mechanisms attributable to the increased myocardial vulnerability to MI/RI in diabetes. In addition to increases in reactive oxygen species (ROS) and oxidative stress, increases in inflammation, reduction in cardiac Akt and STAT3 all occur in the myocardium of diabetes (7). These elements play important roles in diabetics complicated by MI/RI and may be attributed to the increased myocardial sensitivity to MI/RI (8, 9).
Danshen (Salvia miltiorrhiza Bunge), a traditional Chinese medicine that has been widely prescribed to patients with angina pectoris and hyperlipidemia was found to have a preventive effect in type 2 diabetic patients and in type 2 diabetic rats with nephropathy (10, 11). The chemical constituents of Danshen can be classified into two categories: water-soluble (hydrophilic) phenolic compounds and nonpolar (lipophilic) diterpenoidal compounds. Salvianolic acids are the major hydrophilic constituents amongst all. Among salvianolic acids, salvianolic acid A (SAA), salvianolic acid B (SAB), rosmarinus acid, danshensu, caffeic acid, and lithospermic acid are the main phenolic acids. SAA and SAB, in particular, are polyphenolic compounds known to have powerful antioxidant capacities (12).
Recent studies have demonstrated that SAA can exert anti-diabetic effects, preventing diabetic complications by reducing inflammatory response and improving lipid disorders (13), revealing the possible therapeutic effect of SAA on DM (14). Diabetes with MI/RI are not sensitive to pre-, post-conditioning cardioprotective interventions that are otherwise effective in non-diabetic subjects, while the related mechanisms are unclear. SAA can alleviate diabetes complications like vascular disease (14), but few studies support SAA can reduce MI/RI in DM and the mechanism has not been explored.
This review aims to provide a collective understanding of the potential effect of salvianolic acids in protecting against diabetes and myocardial ischemia-reperfusion in recent years and to explore whether salvianolic acid has the potential protective effects in Diabetes that are complicated by MI/RI. It is hopeful that such a collective understanding will help develop new therapeutic interventions for the clinical treatment of diabetic myocardial ischemia-reperfusion.
In 1960, Jennings et al. first reported MIRI, which is a condition that occurs when there is a temporary interruption of blood flow to the heart (ischemia) followed by the restoration of blood flow (reperfusion) (15). Restoring blood flow, such as percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG) is the most effective method to improve patient outcome (16). However, reperfusion itself causes damage to the myocardium, leading to exacerbation of the initial ischemic injury, and as such, even effective restoration of blood flow does not attenuate MIRI (4). Reperfusion triggers a series of tissue responses that contribute to the injury. This includes the production of oxygen free radical and mitochondrial damage, release of inflammatory factors, endoplasmic reticulum stress, and amplification of tissue damage (17). MIRI results from complex pathophysiological mechanisms, including oxidative stress, inflammatory response, endothelial cell dysfunction, mitochondrial dysfunction, calcium overload, apoptosis and autophagy (18).
Reactive oxygen species (ROS) are small reactive molecules that play a significant role in various cellular functions and biological processes, including cell signaling and homeostasis, in almost all eukaryotic cells. Some examples of ROS include superoxide anion radical, hydrogen peroxide (H2O2), hydroxyl radical (OH-), ozone (O3), and singlet oxygen (O2). (19). Under physiological conditions, ROS production is tightly regulated and plays a beneficial role in cell proliferation and metabolism (20). However, when ROS levels becomes excessively high, they can cause oxidative stress, which is a state of imbalance between the production of ROS and the ability of cells to detoxify them or repair the resulting damage (21). Overexpression of ROS and hypoxia in the tissue microenvironment can disrupt normal tissue repair and regeneration. This disruption can contribute to the development of fibrosis, dysfunction, and severity of cardiovascular diseases.
The accumulation of ROS during I/R injury is a major cause of oxidative damage. This phenomenon is also observed in diabetic myocardial injury (22). A study by Liu et al. revealed that I/R-induced apoptosis is mainly a consequence of excessive oxidative stress. Persistent cellular injury including necrosis and apoptosis of cardiomyocytes are the result of intense oxidative stress, which in turn trigger mitochondrial production of ROS in the early stages of ischemia in response to many mildly harmful stimuli to modulate stimulus-induced tolerance to ischemia. During reperfusion, the electron transport chain dysfunction, specifically the dysfunction in complex I (NADH dehydrogenase) and III (coenzyme Q-cytochrome c reductase), leads to excessive release of ROS. This results in the production of superoxide anions (O2-·), which are converted to hydrogen peroxide (H2O2) by the action of superoxide dismutase. In the presence of Fe2+ and Cu+, H2O2 is catalyzed to produce highly reactive hydroxyl radicals (OH), which can cause indiscriminate damage to nucleic acids, proteins, biofilms, and lipid peroxidation. This leads to mitochondrial depolarization, swelling, apoptosis, and cell death (17). Damaged and necrotic cells can activate Toll-like receptor 4 (TLR4) through damage-related molecular pattern (DAMP) activation. This leads to the aggregation of immune cells, which in turn express NADPH oxidase to promote the production of reactive oxygen species and further exacerbate myocardial damage (23). Overall, the overproduction ROS during I/R injury, along with the activation of TLR4 and NADPH oxidase, creates a vicious cycle that intensifies the damage to the myocardium.
Endothelium regulates vascular tone, cell adhesion, thromboresistance, smooth muscle cell proliferation, and vascular wall inflammation by producing and releasing vasoactive molecules. This produces and releases vasoactive molecules such as prostaglandins, nitric oxide (NO), endothelium-dependent hyperpolarizing factors, and endothelium-derived contracting factors (24), which impact vascular tone, cell adhesion, thromboresistance, smooth muscle cell proliferation, and vascular wall inflammation. These molecules help to regulate the degree of vasodilation/contraction, tissue oxygen consumption balance, long-term organ perfusions, vascular structure remodeling, and metabolism (25). The integrity of the endothelial barrier depends on the intercellular junction complex located between adjacent endothelial cells (26). Endothelial dysfunction, characterized by impaired endothelial function, is primarily driven by oxidative stress and inflammation (27). Evidence has shown that endothelial injury is a key mediator of myocardial ischemia/reperfusion injury (26, 28). Additionally, I/R injury itself can lead to endothelial dysfunction, manifested by decreased nitric oxide production, vascular dystonia due to endothelial injury, and prolonged vasoconstriction.
During myocardial I/R, there is disruption of endothelial integrity and decreased microvascular permeability of cardiac myocardium after myocardial I/R (29), which increases the permeability of the endothelial barrier by destroying endothelial barrier function and aggravating the inflammation (30, 31). No reflux phenomenon of myocardium is also seen after I/R, leading to vascular leakage and neutrophil infiltration, and eventually apoptosis of cardiomyocytes and damage to myocardial function (30, 31).
Mitochondria play a crucial role in oxidative stress and cell metabolism, and they are involved in various physiological functions such as endothelial mobilization, aging, proliferation, and growth (32). In cardiomyocytes, mitochondria are responsible for synthesizing about 90% of ATP, which is essential for normal heart functioning or cardiac functional recovery after various injuries (33, 34). Many studies have identified mitochondrial dysfunction as an important prominent mechanism for the progression of myocardial ischemia-reperfusion injury (34, 35). Abnormal mitochondrial fission, decreased mitophagy, and excessive mitochondrial oxidative stress can lead to endothelial dysfunction or death during cardiac reperfusion episodes (36). Mitochondrial dysfunction can lead to cell death through calcium imbalance, overproduction of mitochondrial ROS (mROS), disruption of cellular energy metabolism, impaired ATP production, and the opening of a structure called the mitochondrial permeability transition pore (MPTP), which can ultimately lead to cell death (34, 37). To counteract the effects of ROS and oxidative stress, mitochondria have a complex network of clearance systems (38). Studies have shown that abnormal mitochondrial fission can be an early indicator of mitochondrial dysfunction, and an imbalance between mitochondrial fission and fusion can lead to mitochondrial dysfunction, which in turn can aggravate MIRI damage (28, 34). Another factor contributing to injury is the accumulation of mitochondrial succinate, a metabolite that increases during hypoxia (39). This accumulated succinate is oxidized during reperfusion, resulting in the generation of ROS through a process called reverse electron transport (39). This excessive ROS production further contributes to oxidative stress and tissue damage. During myocardial ischemia, prolonged ischemia induces an increase in mitochondrial fission (40–42). When reperfusion occurs, the uncontrolled production of ROS triggers mitochondrial fission (34, 43, 44). This increased mitochondrial fission reduces mitochondrial membrane potential (MMP), making the MPTP more sensitive and leading to further ROS production. This disruption of the antioxidant balance within the mitochondria can result in the releasing of Cytochrome C (Cyt C) during cardiac microvascular I/R damage, activating caspases and initiating apoptosis through mitochondria-dependent pathways (45–48).
Calcium plays an important role as the second messenger in various cellular processes, including cell proliferation, division, and energy metabolism. However, excessive calcium levels, known as calcium overload, can lead to detrimental effects in cellular function, as proposed by Zimmerman and Hulsmann in 1966 (49). In a stable internal environment, calcium inflow and outflow are dynamically balanced under the regulation of protein channels (50). Maintaining intracellular calcium homeostasis is crucial for the normal function and growth of cardiomyocytes. Calcium overload in cardiomyocytes can exacerbate ischemic damage, which occurs when blood supply to the heart is compromised (51). When calcium overload occurs, Ca2+ dependent protease can promote the conversion of xanthine dehydrogenase to xanthine oxidase, promote the production of reactive oxygen species, and the high concentration of Ca2+ in the cytoplasm increases mitochondrial uptake of Ca2+, which in turn forms calcium phosphate deposition in the mitochondria, and subsequently adversely affects ATP synthesis. Calcium homeostasis cannot be maintained during myocardial ischemia-reperfusion and intracellular calcium overload is a common pathway for irreversible damage of cells subjected to myocardial ischemia-reperfusion (52). During myocardial ischemia, adenosine triphosphate (ATP) production decreases, leading to intracellular acidosis and the activation of Na+/H+ exchange causing a large influx of sodium ions. This sodium influx, coupled with the high calcium concentration, contributes to calcium overload during reperfusion (53). Reperfusion also disrupts mitochondrial membrane potential and accelerates energy expenditure, resulting in mitochondrial calcium overload and excessive production of ROS. The accumulation of calcium ions in cells inhibits mitochondrial ATP synthesis, leading to instability in mitochondrial membrane potential and subsequent damage, such as contraction disorders and apoptosis (54), exacerbating post-hypoxic or post-ischemic cardiomyocytes injuries (40, 55, 56).
The endoplasmic reticulum (ER) regulates the synthesis, folding, and transport of a significant portion of proteins in eukaryotic cells (57). High-quality protein folding can determine cell survival and function as well as normal physiological function. Endoplasmic reticulum homeostasis involves the binding of three ER transmembrane proteins: protein kinase R-like ER kinase (PERK), activated transcription factor 6 (ATF6), and enzyme 1 (IRE1) (58). Such a binding keeps them inactive, and their activation requires specific conditions such as the presence of inositol (58). During environmental injury or disease state, the disruption of endoplasmic reticulum homeostasis can lead to protein misfolding and accumulation of unfolded proteins. This triggers a response called endoplasmic reticulum stress, which activates the unfolded protein response (UPR). The UPR is a cellular mechanism aimed at reducing the burden and damage caused by the ER stress. It helps to restore protein homeostasis within the ER and rebuild the endoplasmic reticulum balance. However, if the endoplasmic reticulum stress becomes chronic or severe, it can promote cell death. Myocardial ischemia is an example that induces ER stress response (59). Activation of the endoplasmic reticulum stress-related pathway induces downstream activation of the apoptotic pathway, thereby promoting the progression of ischemia/reperfusion injury in myocardial tissue (60). Cardiomyocytes express high levels of endoplasmic reticulum stress-related signaling proteins, including transcription factor 6 (ATF6), C/EBP homologous protein (CHOP), glucose regulatory protein 78 (GRP78), etc. Treatments that Inhibit the signaling of endoplasmic reticulum stress can effectively reduce the rate of cell death in conditions like myocardial ischemia-reperfusion injury (61–64). Key cellular events in the pathogenesis of MIRI are summarized in Figure 1.
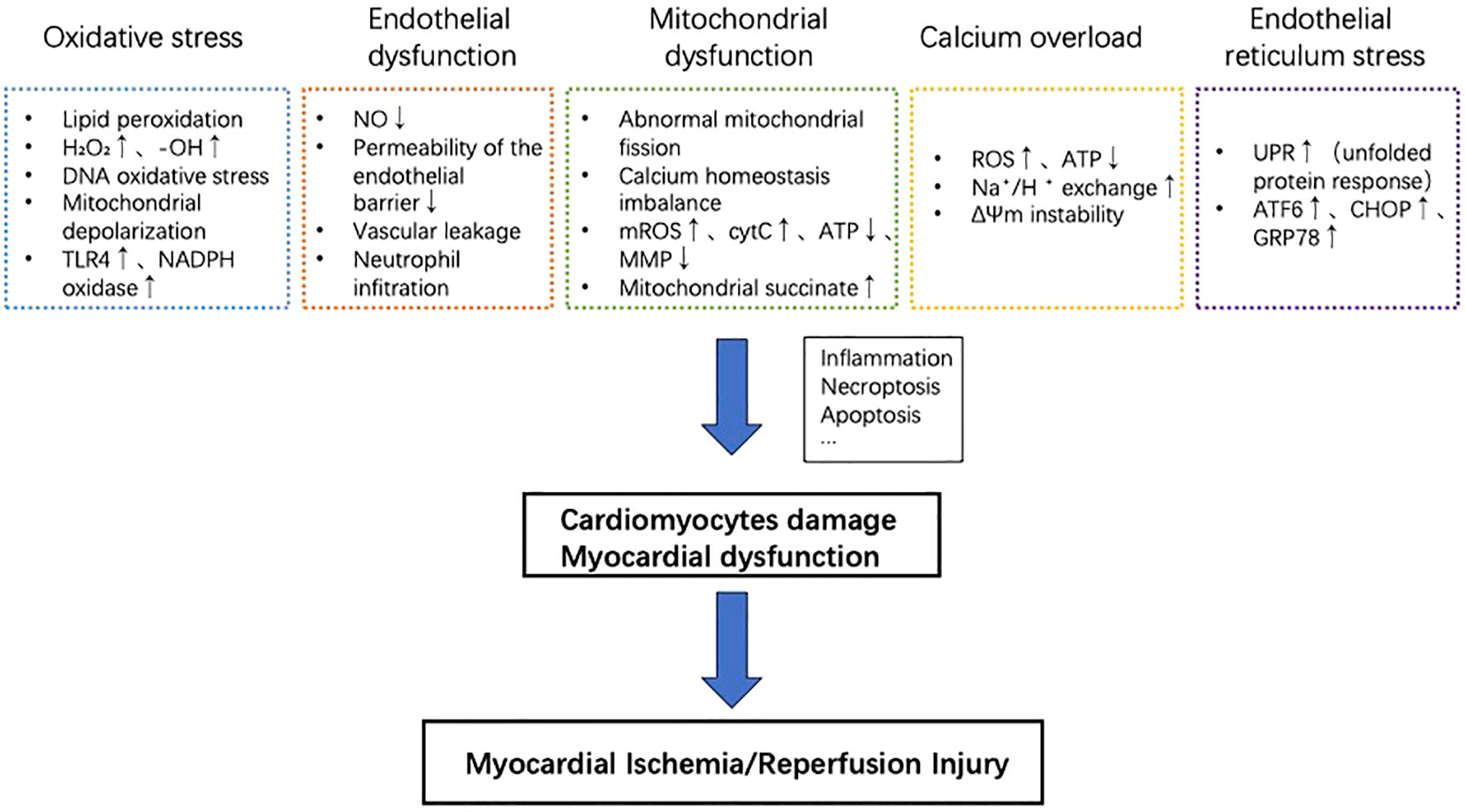
Figure 1 Key cellular events in the pathogenesis of MIRI. Oxidative stress, endothelial dysfunction, mitochondrial dysfunction, calcium overload, and reperfusion injury contribution to myocardial cell damage and cardiac dysfunction. These processes promote inflammation, necrotic apoptosis, apoptosis, and other mechanisms, ultimately exacerbating myocardial ischemia/reperfusion injury.
Ischemic heart disease (IHD) is one of the most common diseases that affects the human lifespan. Percutaneous coronary angioplasty, coronary artery bypass grafting, and other reperfusion methods such as thrombolysis treatment are the most effective treatment methods for myocardial ischemic injury up to date. However, post-ischemic reperfusion itself can cause new heart damage called “ischemia/reperfusion injury”. Although a variety of reperfusion therapies have matured, the development of therapies to reduce ischemia/reperfusion injury has been slow. Modern medical treatment has solved the technical problems of myocardial ischemia injury and blood flow recovery, but a series of complex processes of intracellular environmental changes that are caused by reperfusion after blood flow recovery have not been solved. The endogenous adaptive mechanism that occurs in cardiomyocytes in the face of ischemia-reperfusion or other types of metabolic stress challenges is called the pro-survival cardioprotective mechanism (65). The SAFE pathway, also known as the Survivor Activating Factor Enhancement pathway, is a signaling pathway that plays a crucial role in I/RI. This pathway is initially identified in studies investigating the cardioprotective effects of erythropoietin (EPO) against I/R injury (66, 67). It has been found that EPO activates the Janus Kinase (JAK) and signal transducer and activator of transcription (STAT) signaling pathway, particularly the JAK2/STAT3 pathway, to confer cardioprotection (68). In addition to EPO, other factors and pathways have also been implicated in activating the SAFE pathway. These include cytokines (such as interleukin-6 and interleukin-10), growth factors, and pharmacological agents (such as statins and opioids) (69). These factors can activate JAK/STAT signaling and trigger the downstream protective effects mediated by the SAFE pathway. The Reperfusion Injury Salvage Kinase (RISK) pathway is another signaling pathway that has a cardioprotective effect against I/R injury. It was first identified in studies investigating the cardioprotective effects of Ischemic Preconditioning (IPC), a phenomenon in which brief episodes of ischemia followed by reperfusion protect the heart against subsequent sustained I/R injury. Another cardioprotection mechanism that is relevant to this review is called postconditioning, which involves applying brief episodes of ischemia and reperfusion at the onset of reperfusion after a prolonged period of ischemia. This technique interrupts the initial reperfusion phase to protect the heart against I/R injury. The RISK pathway involves the activation of multiple protein kinases, including phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), and extracellular signal-regulated kinase (ERK) (69–71). Activation of these kinases leads to the phosphorylation and activation of various downstream targets that confer cardioprotection. In addition to the aforementioned pro-survival protective signalings, intracellular signaling molecules are also involved in cardioprotective signaling pathways, such as protein kinase C (PKC), protein kinase A (PKA), protein kinase G (PKG), 5’ amp activated protein kinase (AMPK), p38 mitogen-activated protein kinase (MAPK), extracellular signaling regulatory kinase 1/2 (ERK1/2) (65, 72, 73). However, the interaction between them has not been fully determined (65, 72, 73).
Diabetes is a major risk factor for IHD. Diabetes not only increases the incidence of acute myocardial infarction and myocardial sensitivity to ischemia-reperfusion injury but also alters or diminishes the myocardial response to cardioprotective interventions such as ischemic conditioning that are otherwise effective in subjects without diabetes. In animal models, ischemia preconditioning has been shown to be cardioprotective and reduce myocardial I/R damage. A recent study conducted in the db/db mouse model of type 2 diabetes shows that diabetes disturbs functional adaptation of the non-ischemic remote myocardium after ischemia/reperfusion (74). However, the effects of pretreatment-mediated cardioprotective in diabetic animal models are still controversial and inconclusive. In the study of Tatsumi et al., diabetic myocardial pretreatment stimulation produces a more substantial protective effect compared to regular myocardial pretreatment stimulation (75), while other studies have shown that diabetes attenuates or inhibits pretreatment-mediated cardioprotective effects (76). Diabetes can trigger various histological, biochemical, and physiological changes that contribute to the aggravation of oxidative stress, apoptosis, inflammation, and other pathways by increasing inflammatory factors, which leads to cardiac dysfunction, and exacerbating myocardial ischemia-reperfusion phenomenon (77).
Figure 2 studies conducted in diabetic rodents indicate that high glucose enhance superoxide generation and mitochondrial structural changes that increase the vulnerability of the myocardium to IR injury (78), and treatments that have anti-oxidant property attenuate myocardial IRI through improving mitochondrial homeostasis (28). Thus, excessive oxidative stress and impaired mitochondrial biogenesis in diabetic conditions rendered the diabetic heart more vulnerable to ischemic insults (56, 79). Mechanistically, Nrf2 nuclear translocation triggers Sirt3 upregulation and MnSOD activation, which subsequently reduces mitochondrial levels of ROS (mtROS). However, high glucose levels cause a downregulation of Nrf2 levels in the nucleus, resulting in Sirt3 downregulation and the acetylation of manganese superoxide dismutase (MnSOD), and thus facilitating the production of ROS (80). Energetic stress and mitochondrial ROS formation play critical roles in the pathogenesis of diabetic cardiomyopathy and MIRI (81, 82). AMPK, an important kinase involved in regulating energy homeostasis, plays a role in various metabolic process such as protein metabolism, lipid metabolism, carbohydrate metabolism, autophagy, and mitochondrial homeostasis. It is known that AMPK can sense cellular metabolic conditions and promote mitochondrial biogenesis. In the absence of glucose and ATP, AMPK is activated. Activation of AMPK has been shown to reduce the production of ROS and protect mitochondrial biogenesis. In the presence of high glucose, there is a dual inhibitory effect on AMPK. High glucose reduces the protein level and kinase activity of AMPKα, the catalytic subunit of AMPK. Researchers discovered that high glucose stimulation did not cause an increase in ATP levels, but it did cause an increase in the ratio of AMP/ATP and ADP/ATP (83). This suggests that ATP is not the cause of high glucose inhibition of AMPK signaling. Instead, high glucose promotes the production of ROS in cells (83). Under conditions of persistent hyperglycemia, elevated ROS levels are a causative factor in cell death (84). Under normal physiological conditions, cells have an antioxidant system to remove excess ROS. However, in diabetes, there is a decrease in antioxidant system activity and an increase in ROS production. This impairment between oxidant and antioxidant systems lead to oxidative stress, and results in various forms of damage to cells and tissue (84).
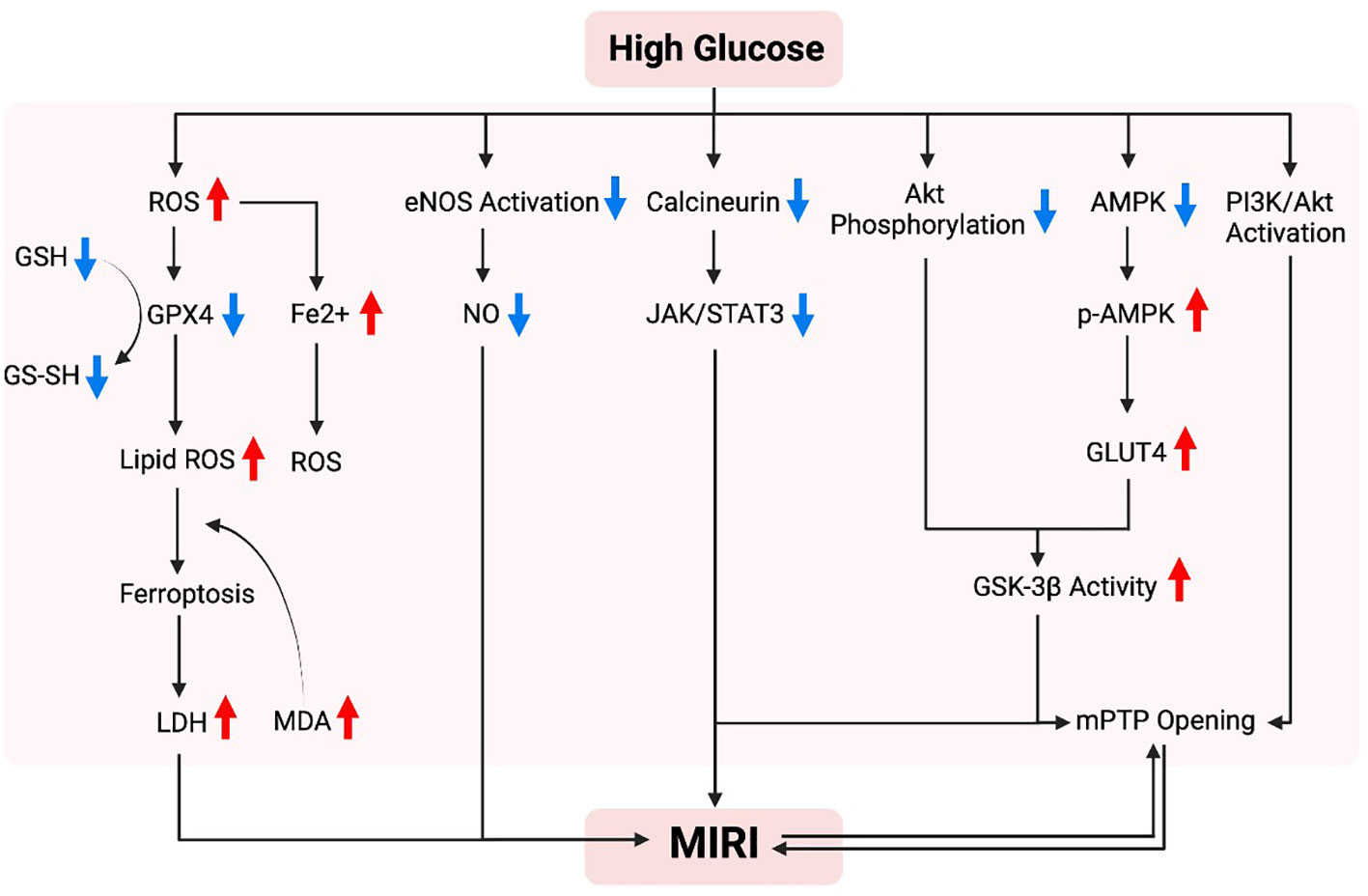
Figure 2 Possible mechanism of hyperglycemia in promoting myocardial ischemia-reperfusion injury. In a high-glycemic environment, there is an increased vulnerability to myocardial ischemia/reperfusion injury, which can be attributed to several mechanisms. These mechanisms include the overproduction of reactive oxygen species (ROS), an excessive burden of oxidative stress, abnormal alterations in mitochondria resulting in cell death due to iron overload, and dysfunction of endothelial nitric oxide synthase (eNOS). Furthermore, impaired protection against such injury is often linked to inadequate activation of pro-survival signaling pathways, including Akt, AMPK, JAK/STAT3, and PI3K/Akt.
Iron death, also known as ferroptosis, is a process in which iron-dependent cell death occurs due to increased lipid peroxidation. It is involved in various pathological processes such as cancer drug resistance, neurodegenerative diseases, IR/I and more (85). Several factors and markers are associated with iron death, including lactate dehydrogenase (LDH) activity, lipid peroxidation by reactive oxygen species (ROS), iron (Fe2+) levels, glutathione (GSH) levels, and malondialdehyde (MDA) levels. Studies have shown that high glucose (HG) conditions can increase ROS production with subsequently increased production of the lipid peroxidation product MDA, which in the presences of increased Fe2+ levels but decreased GSH and GPX4 levels, jointly lead to the induction of iron death (86). HG intake also leads to increased production of ROS, exacerbating oxidative stress increasing the production of GSSG, and reducing GSH content (87). This can result in mitochondrial abnormalities, such as decreased size, loss of mitochondrial ridges, and damage to the outer mitochondrial membrane. Glutathione peroxidase 4 (GPX4) is a key regulator of iron death, and its protein levels are significantly reduced under HG condition. The reduction in GPX4 leads to increased lipid ROS formation, lipid peroxidation, and ultimately cell iron death (86). Studies have found that adding ferroptosis cell death inhibitor can reduce cell death in HG environments, indicating the potential therapeutic importance of targeting this pathway (85). Overall, the process of ferroptosis and the proteins associated with it are strongly linked to glucose and lipid metabolism disorders (88). Herb extracts that antioxidant and anti-inflammatory properties such as Astragaloside IV has been shown to attenuate diabetic heart dysfunction in rats via inhibiting ferroptosis (89). However, studies regarding the relative role of ferroptosis in the diabetic myocardial IRI are rare and not definitive (77, 90, 91).
Endothelial nitric oxide synthase (eNOS) is an enzyme that produces nitric oxide (NO) in the endothelial cells of blood vessels. NO is the key regulator of vascular function and homeostasis, and it plays a crucial role in maintaining the health of the cardiovascular system. Dysfunction of eNOS has been found to be associated with the development of diabetes (92). Studies have shown that eNOS dysfunction is closely linked to a high glucose environment, which is characteristic of diabetes. Restoring normal eNOS function is essential for improving vascular health in individuals with diabetes (93). In fact, upregulation of eNOS expression has been found to have a protective effect in diabetic patients (94). It has been discovered that eNOS uncoupling, which is the loss of balance between NO production and ROS generation, is a significant source of increased ROS production in diabetes. Increased oxidative stress further exacerbates eNOS uncoupling and endothelial dysfunction, contributing to cardiovascular damage (95). The PI3K/Akt/eNOS signaling pathway is also affected/impaired by diabetes (96, 97). This pathway involves phosphatidylinositol 3-kinase (PI3K) and AKT/protein kinase B (PKB/AKT), which respond to external signals and regulate various cellular processes, including metabolism, proliferation, cell survival, growth, and angiogenesis (98). Impairment in this pathway can lead to both cardiovascular damages in diabetes (79, 99). The activation of the PI3K-AKT pathway has been shown to play a crucial role in protecting the heart from myocardial IR/I. Cardiac protective interventions such as Ischemic preconditioning, a process that exposes the tissue to brief periods of ischemia before a more prolonged ischemic event, can activate the PI3K-AKT pathway and provide protection to the heart (73, 100). Additionally, the Akt and JAK/STAT3 signaling pathways have been found to be involved in reducing diabetic heart I/R damage (101). However, both the PI3K-AKT pathway and the JAK/STAT3 signaling pathway are impaired in the myocardium of diabetic subjects (102, 103), rendering the diabetic hearts more vulnerable to ischemia reperfusion injury and less or not sensitive to therapeutic interventions that are otherwise effective in non-diabetic subjects (104–106).
Salvianolic acid is a compound found in the herb salvia, and it has been found to have several beneficial properties, including antioxidant, anti-inflammatory, and antiplatelet properties (107). In the context of myocardial IR/I, salvianolic acid has shown potential cardioprotective effect. Studies have indicated that salvianolic acids, specifically SAA and SAB, can help reduce damage to cardiomyocytes during MIRI in a rat model of ischemia-reperfusion injury (108). However, the exact mechanism by which salvianolic acid exerts its protective effects on MIRI is still not fully understood.
SAA possesses a polyphenolic acid chemical structure (Figure 3), exhibiting strong antioxidant capacity. It has also been found to have antioxidant, anticancer, antifibrotic, anti-inflammatory and antiplatelet aggregation properties (109). In vitro studies have shown that SAA exhibits potent free radical scavenging ability assessed by the methods of 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging assay and 2,2-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid (ABTS(+)) radical cation decolorization assay (110). SAA has been shown to attenuate myocardial functional impairment and cell death caused by oxidative stress and attenuate hydrogen peroxide-induced oxidative stress damage to cells both in vivo in rodent models and in vitro in cultured H9c2 cardiomycytes (111, 112). Experimental evidence has demonstrated that SAA pretreatment upregulates the anti-apoptotic protein Bcl-2 and inhibits pro-apoptotic proteins Bak and Bax, thereby inhibiting apoptosis (113). Additionally, SAA has been found to significantly ameliorate mitochondrial dysfunction caused by myocardial ischemia in an isoprenaline-induced myocardial ischemia a rat model (114). The contractile function of cardiomyocytes, as reflected by their shortening, was dose-dependently improved by SAA after myocardial ischemia-reperfusion (115). Furthermore, SAA pretreatment has been shown to reduce lactate dehydrogenase (LDH) leakage in ischemic myocardium, decrease LDH release from the ex vivo heart, and significantly improve cell viability. SAA also downregulates the expression of cleaved caspase-3 protein, thereby inhibiting apoptosis in cardiomyocytes. These findings suggest that SAA pretreatment before ischemia-reperfusion inhibits cardiomyocytes necrosis and apoptosis, thereby reducing ischemia-reperfusion-induced cardiomyocyte damage (115, 116).
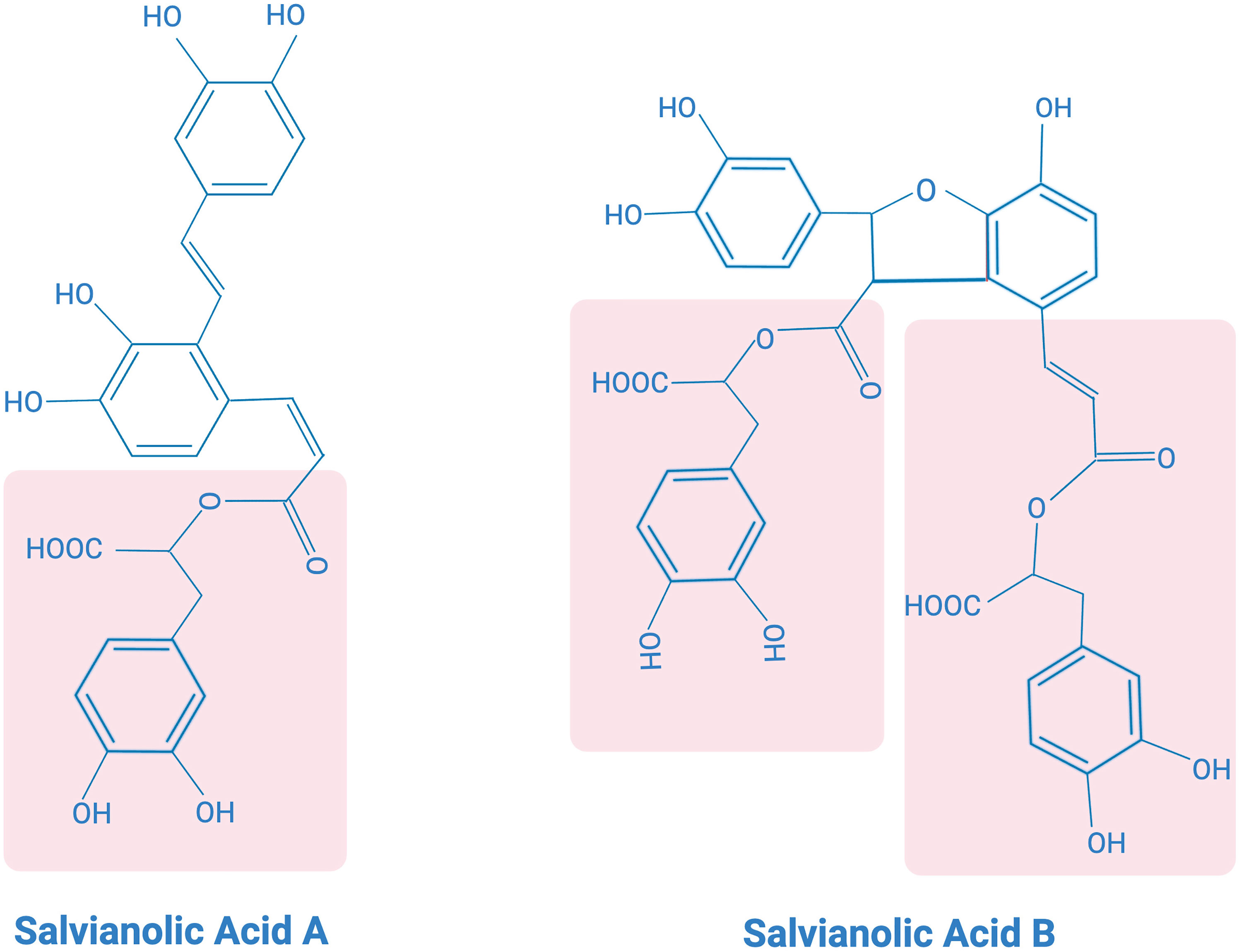
Figure 3 Chemical structures of salvianolic acid (A) and salvianolic acid (B).
SAB has been shown to effectively attenuate cardiovascular injuries by reducing the expression of related inflammatory factors, inhibiting apoptosis, and reducing oxidative stress in experimental settings (117). Numerous studies have demonstrated the cardiomyocyte protective effects of SAB during myocardial ischemia-reperfusion injury (MIRI) and its ability to reduce oxidative stress-induced damage (118). Similar to SAA, SAB also reduces post-ischemic LDH leakage (119). Experiments investigating the cardioprotective effect of SAB on myocardial ischemia-reperfusion injury, based on cell viability and LDH leakage, have shown that SAB inhibit autophagy, enhances cell viability, reduce LDH leakage, and increases the survival rate of cardiomyocytes after I/R (120). There is evidence showing that the cardioprotective effect of SAB on MIRI is dose-dependent, and both high and low doses of SAB have been found to reduce the size of myocardial infarction after treatment. Moreover, SAB effectively reduces cardiomyocyte apoptosis by significantly increasing the ratio of Bcl-2 expression to Bcl-2/Bax and reducing Bax expression (121). During ischemia-reperfusion, a large amount of ROS is released, accompanied with increased lipid peroxidation product malondialdehyde (MDA) (122) and other specific indicators of ROS-induced lipid peroxidation such as 15-F2t-Isoprostane (123, 124). Studies have demonstrated that SAB treatment can reduce high levels of malondialdehyde measured in rat models of testicular ischemia-reperfusion and myocardial ischemia-reperfusion, with no significant side effects observed throughout the treatment (108, 125). Recent research has also shown that SBB attenuates post-ischemic myocardial apoptosis, inhibits ROS production, decreases MDA levels, and enhances superoxide dismutase (SOD) activity through a mechanism that involves the regulation of the TRIM8/GPX1 axis in vivo, making it a potential candidate for the prevention or treatment of MIRI in cultured AC16 cardiomyocytes (118).
In rat models of myocardial IRI, Salvianolic Acid A (SAA) pretreatment has been shown to significantly reduce post-ischemic myocardial infarction concomitant with reduced plasma levels of cTnT, CK-MB, TNF-α and IL-1β compared with untreated I/R group (126). Platelets play a critical role in I/R injury, as activated platelets produce various factors that promote blood clot formation. It has been found that SAA treatment can resist ADP and collagen-induced human blood platelet aggregation and thrombosis by inhibiting the abnormal increase of the phosphorylation of Akt and also inhibits PI3K, and these effects of SAA was comparable to that of the PI3K inhibitor LY294002 both in vitro and in vivo in a mouse model of arterial thrombosis (127). NO is known to play a central role in maintaining cardiovascular homeostasis, and SAA treatment increases rat left ventricle NO content after myocardial I/R (126). Studies have found that SAA can also exerts cardioprotective effects through the ERK1/2 pathway, and this effect is inhibited by the ERK2/098059 inhibitor PD600125 (PD), suggesting that the protective effect of SAA on I/R cardiomyocytes may also depend on the inhibition of the JNK pathway (128). SAA has also been found to inhibit I/R-induced cardiomyocyte apoptosis through the PI3K/Akt, JNK, and ERK1/2 pathways. Among these pathways, ERK1/2 and JNK are regulated by upstream kinases (MAPK kinases), which activate each other in a stepwise manner. Experimental results have shown that inhibition of the p38 MAPK signaling pathway and the JNK signaling pathway can effectively protect and improve MIRI (128). Additionally, Salvianolic Acid B (SAB) has been shown to regulate the PI3K/Akt pathway, and to inhibit apoptosis by downregulating JNK phosphorylation, BCL2-associated X (Bax)/B-cell lymphoma-2 (Bcl-2), and caspase-3 expression (121) In addition, a recent study demonstrated that ubiquitin-proteasome degradation of GPX4 occurs in both MIRI models in rats and in in vitro models of cardiomyocyte hypoxia/reoxygenation, while SAB can reduce this degradation and inhibit ferroptosis and apoptosis of cardiomyocytes during MIRI and H/R and protect the cardiovascular system by the GPX4/ROS/JNK-mediated crosstalk mechanism (129). SAA and SAB have respectively been demonstrated to regulate the Jak/STAT3 signaling pathway in the liver (130) and in the intervertebral discs in rats (130). However, the potential impacts of SAA and SAB on the Jak/STAT3 signaling in the heart especially in the context of MIRI have not been explored thus far, which merits in depth future study given the critical role Jak/STAT3 signaling pathway plays during MIRI (45, 131, 132). The current understandings regarding the impacts of SAA and SAB on the signaling pathways that affect MIRI are summarized respectively in Figure 4.
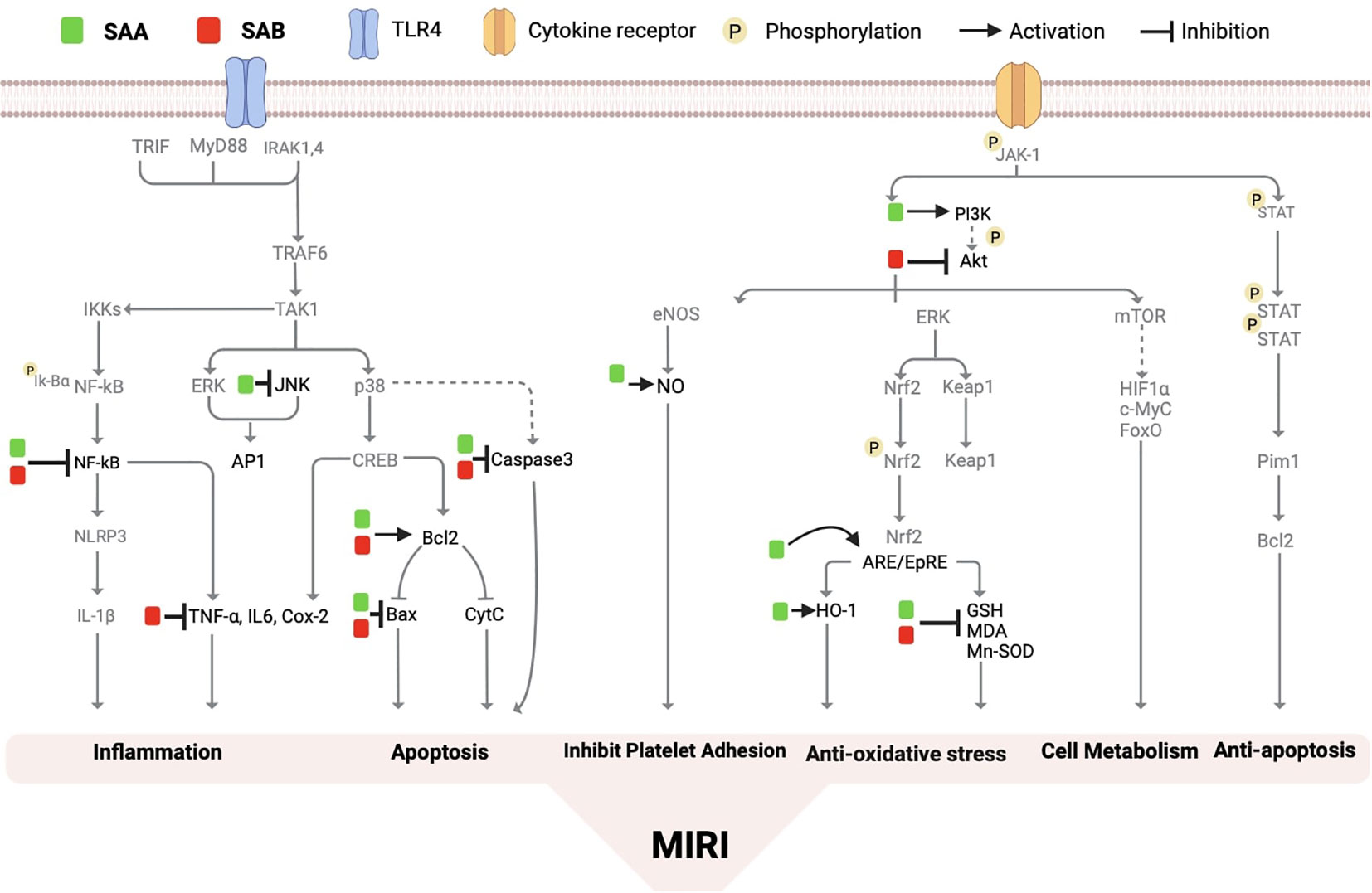
Figure 4 The molecular mechanism of salvianolic acid A and salvianolic acid B on myocardial ischemia/reperfusion injury. Salvianolic acid A and salvianolic acid B have the ability to regulate various molecular pathways including nuclear factor kB (NF-kB), stress-activated protein kinase (Bax, Caspase 3, malondialdehyde (MDA), heme oxygenase-1 (HO-1), bcl2, phosphatidylinositol 3-kinase (PI3K), nitric oxide (NO), antioxidant reaction element (ARE), tumor necrosis factor (TNalpha), protein kinase B (PKB/Akt), interleukin-6 (IL-6), and others. By inhibiting the production of reactive oxygen species, these compounds effectively improve inflammation, apoptosis, autophagy, microcirculation disorders, cell growth, and metabolism.
In this study, we provide a systematic summary of the cardioprotective mechanism of salvianolic acid in diabetic myocardial ischemia-reperfusion injury, as well as various signaling molecules and mechanisms associated with myocardial I/R injury. Both salvianolic acid A and B have the potential to exert cardioprotective effects, either through similar or different mechanisms (Table 1). The relevant signaling pathways involved in myocardial ischemia-reperfusion injury include phosphatidylinositol-3 kinase/Akt (PI3K/Akt), mitogen-activated protein kinases (MAPKs), JANUS kinase/signal transduction and transcriptional activators (JAK/STAT), nuclear factor-κB (NF-κB), and others (96). Studies have revealed that diabetes can further impair the phosphatidylinositol 3-kinase/Akt/eNOS (PI3K/Akt/eNOS) pathway and activate JAK/STAT3 signaling, thereby exacerbating myocardial ischemia-reperfusion injury in diabetic rats which can be attenuated by treatment with SAA (96).
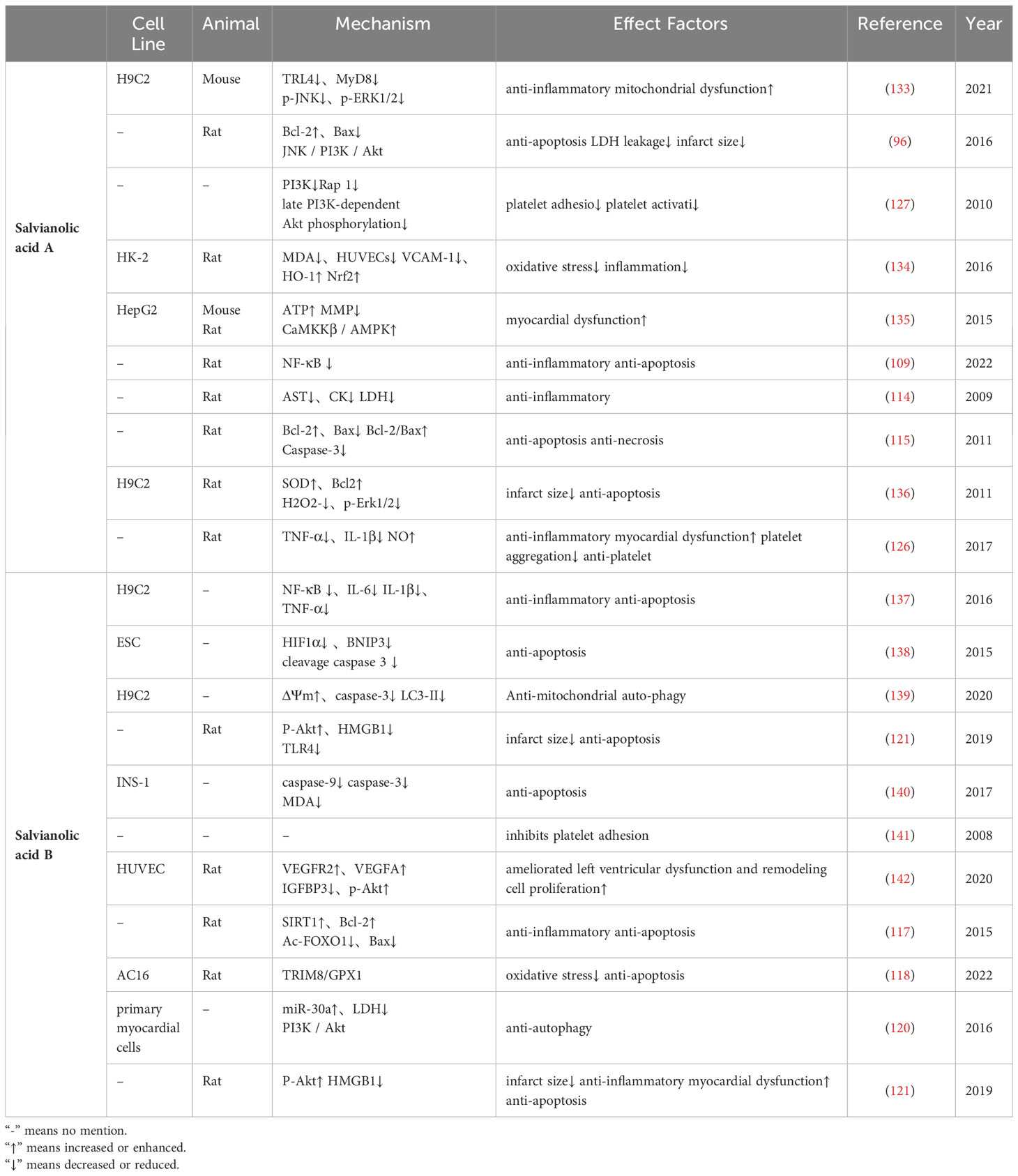
Table 1 Salvianolic Acid potential cardiomyocytes in diabetic myocardial ischemia/reperfusion injury.
As mentioned earlier, SAA pretreatment has been shown to have a protective effect on the myocardium during I/R in non-diabetic rats. Further studies have found that Sal A pretreatment significantly improved cardiac hemodynamic and reduced LDH activity after I/R in diabetic rats, with concomitant reduction in post-ischemic myocardial infarction apoptosis (96). Similarly, SAB has been found to significantly reduce intracellular reactive oxygen species and malondialdehyde (MDA) levels, effectively reduce oxidative stress induced by high glucose rat insulinoma cell line INS-1 cells (140). Clinical studies have observed that patients with antiplatelet therapy appears to have similar effects in patients with diabetic coronary artery disease compared to patients with non-diabetic myocardial ischemia-reperfusion injury (143). Sal B has been shown to inhibit platelet aggregation and platelet adhesion by interacting with collagen receptors (141). In subsequent studies, SAA has also been found to significantly inhibit agonist-induced platelet activation by inhibiting PI3K (127). The nuclear factor E2 related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway is involved in the regulation of MIRI damage (144). Pro-inflammatory cytokines also play a significant role in diabetic vascular damage. VCAM-1, a pro-inflammatory cytokine, is known to be inhibited by Nrf2-mediated upregulation of HO-1 in vascular diseases. SAA has been found to reduce VCAM-1 expression by mediating the Nrf2/HO-1 signaling pathway (134).
Studies have demonstrated that insulin has cardioprotective effects mediated by the Akt signaling pathway, leading to the activation of eNOS through PI3K/Akt activation (145). The impairment of the PI3K/Akt signaling pathway is involved in myocardial I/R damage in diabetic rats, and high glucose further inhibits the PI3K/Akt pathway. Experimental evidence supports the significant increase in SERCA2 activity through JNK/PI3K/Akt signaling, resulting in anti-apoptotic effects and improvement in cardiac contraction and diastolic function in diabetic rats. Chen et al. found that SAA pretreatment significantly increased the level of the anti-apoptotic protein Bcl-2 in diabetic rats through the JNK/Akt signaling pathway, while reducing the levels of pro-apoptotic proteins Bax and cleaved-caspase-3, ultimately increasing the Bcl-2/Bax ratio and protecting against myocardial I/R damage in diabetic rats (96). Similarly, Sal B has been shown to reduce the expression of insulin-like growth factor binding protein 3 (IGFBP3) induced by high glucose, leading to the phosphorylation of extracellular signal-regulating protein kinase and protein kinase B (AKT) activity in rat models of diabetic cardiomyopathy and in cultured HUVECs under hypoxia (142). SAB is also considered a potent inhibitor of the Akt/mTOR pathway, reducing the phosphorylation of Akt and its downstream target mTOR (146, 147). Furthermore, Sal A can regulate glucose metabolism by increasing ATP production with concurrent reduction of mitochondrial membrane potential (MMP), and improving mitochondrial function through Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ)/AMPK signaling pathway in both type 1 and type 2 diabetic mice (135). NF-κB, which induces inflammatory factors involved in cardiomyocyte apoptosis, is also a downstream target of STAT3. Inhibition of NF-κB activity can prevent H9C2 cardiomyocyte apoptosis (148). Studies have found that SAB can inhibit the activation of the MAPK/NF-κB pathway induced by ox-LDL (149). SAB pretreatment has also been reported to reduce NF-κB levels (150), but whether SAB directly targets NF-κB or acts through its upstream pathway Akt/JAK or STAT3 remains unclear (113).
This review highlights the evidence and possible mechanisms by which salvianolic acid may reduce diabetic myocardial I/R damage. Possible mechanisms include modulation of oxidative stress, inflammatory response, mitochondrial dysfunction, ferroptosis and apoptosis through pathways such as PI3K/Akt, JAK/STAT, and NF-κB. A recent study has found that aldehyde dehydrogenase 2 (ALDH2) can activate the PI3K/AKT/mTOR pathway to alleviate ischemia and reperfusion injury in diabetic cardiomyopathy (44). However, it should be noted that studies have shown that Sal B can inhibit the Akt/mTOR pathway (147). There is currently no research showing that Sal B can exert cardioprotective effects by mediating the Akt/mTOR pathway. Although salvianolic acid has been studied in various clinical studies as an active ingredient in salvia, its research on diabetic myocardial I/R damage is relatively limited. Further understanding of the mechanisms underlying salvianolic acid-related myocardial protection will contribute to the development of new protective strategies and discovery of more effective therapies against diabetic myocardial I/R damage in the future.
YJ: Writing – original draft. YC: Writing – original draft. RH: Writing – review & editing. YX: Supervision, Writing – review & editing. ZX: Supervision, Writing – review & editing. WX: Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors’ work were supported by grant of National Natural Science Foundation of China (NSFC, 81970427 and 82270306), Research Grant Council (RGC) GRF grant (17118619) of Hong Kong, China, and Guangdong Basic and Applied Basic Research Foundation (2022A1515011116).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Xiang M, Lu Y, Xin L, Gao J, Shang C, Jiang Z, et al. Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxid Med And Cell Longevity (2021) 2021:1–23. doi: 10.1155/2021/6614009
2. Xiong Z, Xu J, Liu X. Oxymatrine exerts A protective effect in myocardial ischemia/reperfusion−Induced acute lung injury by inhibiting autophagy in diabetic rats. Mol Med Rep (2021) 23(3). doi: 10.3892/mmr.2021.11822
3. Ge L, Cai Y, Ying F, Liu H, Zhang D, He Y, et al. Mir-181c-5p exacerbates hypoxia/reoxygenation-induced cardiomyocyte apoptosis via targeting ptpn4. Oxid Med Cell Longev (2019) 2019:1957920. doi: 10.1155/2019/1957920
4. He J, Liu D, Zhao L, Zhou D, Rong J, Zhang L, et al. Myocardial ischemia/reperfusion injury: mechanisms of injury and implications for management (Review). Exp Ther Med (2022) 23:430. doi: 10.3892/etm.2022.11357
5. Tao L, Huang K, Wang J, Xue Y, Zhou Y, He F, et al. Retinol palmitate protects against myocardial ischemia/reperfusion injury via reducing oxidative stress and inhibiting apoptosis. Am J Transl Res (2019) 11:1510–20.
6. Luo J, Yan D, Li S, Liu S, Zeng F, Cheung CW, et al. Allopurinol reduces oxidative stress and activates nrf2/P62 to attenuate diabetic cardiomyopathy in rats. J Cell Mol Med (2020) 24:1760–73. doi: 10.1111/jcmm.14870
7. Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL, et al. Hyperglycemia abrogates ischemic postconditioning cardioprotection by impairing adipor1/caveolin-3/stat3 signaling in diabetic rats. Diabetes (2016) 65:942–55. doi: 10.2337/db15-0782
8. Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia ZY. Lipopolysaccharide (Lps) aggravates high glucose- and hypoxia/reoxygenation-induced injury through activating ros-dependent nlrp3 inflammasome-mediated pyroptosis in H9c2 cardiomyocytes. J Diabetes Res (2019) 2019:8151836. doi: 10.1155/2019/8151836
9. Zheng HX, Chen J, Zu YX, Wang EZ, Qi SS. Chondroitin sulfate prevents stz induced diabetic osteoporosis through decreasing blood glucose, antioxidative stress, anti-inflammation and Opg/Rankl expression regulation. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155303
10. Hou B, Qiang G, Zhao Y, Yang X, Chen X, Yan Y, et al. Salvianolic acid A protects against diabetic nephropathy through ameliorating glomerular endothelial dysfunction via inhibiting age-rage signaling. Cell Physiol Biochem (2017) 44:2378–94. doi: 10.1159/000486154
11. Zhou AM, Xiang YJ, Liu EQ, Cai CH, Wu YH, Yang LB, et al. Salvianolic acid A inhibits platelet activation and aggregation in patients with type 2 diabetes mellitus. BMC Cardiovasc Disord (2020) 20:15. doi: 10.1186/s12872-019-01316-z
12. Ho JH, Hong CY. Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J BioMed Sci (2011) 18:30. doi: 10.1186/1423-0127-18-30
13. Sun J, Leng P, Li X, Guo Q, Zhao J, Liang Y, et al. Salvianolic acid A promotes mitochondrial biogenesis and mitochondrial function in 3t3-L1 adipocytes through regulation of the ampk-pgc1α Signalling pathway. Adipocyte (2022) 11:562–71. doi: 10.1080/21623945.2022.2116790
14. Ma Q, Yang Q, Chen J, Yu C, Zhang L, Zhou W, et al. Salvianolic acid A ameliorates early-stage atherosclerosis development by inhibiting nlrp3 inflammasome activation in zucker diabetic fatty rats. Molecules (2020) 25(5). doi: 10.3390/molecules25051089
15. Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of A coronary artery in the dog. Arch Pathol (1960) 70:68–78.
16. Schanze N, Bode C, Duerschmied D. Platelet contributions to myocardial ischemia/reperfusion injury. Front Immunol (2019) 10:1260. doi: 10.3389/fimmu.2019.01260
17. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol (2016) 7:113–70. doi: 10.1002/cphy.c160006
18. Korshunova AY, Blagonravov ML, Neborak EV, Syatkin SP, Sklifasovskaya AP, Semyatov SM, et al. Bcl2−Regulated apoptotic process in myocardial ischemia−Reperfusion injury (Review). Int J Mol Med (2021) 47:23–36. doi: 10.3892/ijmm.2020.4781
19. Li R, Jia Z, Trush MA. Defining ros in biology and medicine. React Oxyg Species (Apex) (2016) 1:9–21. doi: 10.20455/ros.2016.803
20. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: A scientific statement from the American heart association. Circ Res (2016) 119:E39–75. doi: 10.1161/RES.0000000000000110
21. Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to dna: formation, measurement, and biological significance. Rev Physiol Biochem Pharmacol (1997) 131:1–87. doi: 10.1007/3-540-61992-5_5
22. Li X, Ma N, Xu J, Zhang Y, Yang P, Su X, et al. Targeting ferroptosis: pathological mechanism and treatment of ischemia-reperfusion injury. Oxid Med Cell Longev (2021) 2021:1587922. doi: 10.1155/2021/1587922
23. Liao X, Song X, Li J, Li L, Fan X, Qin Q, et al. An injectable co-assembled hydrogel blocks reactive oxygen species and inflammation cycle resisting myocardial ischemia-reperfusion injury. Acta Biomater (2022) 149:82–95. doi: 10.1016/j.actbio.2022.06.039
24. Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol (2017) 37:E108–14. doi: 10.1161/ATVBAHA.117.309813
25. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation (2007) 115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859
26. Kong Q, Dai L, Wang Y, Zhang X, Li C, Jiang S, et al. Hspa12b attenuated acute myocardial ischemia/reperfusion injury via maintaining endothelial integrity in A Pi3k/Akt/Mtor-dependent mechanism. Sci Rep (2016) 6:33636. doi: 10.1038/srep33636
27. Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: still A therapeutic target? Nutrients (2019) 11(9). doi: 10.3390/nu11092090
28. Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol (2022) 21:106. doi: 10.1186/s12933-022-01532-6
29. Chiba Y, Morioka K, Muraoka R, Ihaya A, Kimura T, Uesaka T, et al. Effects of depletion of leukocytes and platelets on cardiac dysfunction after cardiopulmonary bypass. Ann Thorac Surg (1998) 65:107–13. doi: 10.1016/S0003-4975(97)01111-9
30. Singhal AK, Symons JD, Boudina S, Jaishy B, Shiu YT. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vasc Dis Prev (2010) 7:1–14. doi: 10.2174/1874120701007010001
31. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med (2011) 17:1391–401. doi: 10.1038/nm.2507
32. Kirkman DL, Robinson AT, Rossman MJ, Seals DR, Edwards DG. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am J Physiol Heart Circ Physiol (2021) 320:H2080–100. doi: 10.1152/ajpheart.00917.2020
33. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. Am J Physiol Heart Circ Physiol (2018) 315:H1341–52. doi: 10.1152/ajpheart.00028.2018
34. Zhao Z, Sun W, Guo Z, Liu B, Yu H, Zhang J. Long noncoding rnas in myocardial ischemia-reperfusion injury. Oxid Med Cell Longev (2021) 2021:8889123. doi: 10.1155/2021/8889123
35. Daiber A, Andreadou I, Oelze M, Davidson SM, Hausenloy DJ. Discovery of new therapeutic redox targets for cardioprotection against ischemia/reperfusion injury and heart failure. Free Radic Biol Med (2021) 163:325–43. doi: 10.1016/j.freeradbiomed.2020.12.026
36. Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou H, et al. Sglt2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the xo-serca2-camkii-coffilin pathways. Theranostics (2022) 12:5034–50. doi: 10.7150/thno.75121
37. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol (2017) 57:535–65. doi: 10.1146/annurev-pharmtox-010715-103335
38. Yi C, Song M, Sun L, Si L, Yu D, Li B, et al. Asiatic acid alleviates myocardial ischemia-reperfusion injury by inhibiting the ros-mediated mitochondria-dependent apoptosis pathway. Oxid Med Cell Longev (2022) 2022:3267450. doi: 10.1155/2022/3267450
39. Zhang H, Yan Q, Wang X, Chen X, Chen Y, Du J, et al. The role of mitochondria in liver ischemia-reperfusion injury: from aspects of mitochondrial oxidative stress, mitochondrial fission, mitochondrial membrane permeable transport pore formation, mitophagy, and mitochondria-related protective measures. Oxid Med Cell Longev (2021) 2021:6670579. doi: 10.1155/2021/6670579
40. Yu J, Wu J, Xie P, Maimaitili Y, Wang J, Xia Z, et al. Sevoflurane postconditioning attenuates cardiomyocyte hypoxia/reoxygenation injury via restoring mitochondrial morphology. Peerj (2016) 4:E2659. doi: 10.7717/peerj.2659
41. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy (2021) 17:1–382. doi: 10.1080/15548627.2020.1797280
42. Jin Z, Ji Y, Su W, Zhou L, Wu X, Gao L, et al. The role of circadian clock-controlled mitochondrial dynamics in diabetic cardiomyopathy. Front Immunol (2023) 14:1142512. doi: 10.3389/fimmu.2023.1142512
43. Cai C, Guo Z, Chang X, Li Z, Wu F, He J, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the Ampkalpha1/Ulk1/Fundc1/mitophagy pathway. Redox Biol (2022) 52:102288. doi: 10.1016/j.redox.2022.102288
44. Tan X, Chen YF, Zou SY, Wang WJ, Zhang NN, Sun ZY, et al. Aldh2 attenuates ischemia and reperfusion injury through regulation of mitochondrial fusion and fission by pi3k/akt/mtor pathway in diabetic cardiomyopathy. Free Radic Biol Med (2023) 195:219–30. doi: 10.1016/j.freeradbiomed.2022.12.097
45. Wu Q, Wang T, Chen S, Zhou Q, Li H, Hu N, et al. Cardiac protective effects of remote ischaemic preconditioning in children undergoing tetralogy of fallot repair surgery: A randomized controlled trial. Eur Heart J (2018) 39:1028–37. doi: 10.1093/eurheartj/ehx030
46. Cheng Y, Liu DZ, Zhang CX, Cui H, Liu M, Zhang BL, et al. Mitochondria-targeted antioxidant delivery for precise treatment of myocardial ischemia-reperfusion injury through A multistage continuous targeted strategy. Nanomedicine (2019) 16:236–49. doi: 10.1016/j.nano.2018.12.014
47. Zhu Q, Li H, Xie X, Chen X, Kosuru R, Li S, et al. Adiponectin facilitates postconditioning cardioprotection through both ampk-dependent nuclear and ampk-independent mitochondrial stat3 activation. Oxid Med Cell Longev (2020) 2020:4253457. doi: 10.1155/2020/4253457
48. Chen CL, Zhang L, Jin Z, Kasumov T, Chen YR. Mitochondrial redox regulation and myocardial ischemia-reperfusion injury. Am J Physiol Cell Physiol (2022) 322:C12–23. doi: 10.1152/ajpcell.00131.2021
49. Zimmerman AN, Hulsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature (1966) 211:646–7. doi: 10.1038/211646a0
50. Talukder MA, Zweier JL, Periasamy M. Targeting calcium transport in ischaemic heart disease. Cardiovasc Res (2009) 84:345–52. doi: 10.1093/cvr/cvp264
51. Kandula V, Kosuru R, Li H, Yan D, Zhu Q, Lian Q, et al. Forkhead box transcription factor 1: role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc Diabetol (2016) 15:44. doi: 10.1186/s12933-016-0361-1
52. Bagur R, Hajnóczky G. Intracellular ca(2+) sensing: its role in calcium homeostasis and signaling. Mol Cell (2017) 66:780–8. doi: 10.1016/j.molcel.2017.05.028
53. Pittas K, Vrachatis DA, Angelidis C, Tsoucala S, Giannopoulos G, Deftereos S. The role of calcium handling mechanisms in reperfusion injury. Curr Pharm Des (2018) 24:4077–89. doi: 10.2174/1381612825666181120155953
54. Park YH, Choi C, Park EM, Kim HS, Park HJ, Bae SC, et al. Over-expression of rice leucine-rich repeat protein results in activation of defense response, thereby enhancing resistance to bacterial soft rot in chinese cabbage. Plant Cell Rep (2012) 31:1845–50. doi: 10.1007/s00299-012-1298-9
55. Mao X, Wang T, Liu Y, Irwin MG, Ou JS, Liao XL, et al. N-acetylcysteine and allopurinol confer synergy in attenuating myocardial ischemia injury via restoring Hif-1alpha/Ho-1 signaling in diabetic rats. PloS One (2013) 8:E68949. doi: 10.1371/journal.pone.0068949
56. Deng F, Wang S, Zhang L, Xie X, Cai S, Li H, et al. Propofol through upregulating caveolin-3 attenuates post-hypoxic mitochondrial damage and cell death in H9c2 cardiomyocytes during hyperglycemia. Cell Physiol Biochem (2017) 44:279–92. doi: 10.1159/000484680
57. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J (2019) 286:241–78. doi: 10.1111/febs.14608
58. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
59. Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, et al. Atf6 decreases myocardial ischemia/reperfusion damage and links er stress and oxidative stress signaling pathways in the heart. Circ Res (2017) 120:862–75. doi: 10.1161/CIRCRESAHA.116.310266
60. Caccioppo A, Franchin L, Grosso A, Angelini F, D'ascenzo F, Brizzi MF. Ischemia reperfusion injury: mechanisms of damage/protection and novel strategies for cardiac recovery/regeneration. Int J Mol Sci (2019) 20 (20). doi: 10.3390/ijms20205024
61. Zhou G, Peng Y, Guo M, Qu C, Luo S, Jiang Y, et al. Esomeprazole inhibits endoplasmic reticulum stress and ameliorates myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun (2022) 627:84–90. doi: 10.1016/j.bbrc.2022.08.013
62. Sun M, Wang R, Xia R, Xia Z, Wu Z, Wang T. Amelioration of myocardial ischemia/reperfusion injury in diabetes: A narrative review of the mechanisms and clinical applications of dexmedetomidine. Front Pharmacol (2022) 13:949754. doi: 10.3389/fphar.2022.949754
63. Chen W, Ma M, Song Y, Hua Y, Jia H, Liu J, et al. Exercise attenuates myocardial ischemia-reperfusion injury by regulating endoplasmic reticulum stress and mitophagy through M(2) acetylcholine receptor. Antioxid Redox Signal (2023). doi: 10.1089/ars.2022.0168 (Epub ahead of print).
64. Xia B, Li Q, Zheng K, Wu J, Huang C, Liu K, et al. Down-regulation of hrd1 protects against myocardial ischemia-reperfusion injury by regulating pparalpha to prevent oxidative stress, endoplasmic reticulum stress, and cellular apoptosis. Eur J Pharmacol (2023) 954:175864. doi: 10.1016/j.ejphar.2023.175864
65. Jovanović A. Cardioprotective signalling: past, present and future. Eur J Pharmacol (2018) 833:314–9. doi: 10.1016/j.ejphar.2018.06.029
66. Rafiee P, Shi Y, Su J, Pritchard KA Jr., Tweddell JS, Baker JE. Erythropoietin protects the infant heart against ischemia-reperfusion injury by triggering multiple signaling pathways. Basic Res Cardiol (2005) 100:187–97. doi: 10.1007/s00395-004-0508-1
67. Bell RM, Botker HE, Carr RD, Davidson SM, Downey JM, Dutka DP, et al. 9th hatter biannual meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res Cardiol (2016) 111:41. doi: 10.1007/s00395-016-0558-1
68. Lu MJ, Chen YS, Huang HS, Ma MC. Erythropoietin alleviates post-ischemic injury of rat hearts by attenuating nitrosative stress. Life Sci (2012) 90:776–84. doi: 10.1016/j.lfs.2012.04.012
69. Comita S, Femmino S, Thairi C, Alloatti G, Boengler K, Pagliaro P, et al. Regulation of stat3 and its role in cardioprotection by conditioning: focus on non-genomic roles targeting mitochondrial function. Basic Res Cardiol (2021) 116:56. doi: 10.1007/s00395-021-00898-0
70. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking A risk for cardioprotection. Heart Fail Rev (2007) 12:217–34. doi: 10.1007/s10741-007-9026-1
71. Lecour S. Activation of the protective survivor activating factor enhancement (Safe) pathway against reperfusion injury: does it go beyond the risk pathway? J Mol Cell Cardiol (2009) 47:32–40. doi: 10.1016/j.yjmcc.2009.03.019
72. Qin Y, Shi Y, Yu Q, Yang S, Wang Y, Dai X, et al. Vitamin B12 alleviates myocardial ischemia/reperfusion injury via the sirt3/ampk signaling pathway. BioMed Pharmacother (2023) 163:114761. doi: 10.1016/j.biopha.2023.114761
73. Wu B, Cao Y, Meng M, Jiang Y, Tao H, Zhang Y, et al. Gabapentin alleviates myocardial ischemia-reperfusion injury by increasing the protein expression of Gaba(A)Rδ. Eur J Pharmacol (2023) 944:175585. doi: 10.1016/j.ejphar.2023.175585
74. Funk F, Kronenbitter A, Isic M, Flocke V, Gorressen S, Semmler D, et al. Diabetes disturbs functional adaptation of the remote myocardium after ischemia/reperfusion. J Mol Cell Cardiol (2022) 173:47–60. doi: 10.1016/j.yjmcc.2022.09.002
75. Tatsumi T, Matoba S, Kobara M, Keira N, Kawahara A, Tsuruyama K, et al. Energy metabolism after ischemic preconditioning in streptozotocin-induced diabetic rat hearts. J Am Coll Cardiol (1998) 31:707–15. doi: 10.1016/S0735-1097(97)00556-1
76. Granfeldt A, Lefer DJ, Vinten-Johansen J. Protective ischaemia in patients: preconditioning and postconditioning. Cardiovasc Res (2009) 83:234–46. doi: 10.1093/cvr/cvp129
77. Tian H, Xiong Y, Zhang Y, Leng Y, Tao J, Li L, et al. Activation of nrf2/fpn1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones (2021) 27:149–64. doi: 10.1007/s12192-022-01257-1
78. Sanchez-Perez P, Mata A, Torp MK, Lopez-Bernardo E, Heiestad CM, Aronsen JM, et al. Energy substrate metabolism, mitochondrial structure and oxidative stress after cardiac ischemia-reperfusion in mice lacking ucp3. Free Radic Biol Med (2023) 205:244–61. doi: 10.1016/j.freeradbiomed.2023.05.014
79. Gao S, Wang R, Dong S, Wu J, Perek B, Xia Z, et al. Inactivation of topk caused by hyperglycemia blocks diabetic heart sensitivity to sevoflurane postconditioning by impairing the pten/Pi3k/Akt signaling. Oxid Med Cell Longev (2021) 2021:6657529. doi: 10.1155/2021/6657529
80. Oh JY, Choi GE, Lee HJ, Jung YH, Chae CW, Kim JS, et al. 17β-estradiol protects mesenchymal stem cells against high glucose-induced mitochondrial oxidants production via nrf2/sirt3/mnsod signaling. Free Radic Biol Med (2019) 130:328–42. doi: 10.1016/j.freeradbiomed.2018.11.003
81. Kaludercic N, Di Lisa F. Mitochondrial ros formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med (2020) 7:12. doi: 10.3389/fcvm.2020.00012
82. Santos-Gallego CG, Requena-Ibanez JA, Picatoste B, Fardman B, Ishikawa K, Mazurek R, et al. Cardioprotective effect of empagliflozin and circulating ketone bodies during acute myocardial infarction. Circ Cardiovasc Imaging (2023) 16:E015298. doi: 10.1161/CIRCIMAGING.123.015298
83. Jiang P, Ren L, Zhi L, Yu Z, Lv F, Xu F, et al. Negative regulation of ampk signaling by high glucose via E3 ubiquitin ligase Mg53. Mol Cell (2021) 81:629–637.E5. doi: 10.1016/j.molcel.2020.12.008
84. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res (2008) 79:341–51. doi: 10.1093/cvr/cvn104
85. Tang X, Li X, Zhang D, Han W. Astragaloside-iv alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of mir-138-5p/sirt1/nrf2. Bioengineered (2022) 13:8240–54. doi: 10.1080/21655979.2022.2049471
86. Huang J, Chen G, Wang J, Liu S, Su J. Platycodin D regulates high glucose-induced ferroptosis of hk-2 cells through glutathione peroxidase 4 (Gpx4). Bioengineered (2022) 13:6627–37. doi: 10.1080/21655979.2022.2045834
87. Shao J, Bai Z, Zhang L, Zhang F. Ferrostatin-1 alleviates tissue and cell damage in diabetic retinopathy by improving the antioxidant capacity of the xc(-)-gpx4 system. Cell Death Discovery (2022) 8:426. doi: 10.1038/s41420-022-01141-y
88. Chen C, Lu C, He D, Na N, Wu Y, Luo Z, et al. Inhibition of hmgb1 alleviates myocardial ischemia/reperfusion injury in diabetic mice via suppressing autophagy. Microvasc Res (2021) 138:104204. doi: 10.1016/j.mvr.2021.104204
89. Li X, Li Z, Dong X, Wu Y, Li B, Kuang B, et al. Astragaloside iv attenuates myocardial dysfunction in diabetic cardiomyopathy rats through downregulation of cd36-mediated ferroptosis. Phytother Res (2023) 37:3042–56. doi: 10.1002/ptr.7798
90. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an Ampk-dependent manner. J Cell Mol Med (2020) 24:6670–9. doi: 10.1111/jcmm.15318
91. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol (2020) 39:210–25. doi: 10.1089/dna.2019.5097
92. Dong J, Ping Y, Wang Y, Zhang Y. The roles of endothelial nitric oxide synthase gene polymorphisms in diabetes mellitus and its associated vascular complications: A systematic review and meta-analysis. Endocrine (2018) 62:412–22. doi: 10.1007/s12020-018-1683-4
93. Hoang HH, Padgham SV, Meininger CJ. L-arginine, tetrahydrobiopterin, nitric oxide and diabetes. Curr Opin Clin Nutr Metab Care (2013) 16:76–82. doi: 10.1097/MCO.0b013e32835ad1ef
94. Li H, Wallerath T, Münzel T, Förstermann U. Regulation of endothelial-type no synthase expression in pathophysiology and in response to drugs. Nitric Oxide (2002) 7:149–64. doi: 10.1016/S1089-8603(02)00111-8
95. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: is there A hyperglycemia-induced imbalance of nox and nos? Int J Mol Sci (2019) 20(15). doi: 10.3390/ijms20153775
96. Chen Q, Xu T, Li D, Pan D, Wu P, Luo Y, et al. Jnk/pi3k/akt signaling pathway is involved in myocardial ischemia/reperfusion injury in diabetic rats: effects of salvianolic acid A intervention. Am J Transl Res (2016) 8:2534–48.
97. Lin F, Yang Y, Wei S, Huang X, Peng Z, Ke X, et al. Hydrogen sulfide protects against high glucose-induced human umbilical vein endothelial cell injury through activating Pi3k/Akt/Enos pathway. Drug Des Devel Ther (2020) 14:621–33. doi: 10.2147/DDDT.S242521
98. Li Z, Gao J, Sun D, Jiao Q, Ma J, Cui W, et al. Lncrna meg3: potential stock for precision treatment of cardiovascular diseases. Front Pharmacol (2022) 13:1045501. doi: 10.3389/fphar.2022.1045501
99. Zhang D, Mei L, Long R, Cui C, Sun Y, Wang S, et al. Riperc attenuates cerebral ischemia injury through regulation of mir-98/pik3ip1/pi3k/akt signaling pathway. Oxid Med Cell Longev (2020) 2020:6454281. doi: 10.1155/2020/6454281
100. Mocanu MM, Bell RM, Yellon DM. Pi3 kinase and not P42/P44 appears to be implicated in the protection conferred by ischemic preconditioning. J Mol Cell Cardiol (2002) 34:661–8. doi: 10.1006/jmcc.2002.2006
101. Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, et al. N-acetylcysteine and allopurinol up-regulated the jak/stat3 and pi3k/akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med (2013) 63:291–303. doi: 10.1016/j.freeradbiomed.2013.05.043
102. Xue R, Lei S, Xia ZY, Wu Y, Meng Q, Zhan L, et al. Selective inhibition of pten preserves ischaemic post-conditioning cardioprotection in stz-induced type 1 diabetic rats: role of the Pi3k/Akt and Jak2/Stat3 pathways. Clin Sci (Lond) (2016) 130:377–92. doi: 10.1042/CS20150496
103. Lei S, Su W, Xia ZY, Wang Y, Zhou L, Qiao S, et al. Hyperglycemia-induced oxidative stress abrogates remifentanil preconditioning-mediated cardioprotection in diabetic rats by impairing caveolin-3-modulated Pi3k/Akt and jak2/Stat3 signaling. Oxid Med Cell Longev (2019) 2019:9836302. doi: 10.1155/2019/9836302
105. Pipicz M, Demjan V, Sarkozy M, Csont T. Effects of cardiovascular risk factors on cardiac stat3. Int J Mol Sci (2018) 19(11). doi: 10.3390/ijms19113572
106. Wang C, Li H, Wang S, Mao X, Yan D, Wong SS, et al. Repeated non-invasive limb ischemic preconditioning confers cardioprotection through Pkc-ℇ/Stat3 signaling in diabetic rats. Cell Physiol Biochem (2018) 45:2107–21. doi: 10.1159/000488047
107. Xu T, Wu X, Chen Q, Zhu S, Liu Y, Pan D, et al. The anti-apoptotic and cardioprotective effects of salvianolic acid A on rat cardiomyocytes following ischemia/reperfusion by dusp-mediated regulation of the Erk1/2/Jnk pathway. PloS One (2014) 9:E102292. doi: 10.1371/journal.pone.0102292
108. Qiao Z, Ma J, Liu H. Evaluation of the antioxidant potential of salvia miltiorrhiza ethanol extract in A rat model of ischemia-reperfusion injury. Molecules (2011) 16:10002–12. doi: 10.3390/molecules161210002
109. Yang Y, Song J, Liu N, Wei G, Liu S, Zhang S, et al. Salvianolic acid A relieves cognitive disorder after chronic cerebral ischemia: involvement of Drd2/Cryab/Nf-κb pathway. Pharmacol Res (2022) 175:105989. doi: 10.1016/j.phrs.2021.105989
110. Sun Y, Zhu H, Wang J, Liu Z, Bi J. Isolation and purification of salvianolic acid A and salvianolic acid B from salvia miltiorrhiza by high-speed counter-current chromatography and comparison of their antioxidant activity. J Chromatogr B Analyt Technol BioMed Life Sci (2009) 877:733–7. doi: 10.1016/j.jchromb.2009.02.013
111. Quan W, Wu B, Bai Y, Zhang X, Yin J, Xi M, et al. Magnesium lithospermate B improves myocardial function and prevents simulated ischemia/reperfusion injury-induced H9c2 cardiomyocytes apoptosis through akt-dependent pathway. J Ethnopharmacol (2014) 151:714–21. doi: 10.1016/j.jep.2013.11.036
112. Li ZM, Xu SW, Liu PQ. Salvia miltiorrhizaburge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin (2018) 39:802–24. doi: 10.1038/aps.2017.193
113. Qin T, Rasul A, Sarfraz A, Sarfraz I, Hussain G, Anwar H, et al. Salvianolic acid A & B: potential cytotoxic polyphenols in battle against cancer via targeting multiple signaling pathways. Int J Biol Sci (2019) 15:2256–64. doi: 10.7150/ijbs.37467
114. Wang SB, Tian S, Yang F, Yang HG, Yang XY, Du GH. Cardioprotective effect of salvianolic acid A on isoproterenol-induced myocardial infarction in rats. Eur J Pharmacol (2009) 615:125–32. doi: 10.1016/j.ejphar.2009.04.061
115. Pan H, Li D, Fang F, Chen D, Qi L, Zhang R, et al. Salvianolic acid A demonstrates cardioprotective effects in rat hearts and cardiomyocytes after ischemia/reperfusion injury. J Cardiovasc Pharmacol (2011) 58:535–42. doi: 10.1097/FJC.0b013e31822de355
116. Zhang J, Liu A, Hou R, Zhang J, Jia X, Jiang W, et al. Salidroside protects cardiomyocyte against hypoxia-induced death: A hif-1alpha-activated and vegf-mediated pathway. Eur J Pharmacol (2009) 607:6–14. doi: 10.1016/j.ejphar.2009.01.046
117. Lv H, Wang L, Shen J, Hao S, Ming A, Wang X, et al. Salvianolic acid B attenuates apoptosis and inflammation via sirt1 activation in experimental stroke rats. Brain Res Bull (2015) 115:30–6. doi: 10.1016/j.brainresbull.2015.05.002
118. Lu B, Li J, Gui M, Yao L, Fan M, Zhou X, et al. Salvianolic acid B inhibits myocardial I/R-induced ros generation and cell apoptosis by regulating the trim8/gpx1 pathway. Pharm Biol (2022) 60:1458–68. doi: 10.1080/13880209.2022.2096644
119. Liu J, Dong W, Gao C, Meng Y. Salvianolic acid B protects cardiomyocytes from ischemia/reperfusion injury by mediating circtrrap/mir-214-3p/sox6 axis. Int Heart J (2022) 63:1176–86. doi: 10.1536/ihj.22-102
120. Li D, Wang J, Hou J, Fu J, Liu J, Lin R. Salvianolic acid B induced upregulation of mir-30a protects cardiac myocytes from ischemia/reperfusion injury. BMC Complement Altern Med (2016) 16:336. doi: 10.1186/s12906-016-1275-x
121. Liu H, Liu W, Qiu H, Zou D, Cai H, Chen Q, et al. Salvianolic acid B protects against myocardial ischaemia-reperfusion injury in rats via inhibiting high mobility group box 1 protein expression through the Pi3k/Akt signalling pathway. Naunyn Schmiedebergs Arch Pharmacol (2020) 393:1527–39. doi: 10.1007/s00210-019-01755-7
122. Zimmerman BJ, Granger DN. Reperfusion injury. Surg Clin North Am (1992) 72:65–83. doi: 10.1016/S0039-6109(16)45628-8
123. Ansley DM, Xia Z, Dhaliwal BS. The relationship between plasma free 15-F2t-isoprostane concentration and early postoperative cardiac depression following warm heart surgery. J Thorac Cardiovasc Surg (2003) 126:1222–3. doi: 10.1016/S0022-5223(03)00794-3
124. Xia Z, Godin DV, Chang TK, Ansley DM. Dose-dependent protection of cardiac function by propofol during ischemia and early reperfusion in rats: effects on 15-F2t-isoprostane formation. Can J Physiol Pharmacol (2003) 81:14–21. doi: 10.1139/y02-170
125. Wei SM, Huang YM. Attenuation effect of salvianolic acid B on testicular ischemia-reperfusion injury in rats. Oxid Med Cell Longev (2022) 2022:7680182. doi: 10.1155/2022/7680182
126. Yuan X, Xiang Y, Zhu N, Zhao X, Ye S, Zhong P, et al. Salvianolic acid A protects against myocardial ischemia/reperfusion injury by reducing platelet activation and inflammation. Exp Ther Med (2017) 14:961–6. doi: 10.3892/etm.2017.4619
127. Huang ZS, Zeng CL, Zhu LJ, Jiang L, Li N, Hu H. Salvianolic acid A inhibits platelet activation and arterial thrombosis via inhibition of phosphoinositide 3-kinase. J Thromb Haemost (2010) 8:1383–93. doi: 10.1111/j.1538-7836.2010.03859.x
128. Yang ZH, Lu YJ, Gu KP, Xiang ZY, Huang HM. Effect of ulinastatin on myocardial ischemia-reperfusion injury through jnk and P38 mapk signaling pathways. Eur Rev Med Pharmacol Sci (2019) 23:8658–64. doi: 10.26355/eurrev_201910_19183
129. Xu X, Mao C, Zhang C, Zhang M, Gong J, Wang X. Salvianolic acid B inhibits ferroptosis and apoptosis during myocardial ischemia/reperfusion injury via decreasing the ubiquitin-proteasome degradation of gpx4 and the ros-jnk/mapk pathways. Molecules (2023) 28(10). doi: 10.3390/molecules28104117
130. Dai S, Liang T, Shi X, Luo Z, Yang H. Salvianolic acid B protects intervertebral discs from oxidative stress-induced degeneration via activation of the Jak2/Stat3 signaling pathway. Oxid Med Cell Longev (2021) 2021:6672978. doi: 10.1155/2021/6672978
131. Cai Y, Ying F, Liu H, Ge L, Song E, Wang L, et al. Deletion of rap1 protects against myocardial ischemia/reperfusion injury through suppressing cell apoptosis via activation of stat3 signaling. FASEB J (2020) 34:4482–96. doi: 10.1096/fj.201901592RR
132. Domingos AE, Seara FAC, Oliveira DF, Maciel L, Barbosa RAQ, Barcellos LC, et al. Mitochondrial dysfunction and cardiac ischemia/reperfusion injury are attenuated by nandrolone: role of jak-stat3 pathway. Steroids (2023) 197:109247. doi: 10.1016/j.steroids.2023.109247
133. Yang Z, Chen Y, Yan Z, Xu TT, Wu X, Pi A, et al. Inhibition of TLR4/MAPKs pathway contributes to the protection of salvianolic acid a against lipotoxicity-induced myocardial damage in cardiomyocytes and obese mice. Front Pharmacol (2021) 12:627123. doi: 10.3389/fphar.2021.627123
134. Wu P, Yan Y, Ma LL, Hou BY, He YY, Zhang L, et al. Effects of the nrf2 protein modulator salvianolic acid A alone or combined with metformin on diabetes-associated macrovascular and renal injury. J Biol Chem (2016) 291:22288–301. doi: 10.1074/jbc.M115.712703
135. Qiang G, Yang X, Shi L, Zhang H, Chen B, Zhao Y, et al. Antidiabetic effect of salvianolic acid A on diabetic animal models via Ampk activation and mitochondrial regulation. Cell Physiol Biochem (2015) 36:395–408. doi: 10.1159/000430258
136. Fan H, Yang L, Fu F, Xu H, Meng Q, Zhu H, et al. Cardioprotective effects of salvianolic acid a on myocardial ischemia-reperfusion injury in vivo and in vitro. Evid Based Complement Alternat Med (2012) 2012:508938. doi: 10.1155/2012/508938
137. Sun B, Li C, Zuo L, Liu P. Protection of SAL b with H9C2 cells. Pharm Biol (2016) 54(5):889–95. doi: 10.3109/13880209.2015.1089911
138. Huang CY, Chen SY, Fu RH, Huang YC, Chen SY, Shyu WC, et al. Differentiation of embryonic stem cells into cardiomyocytes used to investigate the cardioprotective effect of salvianolic acid b through BNIP3 involved pathway. Cell Transplant (2015) 24(3):561–71. doi: 10.3727/096368915X686995
139. Xin GJ, Fu JH, Han X, Li L, Guo H, Meng HX, et al. Salvianolic acid b regulates mitochondrial autophagy mediated by NIX to protect H9c2 cardiomyocytes from hypoxia/reoxygenation injury. Zhongguo Zhong Yao Za Zhi (2020) 45(12):2960–5. doi: 10.19540/j.cnki.cjcmm.20200224.402
140. Tao S, Ren Y, Zheng H, Zhao M, Zhang X, Zhu Y, et al. Salvianolic acid B inhibits intermittent high glucose-induced ins-1 cell apoptosis through regulation of bcl-2 proteins and mitochondrial membrane potential. Eur J Pharmacol (2017) 814:56–62. doi: 10.1016/j.ejphar.2017.08.007
141. Wu YP, Zhao XM, Pan SD, Guo DA, Wei R, Han JJ, et al. Salvianolic acid B inhibits platelet adhesion under conditions of flow by A mechanism involving the collagen receptor alpha2beta1. Thromb Res (2008) 123:298–305. doi: 10.1016/j.thromres.2008.05.020
142. Li CL, Liu B, Wang ZY, Xie F, Qiao W, Cheng J, et al. Salvianolic acid B improves myocardial function in diabetic cardiomyopathy by suppressing Igfbp3. J Mol Cell Cardiol (2020) 139:98–112. doi: 10.1016/j.yjmcc.2020.01.009
143. Otel I, Ledru F, Danchin N. Ischemic heart disease in type 2 diabetes. Metabolism (2003) 52:6–12. doi: 10.1016/S0026-0495(03)00216-6
144. Ge H, Lin W, Lou Z, Chen R, Shi H, Zhao Q, et al. Catalpol alleviates myocardial ischemia reperfusion injury by activating the Nrf2/Ho-1 signaling pathway. Microvasc Res (2022) 140:104302. doi: 10.1016/j.mvr.2021.104302
145. Jonassen AK, Sack MN, Mjøs OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and P70s6 kinase cell-survival signaling. Circ Res (2001) 89:1191–8. doi: 10.1161/hh2401.101385
146. Gong L, Di C, Xia X, Wang J, Chen G, Shi J, et al. Akt/mtor signaling pathway is involved in salvianolic acid B-induced autophagy and apoptosis in hepatocellular carcinoma cells. Int J Oncol (2016) 49:2538–48. doi: 10.3892/ijo.2016.3748
147. Jing Z, Fei W, Zhou J, Zhang L, Chen L, Zhang X, et al. Salvianolic acid B, A novel autophagy inducer, exerts antitumor activity as A single agent in colorectal cancer cells. Oncotarget (2016) 7:61509–19. doi: 10.18632/oncotarget.11385
148. Chen X, Li X, Zhang W, He J, Xu B, Lei B, et al. Activation of ampk inhibits inflammatory response during hypoxia and reoxygenation through modulating jnk-mediated Nf-κb pathway. Metabolism (2018) 83:256–70. doi: 10.1016/j.metabol.2018.03.004
149. Zhang Y, Feng X, Du M, Ding J, Liu P. Salvianolic acid B attenuates the inflammatory response in atherosclerosis by regulating Mapks/ Nf-kappab signaling pathways in Ldlr-/- mice and raw264.7 cells. Int J Immunopathol Pharmacol (2022) 36:3946320221079468. doi: 10.1177/03946320221079468
Keywords: salvianolic acids A, salvianolic acids B, myocardial ischemia reperfusion injury, diabetics introduction, cardioprotection
Citation: Jiang Y, Cai Y, Han R, Xu Y, Xia Z and Xia W (2024) Salvianolic acids and its potential for cardio-protection against myocardial ischemic reperfusion injury in diabetes. Front. Endocrinol. 14:1322474. doi: 10.3389/fendo.2023.1322474
Received: 16 October 2023; Accepted: 21 December 2023;
Published: 12 January 2024.
Edited by:
Leonardo M.R. Ferreira, Medical University of South Carolina, United StatesReviewed by:
Ren Jun Guo, China Academy of Chinese Medical Sciences, ChinaCopyright © 2024 Jiang, Cai, Han, Xu, Xia and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiyi Xia, d2VpeWkueGlhQHBvbHl1LmVkdS5oaw==; d2VpeWlwaWdAZ21haWwuY29t; Zhengyuan Xia, enl4aWFAaGt1Lmhr
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.